

ABSTRACT
The physiological AKT-MTORC1 and AMPK signaling pathways are considered key nodes in the regulation of anabolism-catabolism, and particularly of macroautophagy/autophagy. Indeed, it is reported that these are altered processes in neurodegenerative proteinopathies such as Alzheimer disease (AD), mainly characterized by deposits of β-amyloid (Aβ) and hyperphosphorylated MAPT. These accumulations disrupt the optimal neuronal proteostasis, and hence, the recovery/enhancement of autophagy has been proposed as a therapeutic approach against these proteinopathies. The purpose of the present study was to characterize the modulation of autophagy by MTORC1 and AMPK signaling pathways in the highly specialized neurons, as well as their repercussions on Aβ production. Using a double transgenic mice model of AD, we demonstrated that MTORC1 inhibition, either in vivo or ex vivo (primary neuronal cultures), was able to reduce amyloid secretion through moderate autophagy induction in neurons. The pharmacological prevention of autophagy in neurons augmented the Aβ secretion and reversed the effect of rapamycin, confirming the anti-amyloidogenic effects of autophagy in neurons. Inhibition of AMPK with compound C generated the expected decrease in autophagy induction, though surprisingly did not increase the Aβ secretion. In contrast, increased activity of AMPK with metformin, AICAR, 2DG, or by gene overexpression did not enhance autophagy but had different effects on Aβ secretion: whereas metformin and 2DG diminished the secreted Aβ levels, AICAR and PRKAA1/AMPK gene overexpression increased them. We conclude that AMPK has a significantly different role in primary neurons than in other reported cells, lacking a direct effect on autophagy-dependent amyloidosis.
Abbreviations: 2DG: 2-deoxy-D-glucose; Aβ: β-amyloid; ACACA: acetyl-CoA carboxylase alpha; ACTB: actin beta; AD: Alzheimer disease; AICAR: 5-aminoimidazole-4-carboxamide-1-β-riboside; AKT: AKT kinases group (AKT1 [AKT serine/threonine kinase 1], AKT2 and AKT3); AMPK: adenosine 5‘-monophosphate (AMP)-activated protein kinase; APP: amyloid beta precursor protein; APP/PSEN1: B6.Cg-Tg (APPSwe, PSEN1dE9) 85Dbo/J; ATG: autophagy related; ATP: adenosine triphosphate; BafA1: bafilomycin A1; CA: constitutively active; CGN: cerebellar granule neuron; CoC/compound C: dorsommorphin dihydrochloride; ELISA: enzyme-linked immunosorbent assay; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; Gmax: GlutaMAX™; IN1: PIK3C3/VPS34-IN1; KI: kinase-inactive; MAP1LC3B/LC3: microtubule associated protein 1 light chain 3; MAPT/TAU: microtubule associated protein tau; Metf: metformin; MRT: MRT68921; MTORC1: mechanistic target of rapamycin kinase complex 1; NBR1: NBR1 autophagy cargo receptor; PRKAA: 5ʹ-AMP-activated protein kinase catalytic subunit alpha; PtdIns3K: phosphatidylinositol 3-kinase; Rapa: rapamycin; RPS6KB1/S6K: ribosomal protein S6 (RPS6) kinase polypeptide 1; SCR: scramble; SQSTM1/p62: sequestosome 1; ULK1/2: unc-51 like autophagy activating kinase 1/2; WT: wild type.
KEYWORDS: 2-deoxy-D-glucose, aicar, alzheimer, amyloid accumulation, bafilomycin A1, cultured cerebellar granule neuron, dorsomorphin, metformin, rapamycin, sh-SY5Y
Introduction
The aging process is a complex, progressive decline of physiological functions with several involved molecular mechanisms, constituting a primary risk factor for many diseases. Aberrant accumulation of proteins is a character of many age-associated neuropathologies, such as Alzheimer (AD), Parkinson, Huntington, and prion diseases, and falls into the category of proteinopathies [1]. These pathologic conditions are at least in part the result of a disruption in the regulation of proteostasis, the maintenance of concentration, conformation, and location of every member of the proteome needed for their correct function and proper cellular physiology [2]. Its preservation depends on a delicate balance between all the mechanisms involved in the synthesis, folding, traffic, and degradation of proteins.
Neurons are especially sensitive to the correct maintenance of proteostasis. As post-mitotic cells, neurons cannot dilute harmful protein aggregates or dysfunctional organelles through cellular division [3]. In addition, their polarized and excitable character, with dendrites and axons spanning long distances, require compartmentalization of proteostatic mechanisms and their associated high energy demands to a specific location. Thus, the proper elimination of toxic by-products and recycling activity of the lysosome is crucial to prevent pathological processes. Within degradation systems, autophagy plays an essential role in maintaining physiological conditions in neurons. In particular, its dysfunction is associated with the abnormal protein polymerization/aggregation observed in age-related neurodegenerative diseases [4], such as β-amyloid and MAPT (microtubule associated protein tau) in AD, HTT (huntingtin) in Huntington, or SNCA/synuclein in Parkinson [5–7]. Indeed, several studies have shown that autophagy deficits occur in the early stages of AD [8–11].
Autophagy is the catabolic mechanism used to degrade unnecessary or dysfunctional cellular components, through the action of lysosomes in animal cells, or vacuoles in plants and yeasts [12,13]. Three main subtypes have been described to date: macroautophagy, chaperone-mediated autophagy, and microautophagy (for a review see [14,15]). Among them, macroautophagy (hereafter referred to simply as autophagy) is the best characterized in the nervous system [16,17]. It consists of the formation of a phagophore, sequestering part of the cytoplasm in a double-membraned autophagic vesicle or autophagosome, for subsequent fusion with the lysosome and degradation of its content. The dynamic process of autophagosome generation and degradation by fusion with the lysosome has been termed autophagic flux (for a review see [3,18]). Briefly, in mammalian cells, initiation is carried out by activation of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex by the ULK1/2 (unc-51 like autophagy activating kinase 1/2) complex, leading to primary nucleation of the membrane and recruitment of many ATG (autophagy related) proteins. Membrane elongation then occurs under the direction of two ubiquitin-like conjugation systems, ATG12–ATG5 and MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3). ATG12 binds ATG5 and ATG16L1 to facilitate the recruitment and lipidation of the cytosolic form of LC3 (LC3-I) with phosphatidylethanolamine to become the phagophore-specific and autophagosome membrane-associated form LC3-II [18–20]. Cytoplasmic substrates are recognized by autophagic receptors, such as SQSTM1/p62 (sequestosome 1) or NBR1 (NBR1 autophagy cargo receptor), which also recognize the LC3-II protein [21,22]. Finally, the autophagosome fuses its outer membrane with a lysosome, forming a single membrane autolysosome, whose contents are degraded by acidic lysosomal hydrolases, restoring the lysosome.
Of the several reported physiological signaling pathways involved in the regulation of autophagy, two central nodes in the regulation of the anabolism-catabolism balance stand out: the AKT-mechanistic target of rapamycin kinase complex 1 (MTORC1) and AMP-activated protein kinase (AMPK) pathways. Both integrate several metabolic signals such as the availability of nutrients (amino acids and growth factors via AKT-MTORC1) or energetic status (defined by oxygen and glucose levels via [AMP][ADP]/[ATP]-AMPK; for a review see [23,24]). MTORC1 prevents autophagy through inhibitory phosphorylation of ULK1/2 and class III PtdIns3K complexes, but also of the TFEB (transcription factor EB), which is involved in the synthesis of ATG and lysosomal proteins (for a review see [25]). Thus, the principal strategy for experimental induction of autophagy is inhibition of MTORC1 activity, by nutrient deprivation or the use of compounds such as rapamycin [25–27]. A common indicator of MTORC1 activity is the phosphorylation of its substrate RPS6KB1/S6K (ribosomal protein S6 kinase, polypeptide 1) on Thr389, which in turn phosphorylates RPS6 on Ser240/244. AMPK plays a dual role in the induction of autophagy, as it switches on the ULK1/2 and class III PtdIns3K complexes but also inhibits MTORC1 activity [28,29]. Phosphorylation on Thr172 of the subunit PRKAA (5ʹ-AMP-activated protein kinase catalytic subunit alpha) of AMPK is usually employed as a positive marker of its activity, as well as those of its targets, including ACACA (acetyl-CoA carboxylase alpha) on Ser79. In AD, there are reports of altered MTORC1 and AMPK activity levels, thus disrupting the optimal cellular response to metabolic and stress insults, and worsening neuronal proteostasis.
Therefore, in the present study, we analyzed whether the trigger of autophagy, either by MTORC1 inhibition or AMPK activation, could similarly control the pathological accumulation of β-amyloid. For this purpose, we used double-transgenic APP/PSEN1 mice, which accumulate human Aβ1-40 and Aβ1-42 proteins in a time-dependent fashion similarly in cortex and cerebellum [30]. Thus, we used primary cerebellar granule neurons (CGNs) from APP/PSEN1 mice, as they constitute a highly pure and homogeneous neuronal pool.
We demonstrated that MTORC1 inhibition, either in vivo or ex vivo (primary neuronal cultures), was able to reduce amyloid secretion through moderate autophagy induction in neurons. In contrast, activation of AMPK by several methods (pharmacologically or by overexpression) did not generate a major contribution to autophagy and consequently did not reduce amyloid production. Thus we conclude that AMPK has a more complex modulation of autophagy in neuronal systems, lacking a direct effect on amyloidosis.
Results
Rapamycin reduces β-amyloid levels in mice of a double transgenic AD model
First, we did a comparative analysis (wild type vs. APP/PSEN1) of two autophagy markers (LC3-II and SQSTM1) and studied MTORC1 activity using different phospho-epitopes of RPS6KB1 and the subsequent phosphorylation of its substrate, RPS6. Protein assays of the cerebral cortex from 9-month-old mice by western blot showed that SQSTM1 levels were significantly higher in APP/PSEN1 mice, whereas LC3-II variations were not statistically significant (Figure 1A). Additionally, APP/PSEN1 mouse samples showed increased p-RPS6 levels, and apparent higher levels of p-RPS6KB1 (non-significant), when compared to those of wild type littermates (Figure 1B). These results are in accordance with previous data that described hyperactivation of MTORC1 and the accumulation of autophagic markers in AD [31,32].
