

Abstract
Objective
To explore the phenotypic spectrum of RHOBTB2-related disorders and specifically to determine whether patients fulfill criteria for alternating hemiplegia of childhood (AHC), we report the clinical features of 11 affected individuals.
Methods
Individuals with RHOBTB2-related disorders were identified through a movement disorder clinic at a specialist pediatric center, with additional cases identified through collaboration with other centers internationally. Clinical data were acquired through retrospective case-note review.
Results
Eleven affected patients were identified. All had heterozygous missense variants involving exon 9 of RHOBTB2, confirmed as de novo in 9 cases. All had a complex motor phenotype, including at least 2 different kinds of movement disorder, e.g., ataxia and dystonia. Many patients demonstrated several features fulfilling the criteria for AHC: 10 patients had a movement disorder including paroxysmal elements, and 8 experienced hemiplegic episodes. In contrast to classic AHC, commonly caused by mutations in ATP1A3, these events were reported later only in RHOBTB2 mutation–positive patients from 20 months of age. Seven patients had epilepsy, but of these, 4 patients achieved seizure freedom. All patients had intellectual disability, usually moderate to severe. Other features include episodes of marked skin color change and gastrointestinal symptoms, each in 4 patients.
Conclusion
Although heterozygous RHOBTB2 mutations were originally described in early infantile epileptic encephalopathy type 64, our study confirms that they account for a more expansive clinical phenotype, including a complex polymorphic movement disorder with paroxysmal elements resembling AHC. RHOBTB2 testing should therefore be considered in patients with an AHC-like phenotype, particularly those negative for ATPA1A3 mutations.
Heterozygous variants in RHOBTB2, encoding an atypical Rho GTPase, have recently been reported in the developmental and epileptic encephalopathies.1–3 Affected patients present with developmental delay and onset of epilepsy in the first 3 years of life.2 Given that it is a newly identified genetic disorder, the phenotypic spectrum of RHOBTB2-related disorders (RRD) has not yet been fully delineated. We report a cohort of individuals with RRD whose presentation included a prominent motor phenotype with a complex polymorphic movement disorder. In several cases, the clinical features resembled those reported in patients with alternating hemiplegia of childhood (AHC).4
Methods
Patients were identified through the Neurogenetic Movement Disorders Clinic at Great Ormond Street Hospital and the Epilepsy Genomics service at the National Hospital for Neurology and Neurosurgery, Queen Square. Additional cases were identified through collaborating neurologists and geneticists in Ireland, Denmark, Belgium, Germany, and France. Data were obtained through a retrospective review of clinical records.
Molecular genetic diagnosis was made by whole-exome sequencing, whole-genome sequencing, or gene panel testing. All results were confirmed through accredited diagnostic laboratory services except patient 10, whose diagnosis was made through research triome whole-genome sequencing with Sanger sequencing confirmation in the proband and family (REC reference 13LO168), and patients 4, 5, and 9, who were diagnosed through research whole-exome sequencing. Genetic variants not previously reported in the literature were assessed for pathogenicity following standard American College of Medical Genetics guidelines.5 Seizures were classified according to the 2017 International League Against Epilepsy classification and terminology.6
Standard Protocol Approvals, Registrations, and Patient Consents
This project was approved by the Great Ormond Street Research and Development Office (reference No. 19NM23). Because it involved retrospective collection of fully anonymized data collected during clinical care, written consent from patients' guardians was not sought but verbal assent was. When identifiable videos and photographic images are used, written informed consent for their review and publication was provided by patients or their parents or guardians.
Data Availability
The data relevant to this article are published within the text and tables.
Results
Eleven unrelated patients were identified, of whom 4 were female. Age at the latest clinical review ranged from 2 to 60 years (mean age 16.3 years, median age 9 years).
Molecular Genetic Findings
All patients had heterozygous missense variants in RHOBTB2. Seven of 11 involved the amino acid residue at position 511. All the other identified variants also occurred within exon 9 (figure 1). Three of the variants were novel (c.1531C>G, p.Arg511Gly; c.722C>A, p. Ser241Tyr; and c.717G>C, p. Trp239Cy) and were classified as likely pathogenic according to American College of Medical Genetics guidelines.5 Parental testing was available for 9 of 11 patients and in all cases confirmed de novo status (table 1).
