

Supplemental Digital Content is available in the text.
Keywords: fibroblasts; fibrosis; heart failure; macrophages; MIRN21 microRNA, mouse; myofibroblasts; ventricular remodeling
Abstract
Background:
Cardiac macrophages (cMPs) are increasingly recognized as important regulators of myocardial homeostasis and disease, yet the role of noncoding RNA in these cells is largely unknown. Small RNA sequencing of the entire miRNomes of the major cardiac cell fractions revealed microRNA-21 (miR-21) as the single highest expressed microRNA in cMPs, both in health and disease (25% and 43% of all microRNA reads, respectively). MiR-21 has been previously reported as a key microRNA driving tissue fibrosis. Here, we aimed to determine the function of macrophage miR-21 on myocardial homeostasis and disease-associated remodeling.
Methods:
Macrophage-specific ablation of miR-21 in mice driven by Cx3cr1-Cre was used to determine the function of miR-21 in this cell type. As a disease model, mice were subjected to pressure overload for 6 and 28 days. Cardiac function was assessed in vivo by echocardiography, followed by histological analyses and single-cell sequencing. Cocultures of macrophages and cardiac fibroblasts were used to study macrophage-to-fibroblast signaling.
Results:
Mice with macrophage-specific genetic deletion of miR-21 were protected from interstitial fibrosis and cardiac dysfunction when subjected to pressure overload of the left ventricle. Single-cell sequencing of pressure-overloaded hearts from these mice revealed that miR-21 in macrophages is essential for their polarization toward a M1-like phenotype. Systematic quantification of intercellular communication mediated by ligand-receptor interactions across all cell types revealed that miR-21 primarily determined macrophage-fibroblast communication, promoting the transition from quiescent fibroblasts to myofibroblasts. Polarization of isolated macrophages in vitro toward a proinflammatory (M1-like) phenotype activated myofibroblast transdifferentiation of cardiac fibroblasts in a paracrine manner and was dependent on miR-21 in cMPs.
Conclusions:
Our data indicate a critical role of cMPs in pressure overload–induced cardiac fibrosis and dysfunction and reveal macrophage miR-21 as a key molecule for the profibrotic role of cMPs.
Clinical Perspective.
What Is New?
MicroRNA-21 (MiR-21), an important microRNA in cardiac biology and disease, has its highest expression in cardiac macrophages, where it is the single strongest expressed microRNA.
Genetic deletion of miR-21 specifically in macrophages prevented myocardial remodeling and dysfunction induced by pressure overload in mice.
On a mechanistic basis, miR-21 promotes a proinflammatory phenotype in cardiac macrophages that signal to and activate cardiac fibroblasts through a set of paracrine mediators.
What Are the Clinical Implications?
Interference with the activation of cardiac macrophages represents a promising therapeutic strategy in myocardial remodeling and dysfunction.
Synthetic oligonucleotide inhibitors against miR-21, which are currently undergoing clinical testing against fibrotic disease, may also (or even primarily) work through their effect in macrophages.
Future RNA therapies against miR-21 may be targeted toward macrophages to further increase efficacy and limit potential side effects.
Heart failure constitutes a major global disease burden and remains the leading cause of death in industrialized and in developing countries.1 Despite considerable progress within the past 2 decades, the mortality rate of established heart failure remains high, despite optimized treatment regimens.2 Established therapies of heart failure such as β-blocking agents, angiotensin-converting enzyme inhibitors, and aldosterone antagonists largely focus on the inhibition of neurohumoral activation, an effective, but also limiting principle of therapy.
More recently, evidence has accumulated that inflammation within the myocardium is not only a key element of acute cardiac injury, such as during myocardial infarction, but is also observed in chronic cardiac pathologies such as in chronic heart failure.3 Consequently, the modulation of cardiac inflammation has also been put forward as a potential therapeutic strategy for the treatment of chronic HF.3
A key observation justifying this approach was that the mammalian heart exhibits a surprisingly large fraction of immune cells that reside in the myocardium, replicate locally, and respond to inflammatory stimuli.4 Among the different immune cells in the heart, macrophages are the most abundant cell fraction in healthy myocardium. In contrast to conditions involving acute cardiac injury with major influx of immune cells, tissue-resident macrophages are believed to be the dominating immune cell fraction during cardiac remodeling in chronic heart failure.5
Although the key stimuli, receptors and signaling pathways underlying the activation, plasticity, and replication of macrophages continue to be elucidated,6 the role of noncoding RNAs in these cells has remained largely unknown until now. MicroRNAs (miR) are a class of small noncoding RNAs that have emerged as powerful regulators of gene expression and that have been implicated in many diseases, including cardiovascular disease.7
One of the most intensely studied microRNAs in this context is miR-21, which is upregulated during myocardial disease and the genetic and pharmacological inhibition of which has been found to prevent the progression of myocardial fibrosis and heart failure.8–11 Since its implication in cardiac fibrosis, the profibrotic role of miR-21 has been corroborated in the fibrosis of various other organs and tissues, including liver, kidney, lung, skin, and skeletal muscle.12 Because miR-21 shows relatively high levels of expression in fibroblasts, and because the genetic deletion of miR-21 in mesenchymal cells (which include fibroblasts) likewise prevents fibrosis,10 it is generally assumed that miR-21 exerts its profibrotic action primarily in fibroblasts.
Here, we report that, within the myocardium, miR-21 has its strongest expression in cardiac macrophages, where it is also the single strongest expressed microRNA among all microRNAs. Targeted genetic deletion of miR-21 in macrophages of mice prevented their proinflammatory polarization and subsequently pressure overload–induced cardiac fibrosis and dysfunction. Analysis of intercellular communication, using single-cell sequencing, identified the cardiac fibroblast as the primary recipient cell of intercellular signals that emanate from activated cardiac macrophages and that are controlled by miR-21.
Methods
The authors declare that all data that support the findings of this study are available within the article and its Data Supplement. Materials will be shared on reasonable request. A detailed description of all materials (Table I in the Data Supplement) and methods including RNA sequencing library preparation and accessibility of publicly deposited RNA sequencing data are provided in the Data Supplement.
Experimental Animals and Study Design
All animal studies were performed in accordance with relevant guidelines and regulations of the responsible authorities and approval was obtained from the institutional review board at the Regierung von Oberbayern (ROB-55.2Vet-2532.Vet_02-17-70). Animals were randomly assigned to the experimental groups, and the person performing the surgery and echocardiography was blinded for the genotype.
Transverse Aortic Constriction Murine Model of Heart Failure
Cx3cr1-CreTg/0 and miR-21–floxed mice have been described previously. All mice were bred in C57BL/6 genetic background.13,14
Transverse aortic constriction (TAC) procedure was performed on 8- to 10-week-old male mice as described previously.10 For pre- and postoperative analgesia, the mice were injected intraperitoneally with buprenorphine (0.08 mg/kg) and metamizole (200 mg/kg). Mice were anesthetized in an induction chamber with 4% isoflurane mixed with 0.5 L/min of 100% O2. During the surgical procedure, anesthesia was maintained at 1.5% to 2% isoflurane with 0.5 L/min O2. Partial thoracotomy was performed and a small piece of 6.0 silk suture was ligated between the innominate and left carotid arteries around a 27.5-gauge needle placed parallel to the transverse aorta. In sham surgery, only the chest was opened, but no ligation of the aorta was performed. Cardiac dimensions and function were analyzed by pulse-wave Doppler echocardiography before TAC/sham surgery and, at the end of the experiment, before euthanizing the animals.