Figure 1.
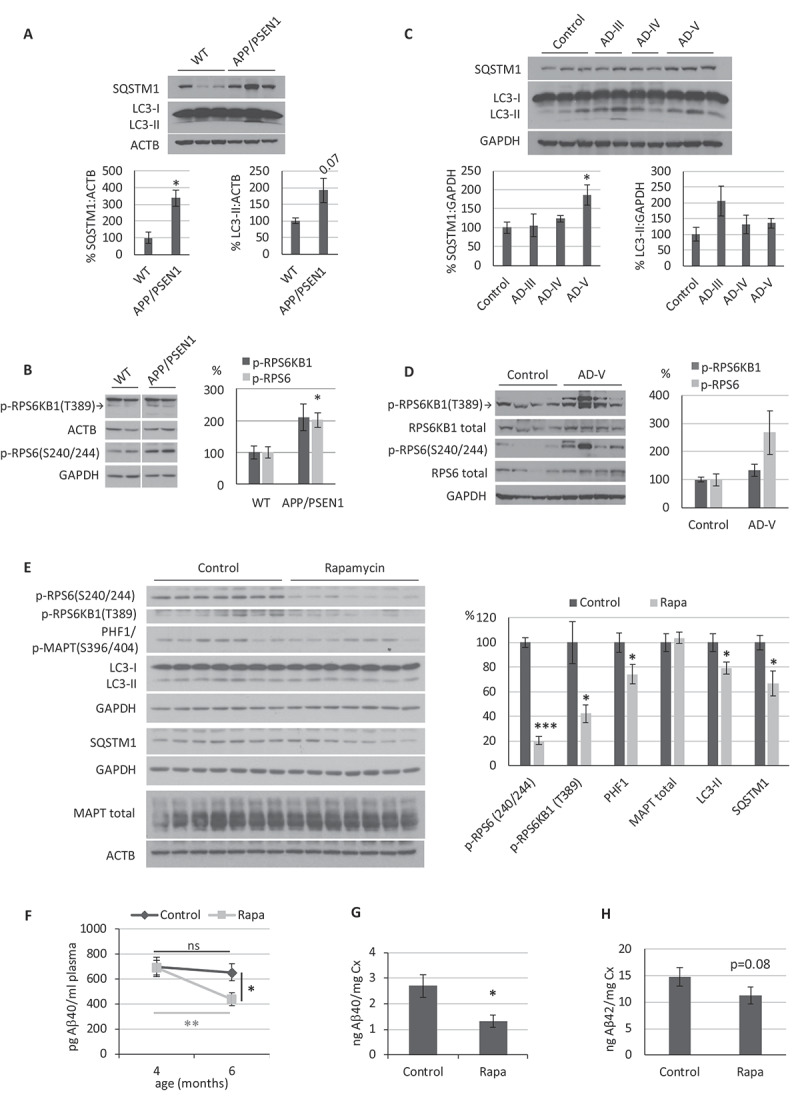
Role of autophagy in the APP/PSEN1 Alzheimer disease (AD) mouse model. (A-D) Western blot analysis of the autophagic markers SQSTM1, LC3 and MTORC1 activity (p-RPS6KB1 T389 and p-RPS6 S240/244) from (A-B) brain cortical samples obtained from 9- month-old APP/PSEN1 and WT mice (n = 3), and from (C-D) hippocampal tissue obtained post mortem from control or Braak stages III, IV and V AD patients (n = 3). (E-H) Analysis of APP/PSEN1 mice intraperitoneally injected with rapamycin every 48 h for 2 months (n = 8 mice treated with vehicle, 9 with rapamycin). (E) Western blot analysis of the autophagic markers SQSTM1 and LC3, MTORC1 activity (p-RPS6KB1 T389 and p-RPS6 S240/244) and MAPT (PHF1: p-MAPT S396/404; MAPT total) in the cerebral cortex. (F) Aβ40 blood plasma levels in samples were measured at the beginning and end of the experiment, (G) Aβ40 and (H) Aβ42 levels in cerebral cortex samples were determined by ELISA. Student’s t-test was performed (ns, non-significant; * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001). Bars represent mean ± standard error of the mean (SEM)
Next, we analyzed hippocampal tissue samples obtained from postmortem brains of AD patients with Braak stages III, IV, and V. Our data showed that AD brains, particularly in samples from Braak stage V, had an increase of SQSTM1 and apparent higher levels of p-RPS6 and LC3-II (non-significant) that were similar to those observed in APP/PSEN1 mice (Figure 1C and D). Thus, these data confirm the reproduction of autophagy-related AD pathology in our APP/PSEN1 mouse model.
To check if the enhancement of autophagic status by MTORC1 inhibition could have a therapeutic effect on our APP/PSEN1 mouse model, we chose the previously described inhibitor of MTORC1 rapamycin. Based on previous studies [33–37], we performed some pilot experiments (data not shown), and we determined an optimal dose of 5 mg/kg rapamycin each 48 h. We treated APP/PSEN1 mice for 2 months, starting at 4 months-of-age since β-amyloid levels exponentially increase and begin to accumulate in APP/PSEN1 mouse brains during this period [38]. Finally, we took blood samples at the beginning and end of the experiment and sacrificed the mice to harvest the brain tissue.
We analyzed cerebral cortex samples by western blot to check that rapamycin was efficiently passing the blood-brain barrier and inhibiting MTORC1 after 2 months of treatment. It was confirmed by lower levels of phosphorylation in the targets p-RPS6KB1(T389) and p-RPS6(S240/244) in rapamycin-treated mice, when compared with APP/PSEN1 mice treated only with vehicle (Figure 1E). Rapamycin treatment led to a reduction in LC3-II, which may be due to an improvement in autophagic flux and degradation rate, as suggested by lowered SQSTM1 levels (Figure 1E). Considering that most of these proteins (SQSTM1, NBR1, or LC3) are not exclusively present in neurons, we decided to use a particularly abundant protein in neurons, such as MAPT/TAU, as an internal control for the effect of rapamycin specifically in neurons. Besides, the phosphorylation of MAPT is increased in AD patients and some amyloidosis mouse models, correlating with the accumulation of amyloid peptide and the degeneration process [30,39,40]. We observed that MAPT-specific phospho-epitope PHF1 showed a lower phosphorylation level in mice treated with rapamycin, without modifying total MAPT levels, which strongly suggests that rapamycin was indeed reaching the neurons (Figure 1E).
Next, we determined the Aβ40 levels in blood plasma by a human-specific Aβ40 (h-Aβ40) ELISA, using samples collected at the beginning and the end of the experiment. The data showed a significant reduction of h-Aβ40 in the blood of APP/PSEN1 mice treated with rapamycin, when compared to the APP/PSEN1 mice treated with vehicle, at the same time point (Figure 1F). We obtained non-significant differences for Aβ42 levels (data not shown). We analyzed the cerebral cortex samples by h-Aβ40-specific ELISA to confirm the observed peripheral anti-amyloid effect of rapamycin. APP/PSEN1 mice treated with rapamycin showed significantly decreased amyloid levels in the brain compared to the APP/PSEN1 mice treated with vehicle (Figure 1G). An apparent decrease was also observed for Aβ42 levels, although it was not significant (p = 0.08) (Figure 1H). Cerebellum samples were also analyzed by h-Aβ40- and h-Aβ42-specific ELISAs, although we observed non-significant differences between groups (Fig. S1).
These results support the initial hypothesis that the MTORC1 inhibitor rapamycin has a therapeutic anti-amyloidogenic effect in our AD mouse model. An increased degradative efficiency, denoted by the lower levels of SQSTM1 and LC3-II (Figure 1E), suggests that the mechanism of action may be autophagy induction and elimination of β-amyloid peptide, even though these proteins are not exclusively present in neurons.
Rapamycin reduces Aβ40 secretion in primary neurons isolated from APP/PSEN1 mice via MTORC1-dependent induction of autophagy
We used cultured cerebellar granule neurons (CGNs) obtained from APP/PSEN1 mice to verify whether the anti-amyloidogenic effect of rapamycin takes place in neurons. Then, we treated them with rapamycin and measured h-Aβ40 secreted to the culture supernatant by ELISA. Our data showed a statistically significant reduction of secreted Aβ40 after 48 h of rapamycin treatment (Figure 2A). In a similar experiment, we analyzed cellular extracts of CGNs from wild type and APP/PSEN1 mice by western blot. Rapamycin efficiently inhibited MTORC1 after 48 h of treatment, downregulating its phosphorylation targets p-RPS6KB1 (T389) and p-RPS6 (S240/244), both in wild type and APP/PSEN1 CGNs (Figure 2B). Observed higher levels of LC3-II and the decrease in autophagy receptors NBR1 and SQSTM1 (Figure 2B) support a higher rate of autophagic degradation in both neuronal genotypes. In contrast, rapamycin treatment did not diminish the levels of APP and BACE1 (Figure 2B). Thus, we hypothesized that in the case of rapamycin-treated CGNs from APP/PSEN1 mice, the lower Aβ40 levels were not due to a reduction in its synthesis, but rather to an increase in autophagic degradation rate, as we confirm in the next two sections.
Figure 2.
