

Abstract
Far from simply lining the inner surface of blood vessels, the cellular monolayer that comprises the endothelium is a highly active organ that regulates vascular tone. In health, the endothelium maintains the balance between opposing dilator and constrictor influences, while in disease, it is the common ground on which cardiovascular risk factors act to initiate the atherosclerotic process. As such, it is the site at which cardiovascular disease begins and consequently acts as a barometer of an individual's likely future cardiovascular health. The vascular endothelium is a very active organ responsible for the regulation of vascular tone through the effects of locally synthesized mediators, predominantly nitric oxide (NO), endothelial NO synthase (eNOS), and superoxide. NO is abundantly evident in normally functioning vasculature where it acts as a vasodilator, inhibits inflammation, and has an antiaggregant effect on platelets. Its depletion is both a sign and cause of endothelial dysfunction resulting from reduced activity of eNOS and amplified production of nicotinamide adenine dinucleotide oxidase, which, in turn, results in raised levels of reactive oxygen species. This cascade is the basis for reduced vascular compliance through an imbalanced regulation of tone with a predominance of vasoconstrictive elements. Further, structural changes in the microvasculature are a critical early step in the loss of normal function. This microvascular dysfunction is known to be highly predictive of future macrovascular events and is consequently a very attractive target for intervention in the hypertensive population in order to prevent cardiovascular events.
Far from simply lining the inner surface of blood vessels, the cellular monolayer that comprises the endothelium is a highly active organ that regulates vascular tone. In health, the endothelium maintains the balance between opposing dilator and constrictor influences while in disease it is the common ground on which cardiovascular risk factors act to initiate the atherosclerotic process. As such, it is the site at which cardiovascular disease begins and consequently acts as a barometer of an individual's likely future cardiovascular health. It is therefore of immense importance in the early detection of cardiovascular disease and may offer a substrate for therapy to prevent the ensuing development of macrovascular events.
Endothelial Dysfunction
Many patients (around 50%) who experience a cardiovascular event do so “out of the blue,” having apparently had previously good cardiovascular health.1 Therefore, the identification of these individuals before the onset of macrovascular disease is of enormous interest, and one area that has received particular attention is that of the microvasculature, in particular, the endothelium.
The microvasculature refers traditionally to the network of arterioles, venules, and capillaries between 15 μm and 300 μm in diameter that constitute the vascular bed and are the main site of vascular resistance.2 Dysfunction of the endothelium, an underpinning mechanism of microvascular disease, was first described in the forearm vasculature in 1990.3 Since then, it has been shown to both precede and predict future cardiovascular events and therefore offers an early opportunity to detect disease.4
Regulation of Endothelial Tone
In health, the endothelium is the source of both endothelium‐derived relaxing (EDRFs) and constricting factors (EDCFs), which exert opposing effects on underlying vascular smooth muscle cells in order to regulate vascular tone.5 The predominant EDRF that acts to cause vasodilation is nitric oxide (NO), while EDCFs including angiotensin II and endothelins have the opposite vasoconstricting effect.5 An imbalance in the homeostasis of these factors toward excessive constriction is a major feature in endothelial dysfunction, and it is the free radical NO that has the major role in this imbalance.
NO in the Regulation of Vascular Tone
NO is synthesized from its precursor L‐arginine by the endothelial form of NO synthase (eNOS) provided that essential cofactors for eNOS activity such as tetrahydobiopterin (BH4) are available.4, 6 Once generated, NO easily diffuses from the endothelium to activate soluble guanylyl cyclase in vascular smooth muscle cells, which, in turn, increases cyclic guanosine monophosphate production, causing relaxation of these smooth muscle cells and vasodilation.4, 6, 7 In addition, NO also has antithrombotic, antiproliferative, and leukocyte adhesion inhibitory effects, thereby suppressing local wall inflammation.4, 6 Therefore, any loss of bioavailability or function of NO is harmful to endothelial health. In fact, in a murine model without the endothelial isoform of NOS, the mice are hypertensive with endothelial dysfunction.8
As a free radical, NO is subject to scavenging both by reactive oxygen species (ROS) and the superoxide anion. Increased levels of superoxide anion in cardiovascular disease results in enhanced NO scavenging to form peroxynitrite (a powerful pro‐oxidant that prevents eNOS bioactivity) thereby reducing NO bioavailability.3, 6 Although superoxide anion is degraded by superoxide dismutase (SOD), the reaction between superoxide and NO is three times faster than its degradation by SOD.6 This oxidative stress in the vascular wall also results from activation of nicotinamide adenine dinucleotide oxidase (NADPH), which augments ROS.9 Increased levels of ROS cause uncoupling of BH4 from eNOS and consequently the generation of superoxide, which reduces NO bioavailability as above.9, 10 Whether eNOS is appropriately coupled is of immense importance because it determines whether the predominant end product of its activity is either NO or superoxide.
