

Abstract
Liddle syndrome, an autosomal dominant form of monogenic hypertension, has been regarded as a rare disorder, which leads to many Liddle syndrome patients being misdiagnosed and experiencing severe complications at an early age. Little is known about the prevalence of Liddle syndrome. In this study, the authors investigated the prevalence of Liddle syndrome confirmed by genetic testing among young hypertension patients of undetermined causes in China. A total of 330 hypertensive patients aged 14 to 40 years after exclusion of common secondary causes of hypertension were enrolled and serum potassium concentrations were measured. Patients with hypokalemia underwent genetic testing of the 13th exon of genes encoding β and γ subunits of the epithelial sodium channel (ENaC). Diagnosis was established by identification of mutations that destroy the PY motif of ENaC. Five patients were diagnosed with Liddle syndrome (prevalence, 1.52%), as well as 12 of their relatives. These patients with Liddle syndrome presented with an earlier onset of hypertension, a stronger family history of hypertension, and higher blood pressure than those with essential hypertension. All patients had hypokalemia and suppressed plasma renin activity. The results demonstrated that Liddle syndrome is an important etiology of hypertension in this young population. Screening of Liddle syndrome should focus on young hypertension patients, particularly those with early penetrance, hypokalemia, and low renin levels after exclusion of common secondary causes.
Liddle syndrome is an autosomal dominant form of monogenic hypertension caused by heterozygous mutations in the genes encoding the β and γ subunits of epithelial sodium channel (ENaC). This disorder was first described in 1963 by Grant Liddle and colleagues.1 Salt‐sensitive hypertension with early penetrance, hypokalemia, metabolic alkalosis, suppression of plasma renin activity (PRA) and aldosterone secretion, and an effective response to ENaC blockers but not spironolactone therapy constitute the typical clinical profile of Liddle syndrome. Because of the similar features of hypertension and hypokalemia with primary aldosteronism, it was termed “pseudoaldosteronism.” However, in contrast to the admittedly dominant role of primary aldosteronism in secondary hypertension,2 Liddle syndrome is generally thought to be a rare disease.
Although more Liddle syndrome cases have been reported worldwide in the past two decades, based on advances in molecular genetics examining this condition, there remains a shortage of data describing the prevalence of Liddle syndrome. Little information was provided in a retrospective study that described the prevalence of the Liddle syndrome phenotype.3 The absence of research on the prevalence of Liddle syndrome confirmed through genetic testing is remarkable given that people with misdiagnosed Liddle syndrome are prone to cardiovascular or cerebrovascular complications at an early age, and especially because Liddle syndrome is a condition easily controlled. In consideration of this clinical significance, we conducted a specific cohort‐based investigation to characterize the prevalence of Liddle syndrome in a Chinese population.
Methods
Study Population
We enrolled 330 consecutive hypertension patients aged 14 to 40 years who were referred to our hypertension center for undetermined causes between January 2010 and December 2014. Patients with hypertension secondary to common causes, including primary aldosteronism, renal disease and renovascular disease, aortic diseases, and obstructive sleep apnea, were excluded. The study was approved by the ethics committee of Fuwai Hospital, Peking Union Medical College, Beijing, China. All participants provided written informed consent.
Criteria for Diagnosis of Liddle Syndrome
Current diagnosis of Liddle syndrome is based on clinical evaluation and genetic testing. Early onset of hypertension, a strong family history of severe hypertension with target organ damage, and a clear‐cut response to ENaC antagonists but not spironolactone treatment are sensitive indicators for clinical assessment of Liddle syndrome. Low levels of serum potassium, PRA, and plasma aldosterone are also of importance. Genetic testing plays a fundamental role in establishing the diagnosis of Liddle syndrome. Identification of pathogenic mutations that destroy the conserved PY motif in the β or γ subunit of ENaC is the most essential indicator of diagnosis.
Screening for Liddle Syndrome
All patients had their medical records reviewed, and an experienced physician inquired and reevaluated the medical history of these patients, with emphasis on their histories of hypertension and hypokalemia, family histories of hypertension, pharmacologic treatment, and control of blood pressure (BP). They all underwent a clinical examination and had their height and weight recorded. BP was measured every 5 minutes with a mercury sphygmomanometer by a skilled physician after the patient was seated for 15 minutes, and the average of three readings was used. Blood was withdrawn after a 12‐hour fast to measure serum sodium and potassium, lipid profile, and fasting glucose.