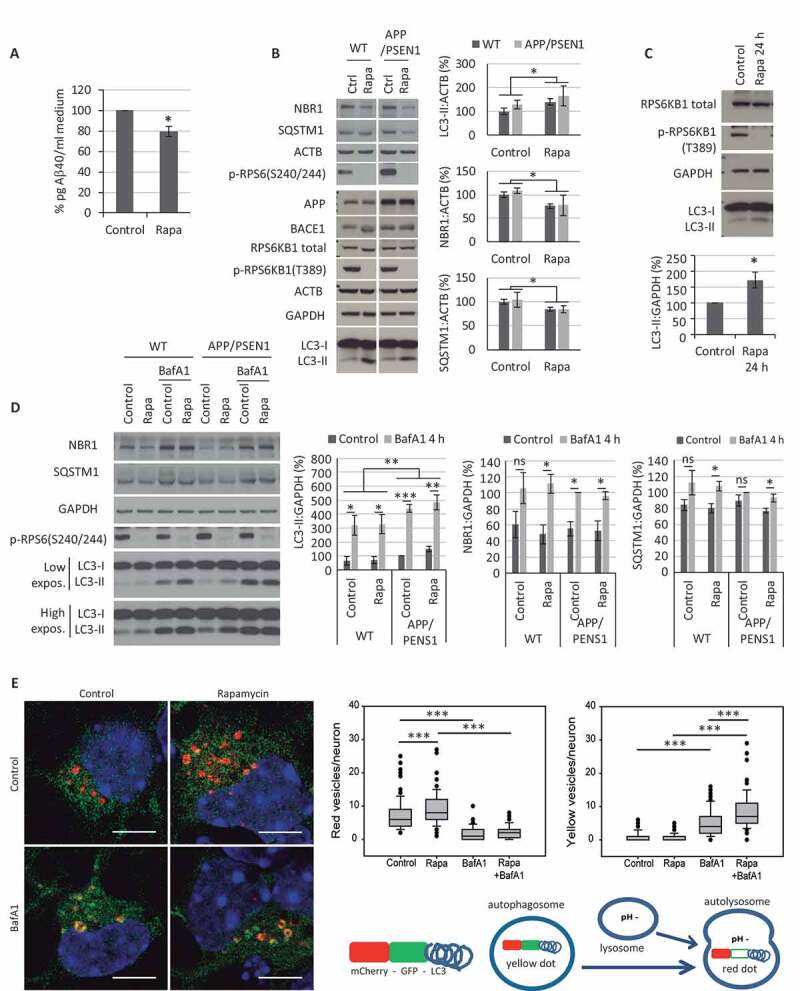
Effect of rapamycin treatment on cerebellar granule neuron (CGN) cultures. (A-B) Effect of 48 h treatment with 200 nM rapamycin (Rapa) in CGNs obtained from APP/PSEN1 mice. (A) Aβ40 levels detected in culture media by ELISA. Student’s t-test (n = 3). (B) Western blot analysis of the autophagic markers NBR1, SQSTM1, and LC3, MTORC1 activity (p-RPS6KB1 T389 and p-RPS6 S240/244), and the amyloidogenic pathway (APP and BACE1) from cellular extracts compared to WT CGN cultures. Two-way ANOVA (n = 3). (C) Western blot analysis of LC3-II levels after 24 h of rapamycin treatment. Student’s t-test (n = 4). (D) Autophagic flux analysis by addition of bafilomycin A1 (BafA1; 100 nM) during the last 4 h of the 24 h rapamycin treatment, and quantification of autophagic markers LC3-II, NBR1 and SQSTM1. Three-way ANOVA (n = 4). Two exposure intensities, low and high, are shown for LC3. (E) CGNs were infected 2 d after plating with lentiviral particles to express a tandem mCherry-GFP tagged LC3 construct. The infected cells were incubated with 200 nM rapamycin for 24 h and/or 10 nM BafA1 for 4 h. Nuclei were stained with DAPI. Scale bar: 5 µm. Formations of mCherry-GFP-LC3 dots (yellow and red) were quantified (100 neurons per experimental condition). Two-way ANOVAs were performed for red (left graph) and yellow (right graph) dots separately. The final scheme represents the color code of each compartment in basal conditions. ns, non-significant; * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Bars represent mean ± SEM (A-D). Box plots represent 10th, 25th, 50th, 75th and 90th percentiles as boxes and error bars, and outliers as dots (E)
Inhibition of MTORC1 by rapamycin slightly increases the autophagic flux in neuronal systems
We proceeded to analyze the autophagic response to rapamycin in neurons further. To estimate the status of autophagic flux, both in constitutive (control) and induced (rapamycin-treated) autophagy, we compared levels of autophagy-specific markers in the presence and absence of the v-ATPase inhibitor BafA1, which blocks lysosomal degradation [26,41,42]. To diminish the off-target effects of sustained partial inhibition of MTORC2 [43], shown by lower levels of p-AKT(S473) (Fig. S2A), we reduced the duration of rapamycin treatment from 48 to 24 h, a time when rapamycin was still able to inhibit MTORC1 and increase the LC3-II levels (Figure 2C). Due to the toxicity of long-term administration of BafA1 (Fig. S2B), it was added only for the final 4 h before harvesting the neurons. In parallel, CGNs from wild type and APP/PSEN1 mice were treated with 200 nM rapamycin for 24 h, and 100 nM BafA1 was added to the culture for the last 4 h. We monitored the marker p-RPS6 (S240/244) as control of rapamycin inhibitory efficiency (Figure 2D). The accumulation of LC3-II, NBR1, and SQSTM1 in the presence of BafA1 confirmed the existence of a proper autophagic flux in neurons from both WT and APP/PSEN1 mice. Also, it corroborated the modulation of NBR1 and SQSTM1 levels by this process (Figure 2D). However, only SQSTM1 showed accumulation in the presence of BafA1 significantly higher when treating the neurons with rapamycin, in both WT and APP/PS1 cultures (Figure 2D). The moderate increase of autophagy with rapamycin was more evident with this technique in the following experiments (Fig. 3A-D and 4A-E).
Figure 3.
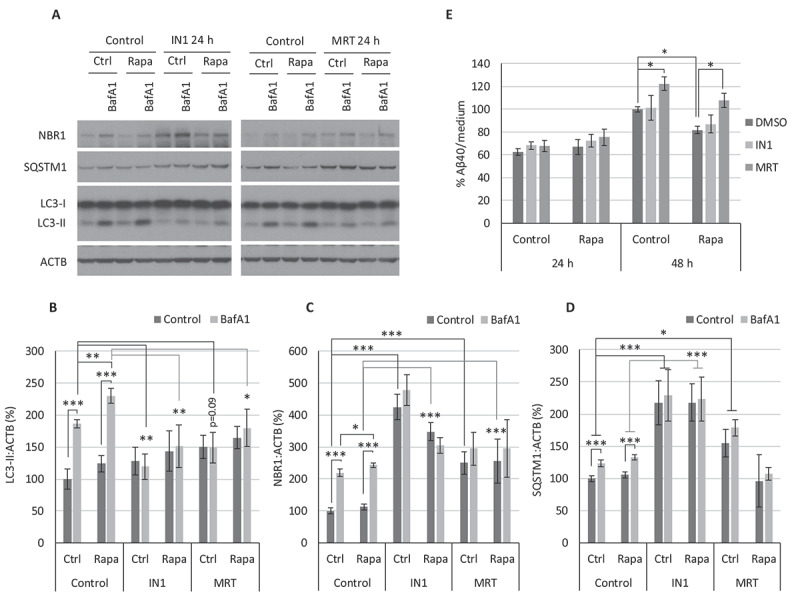
Autophagy prevention with IN1 or MRT reverses the anti-amyloidogenic effect of rapamycin. Cerebellar granule neuron (CGN) cultures were treated for 24 or 48 h with PIK3C3-IN1 (IN1) or MRT68921 (MRT) in the presence or absence of 100 nM rapamycin (Rapa). (A) Western blot analysis of the autophagic markers LC3-II (B), NBR1 (C), and SQSTM1 (D), from cellular extracts after 24 h of treatment with 1 µM IN1 or 1 µM MRT. Two-way ANOVA (n = 3). (E) Aβ40 levels in culture media determined by ELISA after 24 h or 48 h of treatment with IN1 or MRT (24 h: 0.25 µM IN1 or 0.5 µM MRT; 48 h: 0.1 µM IN1 or 0.1 µM MRT). Two-way ANOVA was performed (n = 3). * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Bars represent mean ± SEM
As a complementary strategy, neurons were infected with lentiviral particles to express a tandem mCherry-GFP tagged LC3 construct. Briefly, the fluorescent signal of GFP is more sensitive to the acidic conditions of the lysosome. In contrast, mCherry is more stable, so the colocalization of both GFP and mCherry fluorescent dots corresponds to an autophagosome (yellow dot), but an mCherry signal without GFP indicates the formation of an autolysosome (red dot). The infected cells were incubated with 200 nM rapamycin for 24 h and/or 10 nM BafA1 for 4 h. Formations of mCherry-GFP-LC3 dots (yellow and red) were quantified (Figure 2E). Neurons treated with rapamycin showed a statistically significant increased number of autolysosomes (red dots), which supports the higher autophagic flux rate (Figure 2E, left graph). BafA1 inhibits the lysosomal v-ATPase, preventing its acidification and, therefore, the loss of GFP signal. Thus, the statistically significant increase of yellow vesicles in neurons treated with Rapa+BafA1, compared to BafA1 alone, confirmed a higher autophagic flux with rapamycin (Figure 2E, right graph). Altogether, these results indicate that rapamycin augments LC3-II levels in CGNs as a consequence of a modest increase of autophagic flux, consequently increasing the autophagic degradation rate.
Although our results support only a modest induction of autophagy in CGNs after MTORC1 inhibition, rapamycin has been described as a potent inducer of autophagy, principally in cell lines [44]. To check whether the effect was cell line-dependent, we performed similar autophagic flux assays in 2 neuroblastoma cell lines, SH-SY5Y, and N1E-115. Although rapamycin efficiently inhibited MTORC1 in both cell lines, autophagy induction in the tumorous N1E-115 cells was outstanding compared to the less-proliferating SH-SY5Y, according to variations in LC3-II, NBR1, and SQSTM1 (Fig. S2C and S2D). This result is consistent with the idea that autophagy induction by rapamycin is generally moderate in neuron-like systems. Moreover, downregulation of ATG5 using two interfering RNAs in SH-SY5Y cells confirmed that the lowered levels of SQSTM1 in the presence of rapamycin was indeed mediated by the autophagy degradation process (Fig. S2E).
To rule out that the modest autophagy induction observed with rapamycin in neurons was a particular effect of this chemical compound, we used an alternative, more physiological inhibition of MTORC1: depletion of amino acids, such as glutamine. For this, CGNs were cultured with or without the glutamine analog GlutaMAX™ (Gmax) for 4 or 24 h, showing a modest effect on autophagic flux (Fig. S3A). Similar to rapamycin, we observed a statistically significant decrease of SQSTM1 levels after 24 h of Gmax deprivation (Fig. S3B). These results strongly suggest that the modest induction of autophagy observed in CGNs and SH-SY5Y cells (data not shown) is characteristic of MTORC1 inhibition in neuronal systems.