Figure 1. Pathogenic Mutations Within the RHOBTB2 Gene.

Note that the longest transcript, NM_001160036.1, does not include exons 4 and 5. UTR = untranslated region.
Table 1.
Genetic Variants Found in RRD Cohort

Clinical Features
Movement Disorder
All patients had motor features as part of their clinical phenotype. Severity varied widely, from lack of any head or truncal control (patient 11) to inability to walk (patient 7, apparently due to a combination of hypotonia, dystonia, and ataxia) to mild balance difficulties with near-normal gait (patient 10). Pyramidal signs (such as brisk reflexes or a positive Babinski sign) were evident in 5 of 11 patients, although clear velocity-dependent spasticity was present in only 2 of them. In addition to gait difficulties, 5 of 11 patients had a generalized hyperkinetic choreiform movement disorder (videos 1–3 ). Ten of 11 patients had additional paroxysmal movements: this included hemiplegic episodes in 8, episodic focal dystonia in 7 (videos 4–6 ), episodic ataxia in 4, episodic exacerbation of dyskinesia in 4, and nonepileptic myoclonus in 2 patients. Three patients also exhibited stereotypies. The pattern and nature of paroxysmal movements often varied for each individual over time. For example, in patient 1, dyskinetic episodes were prominent when he was a toddler and then settled with age before re-emerging at 8 years of age. Five families described reliable sensory triggers such as temperature change, which elicited episodes of abnormal movement. This appeared to relate to a sudden change in temperature, for example, coming out of the bath, rather than to extremes of environmental temperature per se. Car journeys also triggered events in some families (of patients 1 and 2), possibly associated with changes in position, movement, or temperature. Two other families (of patients 4 and 5) reported a diurnal variation in episodes of unsteadiness, with increased occurrence in the mornings (tables 2 and 3).
Table 2.
Patients With RRD: Epilepsy, EEG Features, Medications, and Other Neurologic/Systemic Features

Table 3.
RRD Patients: Baseline and Paroxysmal Movement Disorders

Patient 2 as a toddler is sitting in his high chair. While he is laughing, choreiform dyskinesia and rhythmic tremulous movements involving all 4 limbs, together with some dystonic posturing of the feet, is seen.Download Supplementary Video 1 (8.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_1
Patient 1 as a baby with severe generalized hyperkinesia and chorea involving the trunk (leading to truncal instability) and all limbs. He is seen to arch to the right side and also backward.Download Supplementary Video 1 (10MB, mp4) via http://dx.doi.org/10.1212/011543_Video_2
Patient 10 at 18 years of age. Subtle dysmetria (double-tapping), hyperkinesia (possibly including low-amplitude myoclonic jerks) of the extremities, and orofacial dyskinesia are evident.Download Supplementary Video 1 (8MB, mp4) via http://dx.doi.org/10.1212/011543_Video_3
Patient 1, 1 year of age, is seen playing on the floor. Note dystonic posturing of the feet and intermittent eye deviation.Download Supplementary Video 1 (8.4MB, mp4) via http://dx.doi.org/10.1212/011543_Video_4
Patient 2 as a baby is seen after a bath. Parents report that changes of temperature such as bathing often trigger these episodes. Asymmetrical posturing of all 4 limbs and with dystonic tremor are present.Download Supplementary Video 1 (6.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_5
Patient 3, 37 years of age, is seen walking, with asymmetrical dystonic posturing of the upper limbs. Note also his broad-based, slightly asymmetrical and unsteady gait pattern.Download Supplementary Video 1 (2.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_6
Hemiplegic episodes were not reported in infancy. The earliest such episode was seen at 20 months of age (patient 8), although for the adult patients exact age at onset was not recorded. These episodes consisted of unilateral or asymmetrical weakness accompanied by what parents described as a vacant appearance (although without loss of responsiveness to external stimuli), associated with drooling and reduced activity (videos 7 and 8). Hemidystonia was reported in 1 patient (patient 8). Duration varied from minutes to 2 weeks (patients 6 and 7). In patient 6, an EEG undertaken during the longest hemiparetic episode showed contralateral voltage reduction but no epileptiform discharges, rendering nonconvulsive status epilepticus unlikely. In some but by no means all cases, hemiplegic episodes followed an identifiable trigger such as a seizure (patient 7) or a minor head injury (patient 6).
