

Abstract
Blood pressure (BP) in patients with sickle cell disease (SCD) has been reported to be lower than in persons in the general population. Data on arterial stiffness, which is an important risk factor for the progression of BP, are inconclusive for this patient population. Forty‐five adult patients with SCD and 40 controls matched for sex, age, and body mass index were studied. Brachial systolic BP (SBP) and diastolic BP (DBP) were significantly lower in the patient group (SBP 115.1±13.8 mm Hg vs 121.9±11.3 mm Hg and DBP 68.5±8.0 mm Hg vs 80.6±9.1 mm Hg, P<.05, respectively). Augmentation index (AIx), however, was significantly higher in SCD patients compared with healthy controls (24.9±9.6 for patients vs 12.4±10.8 for controls, P<.001), while carotid femoral pulse wave velocity was comparable between the two groups. The study shows that mechanisms other than arterial elasticity are involved in the low BP phenotype of patients with SCD.
Sickle cell disease (SCD) is an autosomal recessive inherited blood disorder that arises from the point mutation of the β‐globin gene resulting in the substitution of valine with glutamine at the sixth position of the beta chain, which leads to the expression of hemoglobin S (HbSS). The high frequency of the sickle cell hemoglobin (HbS) gene in malaria‐endemic regions is caused by a heterozygote advantage against fatal malaria.1 The actual anemia of the disease is caused by hemolysis and the destruction of red blood cells because of their shape. Persistent intravascular hemolysis over decades leads to chronic vasculopathy.2 Many HbSS patients develop pulmonary hypertension. Nonetheless, patients with SCD have lower systemic blood pressure (BP) than those without the disease.3, 4, 5
The physiologic background of this observation is still unknown, while some proposed mechanisms include sodium and water wasting as a result of medullary defect, systemic vasodilation compensating for microcirculatory flow disturbances, increased production of prostaglandins and nitric oxide (NO), and reduced vascular reactivity.6, 7, 8, 9
One of the main components of the BP phenotype is arterial stiffness. Whether it precedes BP rise or comes after long‐time exposure to high BP is not known.10, 11 Arterial stiffness is associated with high BP, but more often with high pulse pressure (PP), which is affected by aortic stiffness and the balance between aortic flow and lumen diameter in the proximal aorta as well as by wave reflection.11 To what extent the BP pattern in SCD is caused by alterations in arterial elasticity is largely unknown.
The aim of the present study is to report BP levels in patients with SCD and healthy matched controls and to investigate the effect of arterial stiffness and renal function on BP levels in this patient population. We also sought to investigate whether aortic pressures follow the pattern of peripheral BP.
Patients and Methods
A total of 45 adult patients with SS‐type SCD aged between 26 and 65 years (mean age, 43 years) followed in our center between October 2012 to March 2013 and in a steady state of disease for at least 6 months (no acute illness and no vaso‐occlusive or acute chest syndrome episode) were enrolled in the study. Patients with S‐β‐thalassemia genotype, diabetes mellitus or other endocrine disease, arterial hypertension, and glomerular filtration rate (GFR) <59 mL/min/1.73 m2 were excluded. Forty healthy individuals matched for sex, age, and body mass index (BMI) were also recruited as controls.
The study protocol conformed to the ethical guidelines of the Declaration of Helsinki and was approved by the institutional review board of Aristotle University of Thessaloniki. All participants gave their written informed consent.
Clinical data were collected using a standardized form that included sociodemographic characteristics, medical history, and current medication.
Study Protocol
All clinical and biochemical measurements were performed from 7:30 am to 11:30 am after an overnight fast. The waist circumference was measured in the standing position at the level of the umbilicus, and the waist‐to‐hip ratio was calculated by dividing waist circumference by hip circumference.
Fasting parameters, including levels of fasting plasma glucose, fasting insulin, triglycerides (TGs), low‐density lipoprotein (LDL), high‐density lipoprotein, and total cholesterol, and basic biochemistry parameters were measured in each patient using standardized procedures.