Autophagy prevention reverses the Aβ-lowering effect of rapamycin
To confirm that an increased autophagic degradation rate mediated the anti-amyloidogenic effect of rapamycin, we proceeded to block the initial formation of autophagosomes. For this purpose, PIK3C3/VPS34-inhibitor 1 (IN1), which specifically inhibits the PIK3C3 subunit of class III PtdIns3K complex without affecting other PIK3s [45,46], and MRT68921 (MRT), a potent specific inhibitor of the ULK complex kinases ULK1/2 [47], were used. We treated CGNs for 24 h with or without rapamycin in the presence or absence of IN1 or MRT inhibitors, and in a parallel set of experiments, we added BafA1 only for the last 4 h (Figure 3A-D). Western blot analysis showed that both inhibitors, IN1 and MRT, were able to reduce autophagosome formation and, therefore, autophagic flux, both in basal (control) and rapamycin-induced autophagy conditions, as LC3-II accumulation with BafA1 diminished, and degradation of NBR1 and SQSTM1 (Figure 3A-D).
In an equivalent set of experiments, we treated APP/PSEN1 CGNs either with 0.25 µM IN1 or 0.5 µM MRT for 24 h; or 0.1 µM IN1 or 0.1 µM MRT for 48 h. We harvested culture media to determine the level of Aβ40 by specific ELISA. We could not observe significant differences with any treatment at 24 h, nor with nontoxic concentrations of IN1 at 48 h (Figure 3E). However, MRT-treated cells had significantly increased levels of Aβ40 after 48 h (Figure 3E). Moreover, after 48 h, a time when rapamycin significantly reduces β-amyloid secretion, MRT was able to revert this effect to control levels (Figure 3E). These results allowed us to conclude that autophagy has an anti-amyloidogenic effect and that the modest enhancement of autophagy by rapamycin mediates its Aβ-lowering effect in neurons.
AMPK activation does not induce autophagy in CGN or SH-SY5Y cell cultures
AMPK stimulates the initial steps of autophagy, favoring activation of the ULK complex and a pro-autophagic configuration of class III PtdIns3K complex [28,29]. Thus, our second aim was to analyze the role of AMPK as an inducer of autophagy using our neuronal systems.
We proceeded to test autophagic flux in CGNs treated with the AMPK activators AICAR or Metf, and we combined these treatments with rapamycin to identify any additive effects (Figure 4A, C–E). AMPK activity was analyzed using an antibody against the phosphorylation site Thr172 (T172) of its subunit PRKAA, as well as the phosphorylation of its targets ACACA (S79) and ULK1 (S555). Autophagy status was monitored as before by measuring the levels of LC3-II, NBR1, and SQSTM1. Analyses were performed after only 4 h of treatment to ensure the maintenance of optimal AMPK activation and diminish the duration of subsequent inhibition of MTORC1 (Fig. S4A and S4B). The BafA1 concentration was reduced to 10 nM to avoid possible off-target and saturating effects, as it tends to lead to higher differences in flux determinations between treatments (Fig. S4C).
Figure 4.
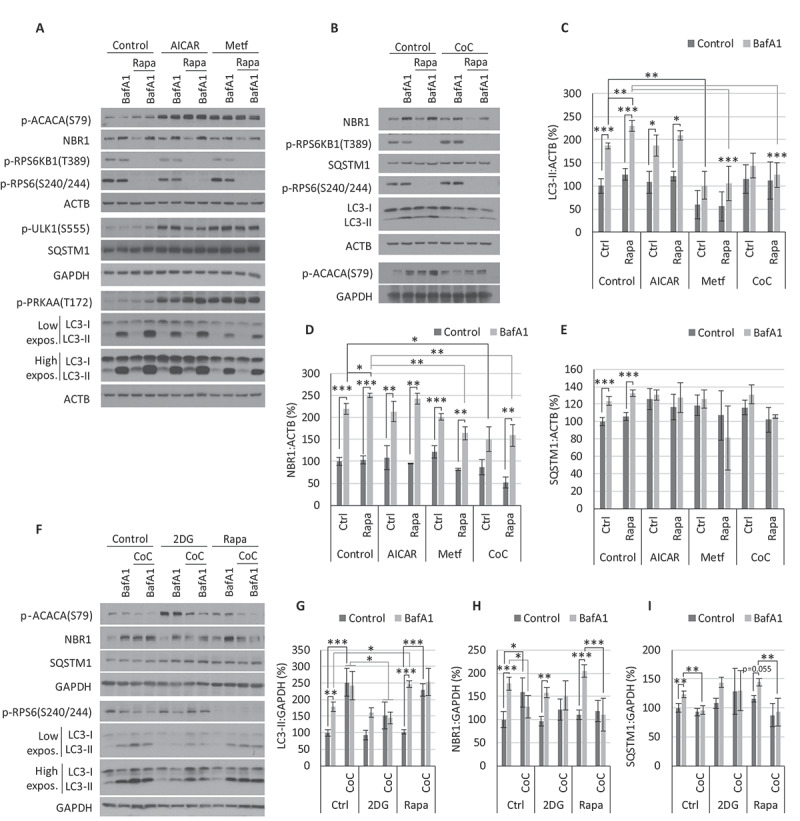
Autophagic flux analysis after AMPK and MTORC1 modulation. (A-E) Cerebellar granule neuron (CGN) cultures were treated for 4 h with 1 mM AICAR, 2.5 mM metformin (Metf) or 5 µM compound C (CoC) in the presence/absence of 200 nM rapamycin (Rapa) and/or 10 nM BafA1. Western blot analysis of the autophagic markers LC3-II (C), NBR1 (D), and SQSTM1 (E). Two-way ANOVA was performed (n = 3). (F-I) Cultures of SH-SY5Y were treated for 4 h with 5 µM CoC in the presence/absence of 10 nM BafA1 and/or 200 nM rapamycin or 20 mM 2DG. Western blot analysis of the autophagic markers LC3-II (G), NBR1 (H), and SQSTM1 (I). Two-way ANOVA was performed (n = 3). * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. p-RPS6KB1 T389 and p-RPS6 S240/244 were employed as indicators of MTORC1 activity; p-PRKAA T172 and p-ACACA S79 of AMPK activity, and p-ULK1 (S555) as an indicator of AMPK activity over the pro-autophagic complex ULK. Two exposure intensities, low and high, are shown for LC3. Bars represent mean ± SEM
Our data showed that AICAR and Metf efficiently activated AMPK, as indicated by phosphorylation on T172 and its targets ACACA (S79) and the pro-autophagic ULK1 (S555) (Figure 4A). However, neither of them increased the autophagic flux, as the changes in LC3-II, NBR1, and SQSTM1 with or without BafA1 were similar or even lower than the controls (Figure 4A, C–E). We added rapamycin, in combination with the AMPK activators, to ensure the lack of autophagy induction was due to an insufficient inhibition of MTORC1. Although the observed NBR1 and LC3-II levels suggested a partial recovery in the presence of rapamycin, they both pointed to an autophagic flux still lower than that observed in neurons treated with rapamycin alone (Figure 4A, C–E). We obtained analogous results in SH-SY5Y cells (Fig. S4D) and after using the AMPK activator 2DG, both in CGNs (data not shown) and SH-SY5Y cells after different treatment periods (Fig. S4B and S4D).
Basal AMPK activity is essential for the achievement of proper autophagy in CGN or SH-SY5Y cell cultures
Since the activation of AMPK led to diminished autophagy efficiency in the neuronal systems assayed, our next query was whether initiating the process required the active form of AMPK [28,29]. Thus, we evaluated autophagic flux after inhibition of AMPK, for which we employed the AMPK inhibitor CoC [48].
Our data revealed that in CGNs, inhibition of AMPK with CoC lowered the accumulation of LC3-II in the presence of BafA1, therefore reducing autophagic flux and degradation rate, as indicated by NBR1 and SQSTM1 levels (Figure 4B-E). Similarly, CoC drastically blocked autophagy flux and degradation rate in SH-SY5Y cells (Figure 4F–I). The inhibition of AMPK by CoC was clear in both neuronal cell systems, evidenced by the level of phosphorylation of ACACA (Figure 4A and F), even prevailing over AMPK activation by 2DG (Figure 4F), AICAR or Metf (Fig. S5A). In the same way, CoC treatment reversed rapamycin-induced autophagy in both CGNs and SH-SY5Y cultures (Figure 4B–I).
Contrary to CGNs, SH-SY5Y cells treated with CoC showed reduced MTORC1 activity and increased LC3-II levels in a time- and concentration-dependent manner (Fig. S5B). This effect has been previously described as an AMPK-independent mechanism in some tumorigenic cell lines [49]. However, constitutive activation of MTORC1 by expression of RHEBQ64L showed no change in the effect of CoC on LC3-II and NBR1 levels or autophagic flux rate (Fig. S5C). Thus, our results support that the autophagic effects of CoC are unrelated to such cell-specific inhibitions of MTORC1.
AMPK activation does not modify the level of Aβ40 secreted by CGNs through autophagy
CGNs obtained from APP/PSEN1 mice were treated with the AMPK activators AICAR, Metf and 2DG, or with the AMPK inhibitor CoC for 24 or 48 h. Culture supernatants were collected, and we determined levels of h-Aβ40 by specific ELISA (Figure 5A). Our data show that both 2DG and CoC reduced Aβ levels after 48 h of treatment similar to rapamycin, whereas Metf caused an even greater drop (Figure 5A). In contrast, AICAR treatment significantly increased secreted amyloid after only 24 h of treatment and remained elevated after 48 h (Figure 5A).
Figure 5.