Patient 1 is seen as a toddler experiencing an episode of generalized weakness. He is quiet, still, and drooling (with tenting of the mouth) but is still alert and responsive to touch. Two brief episodes of upward eye deviation are seen.Download Supplementary Video 1 (7.7MB, mp4) via http://dx.doi.org/10.1212/011543_Video_7
Patient 2 as a baby experiences an episode of generalized weakness. He is noticeably pale, floppy, and weak, with tenting of the mouth, but remains alert. At the end of the video, as he begins to recover some movement, and his color starts to improve.Download Supplementary Video 1 (7.5MB, mp4) via http://dx.doi.org/10.1212/011543_Video_8
Epilepsy
Seven of 11 patients had a diagnosis of epilepsy (mean age at onset 14 months, range 11 week–4 years 6 months), and 2 others had experienced at least 1 seizure (electrographic seizures in patient 4 detected on EEG at 3 years of age, and febrile convulsions in patient 1 at 8 months of age). All of the patients with epilepsy had at least 1 EEG, either ictal or interictal, which showed focal or multifocal epileptiform discharges. Most interictal recordings either were normal or showed only nonspecific slowing. The commonest seizure types were focal seizures with or without impaired awareness and/or evolution to bilateral tonic-clonic seizures, seen in 6 of 7 patients.
Five patients (patients 2, 5, 6, 9, and 11) experienced status epilepticus. For 4 of them (patients 2, 5, 6, and 9), this occurred at between 2 and 5 months of age. For patient 6, but not the others, status epilepticus occurred in the context of a febrile illness. Viral encephalitis was suspected, but no infective agent was found in blood or CSF. She had clustered seizures requiring intubation and ventilation for a period of 6 days (including 2 extubation attempts that failed due to recurrence of seizures). The duration of the episode is not known for patient 5 but was ≈40 minutes in patient 2 and 2 hours in patient 9. Only 1 of these patients, patient 5, went on to have a second episode of status epilepticus, which occurred at 8 years of age. Patient 11 had a single prolonged episode of status epilepticus at 4 years 6 months of age in the context of a severe encephalitis-like episode during which he was comatose for 3 months and experienced hypothermia and autonomic disturbances. After this, he experienced severe and permanent neurodevelopmental impairment.
A number of events clinically suspected to be seizures such as myoclonus, paroxysmal lateral deviation of the head and eyes, and behavioral changes were captured on EEG in some patients (patients 1, 2, 5, 8, 10, and 11) and proved to have no electrographic correlate. For patients 4, 6, and 7, however, ictal EEG was not captured for the majority of event types.
All patients except patients 9 and 10 were taking antiseizure medications at their last review, although in several instances, the main indication was for treatment of abnormal movements rather than epilepsy. The most widely used drug was carbamazepine in 7 of 11 patients, which was felt to be clearly beneficial in at least 2 cases (patient 1 and 5), resulting in reduced frequency of episodes, possibly including reduced paroxysmal movements and epileptic seizures. Topiramate improved seizure control in at least 1 patient (patient 7), whose seizures reduced from 20 per week to 1 per week, and another patient (patient 8, who did not have epilepsy) experienced reduced frequency of abnormal movements on a combination of topiramate and flunarizine. (In another patient [patient 1], however, neither topiramate nor flunarizine showed any benefit for nonepileptic paroxysmal movements.) Two patients (patients 6 and 4) also experienced some improvement with acetazolamide, which was prescribed primarily to treat episodic ataxia. One family (of patient 1) also reported a benefit from a medium-chain triglyceride dietary supplement.7 The effect of other medications was less clear in this patient cohort.