NO in Hypertension
Cardiovascular risk factors such as aging, active and passive smoking, and hypertension have all been linked to reduced levels of NO as a precipitant of endothelial dysfunction.2, 6 In hypertension, sustained elevation of systemic pressure in the microvasculature leads to premature aging and increased turnover of endothelial cells, which are then replaced by regenerated endothelial cells.5 The regenerated endothelium has an impaired ability to release EDRFs, resulting in an imbalance toward EDCFs with consequent vasoconstriction.5, 11, 12 Hypertension has been linked to deficient levels of nitrogen dioxide and increased vascular production of ROS.13 Therefore, the ability of the endothelium to function normally in maintaining balanced vascular tone through its generation of relaxing and constricting factors is essential in preventing the onset of early vascular damage. Conversely, a dysfunctional endothelium leads to functional changes in the microvasculature with a predominant and deleterious constrictive tone.
Hypertension and Endothelial Dysfunction
These functional changes are, however, far from the only mechanism by which the microvasculature is altered in the presence of cardiovascular risk factors. Structural alterations to the vessels themselves are fundamental in accruing damage that will only become clinically apparent with the advent of a macrovascular event. In particular, individuals with hypertension undergo a process of microvascular injury that pre‐dates any form of clinically detectable target organ damage. In fact, endothelial dysfunction has even been demonstrated in normotensive individuals who merely have a family history of hypertension.6 In this regard, it is tempting to consider the microvasculature as the earliest “organ” to show signs of hypertensive damage and therefore the earliest opportunity to intervene to redress this process.
Structural Changes in the Vascular Wall in Hypertension
Resistance vessels are responsible for conducting most of the pressure attenuation necessary between larger conduit arteries and capillaries.2 The microcirculation is the major site of systemic resistance and as such is susceptible to insult from sustained pressure elevation. Vascular beds that are particularly vulnerable to increases in blood pressure are those low impedance networks that are subject to fluctuations in pressure, for instance those seen in the cerebral, ocular, and renal beds.
The concept of structural alterations of the vessel wall in hypertension first emerged from work undertaken by Folkow in the early 1980s.14 He determined that even a small reduction in lumen diameter had a major effect on vessel resistance as a result of the inversely proportional relationship of resistance to the fourth power of the radius (Poiseuille law).14 Following this groundbreaking work, the nature of these vascular wall changes is now well documented.
Remodeling
Hypertension results in structural alterations in microcirculatory beds that can be broadly divided into two types: remodeling and rarefaction. It is remodeling that is responsible for the majority of the chronic elevation in systemic vascular resistance seen in hypertension.2 Data from both human and animal models demonstrate a smaller lumen with rearrangement of the wall constitution in what is described as either eutrophic, hypertrophic, or hypotrophic remodeling.2, 15, 16 Although clearly limited as an isolated specimen and therefore not necessarily applicable to the microvasculature as a whole, evidence from subcutaneous gluteal biopsies has demonstrated inward eutrophic remodeling in hypertension.17 This is a rearrangement of vessel wall components without growth resulting in luminal narrowing and an increased wall‐to‐lumen ratio,15 which, as already mentioned, has a profound effect on resistance.
There is some debate regarding the precise etiology of structural changes in the microvasculature in patients with hypertension. While it is assumed and has been shown that pressure effects are the predominant culprit in establishing hypertensive remodeling, pressure‐independent factors may also have a role. An early animal study using a rat model demonstrated medial hypertrophy after infusion of angiotensin II.18 While treatment with hydralazine prevented the onset of hypertension, it did not prevent medial hypertrophy, leading to the conclusion that a pressor‐independent mechanism may be responsible.18 Specifically, angiotensin II itself is a likely remodeling candidate in hypertension and has been shown as a pressure‐independent mediator of small mesenteric artery medial hypertrophy.18
Rarefaction
The second mechanism by which elevated pressure exerts structural changes in the microvasculature is that of rarefaction. This is most simply thought of as a reduction in the number or combined length of small vessels in a defined volume of tissue.15 Two subforms of rarefaction exist: functional rarefaction, where there is a reduction in the number of vessels perfused rather than an actual reduction in the number of vessels per se, and structural rarefaction, where there is a definite decrease in number of vessels anatomically present.15 Both forms have been demonstrated in skin biopsy samples of hypertensive patients.19 It also seems that a continuum may exist between the subtypes of rarefaction with functional alterations seen to progress to structural with continued pressure effects, as chronic vessel nonperfusion leads ultimately to its structural loss.20 If so, this would render oxidative stress an underpinning mechanism of both main forms of microvascular damage in hypertension, endothelial dysfunction, and deleterious structural alterations of the vessel wall in the form of rarefaction.