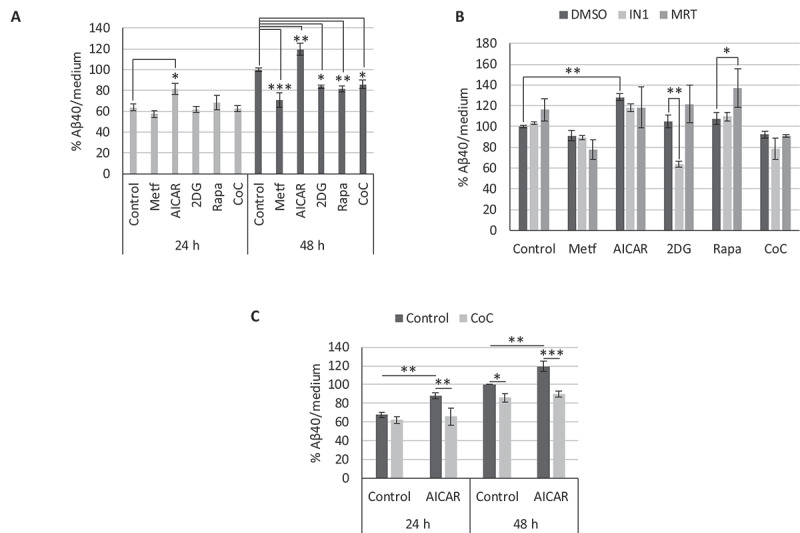
Effect of AMPK modulation on β-amyloid secretion. Levels of h-Aβ40 in culture media of cerebellar granule neurons (CGNs) obtained from APP/PSEN1 mice, after the indicated treatments: (A) CGN cultures were treated with metformin (Metf), AICAR, 2DG, rapamycin (Rapa) or compound C (CoC) for 24 or 48 h (n = 3; one-way ANOVA performed). (B) CGN cultures were treated with Metf, AICAR, 2DG, Rapa or CoC, in combination with 0.25 µM PIK3C3-IN1 (IN1) or 0.5 µM MRT68921 (MRT) for 24 h (n = 3; two-way ANOVA performed). (C) CGN cultures were treated with AICAR in combination with CoC for 24 and 48 h (n = 3; two-way ANOVA performed). Treatment dosages: Metf: 1 mM; AICAR: 0.25 mM; 2DG: 1 mM; Rapa: 100 nM; CoC: 2 µM. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Bars represent mean ± SEM
To check for the role of autophagy in these conflicting results, we proceeded to block the initial formation of autophagosomes by inhibition of the class III PtdIns3K and the ULK complex with IN1 and MRT, respectively. As long-term treatment with combinations of AMPK modulators and autophagy inhibitors led to toxic effects, we only performed experiments lasting 24 h. Contrary to the autophagy inducer rapamycin or control conditions, neither IN1 nor MRT produced an increase in β-amyloid levels when combined with any of the AMPK modulators (Figure 5B).
As the AMPK activator AICAR displayed an opposite response in secreted amyloid compared to Metf and 2DG, we attempted to differentiate those mediated by an off-target effect or by AMPK activity alone. Thus, we treated CGNs from APP/PSEN1 mice with AICAR or Metf in the presence or absence of the AMPK inhibitor CoC, and after 24 or 48 h, we determined the h-Aβ40 levels by specific ELISA. CoC was only able to block the effect of AICAR, as amyloid quantity in the presence of AICAR reverted to control levels at both 24 and 48 h (Figure 5C). On the contrary, CoC did not revert secreted amyloid levels after treatment with Metf or 2DG (data not shown).
These results allow us to conclude that the activation of AMPK mediated the increased amyloid secretion by AICAR. However, this effect was not dependent on autophagy since, similar to Metf, blockage of autophagy by IN1/MRT did not affect amyloid secretion.
Active AMPK overexpression does not induce autophagy but increases levels of Aβ40 secreted by CGNs
To further study the complex role of AMPK in the regulation of autophagy in neurons, we proceeded to modulate its activity through lentiviral-mediated transformation. Constitutively active (CA), kinase-inactive (KI), or wild type forms of PRKAA1 were overexpressed in CGNs (with the expression of a scramble sequence [SCR] as an infection control), and analyzed at 3 d post-infection (Figure 6). Phosphorylation levels of the AMPK targets p-ACACA(S79) and p-ULK1(S555) were increased in the presence of the CA form and, to a lesser extent, with the wild type form of PRKAA1, whereas the KI form led to a modest decrease (Figure 6A). We treated transformed neurons with 10 nM BafA1 for 4 h to analyze autophagy status, though we observed no significant differences between treatments (Figure 6B). To confirm the role of AMPK in β-amyloid secretion, CGNs obtained from APP/PSEN1 mice were infected with the lentiviral constructs containing the CA, KI, or wild type forms of PRKAA1, or the SCR control. Overexpression of all 3 forms of PRKAA1 resulted in a modest increase of amyloid levels (Figure 6C).
Figure 6.
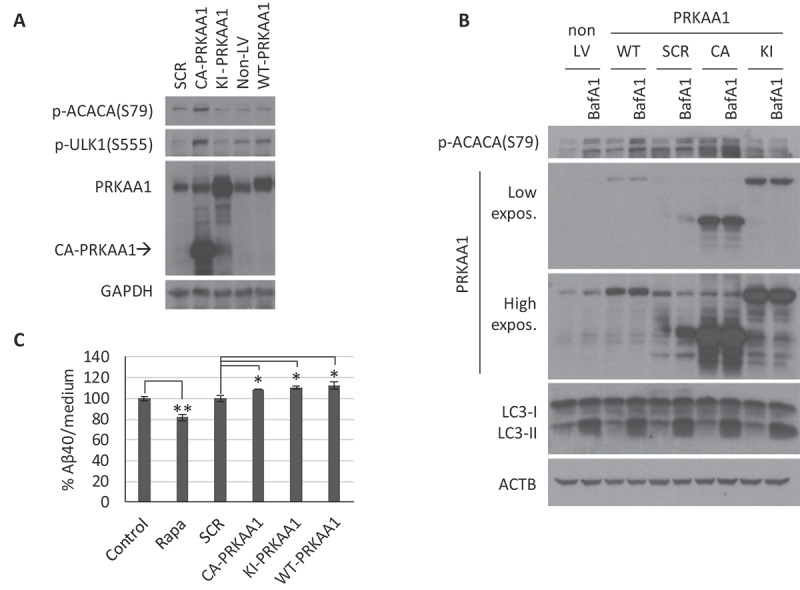
AMPK modulation by gene overexpression in cerebellar granule neurons (CGNs). CGN cultures were infected with lentiviral particles to overexpress the scramble (SCR), constitutive active (CA), kinase-inactive (KI), or WT forms of PRKAA1, and analysis were performed 3 d post-infection. (A) Western blot analysis of PRKAA1 expression levels and AMPK activity with p-ACACA(S79) and p-ULK1(S555). (B) Cells were treated with 10 nM BafA1 for the last 4 h to analyze autophagic flux (LC3-II). Representative western blot of n = 3 independent experiments. Two exposure intensities, low and high, are shown for PRKAA1. (C) Media from APP/PSEN1 CGNs was collected to determine levels of secreted h-Aβ40 by ELISA (n = 3; one-way ANOVA performed; * p ≤ 0.05; ** p ≤ 0.01; Bars represent mean ± SEM)
The data strongly supports the conclusion that in neurons the activation of AMPK (CA or wild type) does not enhance autophagy, and secondly that this activation does not correlate with a reduction of amyloid secretion if anything with an increase of it. Finally, we observed that any of the AMPK modulators did not modify total APP levels, which discards an altered expression of the amyloid precursor as the mechanism underlying the different secreted amyloid levels (Fig. S6).
Discussion
The main aim of this work was to analyze the regulatory mechanisms of autophagy in neurons in physiological conditions, but also to discern whether increasing autophagy may have therapeutic value for the reduction of β-amyloid accumulation, a hallmark of AD. Indeed, AD displays serious imbalances in the proteostatic mechanisms in neurons, especially autophagy (reviewed in [16]). However, there is some controversy regarding whether the enhancement of autophagy can help to degrade excessive amyloid [50,51], or rather would contribute to the generation of acidic compartments where optimal BACE1 activity leads to a higher amyloid production rate [8,52] (reviewed in [53]). Our results support that autophagy can have a therapeutic anti-amyloidogenic effect in neurons, at least when inhibiting MTORC1. However, we also demonstrated that AMPK activation does not lead to this effect.
We showed that the 2-month treatment of APP/PSEN1 mice with the autophagy inducer rapamycin led to a significant reduction of brain Aβ40 levels. Rapamycin was able to cross the blood-brain barrier and target the brain tissue, as seen by a reduction of RPS6KB1 activity. It affected neuronal populations, inferred from the modified phosphorylation levels of the particularly abundant protein in neurons MAPT/TAU [39,40,54]. The lower phosphorylation levels in the MAPT phospho-epitope PHF1 would also point out a reduction in the homeostatic alterations in neurons, characteristic of Alzheimer pathology [55]. Our results with rapamycin are in agreement with other reports describing neuroprotective effects in other AD mice models, such as 3xTg-AD, Tg2576 or PDAPP, through reversion of the MTORC1 hyperactivation characteristic of AD patients, and thus by inducing autophagy [31,56,57], but also by lowering the protein synthesis [56,58]. Oral administration of rapamycin showed anti-amyloidogenic effects in 3xTg-AD and PDAPP mice [31,57,59,60]. Paradoxically, a pro-amyloidogenic effect has been described through inhibition of the alpha-secretase ADAM10 by intraperitoneal administration of 3 mg/kg rapamycin 5 d/week for 2 weeks to Tg2576 mice [34]. Interestingly, the rapamycin analog temsirolimus also showed relevant anti-amyloidogenic effects when intraperitoneally administered to Mo/HuAPP695/PS1dE9 mice at 20 mg/kg every 2 d for 2 months [61]. Nevertheless, according to the immunosuppressive capacity of rapamycin [62], after 2 months of treatment, we observed a diminished ability of small wounds to heal properly. This effect reinforces the need to improve formulations and posology of MTORC1 inhibitors for chronic therapeutic approaches based on autophagy.