All patients with hypokalemia (<3.5 mmol/L) underwent genetic testing for confirmation by screening of the 13th exon of both β‐ENaC and γ‐ENaC. For patients with positive results of genetic testing, plasma aldosterone and PRA were evaluated as the patient kept upright for 1 hour after an overnight fast. When the diagnosis of Liddle syndrome was established in a patient, all family members of the index patient were examined by DNA analysis and measurement of BP, serum potassium, PRA, and plasma aldosterone were performed. In addition, physical examination, echocardiography, blood biochemistry examination, and microalbuminuria measurement were performed in hypertensive individuals. Treatment with amiloride was initiated for mutation carriers with hypertension. After 1 month of amiloride treatment, BP and serum potassium were reassessed in patients with Liddle syndrome.
Statistical Analysis
Statistical analysis was performed using SPSS (SPSS 16.0; SPSS Inc, Chicago, IL). All continuous parameters are expressed as medians and interquartile ranges (IQRs, 25–75th percentile), and all categorical parameters are expressed as proportions. To compare between‐group differences in demographic and clinical characteristics, the nonparametric Wilcoxon two‐sample test was used for continuous variables and the Fisher exact test was used for categorical variables.
Results
General Characteristics of the Study Participants
A total of 330 young hypertension patients of undetermined cause were included in the study. The basic clinical characteristics of the study population are shown in Table 1. The median age was 31.0 (IQR, 26.0–36.0) years and men accounted for 67.6% of the participants. On average, these participants were diagnosed with hypertension around the age of 27 years, and 66.1% of individuals had a family history of hypertension.
Table 1.
Clinical and Biochemical Characteristics of the Total Population
| Characteristic | Study Population (N=330) |
|---|---|
| Age, y | 31.0 (26.0‐36.0) |
| Men, No. (%) | 223 (67.6) |
| Age at hypertension diagnosis, y | 27.0 (20.0‐32.0) |
| Family history of hypertension, No. (%) | 218 (66.1) |
| Systolic blood pressure, mm Hg | 150.0 (140.0‐170.0) |
| Diastolic blood pressure, mm Hg | 100.0 (90.0‐110.0) |
| Hypokalemia, No. (%) | 48 (14.5) |
| Hyperlipidemia, No. (%) | 130 (39.3) |
| Body mass index, kg/m2 | 26.2 (23.4‐29.1) |
| Diabetes mellitus, No. (%) | 17 (5.2) |
| Current smokers, No. (%) | 98 (29.7) |
Prevalence of Liddle Syndrome
Among these 330 young patients, hypokalemia was found in 48 (14.5%). Genetic screening was performed in each of the 48 patients (Figure). A diagnosis of Liddle syndrome was established in five patients (1.52% of the study population). Subsequently, 69 relatives of these patients with Liddle syndrome from four kindreds (one of the five index patients declined further investigation) received evaluation of BP, serum potassium and aldosterone levels, PRA, and genetic testing, and 12 new cases of Liddle syndrome were confirmed. One normotensive child carrying a pathogenic mutation with hypokalemia was identified at the same time.
Figure 1.
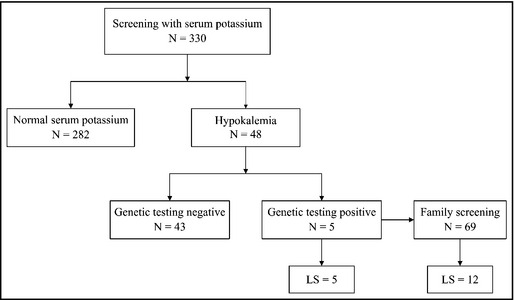
Design of the study. LS indicates Liddle syndrome.
Comparison Between Liddle Syndrome and Essential Hypertension
The clinical features and gene mutations of the patients with Liddle syndrome are summarized in Table 2. Three of the five families have been previously described in detail.4, 5, 6 Although these patients with proved Liddle syndrome were aged from 17 to 78 years, the median age at diagnosis of hypertension was 18 years, relatively younger than those with essential hypertension who developed high BP (Table 3). Predilection for sex was not found among these 17 patients with Liddle syndrome. There were significant differences in family histories for hypertension between Liddle syndrome and essential hypertension (94.1% vs 63.9%, P=.015). Familial aggregation plays a dominant role in Liddle syndrome in a Mendelian fashion. Four of five index patients had a family history of hypertension, and three kindreds were identified, except the one in which clinical assessment and genetic testing were not performed in the other family members. There were fundamental differences between Liddle syndrome and essential hypertension in relation to serum potassium and PRA (P<.001). All patients carrying Liddle's mutations, including the five index patients and 13 relatives, manifested hypokalemia and suppressed PRA, while only three patients had low levels of plasma aldosterone. Markedly, four family members with severe hypertension in two kindreds had died in their 30s. Although BP was much higher in patients with Liddle syndrome (P<.05), BP in these patients seemed easier to control with ENaC blockers and dietary salt restriction, despite previous severity.