Echocardiography
Transthoracic echocardiography was performed on mice to assess cardiac function. Ultrasound gel was applied to the chest of isoflurane-anesthetized mice, and echocardiographic monitoring was performed by using an ultrasound system with a linear transducer with 32 to 55 MHz frequency (Visual Sonics) combined with Vevo 770 software (Visual Sonics). M-mode tracings were recorded, and the dimensions of myocardium at systole and diastole were measured. For speckle-tracking analysis, Vevostrain software (Visual Sonics) was used.
Statistical Analysis
Data denote mean and individual values. Statistical analysis was performed using Graphpad Prism software package (version 8). Data distribution was assessed by the Shapiro-Wilk test for normality. The Bartlett test was performed to test the homogeneity of variance, and the Spearman rank correlation test was performed to test heteroscedasticity. For data sets that did not pass the heteroscedasticity test, values were log-transformed before statistical testing. Differences among multiple means were assessed either by 1-way or 2-way ANOVA followed by the Sidak test. A P value of <0.05 was considered significant.
For detailed methods, see the Expanded Methods in the Data Supplement.
Results
miR-21 Is Highly Expressed in Cardiac Macrophages and Increases in Heart Failure
We initially sought to obtain an inventory of the entire small RNA transcriptomes of the major cell fractions of the mouse heart (Figure 1). To this end, we devised a cell isolation strategy, starting with retrograde perfusion of the isolated heart (Langendorff preparation), followed by fractionation into myocytes, endothelial cells, fibroblasts, and macrophages by using magnetic- and fluorescence-activated cell sorting (see scheme in Figure 1A and Figure I in the Data Supplement). According to these criteria, we detected 307 unique and individual transcripts that were confidently annotated as microRNAs (Figure 1B). In agreement with small RNA transcriptomes obtained in different cell types and tissues,15,16 we likewise observed a 1-sided distribution of the absolute abundance of the individual microRNA molecules, with very few microRNA molecule species constituting the majority of the entire miRNome (Figure 1B). Several of those microRNAs at the upper end of the expression spectrum also exhibited very pronounced cell-type specificity in comparison with the other myocardial cell types (Figure 1C). We found miR-21 to be the highest-expressed microRNA in cardiac macrophages (cMPs), constituting about one-quarter of all microRNA molecules in this cell type (Figure 1B). This is far higher than in other cardiac cell fractions, including fibroblasts (Figure 1B and 1C), a known cell type in which the function of miR-21 is well characterized.
Figure 1.
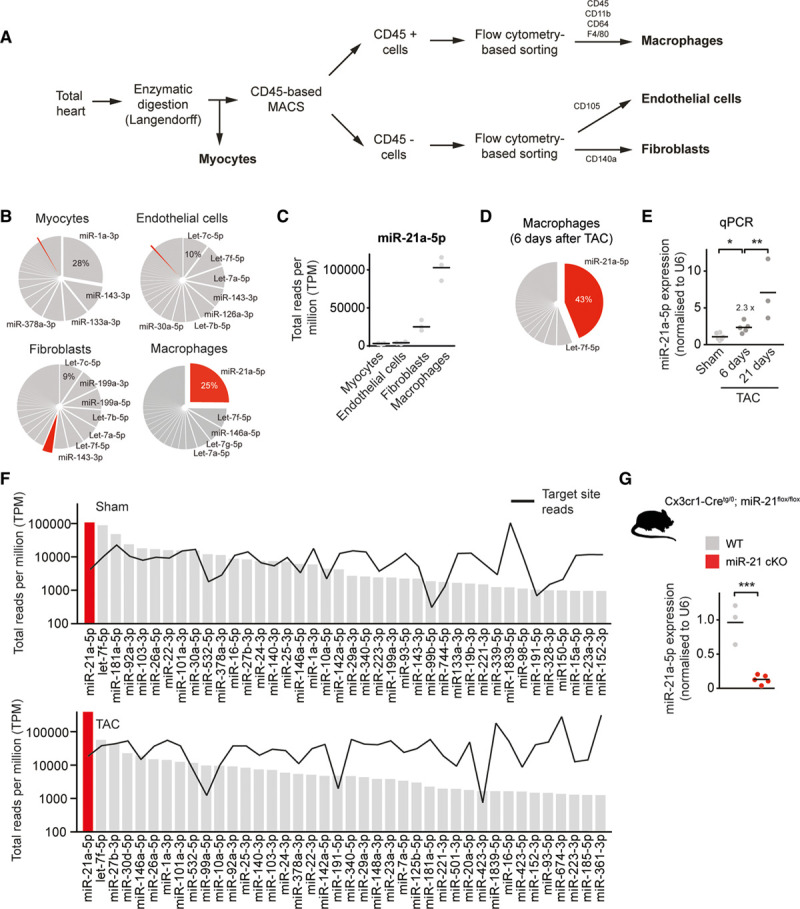
miR-21 is the most abundant and enriched microRNA in cardiac macrophages. A, Scheme for cell isolation. B, MiRNA expression profiles in different myocardial cell types. Pie graph representation of miRNAs in cardiac macrophages and other myocardial cell types (myocytes, fibroblasts, and endothelial cells) isolated from hearts of WT mice. miR-21a-5p is indicated in red. n=3. C, Expression of miR-21a-5p in different myocardial cell types. D, Pie graph representation of miRNAs in cardiac macrophages isolated from hearts of WT mice 6 days after pressure overload (TAC). n=5. E, Relative expression of miR-21a-5p, as determined by SYBR green-based quantitative real-time polymerase chain reaction (qPCR) in cardiac macrophages isolated from hearts 6 or 21 days after pressure overload. Sham n=6; 6 days after TAC n=5; and 21 days after TAC n=3. F, Measured reads of miRNA (bars) and corresponding 3′-untranslated region target pools (black line) in total reads per million (TPM) for the top-expressed miRNAs in cardiac macrophages, isolated from WT mice after sham or TAC surgery. MiR-21 is denoted in red. The y axis is log scale. n=3 per group. G, Macrophage-specific miR-21–deficient mice (miR-21 cKO) were generated by crossing miR-21–floxed mice with a mouse line that expresses Cre recombinase under control of the Cx3cr1 promotor. Diagram shows the expression of miR-21a-5p as determined in macrophages isolated from WT and miR-21 cKO mice. WT n=3; miR-21 cKO n=5. Data denote mean and individual values and were analyzed using 1-way ANOVA (E) or the Student t test (G). *P<0.05, **P<0.01, and ***P<0.001. MACS indicates magnetic-activated cell sorting; miR-21, microRNA-21; miR-21 cKO, macrophage-specific miR-21–deficient mice; miRNA, microRNA; TAC, transverse aortic constriction; and WT, wild type.