The overexpression of mutated APP and PSEN1 in neurons of our AD mouse model led to similar accumulations of Aβ in both cortex and cerebellum [30]. Indeed, some have observed elevated levels of Aβ in the cerebellum of early-onset familial and sporadic AD patients [63,64]. Thus, to study the effect of rapamycin in neurons from our APP/PSEN1 transgenic mouse, we used cultured cerebellar granule neurons (CGNs) from post-natal days 5–7 mice as an isolated neuronal model, which allow us to determine their genotype previously, generate a homogenous pool of neurons and obtain a highly pure culture (96% granule neurons).
We corroborated in primary cell cultures that the pro-autophagic and anti-amyloidogenic effect of rapamycin observed in vivo occurs, at least, in neurons within the brain cell populations. Rapamycin treatment of CGNs from APP/PSEN1 mice led to a decrease in secreted amyloid levels through MTORC1 inhibition and the subsequent induction of autophagy. The reversion of the Aβ-reducing effect of rapamycin when blocking autophagy initiation (using the ULK1/2 inhibitor MRT68921) confirmed the therapeutic role of autophagy enhancement. Moreover, increases in secreted Aβ with this inhibitor in the absence of rapamycin confirmed that basal autophagy could also contribute to amyloid reduction.
On the other hand, our results only support a modest induction of autophagy with rapamycin in both CGN and SH-SY5Y neuronal systems, in contrast to the marked effect observed in the quickly dividing neuroblastoma N1E-115 cells similar to that described in the literature mostly in cell lines and yeast [26]. While some previous studies have defended the ability of rapamycin to induce autophagy in neurons [65–67], others have questioned it [44,68,69]. Our results support the ability of rapamycin to induce autophagy through MTORC1 inhibition since that was analogous to amino acid deprivation. This result was verified after both short- and long-term treatments when MTORC2 activity was also affected [43], although we did not observe any negative effects that have been described for MTORC2 inhibition in autophagy [70]. Thus, a more controlled autophagic status in neurons may constitute one of the great metabolic differences from tumorigenic cell lines with high division rates. Neurons require strict proteostatic regulation according to its post-mitotic, highly polarized, and compartmentalized nature, with high energy demands [3]. Accordingly, other groups have also found peculiarities in autophagy regulation in neuronal cells [44,69,71–73]. Thus, specific analyses of the described regulatory pathways of autophagy in neurons are essential for the development and optimization of neuroprotective strategies based on this process.
The second aim of our work was to analyze the putative contribution of AMPK, a well-described autophagy regulator, in our neuronal system. AMPK constitutes another main metabolic supervisor in almost all cell lineages, and governs the energy status of the cell through sensing AMP/ATP, amino acid and glucose levels, and tracking mitochondrial conditions [24]. AMPK has been described to induce autophagy through the inhibition of MTORC1, but also directly by phosphorylation of the ULK1/2 and the class III PtdIns3K complexes [28,29]. Thus, we wanted to check if it could have a main regulatory role for autophagy in neurons, alone or in combination with MTORC1 inhibition.
Inhibition of AMPK with CoC led to the expected reduction of autophagy flux in both CGNs and SH-SY5Y cells. Surprisingly, increased activity of AMPK (by treatment with 2DG, Metf, or AICAR) did not lead to an enhancement of autophagy, either alone or in combination with rapamycin. The failure to enhance autophagy by these compounds could be due to off-target actions, such as inhibition of the pro-autophagy kinase MAPK8/JNK during Metf treatment [74], though we did not observe this effect with AICAR or 2DG (data not shown). According to our results, there is also some doubt about the impact of these compounds on autophagy [74–76]. Thus, as a second approach, we proceeded to modify AMPK activity by transforming CGNs with lentiviral particles to overexpress constitutive, null, and wild type forms of the kinase, although none of them could modify the autophagic flux.
The AMPK activator AICAR increased the secreted Aβ40 levels by APP/PS1 CGNs, prevented by the inhibition of AMPK with CoC, which supports an AMPK-mediated pro-amyloidogenic effect of AICAR. This result, together with the higher Aβ40 levels when overexpressing constitutive active or WT forms of PRKAA1, points out a pro-amyloidogenic role of AMPK. Metf and 2DG led to a reduction of secreted Aβ40 levels, non-reversed by CoC, so we hypothesize that their anti-amyloidogenic effects are a consequence of an AMPK-independent process. Contrary to the effect of the AMPK inhibitor CoC, the overexpression of the kinase-inactive form of PRKAA1 did not decrease amyloid secretion, which may be related to the remaining activity of the endogenous kinase, as indicated by p-ACACA (S79), which strongly suggest that it was not able to play a dominant-negative effect in these conditions.
Blocking autophagy with inhibitors of ULK1/2 or class III PtdIns3K did not reverse the lowered Aβ40 levels by Metf, 2DG, or CoC, nor the increased Aβ40 levels by AICAR. This result supports that: 1) autophagy does not mediate the pro-amyloidogenic effect of AMPK (according to AICAR, CoC, CA-PRKAA1, and WT-PRKAA1), and 2) autophagy does not mediate the AMPK-independent anti-amyloidogenic effect of Metf and 2DG. Interestingly, the combination of 2DG and PIK3C3-IN1 led to decreased amyloid secretion levels. Whereas the glucose analog 2DG blocks glycolysis to lower ATP levels and activate AMPK [77], inhibition of PIK3C3 switches the cellular respiration from oxidative phosphorylation toward glycolysis, which depends on glucose uptake [78]. We hypothesize that alterations of amyloid secretion by co-treatment with 2DG and PIK3C3-IN1 could be a consequence of both disturbing the glycolytic metabolism affecting the neuronal status, which could also explain the high toxicity observed at long-term treatments of 48 h. Although doing more work to unveil the metabolic processes involved in this effect is needed.
In summary, our work supports that MTORC1 plays an important role in controlling autophagy in neurons, even though quantitatively, the greatest inhibition by rapamycin did not produce an extraordinary increase in autophagy. Still, its possibly therapeutic anti-amyloidogenic effect was measurable both in vivo and ex vivo. Whereas the basal activity of AMPK is essential for the function of autophagy, greater kinase activation is not able to augment the rate of degradation. Moreover, AMPK activation presents a pro-amyloidogenic effect not mediated by autophagy.
According to our data from CGNs and SH-SY5Y, we hypothesize that neurons have a more complex regulation of the initial steps of autophagy. While inhibition of MTORC1 is important to enhance autophagy flux further, more AMPK activity does not enhance this process, clearly different from the generally accepted dogma. Doing more work to define the key elements that control autophagy during AMPK activation in different neuronal systems is needed.
Materials and methods
Animal handling
We employed the double-transgenic mouse strain B6.Cg-Tg (APPSwe, PSEN1dE9) 85Dbo/J, which overexpresses the human genes APP (amyloid beta precursor protein) with the Swedish mutation and exon-9-deleted PSEN1 (presenilin 1; Jackson Laboratory, Bar Harbor; stock no. 005864), hereafter referred to as APP/PSEN1 [30,38]. The genotype of each mouse was confirmed by PCR of DNA isolated from tail biopsies [79]. All animal care and handling strictly followed current Spanish legislation and guidelines, and those of the European Commission (directive 2010/63/EU). The use of wild type and transgenic animals was an absolute requirement for this project; however, experiments were designed to minimize the use of animals. All transgenic mice and non-transgenic littermates were group-housed in standard cages with fiber bedding, under a 12 h/12 h light/dark cycle.
In vivo rapamycin treatment
As previously described [33], a stock solution of 20 mg/ml rapamycin (LC Laboratories, R-5000) in ethanol was conserved at −20ºC, and diluted in the vehicle solution (5% PEG400 [Fluka, 81172], 5% Tween80 [Sigma-Aldrich, P4780] in phosphate-buffered saline, pH 7.4 [PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4]) the day of use. Prior to the experiment, blood plasma samples were collected to measure the β-amyloid levels by ELISA and, accordingly, homogenously distribute 9 mice per group, rapamycin or vehicle-treated. 4-month-old male APP/PSEN1 mice were intraperitoneally injected with 5 mg/kg of rapamycin, or equivalent volumes of vehicle solution, every 48 h for 2 months. Bodyweight was measured weekly and blood plasma samples were also collected at the end of the experiment. Then, mice were sacrificed in CO2 atmosphere and different parts of the brain were conserved at −80ºC.
Cell culture
SH-SY5Y (ATCC, CRL-2266) and N1E-115 (ATCC, CRL-2263) were cultured in Dulbecco’s modified eagle’s medium (DMEM; Gibco, 52100) supplemented with 110 mg/L pyruvate (Merck, 106619), 3.7 g/L sodium bicarbonate (Merck, 106329), 10% fetal bovine serum (FBS; Sigma-Aldrich, F7524), 2 mM glutamine (Merck, 100289), 0.01% streptomycin (PanReac AppliChem, A1852) and 100 U/ml penicillin G (PanReac AppliChem, A1837), at 37ºC in an atmosphere of 5% CO2.
Primary cerebellar granule neuron (CGN) cultures were performed based on previous protocols [80–82] with some modifications. Briefly, after genotyping pups at post-natal day 2–3 (P2, P3), primary CGNs were isolated from post-natal day 5, 6 or 7 (P5-P7) APP/PSEN1 mice, or their wild type littermates. Meninges were carefully removed from cerebella, washed in ice-cold medium (3% bovine serum albumin [BSA; Roche, 1073508001] and 1.2 mM MgCl2 in Hanks Balanced Salt solution [HBSS; Invitrogen, 14170–088]), minced with a razor blade and digested in 0.2% trypsin (Sigma-Aldrich, T4549) with 0.6 mg/ml DNase-I (Roche, 10104159001) for 15 min at 37ºC. A solution containing 20% FBS, 0.2 mg/ml DNase-I, and 2.4 mM MgCl2 in HBSS was added to stop the digestion, and tissue was triturated with sterile glass Pasteur pipettes with 2 different tip diameters. After centrifuging 10 min at 168 x g to remove the FBS, neurons were seeded in Neurobasal (Gibco, 21103–049) supplemented with 1x B27 (Gibco, 17504–044), 2 mM GlutaMAX™ (Gibco, 35050–038), 0.01% streptomycin, 100 U/ml penicillin G and 22 mM KCl, in plates previously coated with 10 µg/ml poly-L-lysine (Sigma-Aldrich, P4707). 1.5 × 106 cells per well were plated in 6-well plates to obtain extracts for western blot, and 105 cells per well in 24-well plates to measure the secreted amyloid levels in the media by ELISA. CGN cultures were incubated at 37ºC in an atmosphere of 5% CO2, and experiments performed until DIV 4 (4 d in vitro), in a serum-free medium, to prevent the consecutive boost of glial cells (in our hands: 96% neurons, 4% glia).