25 OH D3, ferritin, cortisol, parathyroid hormone (PTH), thyroid‐stimulating hormone (TSH), osteocalcin calcium, and phosphate were also measured.
Homeostatic model assessment for insulin resistance (HOMA‐IR) and β‐cell function (HOMA‐β) were calculated according to known formulas.
Oral Glucose Tolerance Test
A 75‐g oral glucose tolerance test was performed with plasma glucose and insulin sampling performed at 0, 30, 60, 90, and 120 minutes.
Pulse Wave Velocity
Aortic (carotid‐to‐femoral [cf]) pulse wave velocity (PWV) was calculated from measurements of common carotid and femoral artery waveforms using an automatic applanation tonometry‐based device, the SphygmoCor Vx system (AtCor, Itasca, IL), as described. Briefly, electrocardiogram‐gated pulse waveforms were obtained sequentially over the common carotid and femoral arteries. PWV was calculated as the distance between recording sites measured over the surface of the body, divided by the time interval between the feet of the pressure waves. All of the measurements were performed by the same observer, who was blinded to the patient's clinical data. Central artery waveforms derived from the radial artery waveform and pressure by using a transfer function validated previously during catheterization studies.12 The point at which the central aortic pressure becomes augmented by wave reflection is recognized by a computer program, and the degree of increase is expressed as the aortic augmentation, which is quantified either in absolute term or as a percentage of aortic pulse pressure (aortic augmentation index [AIx]).
Glomerular Filtration Rate
We calculated eGFR according to the four‐variable Modification of Diet in Renal Disease (MDRD) equation and Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI).13
BP Measurements
BP measurements were performed in the morning with the patients in the seated position following a 5‐minute quiet resting period. BP was measured in both arms with a mercury sphygmomanometer using an appropriately sized cuff. Values for systolic BP (SBP) and diastolic BP (DBP) were defined by Korotkoff phase I and V, respectively. The average of the second and third measurements on both the right and left arms was used in the analysis.
Statistical Analysis
Normality of distribution was tested by Kolmogorov‐Smirnov test. Parametric data were expressed as mean±standard deviation and nonparametric as median (range).
Independent samples t test or Mann‐Whitney test was performed to evaluate differences in measured parameters between the patient population and the control group. We tested differences in our measurements of interest between the patient population and the control group using when variables were parametric and Mann‐Whitney test if otherwise.
Pearson's correlation coefficient r was used to test the relation of AIx, PWV, HOMA‐IR, and Belfiore with the under‐study parameters, while Spearman test was used to find correlations with the HOMA‐β index.
To test which parameters affect arterial stiffness and central BP in our patient population, we used forward stepwise‐regression analysis and created models using AIx, PWV, central SBP, and central DBP as dependent variables. The existence of SCD was marked as 0 for controls and 1 for patients with SCD.
We tested for the magnitude of multicollinearity in our models using the variance inflation factor (VIF). A VIF >2 for a variable was considered high.
P<.05 was considered statistically significant. The analysis was performed using Predictive Analytics Software (PASW version 18.0; Chicago, IL).
Results
BP, Arterial Stiffness, and GFR in the Two Groups
Patients were matched with controls in regards to age (43.3±9.8 years and 39.8±11.5 years, P>.05), BMI (24.1±3.6 kg/m2 and 24.4±3.2 kg/m2, P>.05), and sex (15/30 and 17/23 for female/male, respectively; P>.05).
Brachial SBP and DBP were significantly lower in the patient group compared with the control group (SBP 115.1±13.8 mm Hg vs 121.9±11.3 mm Hg and DBP 68.5±8.0 mm Hg vs 80.6±9.1 mm Hg, P<.05, respectively). Brachial mean arterial pressure (MAP) was also significantly lower in the SCD group (Table 1B, Figure 1 and Figure 2). Similar results were observed for central DBP (70.0±8.3 mm Hg vs 79.8±9.4 mm Hg, P<.001, for patients and controls, respectively). Central MAP was also lower in the patient group (Figure 3).
Table 1.