Other Paroxysmal Events
Eight patients (patients 1, 2, 4–6, and 9–11) had a history of an acute encephalitis-like illness, but the forms this took were variable. In 5 cases (patients 2, 5, 6, 9, and 11), the episode included convulsive status epilepticus, as described above. Two of these episodes (patients 6 and 11) also included thermoregulatory changes—hypothermia in patient 11 and hyperthermia in patient 6—and skin changes, described as flushing in patient 11 but simply as rash in patient 6. Patient 6 in addition went on to have 3 further episodes of loss of consciousness between 3 and 4 years of age, 2 of which immediately followed minor head trauma. Vomiting occurred on at least 1 occasion. Unconsciousness lasted for ≈24 hours on the first occasion and a few hours on the subsequent times but was followed by hemiplegia, which took up to 2 weeks to resolve. EEG was captured during unconsciousness on 2 of these occasions and did not show any epileptiform activity, although on the first occasion voltage was reduced in the hemisphere contralateral to the hemiplegia.
At the age of 3 years, patient 4 had an episode of hemiplegia lasting several hours associated with contralateral electrographic seizure activity in the midtemporal region. MRI of the brain in the acute phase showed changes consistent with acute ischemia in the affected hemisphere but subsequently normalized. Patient 10, also at the age of 3 years, experienced an episode of vomiting and drowsiness severe enough to require brief mechanical ventilation. EEG during the episode was encephalopathic but did not show seizure activity. Patient 1 had 3 separate episodes of behavioral and neurodevelopmental regression associated with increased frequency of paroxysmal abnormal movements and increased vasomotor skin changes at 13 months, 28 months, and 7 years of age. His EEG initially showed only mild slowing, but epileptiform discharges—without clinical correlate—appeared only from the age of 7 years on.
Paroxysmal abnormal eye movements were reported in patients 1, 2, 5, 8, and 11. In patients 1, 2, 5, and 8, these took the form of brief lateral deviation of both eyes, sometimes accompanied by turn of the head in the ipsilateral direction and/or by tongue protrusion. These events were captured on EEG and had no electrographic ictal correlate. Events could occur multiple times per day but were only seconds in duration. Patient 11 had episodes of nystagmus.
One patient (patient 1) experienced recurrent episodes of respiratory disturbance starting at the age of 7 years. These consisted of shallow or irregular breathing or sometimes complete apnea requiring bag-valve-mask ventilation, accompanied by tachycardia and pupillary dilation, lasting for up to 20 minutes. Ventilation was not technically challenging, and there was usually no laryngeal dystonia. The child often remained able to move his head and appeared to be aware of people around him, but his limbs and trunk were paralyzed and flaccid. Episodes could occur from sleep or waking, and there were no obvious triggers. There was no postevent drowsiness. The nature of these episodes remains unclear.