Both CGNs and SH-SY5Y were treated with rapamycin (Rapa; LC Laboratories, R-5000), bafilomycin A1 (BafA1; Santa Cruz Biotechnology, sc-201550), PIK3C3/VPS34-IN1 (IN1; MRC Protein Phosphorylation and Ubiquitylation Unit, VPS34-IN1 [45]) or MRT68921 (MRT; MRT0068921 was kindly provided by Dr. Barbara Saxty, MRC Technology [47]), whose solvent dimethyl sulfoxide (DMSO, PanReac AppliChem, A3672) was employed as a negative control. 2-deoxy-D-glucose (2DG; Sigma-Aldrich, D6134), metformin (Metf; Calbiochem, 317240), 5-aminoimidazole-4-carboxamide-1-β-riboside (AICAR; Calbiochem, 123040) and compound C (CoC, dorsomorphin dihydrochloride; Tocris, 3093) were dissolved in MilliQ water.
Autophagic flux
Autophagic flux rate was measured after blocking autophagosome-lysosome fusion and clearance by addition of the v-ATPase inhibitor BafA1 (or equal volume of DMSO as control) for the last 4 h before harvesting the cells. LC3-II levels were measured by western blot on methanol-activated PVDF (Millipore) membranes.
Gel electrophoresis and western blots
Treated cells were scraped off the plates in 100 µl of SDS-lysis buffer (50 mM Tris pH 7.6, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% SDS). The homogenate was heated at 95ºC with vigorous shaking for 15 min and sonicated for 30 s. Samples were centrifuged at 16,000 x g for 20 min and the supernatants were stored at −20ºC.
Frozen cerebral cortex samples were lysed with a tissue homogenizer in 3 µl/mg ice-cold Triton-lysis buffer (20 mM HEPES [Sigma-Aldrich, H3375], 100 mM NaCl, 100 mM NaF, 1 mM Na3VO4, 5 mM EDTA, 1% Triton X-100 [Merck, 108603], 1x Complete Protease Inhibitor Cocktail [Roche, 1697498], 1 µM okadaic acid [Calbiochem, 459618]). Samples were centrifuged at 16,000 x g for 20 min at 4ºC and the supernatant was diluted 1:10 and stored at −20ºC.
Protein concentration was measured using the DC Protein Assay (Bio-Rad, 5000111), following the manufacturer’s protocol. Samples of 20 µg protein were mixed with 5x loading buffer (10% SDS, 5% beta-mercaptoethanol, 325 mM Tris HCl, pH 6.8, 25% glycerol, and 0.5% bromophenol blue) to a final concentration of 1x, and heated for 5 min at 100ºC. 20 µg protein samples were resolved by SDS/PAGE, and then transferred to nitrocellulose (Whatman) or PVDF (Millipore) membranes. After blocking with 5% BSA for 1 h, the membranes were probed overnight at 4ºC with the primary antibody and incubated with a secondary horseradish peroxidize-conjugated antibody. Washing steps and solutions were made in 0.1% Tween 20 (Merck, 822184) – TBS (100 mM Tris, pH 8, 150 mM NaCl), and antibody binding was detected using Western lightingTM Plus ECL (Perkin-Elmer, NEL105). GAPDH or ACTB/β-actin were used as an internal control. The relative quantity of protein levels in western blots was measured using ImageJ software (ImageJ, Fiji) [83].
The following antibodies and the corresponding dilutions were used: APP A4-22C11 (1:500; Millipore, MAB348), APP-6E10 (1:1000; Covance, SIG-39300), BACE1 (1:1000; Cell Signaling Technology, 5606), p-MAPT/TAU S396/404 (PHF1, 1:200; gift from P Davies Sift), MAPT/TAU5 (1:1000; Millipore, 577801), cleaved CASP3 (1:1000; Cell Signaling Technology, 9661), LC3B (1:4000; Sigma-Aldrich, L7543), ATG5 (1:1000; Sigma-Aldrich, A2859), NBR1 (1:500; Santa Cruz Biotechnology, sc-130380), SQSTM1 (1:4000; Novus Biologicals, H00008878-M01), p-RPS6KB1 T389 (1:1000; Cell Signaling Technology, 9205), RPS6KB1 (1:500; Santa Cruz Biotechnology, sc-230), p-RPS6 S240/244 (1:1000; Cell Signaling Technology, 2215), RPS6 (1:1000; Cell Signaling Technology, 2217), p-PRKAA/AMPKα T172 (1:1000; Cell Signaling Technology, 2535), PRKAA1/AMPKα1 (1:1000; Cell Signaling Technology, 5832), p-AKT S473 (1:1000; Cell Signaling Technology, 9271), AKT (1:1000; Cell Signaling Technology, 9272), p-ACACA S79 (1:1000; Cell Signaling Technology, 3661), p-ULK1 S555 (1:1000; Cell Signaling Technology, 5869), GAPDH (1:2000; Cell Signaling Technology, 2118), ACTB (1:40,000; Sigma-Aldrich, A5441), anti-rabbit IgG-HRP (1:5000; Santa Cruz Biotechnology, sc-2004) and anti-mouse IgG-HRP (1:5000; Santa Cruz Biotechnology, sc-2005).
Aβ quantification by ELISA
The Aβ levels of cerebral cortex samples or of the medium of the cultured neurons were measured using the Human Aβ40 (Invitrogen™, KHB3482) or Aβ42 ELISA kit (Invitrogen™, KHB3442), following the manufacturer’s instructions. Plate absorbance was read with an Opsys MR microplate reader (Dynex Technologies) at 450 nm [38].
Lentiviral transformation
Cells were incubated with lentivirus (MOI = 10) for 24 h and experiments were performed from 48–72 h post-infection. The lentiviral constructs employed were: pMD2.G (Addgene, 12259; Didier Trono Lab) and pCMVR8.74 (Addgene, 22036; Didier Trono Lab) [84]; RHEBQ64L (Addgene, 21050; Peter Vogt Lab) [85]; FUW mCherry-GFP-LC3 (Addgene, 110060; Anne Brunet Lab) [86]. Scramble (SCR), rPRKAA1/rAMPKalpha1(aa1-312; CA-PRKAA1) and rPRKAA1D157A (KI-PRKAA1) were a kind gift from Rachel Hertz [87]. Expression of wild type PRKAA1 was performed by coinfection of pLenti CMV rtTA3 Blast (w756-1; Addgene, 26429; Eric Campeau Lab) and FUW TetO HA-AMPKa1 WT (Addgene, 69824; Anne Brunet Lab); and treatment with 2.5 µg/ml doxycycline (Sigma-Aldrich, D9891) for 24 or 48 h.
Immunofluorescence study
Cerebellar granule neurons were cultured and infected with the lentiviral construct containing FUW mCherry-GFP-LC3 (Addgene, 110060; Anne Brunet Lab) 2 d after plating. The infected cells were incubated with 200 nM rapamycin for 24 h and/or with bafilomycin A1 for 4 h. After that, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, 16005) in PBS and stained with DAPI (2-[4-amidinophenyl]-1H -indole-6-carboxamidine; Merck, 268298) to visualize the nuclei. Fluorescence signal was observed in confocal microscope (Zeiss) and images obtained with the system Zeiss ZEN 2008. Data are shown as number of red and yellow organelles per neuron (100 cells were quantified per experimental condition).
Statistical analysis
Student’s t-tests or ANOVA followed by Holm–Sidak post-hoc test were performed with SigmaPlot 12.5. software (Systat Software Inc., Version 12.5), according to the number of variables, experimental groups and data distribution. Differences were considered statistically significant when p ≤ 0.05 (*), p ≤ 0.01 (**) or p ≤ 0.001 (***). All experiments were independently replicated 3 times, unless otherwise specified in the legend. Data from analysis by western blot and ELISA are represented as mean ± standard error of the mean (SEM) on bar charts. Data from the immunofluorescence study are represented as box plots, which show the 25th, 50th, 75th percentiles as boxes, the 10th and 90th percentiles as error bars, and outliers as dots.
Supplementary Material
Acknowledgments
We are grateful to Dr. Rachel Hertz for giving us the lentiviral constructs of constitutively active and kinase-inactive PRKAA1. The compound MRT0068921 was kindly provided by the company Medical Research Council Technology under a specific MTA agreement.This work was supported by grants from the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED; an initiative of the ISCIII), and from a Collaborative Project ISCIII-CIBERNED Ref: PI2016/01. In addition, it was supported in part by grants from the Spanish Ministerio de Economía y Competitividad (MINECO/FEDER- Proyectos I+D+i Retos 2015, SAF2015-70368-R), Proyectos I+D+i-RETOS- RTI2018-096303-B-C31, and from 2017-CAM-Biomedicina project, (B2017/BMD-3700). Institutional grants from the Fundación Ramón Areces and Banco Santander to the CBMSO are also acknowledged by FW (CBMSO). The professional editing service NB Revisions was used for the technical preparation of the text prior to submission. We are grateful to Fernando Puente-Sánchez for his help in the revision process.
Funding Statement
This work was supported by the Consejería de Educación e Investigación, Comunidad de Madrid [B2017/BMD-3700]; Ministerio de Ciencia, Innovación y Universidades [RTI2018-096303-B-C31]; Ministerio de Economía y Competitividad [SAF2015-70368-R]; Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas [PI2016/01].
Supplemental Material
Supplemental data for this article can be accessed here.