Table 2.
Clinical and Biochemical Characteristics of Patients Carrying Liddle Syndrome Mutations
| Patient a | Sex | Age, y | Age at HT Diagnosis, y | BP Before Amiloride Treatment, mm Hg | pk+ Before Amiloride Treatment, mmol/L | PRA, ng/mL·h | ALD, ng/mL | BP After Amiloride Treatment, mm Hg | sk+ After Amiloride Treatment, mmol/L | Complications | Gene/Protein mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I‐1 | F | 17 | 17 | 192/120 | 2.68 | 0.02 | 0.15 | 130/70 | 4.1 | – | SCNN1B: c.1852C>T/βp.P618S |
| I‐2 | F | 41 | 25 | 190/110 | 2.90 | 0.03 | 0.18 | 126/82 | 4.35 | – | |
| I‐3 | F | 71 | 41 | 176/100 | 3.05 | 0.27 | 0.06 | 120/76 | 3.87 | – | |
| II‐1 | M | 14 | 14 | 192/120 | 2.46 | 0.27 | 0.15 | 134/78 | 4.01 | – | SCNN1B: c.1847C>T/βp.P616L |
| II‐2 | F | 24 | 18 | 196/120 | 2.78 | 0.03 | 0.14 | 120/80 | 4.45 | – | |
| II‐3 | M | 28 | 20 | 172/94 | 2.78 | 0.88 | 0.32 | 126/80 | 4.04 | – | |
| II‐4 | F | 30 | 18 | 166/106 | 2.32 | 0.38 | 0.42 | 136/76 | 3.96 | – | |
| II‐5 | M | 30 | 15 | 180/110 | 2.64 | 0.82 | 0.30 | 128/80 | 4.71 | – | |
| II‐6 | M | 36 | 19 | 186/100 | 2.41 | 0.46 | 0.17 | 130/86 | 4.52 | – | |
| II‐7 | M | 58 | 21 | 200/100 | 2.58 | 0.83 | 0.13 | 126/76 | 4.01 | Left ventricular hypertrophy, left atrial dilation, tachycardia | |
| II‐8 b | M | – | – | – | – | – | – | – | – | Dead from stroke | |
| II‐9 b | F | – | – | – | – | – | – | – | – | Dead from stroke | |
| II‐10 b | M | – | – | – | – | – | – | – | – | Dead from stroke | |
| III‐1 | F | 18 | 18 | 216/118 | 2.50 | 0.16 | 0.19 | 126/80 | 4.24 | – | SCNN1G: c.1711G>T/γp.E571* |
| III‐2 | F | 31 | 18 | 140/110 | 2.62 | 0.38 | 0.50 | 138/76 | 3.92 | – | |
| III‐3 | M | 54 | 19 | 210/120 | 2.00 | 0.05 | 0.18 | 130/74 | 4.22 | Stroke | |
| III‐4 | F | 58 | 20 | 194/116 | 2.76 | 0.12 | 0.23 | 126/80 | 4.59 | Severe aortic stenosis, chest pain | |
| III‐5 | F | 78 | 31 | 176/106 | 2.83 | 0.66 | 0.13 | 134/78 | 4.22 | – | |
| III‐6 | M | 7 | – | 120/72 | 3.32 | 0.57 | 0.21 | – | – | – | |
| III‐7 b | M | – | – | – | – | – | – | – | – | Dead from stroke | |
| IV‐1 | M | 17 | 13 | 180/110 | 3.20 | 0.12 | 0.12 | 130/70 | 4.02 | SCNN1B: c.1696C>T/βp.R566* | |
| V‐1 | M | 24 | 17 | 160/100 | 3.17 | 0.01 | 0.01 | 110/80 | 4.14 | Mild renal function impairment | SCNN1B: c.1854dupC/βp.N619Qfs*3 |
Abbreviations: ALD, aldosterone (reference value, 0.065–0.296 ng/mL); BP, blood pressure; f, female; HT, hypertension; m, male; PRA, plasma renin activity (reference value, 0.93–6.56 ng/mL·h); sk+, serum potassium (reference value 3.5–5.5 mmol/L). aAll patients carrying Liddle syndrome mutations identified in the five pedigrees, except IV for which data were incomplete. bThese patients died from stroke before our family screening and were classified as patients with Liddle syndrome according to their histories of hypertension and relationship with the identified patients. Each Greek letter represents one pedigree and the bold text shows probands.