Biochemistry and Blood Parameters (A); Blood Pressure, Arterial Stiffness, and Glomerular Filtration Rate (B); Bone Homeostasis and Hormones (C); and Glucose Homeostasis (D)
| Variable | SCD | Controls | P Value |
|---|---|---|---|
| (A) | |||
| Cholesterol total | 146.1±42.6 | 197.5±36.7 | <.001 |
| Triglycerides | 113.6±57.6 | 82.0±38.0 | .006 |
| LDL | 82.8±35.1 | 131.8±32.1 | <.001 |
| HDL | 39.1±10.5 | 50.0±13.4 | <.001 |
| Ferritin | 170.3 (1439.9) | 30.7 (175.8) | <.001 |
| Ferrum | 95.2±36.3 | 94±44.4 | NS |
| Creatinine | 0.60±0.15 | 0.75±0.14 | <.001 |
| Urea | 22±8.2 | 26.6±8.4 | .02 |
| Sodium | 139.5±3.7 | 136.0±1.4 | <.001 |
| Potassium | 4.4±0.4 | 4.1±0.4 | <.001 |
| Hemoglobin, g/dL | 10.8±1.1 | 13.1±1.4 | <.001 |
| Hematocrit, % | 29.7±3.5 | 38.5±3.2 | <.001 |
| MCV, fL | 72.9±6.9 | 85.2±3.5 | <.001 |
| MCHC, g/dL | 36.5±1.2 | 34.4±1.1 | <.001 |
| (B) | |||
| Branchial SBP | 115.1±13.8 | 121.9±11.3 | .02 |
| Branchial DBP | 68.5±8.0 | 80.6±9.1 | <.001 |
| Branchial MAP | 91.8±10.1 | 101.3±9.0 | <.001 |
| Branchial PP | 46.9±10.2 | 41.0±9.9 | .025 |
| Heart rate | 76.4±10.5 | 73.8±10.7 | NS |
| Central SBP | 105.4±17.7 | 108.4±10.0 | NS |
| Central DBP | 70.0±8.3 | 79.8±9.4 | <.001 |
| Central MAP | 87.7±11.5 | 94.1±9.2 | .009 |
| Central PP | 35.4±15.5 | 28.5±6.1 | .01 |
| AIx | 24.9±9.6 | 12.4±10.8 | <.001 |
| PWV, m/s | 7.2±1.4 | 7.1±0.8 | NS |
| MDRD, mL/min/1.73 m2 | 138.0±37.3 | 110.6±20.0 | <.001 |
| (C) | |||
| 25 (OH) D3 | 13.1±8.4 | 32.4±14.2 | <.001 |
| PTH | 32.7±20.4 | 31.0±14.9 | NS |
| Calcium | 9.0±0.6 | 9.0±0.4 | NS |
| Phosphate | 3.5±0.5 | 3.6±0.6 | NS |
| Osteocalcin | 14.1±5.7 | 3.6±0.6 | <.001 |
| TSH | 2.1±1.4 | 1.7±0.7 | NS |
| Cortisol | 316±145.5 | 379.9±244.2 | NS |
| (D) | |||
| Fasting glucose, mg/dL | 81.4±12.5 | 78.9±7.9 | NS |
| Fasting insulin, μU/mL | 5.1±2.7 | 11.3±6.6 | <.001 |
| HOMA‐IR, % | 18.6±10.2 | 41.8±25.7 | <.001 |
| QUICKI | 0.38 (0.5) | 0.35 (0.11) | <.001 |
| HOMA‐β, % | 101.1 (1983.6) | 274.1 (4351.1) | <.001 |
Abbreviations: AIx, augmentation index; DBP, diastolic blood pressure; HDL, high‐density lipoprotein; HOMA‐β, homeostatic model assessment for β‐cell function; HOMA‐IR, homeostatic model assessment for insulin resistance; MAP, mean arterial pressure; LDL, low‐density lipoprotein; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; MDRD, Modification of Diet in Renal Disease; PP, pulse pressure; PTH, parathyroid hormone; PWV, pulse wave velocity; SBP, systolic blood pressure; SCD, TSH, thyroid‐stimulating hormone; QUICKI, All blood pressures are measured in mm Hg.