To investigate whether macrophage-based miR-21 also played a role in the development of cardiac disease, we tested it in a disease model for left ventricular pressure overload induced by TAC. Expression of miR-21 in cMPs further increased in a progressive manner 6 and 21 days after the initiation of pressure overload (Figure 1D and 1E). Effective target mRNA regulation through a microRNA requires that its concentration in each cell type matches that of its target sites (ie, the entire targetome in a cell).17,18 We therefore determined the absolute abundance of all mRNAs that contain canonical target sites for the top 40 expressed microRNAs and set it in relation to the absolute copy numbers of the respective microRNA (Figure 1F). Only few microRNAs matched their targetome in cMPs, and miR-21 also exerted the highest microRNA:target ratio among all macrophage microRNAs, both under sham and disease conditions, suggesting a strong regulatory effect of this miR-21 in cMPs (Figure 1F). To further examine the role of miR-21 in cMPs in vivo, we generated macrophage-specific miR-21–deficient mice (miR-21 cKO) by crossing miR-21–floxed mice with a mouse line that expresses Cre recombinase under the control of the Cx3cr1 promotor. This promotor is specifically active in macrophages.13 Cx3cr1-Cretg/0; miR-21flox/flox mice (miR-21 cKO) exhibited >90% decreased levels of miR-21 in cMPs, with preserved miR-21 levels in other cardiac cell types, myeloid and lymphoid cells (Figure 1G and Figure II in the Data Supplement). MiR-21 cKO mice developed normally and exhibited normal cardiac function, as determined by echocardiography, compared with wild-type (WT) littermate controls (data not shown). Thus, we conclude that miR-21 cKO mice are a well-suited model to study the function of miR-21 in cMPs.
Genetic Deletion of miR-21 Specifically in MPs Improves Heart Function After TAC
To investigate the contribution of macrophage miR-21 to pathological cardiac remodeling and dysfunction, we subjected WT and miR-21 cKO mice to TAC and determined the extent of fibrosis and cardiac hypertrophy (Figure 2A). Staining of extracellular matrix proteins in myocardial cross sections indicated that fibrosis in the miR-21 cKO group had been significantly prevented, compared with TAC-treated WT mice (Figure 2B). MiR-21 cKO mice were also protected from TAC-induced cardiac hypertrophy at both organ and cellular levels (Figure IIIA in the Data Supplement and Figure 2C). Consistent with these findings, the expression of hypertrophy-related genes (Acta1, Myh7, Nppa) and fibrosis-related genes (Col1a1, Col1a2) was significantly reduced in TAC-treated miR-21 cKO mice, compared with WT littermates (Figure 2D). Immunofluorescent staining indicated a significantly reduced number of macrophages in TAC-treated miR-21 cKO mice compared with TAC-treated WT mice (Figure IIIB in the Data Supplement). However, the number of peripheral blood mononuclear cells remained unchanged (Figure IIIC in the Data Supplement).
Figure 2.
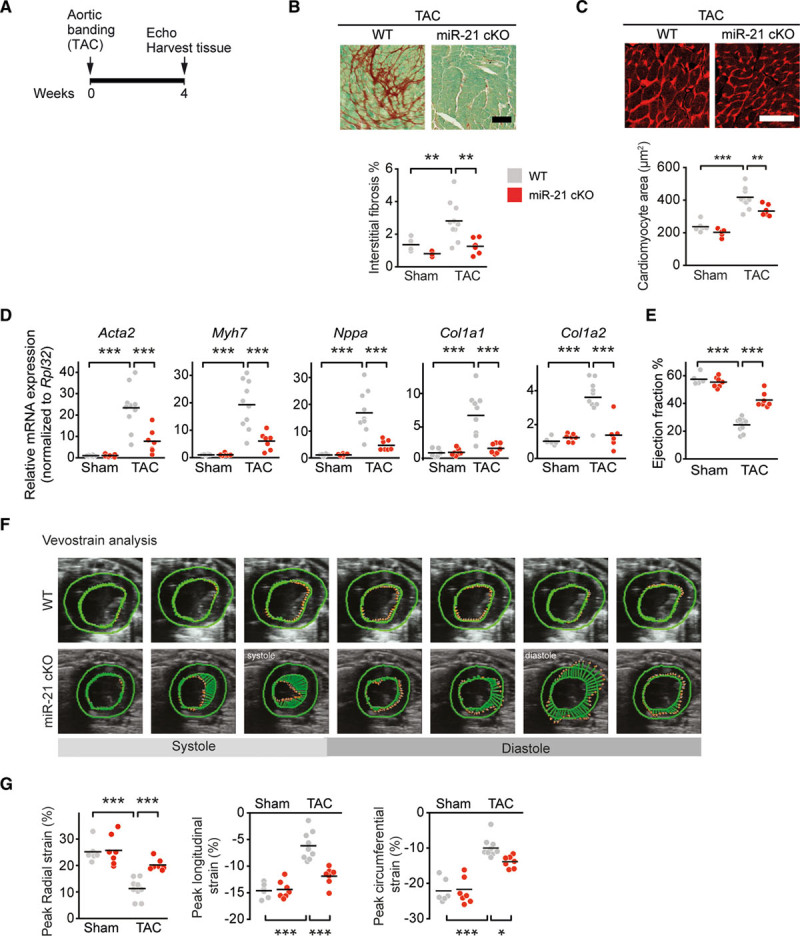
Macrophage-specific deletion of miR-21 prevents pressure overload–induced myocardial remodeling in mice. A, Experimental strategy. Eight-week-old wild-type and miR-21 cKO mice were subjected to either TAC or sham surgery and their hearts were harvested 4 weeks later. Cardiac function was monitored by echocardiography (Echo). B, Sirius red/fast green staining of representative myocardial sections and quantification of extracellular matrix. C, Representative myocardial sections stained with wheat germ agglutinin (WGA) and quantification of cardiac myocyte area. D, Quantitative polymerase chain reaction assessment of mRNA levels of collagens Col1a1 and Col1a2, Acta2, Myh7, and Nppa. E, Echocardiographic determination of cardiac function, as assessed by left ventricular ejection fraction. F, Representative speckle tracings to assess cardiac strain, measured using Vevo strain software at systole and diastole. G, Quantification of peak radial strain (%), peak longitudinal strain (%), and peak circumferential strain (%). WT sham n=6; miR-21 cKO sham n=7; WT TAC n=10; miR-21 cKO TAC n=7. Scale bars in B and C represent 100 µm. Data denote mean and individual values and were analyzed by using 2-way ANOVA with the Sidak posttest (B–E, G). *P<0.05, **P<0.01, and ***P<0.001. miR-21 indicates microRNA-21; miR-21 cKO, macrophage-specific miR-21–deficient mice; TAC, transverse aortic constriction; and WT, wild type.