Table 3.
Clinical Characteristics of Patients With LS and EH
| LS a (n=17) | EH (n=325) | P Value | |
|---|---|---|---|
| Age, y | 30.0 (21.0–56.0) | 31.0 (26.0–36.0) | .716 |
| Men, No. (%) | 7 (41.2) | 221 (68) | .033 |
| Age of hypertension onset, y | 18.0 (17.0–21.0) | 31.0 (26.0–36.0) | .001 |
| Family history of hypertension, No. (%) | 16 (94.1) | 214 (63.9) | .015 |
| SBP, mm Hg | 186.0 (174.0–195.0) | 150.0 (140.0–170.0) | <.001 |
| DBP, mm Hg | 110.0 (100.0–119.0) | 100.0 (90.0–110.0) | .02 |
| Serum K+, mmol/L | 2.68 (2.48–2.87) | 3.96 (3.65–4.19) | <.001 |
| Hypokalemia, No. (%) | 17 (100) | 43 (13.2) | <.001 |
Abbreviations: DBP, diastolic blood pressure; EH, essential hypertension; SBP, systolic blood pressure. aAll patients identified as having Liddle syndrome (LS) including the five index patients and their relatives were included.
Gene Mutations Associated With Liddle Syndrome
Five pathogenic mutations associated with Liddle syndrome were identified in the five index patients and their relatives, involving mutations in both β and γ subunits of ENaC. Remarkably, among these five mutations, three mutations, including βp.P616L, βp.N619Qfs*3, and γp.E571*, have never been reported before this screening work. This increases the number of possible mutations in Liddle syndrome to 27. These five pathogenic mutations had been missense, nonsense, and frameshift mutations, disturbing or truncating the conserved PY motif of β‐ or γ‐ENaC. A definite relationship between genotype and phenotype was not identified, ie, the implicated subunits of ENaC and mutation types presented no overt relationship with the phenotypes.
Discussion
To the best of our knowledge, this is the first study to estimate the prevalence of Liddle syndrome in young hypertensive patients of undetermined reason in China, and the first study to describe the prevalence of Liddle syndrome using genetic testing as the confirmatory test. This study indicated that the prevalence of Liddle syndrome in hypertensive patients of unascertained reason aged 14 to 40 years was 1.52%, demonstrating that Liddle syndrome is not quite as rare as previously thought and is an important etiology of hypertension, especially in young populations. In 2010, Tapolyai and colleagues3 reported a high prevalence of 6% for likely Liddle syndrome in a group of elderly US veterans. Biochemical abnormalities were used as screening criteria in that study. Liddle syndrome is a condition of monogenic hypertension inherited in Mendelian fashion. Although only five patients were confirmed to have Liddle syndrome among the 330 study participants in this study, 12 more patients with Liddle syndrome were identified through screening family members of the index cases. Therefore, the actual prevalence of Liddle syndrome might be underestimated in this study.