Figure 1.
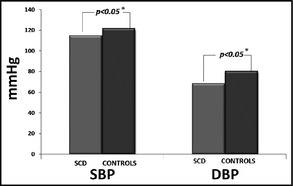
Mean systolic blood pressure (SBP) and diastolic blood pressure (DBP) in the sickle cell disease (SCD) group and the control group. *Indicates statistical significance.
Figure 2.
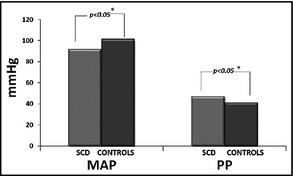
Mean arterial pressure (MAP) and pulse pressure (PP) in the sickle cell disease (SCD) group and the control group. *Indicates statistical significance
Figure 3.
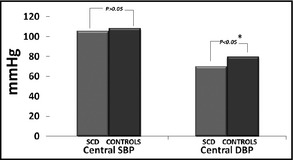
Mean central systolic blood pressure (SBP) and diastolic blood pressure (DBP) in the sickle cell disease (SCD) group and the control group. *Indicates statistical significance.
On the contrary, brachial and central PP were higher in the patient group compared with normal controls (46.9±10.2 mm Hg vs 41.0±9.9 mm Hg and 35.4±15.5 mm Hg vs 28.5±6.1 mm Hg, respectively). Heart rate did not differ between the two groups (Table 1B).
AIx, however, was significantly higher in SCD patients compared with controls (24.9%±9.6% for patients vs 12.4%±10.8% for controls, P<.001), while PWV was comparable between the two groups (Table 1B). PWV difference between groups remained not significant even after brachial DBP adjustment taking into consideration possible different distending pressures that may affect stiffness measurements (Figure 4). In terms of renal function, eGFR was significantly higher in patients with SCD compared with controls (138.0±37.3 mL/min/1.73 m2 vs 110.6±20.0 mL/min/1.73 m2 for patients and controls, respectively).
Figure 4.
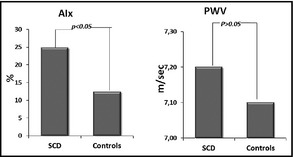
Indexes of arterial stiffness in the sickle cell disease (SCD) group and the control group. AIx indicates augmentation index, PWV, pulse wave velocity.
Table 1C and ID show bone homeostasis hormones and glucose homeostasis parameters, respectively, for the two groups.
Simple Correlations of the Under‐Study Parameters in the SCD Group
Table 2 shows simple correlations of the under‐study parameters.
Table 2.
Simple Correlations of Arterial Stiffness Indices With the Under‐Study Parameters in Patients With Sickle Cell Anemia
| Variables | AIx | PWV | Aortic SBP | Aortic DBP | Aortic PP | MDRD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r | P Value | r | P Value | r | P Value | r | P Value | r | P Value | r | P Value | |
| Aortic SBP | 0.34 | .027 | 0.57 | <.001 | 1 | NS | −0.14 | NS | ||||
| Aortic DBP | 0.011 | NS | 0.32 | .04 | 0.47 | .002 | 1 | NS | −0.37 | .03 | ||
| Aortic PP | 0.38 | .01 | 0.48 | .001 | 0.89 | <.001 | 0.02 | NS | 1 | NS | 0.02 | NS |
| BMI | 0.13 | NS | 0.15 | NS | 0.024 | NS | 0.26 | NS | 0.48 | .001 | −0.38 | .02 |
| Age | 0.34 | .027 | 0.67 | <.001 | 0.51 | .001 | 0.30 | NS | 0.43 | .005 | −0.39 | .02 |
| Branchial SBP | 0.22 | NS | 0.60 | <.001 | 0.70 | <.001 | 0.60 | <.001 | 0.47 | .02 | −0.32 | .05 |
| Branchial DBP | −0.01 | NS | 0.34 | .03 | 0.36 | .02 | 0.91 | <.001 | −0.06 | NS | −0.31 | NS |
| HR | −0.07 | NS | −0.07 | NS | −0.17 | NS | −0.065 | NS | −0.15 | NS | −0.21 | NS |
| Waist | −0.06 | NS | 0.42 | .007 | 0.18 | NS | 0.54 | .001 | −0.06 | NS | −0.31 | NS |
Abbreviations: AIx, augmentation index; BMI, body mass index; DBP, diastolic blood pressure; HR, heart rate; MDRD, Modification of Diet in Renal Disease; NS, not significant; PP, pulse pressure; PWV, pulse wave velocity; SBP, systolic blood pressure.