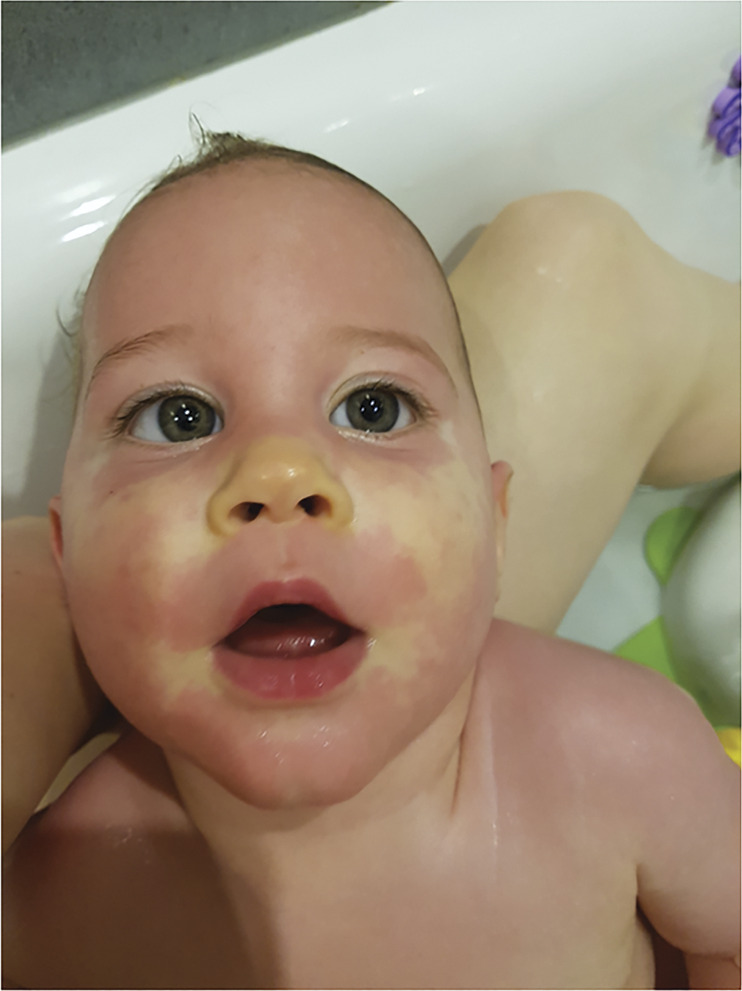
Other reported episodic phenomena included striking episodes of flushing or pallor, sometimes associated with increased abnormal movements, in 4 individuals (patients 1, 2, 5, and 11; figure 2).
Figure 2. Patient 2, 1 Year of Age, Demonstrating an Episode of Patchy Erythema and Pallor During a Bath.
Other Comorbid Conditions
All patients had intellectual disability. Formal IQ testing was not available for most patients, but for 10 of 11, intellectual disability was clinically assessed as moderate or severe, with either absent or significantly impaired verbal communication and comprehension. Patient 10, however, had a milder impairment compared to the rest of the cohort. Only 2 patients experienced developmental regression. In patient 11, there was a severe and lasting regression at the age of 54 months, following a prolonged encephalopathic episode accompanied by status epilepticus and resulting in near-total and permanent loss of developmental abilities. In patient 1, regression occurred on 3 occasions, each time coinciding with exacerbation of the movement disorder. In 3 other patients (patients 5, 6, and 9), developmental slowing was noted after the onset of epilepsy, which in all these cases was very early (≤3 months of age). Milestones (such as social smiling, visual attentiveness, and partial head control) before this age had been apparently normal but were delayed afterward. Patient 6 also experienced a further period of developmental stagnation at 3 years of age at a time of increased seizure frequency.
Marked emotional lability was reported in 2 patients (patients 2 and 10), irritability in 1 patient (patient 1), and challenging behavior in 3 more individuals (patients 3, 5, and 8). Gastrointestinal symptoms, including gastroesophageal reflux, constipation, and episodes of abdominal discomfort, were described in 4 patients (patients 1, 2, 6, and 7).
Brain MRI Findings
MRI brain results were available for 9 of 11 patients. Three patients had normal scans, and 1 other patient (patient 11) had a normal scan at 4 years of age with cerebrocerebellar atrophy apparent on a scan a year later, after a period of significant developmental regression. Two patients (patients 1 and 9) had a thin corpus callosum. Other findings found in 1 patient each included mild static cerebellar atrophy (patient 4), mild dilation of the temporal horns (patient 10), nonspecific occipital lobe changes (patient 6), delayed myelination (patient 9), and meningeal enhancement seen on a scan performed at the time of an episode of status epilepticus (patient 9).