Early penetrance is one of the most remarkable characteristics of Liddle syndrome. Most reported Liddle syndrome patients developed high BP before the age of 30 years, the youngest reported case being a 10‐month‐old baby.7 The mean age at onset of hypertension of the five index cases in this study was 16 years, and all 17 patients with Liddle syndrome developed hypertension at around 20 years. It is suggested that Liddle syndrome should be considered by clinicians when evaluating reasons underlying elevated BP in those with early onset of hypertension. Despite following Mendelian inheritance, patients without family histories of hypertension should not be overlooked. Some sporadic cases of Liddle syndrome have been previously reported.8, 9, 10 We recently identified a young patient with uncontrolled hypertension as having Liddle syndrome, while his parents were normotensives without carrying the Liddle mutation.6
Similar to many other disorders of a Mendelian form, Liddle syndrome displays phenotypic variability. BP and the serum potassium concentration might be two concerning clinical signs of Liddle syndrome with respect to heterogeneity.11, 12 Liddle syndrome cases with severe hypertension to normotension or severe hypokalemia to normal levels of serum potassium are reported.13, 14, 15, 16 The phenotype can vary greatly among patients, even for patients of the same pedigree or with the same mutation.17 However, our results did not show distinct variability in BP and serum potassium levels. All 18 confirmed mutation carriers, including 13 relatives of the probands identified through genetic screening, manifested overt hypokalemia and hypertension no lower than grade II, except for a 7‐year‐old child. Accordingly, we speculate that hypertension and hypokalemia may be two of the most prominent clinical features of Liddle syndrome. In addition, the serum potassium level may play an important role in clinical evaluation of this disorder. It should be kept in mind that the mild phenotype is not rare in Liddle syndrome patients. Even in the original pedigree, people carrying the causal mutation may present with a normal level of serum potassium.17 With respect to PRA and plasma aldosterone, our results showed that PRA was suppressed in all mutation carriers, while plasma aldosterone levels of most carriers were normal. Low‐renin hypertension is a relatively common form of hypertension in adults, and essential hypertension accounts for a large proportion of cases.18 A normal or even low level of plasma aldosterone with a low renin state is uncommon in essential hypertension, and thus suggests the possibility of Liddle syndrome.19
Great advances have been made in the pathogenesis of Liddle syndrome since the first Liddle syndrome mutations in β‐ENaC and γ‐ENaC were identified by Shimkets and colleagues11 in 1994 and Hansson and colleagues20 in 1995. The PY motif, a conserved proline‐rich sequence in the cytosolic C‐terminus of the ENaC subunit, is the binding site of a specific ubiquitin ligase, Need4‐2.21 The interaction between Need4‐2 and the PY motif catalyzes ubiquitination of ENaCs, resulting in their internalization and degradation. The gain‐of‐function mutations associated with Liddle syndrome can delete or alter the PY motif, impeding its combination with Need4‐2, and lead to an abnormal turnover of ENaCs and constitutive activation of the ENaC.22 Volume expansion because of an imbalance in sodium homeostasis in the distal nephron, and possibly other tissues,23 suppresses renin and aldosterone secretion and leads to severe hypertension. However, this traditional theory has recently been challenged.24
DNA analysis could provide direct evidence of mutations associated with Liddle syndrome. In addition, genetic testing has been a rapid and practical tool to confirm the diagnosis of Liddle syndrome. Genetic testing should be considered in young hypertension patients with early penetrance, especially those with hypokalemia, a low renin level, and undetermined causes of elevated BP. One of the greatest advantages of genetic testing is family screening, enabling clinicians to identify people with potential Liddle syndrome. Once diagnosis of Liddle syndrome is established, it provides patients with the possibility to avoid severe complications, as this disease is easy to control with ENaC antagonists and dietary salt restriction.
Limitations
Our study has several limitations. First, this was a single‐center study and the participants were not a random sample of all young hypertension patients. Second, genetic testing was performed only in hypertensive patients with hypokalemia, and we may have excluded some absolute normokalemia patients with Liddle syndrome. As a result, the prevalence of Liddle syndrome described in this study may be underestimated.
Conclusions
This study suggests that Liddle syndrome is not as rare a disorder as previously thought. The study highlights the need to screen for Liddle syndrome among young hypertension patients, especially those with early penetrance, hypokalemia, and low renin level after exclusion of common secondary causes. Genetic testing provides the opportunity to achieve accurate and rapid diagnosis of Liddle syndrome. Potentially, a greater number of people with Liddle syndrome could be identified through family screening, and tailored treatment with high efficacy makes it possible for these individuals to avoid severe complications. It is possible that the prevalence of Liddle syndrome is greater than what we estimated, as we did not perform genetic testing of the nonhypokalemia participants. Therefore, absolutely uniform evaluation should be performed in each participant with or without hypokalemia in future investigations.
Acknowledgments and Disclosures
This work was supported by PUMC Graduate Innovation Fund (10023‐1002‐1001) with a grant to Dr Yang, PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (33320140036), and the Capital Medical Development Scientific Research Fund (2014‐4‐4035) with a grant to Dr Liu. The authors declare no conflicts of interest.
J Clin Hypertens (Greenwich). 2015;17:902–907. DOI: 10.1111/jch.12598. © 2015 Wiley Periodicals, Inc.