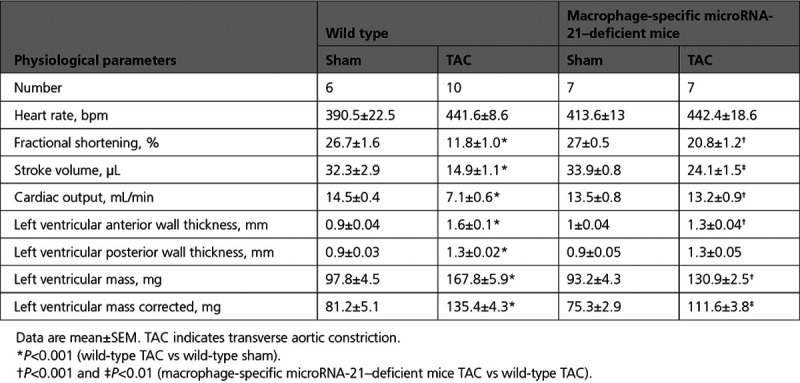
Echocardiography performed before surgery and at days 14 and 28 after sham or TAC surgery was used to assess cardiac function. Both WT and miR-21 cKO mice in the sham groups were healthy and unremarkable, in contrast to the typical impairment of cardiac function that was evident in TAC-treated WT mice (Figure 2E and 2F). TAC-treated miR-21 cKO mice displayed significantly better left ventricular ejection fraction, compared with WT mice (Figure 2E). This was also reflected by other echocardiography parameters such as left ventricular anterior wall thickness in TAC-treated miR-21 cKO mice compared with WT littermates (Table). In addition, we measured left ventricular function by assessing left ventricular strain using speckle-tracking analysis of 2-dimensional echocardiography (Figure 2F). Also, this analysis indicated significantly better left ventricular function with higher levels of radial strain in miR-21 cKO mice, almost reaching levels typically observed in the sham-treated mice (Figure 2G).
Table.
Physiological Parameters in Pressure Overload–Induced Mice After Macrophage-Targeted Deletion of MicroRNA-21
Together, these findings suggest a crucial role for macrophage miR-21 in cardiac remodeling and dysfunction.
Single-Cell Sequencing Analysis Revealed That miR-21 Depletion in Macrophages Favors M2 polarization
To better understand the mechanism through which miR-21 exerts its function in cMPs and how this then determines responses in other myocardial cell fractions, we performed single-cell sequencing by using the nonmyocyte cell population isolated from either WT or miR-21 cKO mice 6 days after pressure overload (Figure 3A). Transcriptional profiles of single cells were obtained using the 10X Chromium platform. We analyzed 10 159 WT Sham, 6230 WT, and 8473 miR-21–deficient cells from TAC hearts that passed quality control and for which an average of ≈2000 median genes per cell were detected (in total, 19 751 genes were detected across cell types; Figure 3B).
Figure 3.
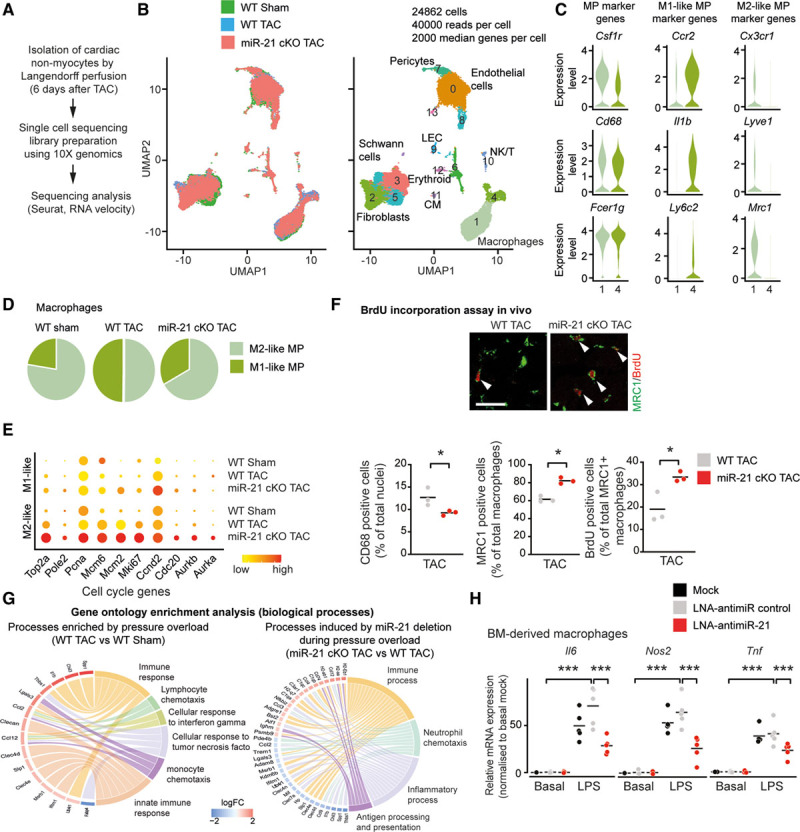
Single-cell sequencing reveals accumulation of M2-like macrophages in the heart after specific deletion of miR-21 in cardiac macrophages. A, Experimental workflow of single-cell sequencing of noncardiomyocyte cells isolated from the hearts of wild-type and miR-21 cKO mice 6 days after TAC. Barcoded cDNA libraries were generated using the 10X Chromium Single Cell 3′-Library kit, sequencing was performed using HiSeq4000 and analyzed using Cell Ranger and Seurat packages. B, Uniform manifold approximation and projection (UMAP) plots for dimensionality reduction of the distribution of wild-type and miR-21 cKO single-cell transcriptomes. C, Violin plots showing expression of marker genes for macrophages, M2-like and M1-like macrophages. D, Pie graphs depicting the proportions of different macrophage clusters. E, Dot plot showing expression of exemplary cell cycle genes. F, Proliferation of cardiac macrophages as assessed by BrdU pulse labeling in wild-type and miR-21 cKO mice 3 days after pressure overload. Top, Representative immunofluorescence staining for MRC1 as a marker for M2-like macrophages and BrdU. Bottom, Quantification of macrophages (left), the relative fraction of M2-like macrophages (middle), and BrdU incorporation into M2-like macrophages (right). n=3 mice per group with >2000 cells assessed per animal. Scale bar, 25 µm. G, Gene ontology enrichment analysis of top 200 deregulated genes on biological processes in cardiac macrophages. Chord diagrams show genes associated with top overrepresented gene ontology terms. H, Expression of proinflammatory genes (Tnf, Il6, Nos2) in bone marrow–derived macrophages, transfected with LNA-anti–miR-21 or LNA-anti–miR control, and incubated with and without lipopolysaccharide (LPS). n=5 independent experiments, with 3 replicates each. Data denote mean and individual values and were analyzed by using Student t test (F) or 2-way ANOVA with Sidak posttest (H). *P<0.5 and ***P<0.001. BM indicates bone marrow; BrdU, 5′-bromo-2′-deoxyuridine; CM, cardiomyocyte; LEC, lymphatic endothelial cell; LNA, locked nucleic acid; miR-21 indicates microRNA-21; miR-21 cKO, macrophage-specific miR-21–deficient mice; MP, macrophage; NK/T, natural killer/T cell; TAC, transverse aortic constriction; and WT, wild type.