AIx significantly and positively correlated with aortic SBP, aortic PP, and age. PWV significantly and positively correlated with all aortic and brachial BP measurements, but also with age, waist circumference, total cholesterol, TGs, and LDL.
Regression Analyses
Stepwise forward linear regression analyses were used to identify determinants of arterial stiffness, eGFR, and central BP.
cf‐PWV was significantly determined by age (β=0.063, P=.001; R 2=0.35, P=.001) (adjustments for aortic SBP, HOMA‐IR, eGFR, heart rate, BMI, waist, total cholesterol, TGs, 25 OH D3, ferritin, cortisol, PTH, TSH, calcium, phosphate, and SCD).
AIx was significantly determined by the existence of SCD (β=9.8, P=.02), age (β=0.72, P=.003), and waist (β=−0.4, P=.04; R 2=0.51, P=.001) (adjustments for aortic SBP, HOMA‐IR, estimated glomerular filtration rate (eGFR), heart rate, BMI, total cholesterol, TGs, 25 OH D3, ferritin, cortisol, PTH, TSH, calcium, and phosphate).
eGFR was determined by ferritin (β=0.045, P=.006) and was negatively associated with central DBP (β=−0.96, P=.04) and age (β=−0.8, P=.049; R 2=0.42, P<.001) (adjustments for sex, aortic SBP, PWV, BMI, total cholesterol, TGs, 25 OH D3, calcium, HOMA‐IR, and SCD).
Age was the sole most significant determinant of central SBP (β=0.63, P<.001; R 2=0.38, P<.001) (adjustments for aortic DBP, HOMA‐IR, eGFR, heart rate, BMI, total cholesterol, TGs, 25 OH D3, ferritin, cortisol, PTH, TSH, calcium, phosphate, and SCD).
Central DBP was determined by waist circumference (β=0.03, P=.017) and central PP (β=0.045, P=.03; R 2=0.34, P=.001) (adjustments for aortic SBP, HOMA‐IR, eGFR, heart rate, BMI, total cholesterol, TGs, 25 OH D3, ferritin, cortisol, PTH, TSH, calcium, phosphate, and SCD).
Discussion
The main finding of the present study was that despite their lower peripheral and central BP levels, patients with SCD had higher peripheral and central PP levels than age‐ and sex‐matched controls.
In addition, SCD along with age and waist circumference significantly determined AIx in the study population. Finally, eGFR was higher in SCD patients and significantly positively affected by ferritin and negatively associated with age and central DBP in the study population.
The first to describe BP levels in SCD were Johnson and colleagues, who in 1981 showed that SCD patients had lower BPs than controls. The authors attribute this paradox phenomenon to the renal tubular defect responsible for the increased sodium and water excretion in these patients.3 Several studies conducted thereafter nearly all confirmed this observation.
Pegelow and colleagues4 showed that in a cohort of 3317 SCD patients, BP in patients was lower than values reported by age‐, ssex‐, and race‐matched patients in the National Health and Nutrition Examination Surveys I and II. However, they show that in SCD patients, this relative systemic hypertension that still falls within the population norms predicts early mortality.
Moreover, Johnson and colleagues14 support this concept with their multivariate analysis showing that SBP percentiles together with oxygen desaturation while asleep and age were independent predictors of left ventricular mass.