Is Microcirculation a Culprit or Victim of Hypertension?
While the evidence largely points to hypertension impacting the microvasculature to cause these deleterious changes in wall structure and function, there are examples that suggest a case for the reverse order of events. For instance, in animal models of hypertension, elevated levels of ROS and arteriolar rarefaction occur even in parts of the microcirculation not exposed to high levels of blood pressure.21 In spontaneously hypertensive rats, the use of an antioxidant to prevent oxidative stress not only prevents rarefaction but also the development of hypertension itself.22 In the human model, rarefaction has been demonstrated in the microvessels of those with a family history of hypertension but who are themselves normotensive.23 In reality, there may well be underlying microvasculature changes in some individuals that at least predispose them to the development of incident hypertension. Indeed, there may be a cyclical process of damage and hypertension that is self‐perpetuating (Figure); however, the majority of available evidence points to the microcirculation being altered in response to sustained elevation in pressure after the onset of essential hypertension.24
Figure 1.
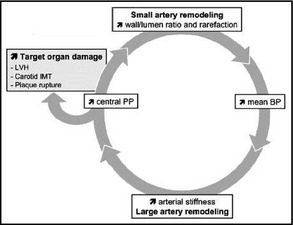
Cycle of microvascular damage in hypertension. LVH indicates left ventricular hypertrophy; IMT, intima‐media thickness; PP, pulse pressure; BP, blood pressure.
Antihypertensive Therapy Class Effects on Endothelial Function
The importance of intervening with therapy at such an early stage in the vascular complications of hypertension is to prevent progression of this microvascular disease to macrovascular disease manifested clinically as cardiovascular events. What should be highlighted, however, is that a reduction in blood pressure by itself will not necessarily translate into an improvement in endothelial function. Although angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers, and third‐generation β‐blockers all restore endothelial function, older β‐blockers have not been shown to have this restorative effect.5, 25 In broad terms, agents that reduce cardiac output such as diuretics and first‐generation β‐blockers, while improving cardiovascular outcomes, do not seem to have an impact on endothelial function, while those that primarily cause vasodilation do.15
Conclusions
The vascular endothelium is a very active organ responsible for the regulation of vascular tone through the effects of locally synthesized mediators, predominantly NO, eNOS, and superoxide. NO is abundantly evident in normally functioning vasculature where it acts as a vasodilator, inhibits inflammation, and has an anti‐aggregant effect on platelets.9 Its depletion is both a sign and cause of endothelial dysfunction resulting from reduced activity of eNOS and amplified production of NADPH oxidase, which, in turn, results in raised levels of ROS. This cascade is the basis for reduced vascular compliance through an imbalanced regulation of tone with a predominance of vasoconstrictive elements. Further, structural changes in the microvasculature are a critical early step in the loss of normal function. This microvascular dysfunction is known to be highly predictive of future macrovascular events26 and is consequently a very attractive target for intervention in the hypertensive population in order to prevent cardiovascular events.
Conflict of Interest
None.
J Clin Hypertens (Greenwich). 2015;17:651–654. DOI: 10.1111/jch.12546. © 2015 Wiley Periodicals, Inc.