Analysis of the single-cell RNA sequencing data set (including cells from both WT and miR-21 cKO mice, pooled together and analyzed by the Seurat package) showed 2 clusters of macrophages (1 and 4), 3 clusters of fibroblasts (2, 3, and 5), 3 clusters of endothelial cells (0, 8, and 13), pericytes (7), lymphatic endothelial cells (9), and other immune cells (10 and 6; Figure 3B and Figure IV in the Data Supplement). Using known genetic markers, subset analysis of macrophages further classified this cell type into 2 different cell populations: M2-like macrophages and M1-like proinflammatory macrophages19–23 (Figure 3C). Comparative sequencing analysis revealed a relative increase in the fraction of M2-like macrophages on deletion of miR-21 in macrophages compared with those isolated from a WT background (Figure 3D). In accordance with this, cell cycle–related genes in M2-polarized macrophages were highly upregulated in miR-21 cKO mice, indicating proliferation of this subpopulation (Figure 3E). To independently assess macrophage subsets and cell cycle activation, we performed pulse labeling with 5′-bromo-2′-deoxyuridine followed by immunostaining. We chose day 3 after TAC where macrophage proliferation has been reported to peak after TAC.24 Consistent with the single-cell sequencing analyses, we found decreased total macrophage numbers, a relative increase in the M2-like fraction and its cell cycle activity in left ventricular myocardium from miR-21 cKO mice compared with controls (Figure 3F). Gene ontology analysis of the macrophage clusters indicates that inflammation-associated genes that had been upregulated in WT mice after pressure overload were largely repressed in miR-21–deficient mice (Figure 3G and Table II in the Data Supplement). These findings were corroborated with corresponding analyses performed separately on individual macrophage subclusters. They confirmed a repression of disease-associated, proinflammatory pathways in M1-like macrophages (cluster 4) on deletion of miR-21 (Figure VA, Upper and Table II in the Data Supplement). Likewise, M2-like macrophages (cluster 1) showed a pronounced reversal of the pressure overload–associated changes on deletion of miR-21 (Figure VA, Lower and Table II in the Data Supplement).
We then aimed to corroborate these findings in a more defined setting in vitro. We assessed the extent of proinflammatory signaling in cultured bone marrow–derived macrophages that had been transfected either with LNA (locked nucleic acid)-anti-miR-control or LNA-anti–miR-21 after stimulation with lipopolysaccharide, a potent proinflammatory stimulus. Consistent with the repression of proinflammatory pathways in the single-cell data obtained from miR-21 cKO mice, the expression of proinflammatory-related genes (Il6, Nos2, Tnf) was significantly reduced in LNA-anti–miR-21–treated compared with LNA-anti–miR-control–treated, bone marrow–derived macrophages (Figure 3H and Figure VI in the Data Supplement).
Altogether, our data suggest that miR-21 is a key molecule necessary for the proinflammatory activation of macrophages in the mammalian heart.
Ligand-Receptor–Pairing Analysis Identifies the cMP as Primary Paracrine Inducer of Fibroblast Activation
Our phenotypic characterization of miR-21 cKO mice (Figure 2) had revealed a prominent effect of macrophage miR-21 on myocardial remodeling during pressure overload, including a significant prevention of myocardial fibrosis (Figure 2B). We therefore asked whether the cMP, on disease induction, would directly signal to cardiac fibroblasts. Recent advances in bioinformatic analysis of single-cell transcriptomic data sets from complex multicellular tissues allow systematic mapping of paracrine intercellular communication based on ligand-receptor pairing.25
We thereby analyzed ≈2500 validated ligand-receptor interaction pairs within and between cell types and created a cellular interaction map, first for homeostatic conditions (Figure VB in the Data Supplement). Next, we sought to identify changes of cellular interaction that occur under TAC conditions, examining all potential ligand-receptor interaction pairs (Figure 4A). On induction of pressure overload, the dominant changes in interaction were observed between M1-like macrophages and activated fibroblasts of the Postn+ subtype (Figure 4A and Figure IV in the Data Supplement). Among the paracrine interactions from M1-like MPs, signaling to Postn+ fibroblasts was most significantly repressed in TAC-treated miR-21–deficient versus TAC-treated WT mice (Figure 4A, Right).
Figure 4.
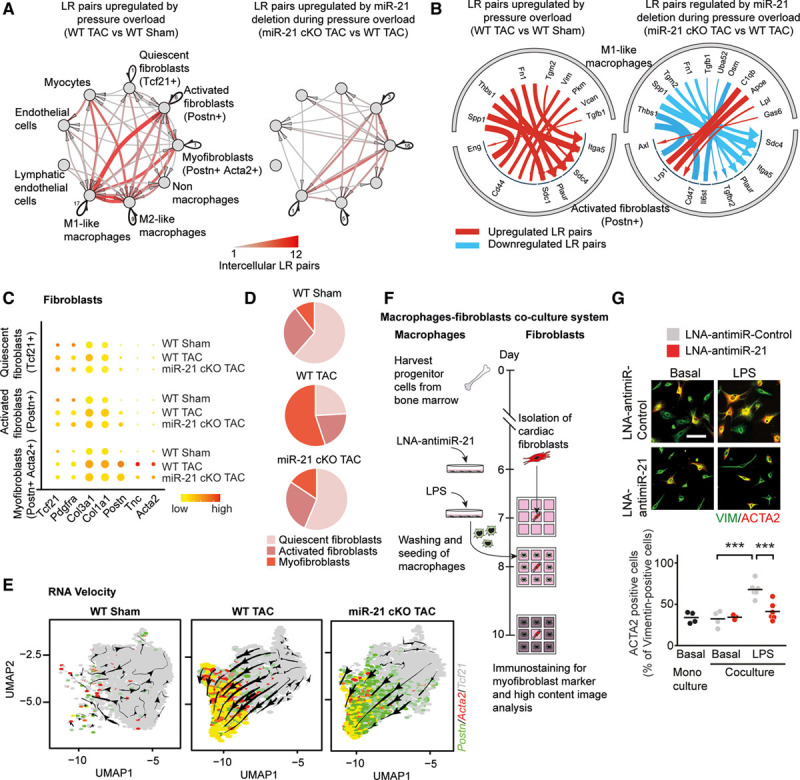
Analysis of ligand-receptor pairing in single-cell transcriptomics data sets identifies the cardiac macrophage as primary paracrine inducer of fibroblast activation. A, Network plot depicting significantly upregulated paracrine and autocrine interactions among different cardiac cell fractions in WT and miR-21 cKO mice. Arrows indicate that the direction of communication, line thickness, and color saturation are proportional to the number of intercellular ligand-receptor pairs; autocrine signaling is depicted in black. Ligands and receptors are considered to be expressed when detected in at least 20% of a cell population. B, Detailed view of paracrine interactions from M1-like macrophages toward activated fibroblast (Postn+) cells depicting the individual ligand-receptor pairs. C, Dot plot showing expression of fibroblast-related genes. D, Pie graphs depicting the proportions of different fibroblast clusters. E, RNA velocity analysis of fibroblast clusters detected by single-cell sequencing. Underlying feature plots label cells that express Tcf21 (gray), Postn (green), Acta2 (red), or both Postn and Acta2 (yellow). F, Experimental strategy for coculture of adult mouse cardiac fibroblasts (AMCF) and bone marrow–derived macrophages. Macrophages were transfected with either LNA-anti–miR-21 or LNA-anti–miR-control, stimulated with LPS, washed and seeded around freshly isolated AMCFs for 48 hours. Monocultures of AMCF served as negative controls. Immunofluorescence staining of AMCFs was performed using antibodies against vimentin (marker for fibroblasts) and α-smooth muscle actin (ACTA2; marker for myofibroblasts). G, Percentage of α-smooth muscle actin–positive cells as a measure of myofibroblast formation. Scale bar, 25 µm. n=4 to 6 independent experiments performed in triplicates. Data are mean and individual values and were analyzed by using 2-way ANOVA with the Sidak posttest. ***P<0.001. LNA indicates locked nucleic acid; LPS, lipopolysaccharide; LR, ligand-receptor; miR-21 indicates microRNA-21; miR-21 cKO, macrophage-specific miR-21–deficient mice; TAC, transverse aortic constriction; UMAP, uniform manifold approximation and projection; and WT, wild type.