Morover, Gordeuk and colleagues showed that in SCD patients, SBP 120 mm Hg to 139 mm Hg and DBP 70 mm Hg to 89 mm Hg defines a category of relative systemic hypertension and is associated with increased risk for pulmonary hypertension and renal dysfunction. As a result of this low normal BP, higher BP levels that are within the normal range for the general population may be indicative of underlying renal disease or other comorbid medical conditions.
Therefore, they propose a new threshold for BP ≤120/70 mm Hg in patients with SCD.15
Although the exact mechanism of the lower BP is unknown, contributing factors could include salt‐losing sickle cell nephropathy16; lower peripheral resistance; alteration of circulating levels of catecholamine, renin, aldosterone and prostaglandin; or changes in the sensitivity of receptors to these agents.17, 18 It is also possible that low BP is a compensation mechanism that is developed to overcome the detrimental effects of vaso‐occlusive disease.
Another hypothesis for the resistance of hypertension in SCD is the activation of the NO system. One study showed that higher urinary excretion of NO metabolites was higher in SCD patients than in nonanemic control patients,19 and another study showed a decreased response of peripheral vessels to blockade of the NO system, both suggestive of chronic NO activation.20, 21
Later studies showed that hemolysis consumes NO and induces oxidative stress and thus induces endothelial dysfunction, cancelling NO abundance as a mechanism for the reduced BP.22, 23
A different plausible explanation for the adverse cardiovascular events of low normal BP in SCD is that they might eventually have true hypertension since data in children indicate a high percentage of masked hypertension in this patient population. Two studies have demonstrated this altered BP pattern that not only misclassifies these patients as being normotensive, but also may lead to detrimental side effects such as microalbuminuria, left ventricular hypertrophy, and stroke, as well as other consequences of untreated high BP.24, 25
When evaluating BP in SCD, we should consider that, as a group, patients with anemia have lower than expected SBP and DBP.3, 4 However, it has been reported that BP in SCD is higher than expected given the severity of anemia, further building the case of the existence of relative hypertension in this patient population.5
In addition, one of the parameters that may confound the relationship between SCD patients and controls in relation to their BP status may be the smaller stature of SCD patients; however, in our population, BMI and waistline were comparable between the two groups.
Arterial stiffness and BP are highly interrelated conditions.10 Two studies investigated the topic of arterial stiffness in SCD patients with conflicting results.26, 27
In the first study, Lemogoum and colleagues27 showed that cf‐PWV and carotid‐brachial PWV, as well as aortic AIx, was lower in SCD patients compared with controls and they were both negatively associated with hemoglobin SS type. In contrast, Belizna and colleagues26 showed that 49 patients with SCD had higher carotid stiffness than 47 matched controls and suggest an association between arterial stiffness with stroke. The latter is in accordance with the results of the present study indicating that low levels of BP in patients with SCD may not be a consequence of higher arterial elasticity rather than other mechanisms that remain to be completely clarified.
SCD patients in our study, despite both their lower brachial and central SBP and DBP, demonstrated higher brachial, central PP, and AIx, all indicative of increased arterial stiffness; however, PWV was comparable in the two groups,which does not allow definite conclusions.
Another issue in SCD that the present study addresses is renal disease. It was as early as 1955 that Etteldorf and colleagues28 found GFR in children with SCD to be abnormally high while it normalized during adolescence and further declined with age.
In 1992, Falk and colleagues29 demonstrated that 25% of patients with SCD have proteinuria. It has been proposed that this nephropathy may be the consequence of a toxin such as iron administered during blood transfusions. The fundamental lesion is focal segmental glomerulosclerosis in the setting of glomerular hypertrophy.
Haymann and colleagues30 suggested that red blood cell (RBC) sickling‐induced vaso‐occlusion is not causative in glomerular hyperfiltration since it does not cosegregate with vaso‐occlusive clinical complications of SCD. Instead, in addition to younger age, markers of chronic hemolysis such as lower hemoglobin levels, greater hemoglobin F levels, and higher reticulocyte count are independent risk factors for glomerular hyperfiltration in SCD.