References
- 1. Minson CT, Green DJ. Measures of vascular reactivity: prognostic crystal ball or Pandora's box? J Appl Physiol. 2008;105:398–399. [DOI] [PubMed] [Google Scholar]
- 2. Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and microvascular remodelling. Cardiovasc Res. 2008;78:274–285. [DOI] [PubMed] [Google Scholar]
- 3. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–1992. [DOI] [PubMed] [Google Scholar]
- 4. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. [DOI] [PubMed] [Google Scholar]
- 5. Tang EH, Vanhoutte PM. Endothelial dysfunction: a strategic target in the treatment of hypertension? Pflugers Arch. 2010;459:995–1004. [DOI] [PubMed] [Google Scholar]
- 6. Flammer AJ, Lüscher TF. Human endothelial dysfunction: EDRFs. Pflugers Arch. 2010;459:1005–1013. [DOI] [PubMed] [Google Scholar]
- 7. Fernhall B, Agiovlasitis S. Arterial function in youth: window into cardiovascular risk. J Appl Physiol. 2008;105:325–333. [DOI] [PubMed] [Google Scholar]
- 8. Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch Pharm Res. 2009;32:1103–1108. [DOI] [PubMed] [Google Scholar]
- 9. Schiffrin EL. Oxidative stress, nitric oxide synthase, and superoxide dismutase: a matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension. 2008;51:31–32. [DOI] [PubMed] [Google Scholar]
- 10. Dixon LJ, Morgan DR, Hughes SM, et al. Functional consequences of endothelial nitric oxide synthase uncoupling in congestive cardiac failure. Circulation. 2003;107:1725–1728. [DOI] [PubMed] [Google Scholar]
- 11. Michel FS, Man GS, Man RYK, Vanhoutte PM. Hypertension and the absence of EDHF‐mediated responses favour endothelium‐dependent contractions in renal arteries of the rat. Br J Pharmacol. 2008;155:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stankevicius E, Martinez AC, Mulvany MJ, Simonsen U. Blunted acetylcholine relaxation and nitric oxide release in arteries from renal hypertensive rats. J Hypertens. 2002;20:1571–1579. [DOI] [PubMed] [Google Scholar]
- 13. Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D‐dependent NAD(P)H oxidase‐sensitive pathways. J Hypertens. 2001;19:1245–1254. [DOI] [PubMed] [Google Scholar]
- 14. Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504. [DOI] [PubMed] [Google Scholar]
- 15. Levy BI, Schiffrin EL, Mourad J, et al. Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation. 2008;118:968–976. [DOI] [PubMed] [Google Scholar]
- 16. AgabitiRosei E, Heagerty AM, Rizzoni D. Effects of antihypertensive treatment on small artery remodelling. J Hypertens. 2009;27:1107–1114. [DOI] [PubMed] [Google Scholar]
- 17. Intengan HD, Deng LY, Li JS, Schiffrin EL. Mechanics and composition of human subcutaneous resistance arteries in essential hypertension. Hypertension. 1999;33:569–574. [DOI] [PubMed] [Google Scholar]
- 18. Griffin S, Brown W, MacPherson F, et al. Angiotensin II causes vascular hypertrophy in part by a non‐pressor mechanism. Hypertension. 1991;17:626–635. [DOI] [PubMed] [Google Scholar]
- 19. Serne EH, Gans ROB, ter Maaten JC, et al. Impaired skin capillary recruitment in essential hypertension is caused by both functional and structural capillary rarefaction. Hypertension. 2001;38:238–242. [DOI] [PubMed] [Google Scholar]
- 20. Prewitt RL, Chen II, Dowell R. Development of microvascular rarefaction in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 1982;243:H243–H251. [DOI] [PubMed] [Google Scholar]
- 21. Boegehold MA, Johnson MD, Overbeck HW. Pressure‐independent arteriolar rarefaction in hypertension. Am J Physiol Heart Circ Physiol. 1991;261:H83–H87. [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi N, DeLano FA, Schmid‐Schonbein GW. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2005;25:2114–2121. [DOI] [PubMed] [Google Scholar]
- 23. Antonios TFT, Rattray FM, Singer DRJ, et al. Rarefaction of skin capillaries in normotensive offspring of individuals with essential hypertension. Heart. 2003;89:175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laurent S, Briet M, Boutouyrie P. Large and small artery cross‐talk and recent morbidity‐mortality trials in hypertension. Hypertension. 2009;54:388–392. [DOI] [PubMed] [Google Scholar]
- 25. Mancini GBJ, Henry GC, Macaya C, et al. Angiotensin‐converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease: the TREND (Trial on Reversing ENdothelial Dysfunction) study. Circulation. 1996;94:258–265. [DOI] [PubMed] [Google Scholar]
- 26. Touyz RM. Vascular remodeling, retinal arteries, and hypertension. Hypertension. 2007;50:603–604. [DOI] [PubMed] [Google Scholar]