The remarkable extent of these perturbations made us hypothesize that it is the activated macrophage that mediates profibrotic activation of the cardiac fibroblast during pressure overload–induced remodeling. We then examined in more detail which ligand-receptor pairs directly promote the fibrotic response in a miR-21–dependent manner (Figure 4B). In TAC-treated WT mice, the most upregulated candidate ligands with predicted receptor signaling to activated fibroblasts were ligands with documented profibrotic activity.26–30 These fibroblast-activating signaling pathways were found to be downregulated in cMPs of TAC-treated miR-21–deficient mice (5 of 8 passed the threshold of regulation applied in Figure 4B; see also Table III in the Data Supplement). Quantitative real-time polymerase chain reaction of RNA isolated from left ventricular myocardium of miR-21 cKO mice and WT littermates subjected to TAC confirmed the reduced expression of the identified ligand genes (Figure VII in the Data Supplement).
We also found 1 signaling pathway to be activated in the latter group, namely Lrp1-dependent signaling (Figure 4B). Lrp1, a member of the low-density lipoprotein receptor family of proteins, has been reported as a potent integrator of diverse antifibrotic signals such as ApoE (apolipoprotein E) and C1qb.31 Together, these data indicate that, on pressure overload of the heart, the cMP sends a powerful array of profibrotic mediators to the cardiac fibroblast.
To study the effects of these signals in the cardiac fibroblast, we assessed the dynamic transcriptional response of this cell population on pressure overload and the relevance of miR-21 therein (Figure 4C–4E). Quantitative analysis of the transcriptomes of the 3 major fibroblast subclusters32 (Figure IV in the Data Supplement) indicated that deletion of macrophage miR-21 specifically repressed myofibroblast transformation (Figure 4C and 4D). RNA velocity analysis of the single-cell sequencing data showed that the fraction of activated fibroblasts (denoted by Postn) was directed toward myofibroblasts (denoted by Acta2 and Tcf21 abundance) on pressure overload (Figure 4E). This transcriptional response toward a prospective myofibroblast phenotype was largely abolished in miR-21 cKO mice (Figure 4E). In good agreement with this, miR-21 deficiency in macrophages leads to fewer myofibroblasts than in the WT background (Figure 4D).
To obtain further proof that macrophage miR-21 is a driver of myofibroblast differentiation, we analyzed this process in a defined coculture setting of activated macrophages and cardiac fibroblasts (Figure 4F). Bone marrow–derived macrophage progenitor cells were differentiated to mature macrophages, transfected with either LNA-anti–miR-21 or LNA-anti–miR-control for 24 hours and stimulated with lipopolysaccharide. The mature macrophages were then added to the surrounding 8 wells of 9-well coculture slides, such that a ratio of fibroblast to macrophages of 3:1 was achieved, closely mimicking cell ratios in myocardium in vivo.10,33 Automated, high-content image acquisition of Acta2-stained cells (Metamorph) was then used as a parameter for myofibroblast formation (Figure 4G). Unstimulated, quiescent macrophages did not induce myofibroblast transformation in this assay, and the inhibition of miR-21 had no detectable effect under these conditions (Figure 4G). In contrast, proinflammatory polarization of cultured macrophages with lipopolysaccharide conditioned their secretome to promote strong myofibroblast transformation. Consistent with the data obtained from the miR-21 cKO mice, macrophage-dependent myofibroblast transformation was significantly reduced on pretreatment of the macrophages with LNA-anti–miR-21(Figure 4G). This finding further corroborates the dependency of cMPs on miR-21 to exert paracrine, profibrotic signaling to cardiac fibroblasts.
Discussion
This study reports on a key regulatory role for miR-21 in cMPs, where it controls paracrine, profibrotic signaling toward cardiac fibroblasts, and thus determines cardiac remodeling and overall cardiac function.
MiR-21 has a well-documented role in tissue fibrosis of the heart,8,10,11 and other organs, as well,34–36 and its therapeutic inhibition showed efficacy in a broad panel of preclinical models of tissue fibrosis. Consequently, a synthetic oligonucleotide inhibitor of miR-21 (RG-012) has been developed that passed phase I clinical testing37 and is currently tested in a phase II clinical study in kidney fibrosis.38 Until recently, the regulatory role of miR-21 in fibroblasts has been regarded as the primary cause underlying the pathological role of miR-21 in tissue fibrosis and disease and, hence, the therapeutic effects of anti–miR-21. Our results of quantitative small RNA sequencing from purified myocardial cell fractions revealed an enormous concentration of miR-21 in the cMP, where it is the single highest expressed microRNA. Also, this cell type displays the highest miR-21 expression among all myocardial cell types. This key finding was the starting point of the present study and prompted us to delineate the contribution of cMP miR-21 to tissue fibrosis and cardiac function.
The effects we report to be mediated by macrophage miR-21 are profound and go far beyond primary immune functions. Four major findings of our study support the central role of cMP miR-21 in the myocardium: (1) Starting from a very high cellular concentration, miR-21 was further rapidly upregulated in cMPs in early stages of disease. (2) Genetic deletion of miR-21 in cMPs inhibited polarization of cMPs toward a proinflammatory (M1-like) phenotype and promoted cell cycle activity of M2-like macrophages. (3) Loss of miR-21 in cMPs prevented myofibroblast transformation, myocardial fibrosis, and eventually cardiac dysfunction in a murine disease model. (4) Mechanistically, miR-21 controls paracrine, profibrotic macrophage-to-fibroblast signaling.
cMP as a Therapeutic Target
Our data provide further evidence that the previously underappreciated cMP exerts a critical role in myocardial disease. Bone marrow, that is, monocyte-derived macrophages and their massive tissue infiltration in conditions such as acute ischemia are a comparably well-studied cell population.39 In contrast, much less is known about the so-called tissue resident macrophage that has come into focus in the past few years.40 Recent studies have suggested that this cell population appears to play a key role in the immunologic response to nonischemic cardiac dysfunction.24 Instrumental to the investigation of these cells were Cre-transgenic mouse lines that permitted lineage tracing during development, but also highly specific targeting of tissue resident macrophages including those in the heart.41,42 In the present study, we used a Cx3cr1-Cre line and achieved a nearly 90% reduction of miR-21 in the cMPs. Such strong inhibition of miR-21 prevented proinflammatory polarization of cMPs during cardiac disease. In agreement with other studies,43 our study suggests therapeutic modulation of cMP polarization as a promising strategy in conditions such as cardiac remodeling observed during the progression of heart failure.44
Remarkably, miR-21 deficiency in cMPs had a strong impact on the activation and myofibroblast transformation of the cardiac fibroblast. Our data suggest that the proinflammatory state of the cMP and its fibroblast-activating secretome are necessary for the activation of cardiac fibroblasts to promote myocardial fibrosis. This notion is supported by several studies that have likewise suggested a critical role for cardiac immune cells for tissue fibrosis.6 One such study has investigated miR-21 and suggested that the macrophage determines fibroblast activation through the secretion of miR-21–containing exosomes.45 To become functionally relevant in the recipient cell, intercellular transfer of a microRNA necessitates a quantitative increase of the microRNA to this cell type. Our model of macrophage-specific genetic deletion allows us to test for this hypothesis by quantitative determination of the concentrations of miR-21 in the fibroblast population on macrophage-specific deletion of miR-21. Under basal conditions, we found the fibroblast concentrations of miR-21 to be preserved, arguing against a major role of exosome-based transfer of miR-21 in this setting. Follow-up studies appear warranted to determine whether this also applies to disease states where exosome transfer may be altered or whether this mechanism is specific to certain organs, such as the fibroblasts in tendon tissue studied by Cui et al.45 In any case, our quantitative miRNome data on miR-21 concentrations in fibroblasts predict that very high numbers of exosome-associated miR-21 molecules need to be received by and operative in recipient fibroblasts to further augment the high endogenous miR-21 concentrations to a functionally relevant extent.