Hemolysis causes hyperfiltration through the induction of anemia, which causes increased renal plasma flow as a consequence of increased cardiac output.
Additionally, higher hemolysis rate leads to increased tissue iron deposition, which may be a cause of hyperfiltration.30
In our study, patients had higher GFR than controls, and hyperfiltration (MDRD‐GFR >130 mL/min per 1.73 m2 in women and >140 in men) was found in 51%.
Our multivariate analysis showed that ferritin significantly affected GFR, along with a negative association with age and aortic DBP. Our population consisted of relatively young individuals with a mean age of 40 years; therefore, our results can only be extrapolated to this age group.
Previous studies have found similar results, associating hyperfiltration with young age and chronic kidney disease that leads to end‐stage chronic kidney disease in older patients.30
Study Limitations
Our study has some limitations. First, the fact that BP was measured in the office and not ambulatory gives only a snapshot of what happens with BP given that SCD patients present desaturations in their sleep and possibly obstructive sleep apnea. They are also a group with a high incidence of masked hypertension.
In addition, the cross‐sectional design does not allow for conclusions regarding cardiovascular endpoints or target organ damage in our study group. Further, the fact that patients with a small age range were included in the study makes it difficult to extrapolate the results to the whole SCD population.
Conclusions
A more detailed examination of BP in patients with SCD is in order. This should include a 24‐hour BP measurement especially in those who have signs of target organ damage in order to identify cases of masked hypertension. Since there are still no separate/specific criteria by the European Society of Hypertension or the Eighth Joint National Committee for defining BP in SCD patients as a special population, this issue should be carefully addressed by opinion makers. More studies are needed to further evaluate the benign (or not) character of low BP in patients with SCD.
Disclosures
The authors report no specific funding in relation to this research and no conflicts of interest to disclose.
Acknowledgment
We thank Dr Spyros Gerou, MD, head of the “Analysis” laboratory, for his kind contribution in the measurement of the biochemistry parameters.
J Clin Hypertens (Greenwich). 2015:726–731. DOI: 10.1111/jch.12572. © 2015. Wiley Periodicals, Inc.
References
- 1. Aidoo M, Terlouw DJ, Kolczak MS, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002;359:1311–1312. [DOI] [PubMed] [Google Scholar]
- 2. Vichinsky E. Overview of the clinical manifestations of sickle cell disease. Uptodate 2014.
- 3. Johnson CS, Giorgio AJ. Arterial blood pressure in adults with sickle cell disease. Arch Intern Med. 1981;141:891–893. [PubMed] [Google Scholar]
- 4. Pegelow CH, Colangelo L, Steinberg M, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. 1997;102:171–177. [DOI] [PubMed] [Google Scholar]
- 5. Rodgers GP, Walker EC, Podgor MJ. Is “relative” hypertension a risk factor for vaso‐occlusive complications in sickle cell disease? Am J Med Sci. 1993;305:150–156. [DOI] [PubMed] [Google Scholar]
- 6. Allon M, Lawson L, Eckman JR, et al. Effects of nonsteroidal antiinflammatory drugs on renal function in sickle cell anemia. Kidney Int. 1988;34:500–506. [DOI] [PubMed] [Google Scholar]
- 7. de Jong PE, Statius van Eps LW. Sickle cell nephropathy: new insights into its pathophysiology. Kidney Int. 1985;27:711–717. [DOI] [PubMed] [Google Scholar]
- 8. Hatch FE, Crowe LR, Miles DE, et al. Altered vascular reactivity in sickle hemoglobinopathy. A possible protective factor from hypertension. Am J Hypertens. 1989;2:2–8. [DOI] [PubMed] [Google Scholar]