References
- 1. Liddle GW, Bledsoe T, Coppage W. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. 1963;76:199–213. [Google Scholar]
- 2. Rossi GP. Prevalence and diagnosis of primary aldosteronism. Curr Hypertens Rep. 2010;12:342–348. [DOI] [PubMed] [Google Scholar]
- 3. Tapolyai M, Uysal A, Dossabhoy NR, et al. High prevalence of Liddle syndrome phenotype among hypertensive US veterans in northwest Louisiana. J Clin Hypertens (Greenwich). 2010;12:856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang LP, Gao LG, Zhou XL, et al. Genetic diagnosis of Liddle's syndrome by mutation analysis of SCNN1B and SCNN1G in a Chinese family. Chin Med J. 2012;125:1401–1404. [PubMed] [Google Scholar]
- 5. Gao L, Wang L, Liu Y, et al. A family with Liddle syndrome caused by a novel missense mutation in the PY motif of the beta‐subunit of the epithelial sodium channel. J Pediatr. 2013;162:166–170. [DOI] [PubMed] [Google Scholar]
- 6. Yang KQ, Lu CX, Xiao Y, et al. A novel frameshift mutation of epithelial sodium channel beta‐subunit leads to Liddle syndrome in an isolated case. Clin Endocrinol (Oxf). 2015;82:611–614. [DOI] [PubMed] [Google Scholar]
- 7. Assadi FK, Kimura RE, Subramanian U, Patel S. Liddle syndrome in a newborn infant. Pediatr Nephrol. 2002;17:609–611. [DOI] [PubMed] [Google Scholar]
- 8. Nakano Y, Ishida T, Ozono R, et al. A frameshift mutation of beta subunit of epithelial sodium channel in a case of isolated Liddle syndrome. J Hypertens. 2002;20:2379–2382. [DOI] [PubMed] [Google Scholar]
- 9. Uehara Y, Sasaguri M, Kinoshita A, et al. Genetic analysis of the epithelial sodium channel in Liddle's syndrome. J Hypertens. 1998;16:1131–1135. [DOI] [PubMed] [Google Scholar]
- 10. Yamashita Y, Koga M, Takeda Y, et al. Two sporadic cases of Liddle's syndrome caused by de novo ENaC mutations. Am J Kidney Dis. 2001;37:499–504. [PubMed] [Google Scholar]
- 11. Shimkets RA, Warnock DG, Bositis CM, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. [DOI] [PubMed] [Google Scholar]
- 12. Botero‐Velez M, Curtis JJ, Warnock DG. Brief report: Liddle's syndrome revisited–a disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994;330:178–181. [DOI] [PubMed] [Google Scholar]
- 13. Hiltunen TP, Hannila‐Handelberg T, Petajaniemi N, et al. Liddle's syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens. 2002;20:2383–2390. [DOI] [PubMed] [Google Scholar]
- 14. Tamura H, Schild L, Enomoto N, et al. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97:1780–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Findling JW, Raff H, Hansson JH, Lifton RP. Liddle's syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab. 1997;82:1071–1074. [DOI] [PubMed] [Google Scholar]
- 16. Furuhashi M, Kitamura K, Adachi M, et al. Liddle's syndrome caused by a novel mutation in the proline‐rich PY motif of the epithelial sodium channel beta‐subunit. J Clin Endocrinol Metab. 2005;90:340–344. [DOI] [PubMed] [Google Scholar]
- 17. Rossi E, Farnetti E, Nicoli D, et al. A clinical phenotype mimicking essential hypertension in a newly discovered family with Liddle's syndrome. Am J Hypertens. 2011;24:930–935. [DOI] [PubMed] [Google Scholar]
- 18. Warnock DG. Liddle syndrome: genetics and mechanisms of Na+ channel defects. Am J Med Sci. 2001;322:302–307. [DOI] [PubMed] [Google Scholar]
- 19. Jain G, Ong S, Warnock DG. Genetic disorders of potassium homeostasis. Semin Nephrol. 2013;33:300–309. [DOI] [PubMed] [Google Scholar]
- 20. Hansson JH, Nelson‐Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82. [DOI] [PubMed] [Google Scholar]
- 21. Schild L. The epithelial sodium channel and the control of sodium balance. Biochim Biophys Acta. 2010;1802:1159–1165. [DOI] [PubMed] [Google Scholar]
- 22. Ellison DH. Ubiquitylation and the pathogenesis of hypertension. J Clin Invest. 2013;123:546–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Huysse JW, Amin MS, Yang B, Leenen FH. Salt‐induced hypertension in a mouse model of Liddle syndrome is mediated by epithelial sodium channels in the brain. Hypertension. 2012;60:691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem. 2010;285:23527–23531. [DOI] [PMC free article] [PubMed] [Google Scholar]