Another study reported on the release of transforming growth factor β1 by platelets and its control by miR-21,46 suggesting that intravenous anti–miR-21 may also act antifibrotic through its action in platelets rather than in myocardial cells. Our study design does not allow us to specifically determine the potential contribution of miR-21 in platelets (where it has rather moderate levels compared with many other cell types), nor did it study conditions of platelet activation such as thrombus formation or include systemic manipulation of miR-21 by anti–miR-21 oligonucleotides as in the study by Barwari et al.46 Nevertheless, our study provides genetic proof that miR-21 in cMPs and its control of polarization is essential for disease-associated fibrosis to occur.
What Are the Consequences of These Findings for the Application and Further Development of Anti–miR-21 Molecules for Therapeutic Purposes?
Our data suggest that, provided sufficient delivery to this cell type can be achieved, the cMP may provide for a superior target cell to aim for cell type–specific delivery. This notion seems particularly relevant in the context of previous studies that indicated a protective effect of miR-21 on cardiac myocytes in addition to its pathological role in cardiac nonmyocyte cell types.10,47 Such delivery to macrophages does not necessarily need to be confined to the myocardium. Rather, our Cx3cr1-Cre–based approach and the mild phenotypic effects of global genetic deletion of miR-21 predict few systemic effects apart from those in the diseased tissue. Furthermore, concomitant inhibition of miR-21 in macrophages and fibroblasts may warrant further investigation. In fibroblasts, miR-21 likewise has high concentrations, and we and others have delineated a pathological role in myofibroblast transformation therein.8,10,48 We are tempted to speculate that such a bispecific approach may further enhance the antifibrotic effect of anti–miR-21.
The use of a cell type–specific, transgenic Cre line requires the interpretation of the pertinent data regarding the entirety of the targeted organs and cells therein. Here, we made use of the Cx3cr1-Cre strain,13 which has become a standard in the field to target tissue macrophages. This model has been shown to target both MHCIIlow and MHCIIhigh macrophages (representing M1- and M2-like macrophages) by Molawi et al.49 However, some activity has also been reported in mast cells and classical dendritic cells,50 both of which are (very) rare cells in the murine myocardium and have not been followed up in this study. Likewise, we cannot formally exclude the involvement of tissue macrophages in other organs besides the heart, yet 2 observations argue against this: first, the transcriptional response of the cMPs occurs early and in the absence of heart failure and overt systemic disease, arguing for a local, intramyocardial activation of the myocardial macrophage. Second, the TAC model does not show fibrosis of major extracardiac organs and we were able to reconstitute macrophage-mediated fibroblast activation with isolated primary cells in vitro, a system that, per design, is devoid of external signals and triggers.
The pivotal finding that miR-21 in cMPs is essential to activate and transform cardiac fibroblasts gives rise to new questions that warrant further studies. As for most heart failure–related cellular pathologies, it remains to be determined what triggers the activation of the cMP in this model of pressure overload–induced cardiac remodeling and failure. Signaling of pathological mechanical strain is an obvious candidate as are danger signals (eg, damage-associated molecular patterns).
Conclusions
In summary, our data demonstrate an important role of macrophage miR-21 for promoting the proinflammatory polarization of this cell type in disease and, subsequently, in cardiac fibrosis and dysfunction. This study also provides the first, definitive evidence that macrophage miR-21 controls myocardial fibrosis through intercellular communication with fibroblasts, the primary recipient of intercellular communications emanating from activated macrophages in the heart. Our data suggest that inhibition of miR-21 in macrophages holds therapeutic promise toward the treatment of fibrotic myocardial disease.
Acknowledgments
The authors thank L. Koblitz for primary cell isolations, J. Kerler and A. Bomhard for performing mouse surgery and echocardiography, S. Brummer for cardiac histology, V. Philippi for supervision of experimental animal studies, K. Eyerich for access to hematology analysis, and B. Laggerbauer for critical reading of the manuscript.
Sources of Funding
This work was supported by the European Union (ERA-CVD MacroERA [01KL1706] to Dr Engelhardt). A.P. Schön was supported by the Otto-Hess-Scholarship of the German Cardiac Society (DGK). Dr Engelhardt is supported by Deutsche Forschungsgemeinschaft (DFG) through TRR267 and the Research Training Group GRK2338 (P09). Dr Schulz is supported by the BMBF (German Ministry of Education and Research; grant 81Z0600204).
Disclosures
Drs Ramanujam and Engelhardt have filed an intellectual property right on the use of miR-21 as a target in fibrotic disease. The other authors report no conflicts.
Supplemental Materials
Expanded Methods
Data Supplement Figures I–VII
Data Supplement Tables I–III
References 51–62
Supplementary Material
Footnotes
D. Ramanujam and A.P. Schön contributed equally.
Sources of Funding, see page 1524
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.120.050682.
Contributor Information
Deepak Ramanujam, Email: deepak.ramanujam@tum.de.
Anna Patricia Schön, Email: patricia.schoen@tum.de.
Christina Beck, Email: christina.beck92@tum.de.
Paula Vaccarello, Email: paula.vaccarello@tum.de.
Giulia Felician, Email: giulia.felician@tum.de.
Anne Dueck, Email: anne.dueck@tum.de.
Dena Esfandyari, Email: dena.esfandyari@tum.de.
Gunter Meister, Email: gunter.meister@vkl.uni-regensburg.de.
Thomas Meitinger, Email: thomas.meitinger@mri.tum.de.
Christian Schulz, Email: Christian.schulz@med.uni-muenchen.de.