- 9. ter Maaten JC, Serne EH, Bakker SJ, et al. Effects of insulin on glucose uptake and leg blood flow in patients with sickle cell disease and normal subjects. Metabolism. 2001;50:387–392. [DOI] [PubMed] [Google Scholar]
- 10. Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension. 2014;64:210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitchell GF. Arterial stiffness and hypertension. Hypertension. 2014;64:210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pauca AL, O'Rourke MF, Kon ND. Prospective evaluation of a method for estimating ascending aortic pressure from the radial artery pressure waveform. Hypertension. 2001;38:932–937. [DOI] [PubMed] [Google Scholar]
- 13. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson MC, Kirkham FJ, Redline S, et al. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood. 2010;116:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gordeuk VR, Sachdev V, Taylor JG, et al. Relative systemic hypertension in patients with sickle cell disease is associated with risk of pulmonary hypertension and renal insufficiency. Am J Hematol. 2008;83:15–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radel E. Advances in the treatment of adolescents with sickle hemoglobinopathies. Adolesc Med. 1994;5:271–292. [PubMed] [Google Scholar]
- 17. Grell GA, Alleyne GA, Serjeant GR. Blood pressure in adults with homozygous sickle cell disease. Lancet. 1981;2:1166. [DOI] [PubMed] [Google Scholar]
- 18. Oguanobi NI, Onwubere BJ, Ibegbulam OG, et al. Arterial blood pressure in adult Nigerians with sickle cell anemia. J Cardiol. 2010;56:326–331. [DOI] [PubMed] [Google Scholar]
- 19. Nahavandi M, Wyche MQ, Perlin E, et al. Nitric oxide metabolites in sickle cell anemia patients after oral administration of hydroxyurea; hemoglobinopathy. Hematology. 2000;5:335–339. [DOI] [PubMed] [Google Scholar]
- 20. Belhassen L, Pelle G, Sediame S, et al. Endothelial dysfunction in patients with sickle cell disease is related to selective impairment of shear stress‐mediated vasodilation. Blood. 2001;97:1584–1589. [DOI] [PubMed] [Google Scholar]
- 21. Eberhardt RT, McMahon L, Duffy SJ, et al. Sickle cell anemia is associated with reduced nitric oxide bioactivity in peripheral conduit and resistance vessels. Am J Hematol. 2003;74:104–111. [DOI] [PubMed] [Google Scholar]
- 22. de MM, Aggoun Y, Niakate A, et al. Endothelial‐dependent vasodilation is impaired in children with sickle cell disease. Haematologica. 2007;92:1709–1710. [DOI] [PubMed] [Google Scholar]
- 23. Morris CR, Kuypers FA, Larkin S, et al. Arginine therapy: a novel strategy to induce nitric oxide production in sickle cell disease. Br J Haematol. 2000;111:498–500. [DOI] [PubMed] [Google Scholar]
- 24. Becker AM, Goldberg JH, Henson M, et al. Blood pressure abnormalities in children with sickle cell anemia. Pediatr Blood Cancer. 2014;61:518–522. [DOI] [PubMed] [Google Scholar]
- 25. Shatat IF, Jakson SM, Blue AE, et al. Masked hypertension is prevalent in children with sickle cell disease: a Midwest Pediatric Nephrology Consortium study. Pediatr Nephrol. 2013;28:115–120. [DOI] [PubMed] [Google Scholar]
- 26. Belizna C, Loufrani L, Ghali A, et al. Arterial stiffness and stroke in sickle cell disease. Stroke. 2012;43:1129–1130. [DOI] [PubMed] [Google Scholar]
- 27. Lemogoum D, Van BL, Najem B, et al. Arterial stiffness and wave reflections in patients with sickle cell disease. Hypertension. 2004;44:924–929. [DOI] [PubMed] [Google Scholar]
- 28. Etteldorf JN, Smith JD, Tuttle AH, Diggs LW. Renal hemodynamic studies in adults with sickle cell anemia. Am J Med. 1955;18:243–248. [DOI] [PubMed] [Google Scholar]
- 29. Falk RJ, Scheinman J, Phillips G, et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin‐converting enzyme. N Engl J Med. 1992;326:910–915. [DOI] [PubMed] [Google Scholar]
- 30. Haymann JP, Stankovic K, Levy P, et al. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5:756–761. [DOI] [PMC free article] [PubMed] [Google Scholar]