Disclosure statement
The authors declare that they have no competing interests. The Funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- [1].Sweeney P, Park H, Baumann M, et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener. 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yerbury JJ, Ooi L, Dillin A, et al. Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem. 2016. May;137(4):489–505. [DOI] [PubMed] [Google Scholar]
- [3].Ariosa AR, Klionsky DJ.. Autophagy core machinery: overcoming spatial barriers in neurons. J Mol Med (Berl). 2016. August 20;94:1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008. February 28;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li Q, Liu Y, Sun M. Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol. 2017 Apr;37(3):377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ruegsegger C, Saxena S. Proteostasis impairment in ALS. Brain Res. 2016. October 1;1648(Pt B):571–579. [DOI] [PubMed] [Google Scholar]
- [7].Tagliaferro P, Burke RE. Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis. 2016;6(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005. October 10;171(1):87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005. February;64(2):113–122. [DOI] [PubMed] [Google Scholar]
- [10].Cuervo AM. Autophagy in neurons: it is not all about food. Trends Mol Med. 2006. October;12(10):461–464. [DOI] [PubMed] [Google Scholar]
- [11].Zhu XC, Yu JT, Jiang T, et al. Autophagy modulation for Alzheimer’s disease therapy. Mol Neurobiol. 2013. December;48(3):702–714. [DOI] [PubMed] [Google Scholar]
- [12].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011. November 11;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [13].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013. July;15(7):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014. January;24(1):92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016. June;2(85):685–713. [DOI] [PubMed] [Google Scholar]
- [16].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017. March 08;93(5):1015–1034. [DOI] [PubMed] [Google Scholar]
- [17].Martinez-Vicente M. Autophagy in neurodegenerative diseases: from pathogenic dysfunction to therapeutic modulation. Semin Cell Dev Biol. 2015. April;40:115–126. [DOI] [PubMed] [Google Scholar]
- [18].Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012. January 10;22(1):R29–34. [DOI] [PubMed] [Google Scholar]
- [19].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000. November 01;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004. June 01;117(Pt 13):2805–2812. [DOI] [PubMed] [Google Scholar]
- [21].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011. March;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009. February 27;33(4):505–516. [DOI] [PubMed] [Google Scholar]
- [23].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017. March 09;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016. July 15;48(7):e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meijer AJ, Lorin S, Blommaart EF, et al. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids. 2015. October;47(10):2037–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011. July 20;12(8):437–452. [DOI] [PubMed] [Google Scholar]
- [28].Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013. January 17;152(1–2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. February;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ordonez-Gutierrez L, Fernandez-Perez I, Herrera JL, et al. AbetaPP/PS1 transgenic mice show sex differences in the cerebellum associated with aging. J Alzheimers Dis. 2016. September 6;54(2):645–656. [DOI] [PubMed] [Google Scholar]
- [31].Caccamo A, Majumder S, Richardson A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010. April 23;285(17):13107–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Caccamo A, Maldonado MA, Majumder S, et al. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011. March 18;286(11):8924–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hartman AL, Santos P, Dolce A, et al. The mTOR inhibitor rapamycin has limited acute anticonvulsant effects in mice. PLoS One. 2012;7(9):e45156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang S, Salemi J, Hou H, et al. Rapamycin promotes beta-amyloid production via ADAM-10 inhibition. Biochem Biophys Res Commun. 2010. July 30;398(3):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zeng LH, Xu L, Gutmann DH, et al. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008. April;63(4):444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cortes CJ, Qin K, Cook J, et al. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci. 2012. September 5;32(36):12396–12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ozcelik S, Fraser G, Castets P, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8(5):e62459.23667480 [Google Scholar]
- [38].Ordonez-Gutierrez L, Anton M, Wandosell F. Peripheral amyloid levels present gender differences associated with aging in AbetaPP/PS1 mice. J Alzheimers Dis. 2015;44(4):1063–1068. [DOI] [PubMed] [Google Scholar]
- [39].Avila J, Lucas JJ, Perez M, et al. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004. April;84(2):361–384. [DOI] [PubMed] [Google Scholar]
- [40].Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017. December 22;12(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998. February;23(1):33–42. [DOI] [PubMed] [Google Scholar]
- [42].Yoshimori T, Yamamoto A, Moriyama Y, et al. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991. September 15;266(26):17707–17712. [PubMed] [Google Scholar]
- [43].Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006. April 21;22(2):159–168. [DOI] [PubMed] [Google Scholar]
- [44].Tsvetkov AS, Mitra S, Finkbeiner S. Protein turnover differences between neurons and other cells. Autophagy. 2009. October;5(7):1037–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bago R, Malik N, Munson MJ, et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J. 2014. November 01;463(3):413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bilanges B, Vanhaesebroeck B. Cinderella finds her shoe: the first Vps34 inhibitor uncovers a new PI3K-AGC protein kinase connection. Biochem J. 2014. December 01;464(2):e7–10. [DOI] [PubMed] [Google Scholar]
- [47].Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015. November 27;290(48):28726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001. October;108(8):1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vucicevic L, Misirkic M, Janjetovic K, et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011. January;7(1):40–50. [DOI] [PubMed] [Google Scholar]
- [50].Hung SY, Huang WP, Liou HC, et al. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009. 5;May(4):502–510. [DOI] [PubMed] [Google Scholar]
- [51].Boland B, Smith DA, Mooney D, et al. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010. November 26;285(48):37415–37426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gowrishankar S, Yuan P, Wu Y, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015. July 14;112(28):E3699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Benito-Cuesta I, Diez H, Ordonez L, et al. Assessment of autophagy in neurons and brain tissue. Cells. 2017. August 23;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ordonez-Gutierrez L, Re F, Bereczki E, et al. Repeated intraperitoneal injections of liposomes containing phosphatidic acid and cardiolipin reduce amyloid-beta levels in APP/PS1 transgenic mice. Nanomed. 2015. February;11(2):421–430. [DOI] [PubMed] [Google Scholar]
- [55].Barage SH, Sonawane KD. Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015. August;52:1–18. [DOI] [PubMed] [Google Scholar]
- [56].Caccamo A, De Pinto V, Messina A, et al. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014. June 4;34(23):7988–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(4):e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Caccamo A, Branca C, Talboom JS, et al. Reducing ribosomal protein S6 Kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J Neurosci. 2015. October 14;35(41):14042–14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Majumder S, Richardson A, Strong R, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lin A-L, Zheng W, Halloran JJ, et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow and Metab. 2013. September;33(9):1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jiang T, Yu JT, Zhu XC, et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res. 2014;81:54–63. [DOI] [PubMed] [Google Scholar]
- [62].Richardson A, Galvan V, Lin AL, et al. How longevity research can lead to therapies for Alzheimer’s disease: the rapamycin story. Exp Gerontol. 2015;68:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sepulveda-Falla D, Matschke J, Bernreuther C, et al. Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer’s disease. Brain Pathol. 2011. July;21(4):452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jacobs HIL, Hopkins DA, Mayrhofer HC, et al. The cerebellum in Alzheimer’s disease: evaluating its role in cognitive decline. Brain. 2018. January 1;141(1):37–47. [DOI] [PubMed] [Google Scholar]
- [65].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008. July 2;28(27):6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Menzies FM, Huebener J, Renna M, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010. January;133(Pt 1):93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rubinsztein DC, Nixon RA. Rapamycin induces autophagic flux in neurons. Proc Natl Acad Sci U S A. 2010. December 7;107(49):E181;author reply E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Roscic A, Baldo B, Crochemore C, et al. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011. October;119(2):398–407. [DOI] [PubMed] [Google Scholar]
- [69].Kruger U, Wang Y, Kumar S, et al. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012. October;33(10):2291–2305. [DOI] [PubMed] [Google Scholar]
- [70].Renna M, Bento CF, Fleming A, et al. IGF-1 receptor antagonism inhibits autophagy. Hum Mol Genet. 2013. November 15;22(22):4528–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tsvetkov AS, Arrasate M, Barmada S, et al. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013. 9;Sep(9):586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Moruno Manchon JF, Uzor NE, Finkbeiner S, et al. SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy. 2016. August 02;12(8):1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Du L, Hickey RW, Bayir H, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009. January 23;284(4):2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dai YL, Huang SL, Leng Y. AICAR and metformin exert AMPK-dependent effects on INS-1E pancreatic beta-cell apoptosis via differential downstream mechanisms. Int J Biol Sci. 2015;11(11):1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Che Q, Lin L, Ai Q, et al. Caloric restriction mimetic 2-deoxyglucose alleviated lethal liver injury induced by lipopolysaccharide/D-galactosamine in mice. Biochem Biophys Res Commun. 2015. April 10;459(3):541–546. [DOI] [PubMed] [Google Scholar]
- [76].Shen C, Ka SO, Kim SJ, et al. Metformin and AICAR regulate NANOG expression via the JNK pathway in HepG2 cells independently of AMPK. Tumour Biol. 2016. August;37(8):11199–11208. [DOI] [PubMed] [Google Scholar]
- [77].Zhang D, Li J, Wang F, et al. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014. December 28;355(2):176–183. [DOI] [PubMed] [Google Scholar]
- [78].Bilanges B, Alliouachene S, Pearce W, et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat Commun. 2017. November 27;8(1):1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Jankowsky JL, Slunt HH, Ratovitski T, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol eng. 2001. June;17(6):157–165. [DOI] [PubMed] [Google Scholar]
- [80].Kramer D, Minichiello L. Cell culture of primary cerebellar granule cells. Methods Mol Biol. 2010;633:233–239. [DOI] [PubMed] [Google Scholar]
- [81].Lee HY, Greene LA, Mason CA, et al. Isolation and culture of post-natal mouse cerebellar granule neuron progenitor cells and neurons. J Vis Exp. 2009;16:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Levi G, Aloisi F, Ciotti MT, et al. Autoradiographic localization and depolarization-induced release of acidic amino acids in differentiating cerebellar granule cell cultures. Brain Res. 1984. January 2;290(1):77–86. [DOI] [PubMed] [Google Scholar]
- [83].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012. June 28;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Naldini L, Blomer U, Gage FH, et al. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996. October 15;93(21):11382–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jiang H, Vogt PK. Constitutively active Rheb induces oncogenic transformation. Oncogene. 2008. September 25;27(43):5729–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Leeman DS, Hebestreit K. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science (New York, NY). 2018. March 16;359(6381):1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Za’tara G, Bar-Tana J, Kalderon B, et al. AMPK activation by long chain fatty acyl analogs. Biochem Pharmacol. 2008. November 15;76(10):1263–1275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.