References
- 1.Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, et al. Global, regional, and national Burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25 10.1016/j.jacc.2017.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pellicori P, Khan MJI, Graham FJ, Cleland JGF. New perspectives and future directions in the treatment of heart failure. Heart Fail Rev.. 2020;25:147–159. doi: 10.1007/s10741-019-09829-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res. 2016;119:159–176. doi: 10.1161/CIRCRESAHA.116.308030 [DOI] [PubMed] [Google Scholar]
- 4.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. doi: 10.1038/nri3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462 10.1016/j.physbeh.2017.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511 [DOI] [PubMed] [Google Scholar]
- 9.Thum T, Chau N, Bhat B, Gupta SK, Linsley SP, Bauersachs J, Engelhardt S. Comparison of different miR-21 inhibitor chemistries in a cardiac disease model. J Clin Invest. 2011;121:21–22 10.1172/JCI45938.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramanujam D, Sassi Y, Laggerbauer B, Engelhardt S. Viral vector-based targeting of miR-21 in cardiac nonmyocyte cells reduces pathologic remodeling of the heart. Mol Ther. 2016;24:1939–1948. doi: 10.1038/mt.2016.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C, Dueck A, Thum T, Laugwitz KL, Maegdefessel L, et al. AntimiR-21 prevents myocardial dysfunction in a pig model of ischemia/reperfusion injury. J Am Coll Cardiol. 2020;75:1788–1800. doi: 10.1016/j.jacc.2020.02.041 [DOI] [PubMed] [Google Scholar]
- 12.Liu RH, Ning B, Ma XE, Gong WM, Jia TH. Regulatory roles of microRNA-21 in fibrosis through interaction with diverse pathways (review). Mol Med Rep. 2016;13:2359–2366. doi: 10.3892/mmr.2016.4834 [DOI] [PubMed] [Google Scholar]
- 13.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest. 2010;120:3912–3916. doi: 10.1172/JCI43604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci U S A. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wessels HH, Lebedeva S, Hirsekorn A, Wurmus R, Akalin A, Mukherjee N, Ohler U. Global identification of functional microRNA-mRNA interactions in Drosophila. Nat Commun. 2019;10:1626. doi: 10.1038/s41467-019-09586-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, Baccarini A, Lazzari G, Galli C, Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372 [DOI] [PubMed] [Google Scholar]
- 18.Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell. 2016;56:347–359 10.1016/j.molcel.2014.09.018.Endogenous [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity.. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi: 10.3389/fimmu.2019.01084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8 10.7554/elife.43882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberger T, Esfandyari D, Messerer D, Percin G, Schleifer C, Thaler R, Liu L, Stremmel C, Schneider V, Vagnozzi RJ, et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat Commun.. 2020;11:4549. doi: 10.1038/s41467-020-18287-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, Alaiti MA, Sweet DR, Zhou L, Qing Y, Gerson SL, et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc Natl Acad Sci U S A. 2018;115:E4661–E4669. doi: 10.1073/pnas.1720065115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Kloppman E, Lizio M, Satagopam VP, Itoh M, Kawaji H, Carninci P, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun. 2015;6:7866. doi: 10.1038/ncomms8866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirk JA, Cingolani OH. Thrombospondins in the transition from myocardial infarction to heart failure. J Mol Cell Cardiol. 2016;90:102–110. doi: 10.1016/j.yjmcc.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner JUG, Dimmeler S. Cellular cross-talks in the diseased and aging heart. J Mol Cell Cardiol. 2020;138:136–146. doi: 10.1016/j.yjmcc.2019.11.152 [DOI] [PubMed] [Google Scholar]
- 28.Shinde AV, Frangogiannis NG. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ. 2018;25:453–456. doi: 10.1038/s41418-017-0028-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898 [DOI] [PubMed] [Google Scholar]
- 30.Wight TN, Kang I, Merrilees MJ. Versican and the control of inflammation. Matrix Biol. 2014;35:152–161. doi: 10.1016/j.matbio.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potere N, Del Buono M, Niccoli G, Crea F, Toldo S, Abbate A. Developing LRP1 agonists into a therapeutic strategy in acute myocardial infarction. Int J Mol Sci. 2019;20:544. doi:10.3390/ijms20030544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol.. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Jiao J, Cermelli S, Muir K, Jung KH, Zou R, Rashid A, Gagea M, Zabludoff S, Kalluri R, et al. miR-21 inhibition reduces liver fibrosis and prevents tumor development by inducing apoptosis of CD24+ progenitor cells. Cancer Res. 2015;75:1859–1867. doi: 10.1158/0008-5472.CAN-14-1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, Li J, Tran PT, Kaimal V, Huang X, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4:121ra18. doi: 10.1126/scitranslmed.3003205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ClinicalTrials.gov. Unique identifier: NCT02855268. Study of lademirsen (SAR339375) injections in patients with Alport syndrome (HERA). Accessed June 20, 2020. https://clinicaltrials.gov/ct2/show/NCT02855268
- 38.ClinicalTrials.gov. Unique identifier: NCT03373786. A study of RG-012 in subjects with Alport syndrome. Accessed June 20, 2020. https://clinicaltrials.gov/ct2/show/NCT03373786
- 39.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124:263–278. doi: 10.1161/CIRCRESAHA.118.314028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N, Kovacic JC. The macrophage in cardiac homeostasis and disease: JACC Macrophage in CVD Series (Part 4). J Am Coll Cardiol. 2018;72:2213–2230. doi: 10.1016/j.jacc.2018.08.2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- 42.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L, Wijnands E, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. 2013;128:1420–1432. doi: 10.1161/CIRCULATIONAHA.112.001357 [DOI] [PubMed] [Google Scholar]
- 44.Ardura JA, Rackov G, Izquierdo E, Alonso V, Gortazar AR, Escribese MM. Targeting macrophages: friends or foes in disease? Front Pharmacol. 2019;10:1255. doi: 10.3389/fphar.2019.01255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui H, He Y, Chen S, Zhang D, Yu Y, Fan C. Macrophage-derived miRNA-containing exosomes induce peritendinous fibrosis after tendon injury through the miR-21-5p/Smad7 pathway. Mol Ther Nucleic Acids. 2019;14:114–130. doi: 10.1016/j.omtn.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barwari T, Eminaga S, Mayr U, Lu R, Armstrong PC, Chan M V., Sahraei M, Fernández-Fuertes M, Moreau T, Barallobre-Barreiro J, et al. Inhibition of profibrotic microRNA-21 affects platelets and their releasate. JCI Insight. 2018;3:e123335. doi: 10.1172/jci.insight.123335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem.. 2010;285:20281–20290. doi: 10.1074/jbc.M110.109207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211:2151–2158. doi: 10.1084/jem.20140639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakajima H, Nakajima HO, Salcher O, Dittiè AS, Dembowsky K, Jing S, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–579. doi: 10.1161/01.res.86.5.571 [DOI] [PubMed] [Google Scholar]
- 52.Ganesan J, Ramanujam D, Sassi Y, Ahles A, Jentzsch C, Werfel S, Leierseder S, Loyer X, Giacca M, Zentilin L, et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation. 2013;127:2097–2106. doi: 10.1161/CIRCULATIONAHA.112.000882 [DOI] [PubMed] [Google Scholar]
- 53.Hauptmann J, Schraivogel D, Bruckmann A, Manickavel S, Jakob L, Eichner N, Pfaff J, Urban M, Sprunck S, Hafner M, et al. Biochemical isolation of Argonaute protein complexes by Ago-APP. Proc Natl Acad Sci U S A. 2015;112:11841–11845. doi: 10.1073/pnas.1506116112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Wang R, Zhang S, Song S, Jiang C, Han G, Wang M, Ajani J, Futreal A, Wang L. iTALK: an R package to characterize and illustrate intercellular communication. BioRxiv. 2019:507871. 10.1101/507871 [DOI] [Google Scholar]
- 60.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3 [DOI] [PubMed] [Google Scholar]
- 61.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.