Discussion
RHOBTB2 is a relatively newly recognized disease-causing gene. It encodes a Rho GTPase, one of a family of proteins involved in regulating actin dynamics, and is believed to play a key role in cell morphology and, more specifically, dendritic arborization. It is highly expressed in brain, especially the cortex and basal ganglia.8
Pathogenic mutations in RHOBTB2 have previously been reported in 13 patients.2,3 All reported variants are de novo missense changes, suggesting either complete or very high penetrance. Several of the reported variants are recurrent mutations, an observation further confirmed in our study, in which 7 of the 9 mutations are previously described. Moreover, all known pathogenic variants are found clustered in exon 9, which encodes the BTB1 protein domain and part of BTB2. In our cohort, 7 of 11 patients carried a pathogenic variant affecting residue Arg511. This residue was also identified as the most frequent mutational hotspot in previously reported cohorts.2,3 We did not, however, identify a significant difference in phenotype between patients who had a mutation involving this residue and those with mutations affecting other sites. Exploration of any genotype-phenotype correlation would require analysis of a larger cohort, which may be expected to become available as more patients are diagnosed with RRD. Although our most mildly affected patient (patient 10) has a mutation slightly outside the main cluster of reported pathogenic variants, so too does patient 11, who has an exceptionally severe phenotype (figure 1).
Developmental and epileptic encephalopathy was a universal feature in previously reported cases, with all having onset of seizures by 4 years of age and 11 patients presenting before 1 year of age. Therefore, this disorder was designated early infantile epileptic encephalopathy type 64 (Mendelian Inheritance in Man 618004), although it is clear that the clinical phenotype is more extensive. Epilepsy is not always present, and besides epilepsy, RRD can also present with a number of other comorbid conditions, including intellectual disability, neurobehavioral features, acute encephalopathic episodes, and, as demonstrated by our data, a prominent movement disorder.
Eleven of 13 of the previously reported cases were deemed to have a movement disorder of some description, including dystonia, dyskinesia, paroxysmal disorders, and stereotypies, but this has not been described in detail. All 11 patients in our cohort display a complex, polymorphic movement disorder, usually with both a paroxysmal and nonparoxysmal element. This was, in most cases, a more salient and disabling feature than their epilepsy.
All patients within our cohort had a complex motor phenotype, with at least 2 types of movement disorder. Most patients experienced a hyperkinetic movement disorder with dystonia and/or dyskinesia, but other abnormal movements such as myoclonus were also seen. Severity of functional impact varied widely: 3 of 11 patients were nonambulant; 2 could walk only with support; and 1 had only mild gait disturbance. Gait difficulties in RRD may arise as from a combination of delayed motor development and an underlying movement disorder.
The paroxysmal movement disorders were also variable but notably in the majority included paroxysmal hemiplegia. This differed somewhat from classic AHC caused by mutations in ATP1A39 in that hemiplegic episodes first appeared later in childhood, from 20 months on rather than by 18 months as in the accepted criteria of AHC4 (table 4). The hemiplegic spells appeared to share many features with episodes that were characterized by symmetrical weakness, affecting both sides of the body, often accompanied by pallor, quietness, and a vacant appearance but continued responsiveness to external stimuli. In several individuals (patients 1, 2, and 10), these symmetrical episodes appeared some time before the bilateral asymmetrical episodes. As described in classic AHC, other paroxysmal phenomena were also evident in this cohort, including exacerbations of dyskinesia, episodic ataxia, and focal dystonia, often associated with reproducible sensory triggers. Unlike in ATP1A3-related AHC, episodes did not reliably terminate with sleep. Some disordered eye movements were observed, including frequent lateral deviation of both eyes in patients 1, 2, 5, and 8, but these did not particularly resemble the specific eye movement disorders such as monocular nystagmus that are reported in ATP1A3-related disease.
Table 4.
Comparison Between Classic AHC and Alternating Hemiplegia Occurring in RRD

The majority of our cohort did have epilepsy, with focal seizures being the commonest. Epilepsy was not in general the most debilitating feature of the disorder. Four of 7 patients with epilepsy were seizure-free (on or off medication) at the time of their last review. It is plausible that some of the seizures described, including myoclonic, gelastic, and atypical absence episodes, were not always truly epileptic events, because very similar events in other patients were found to have no EEG correlate. In most cases, it was often hard or impossible to distinguish clinically between epileptic and nonepileptic paroxysmal events. Frequently, episodes that clinicians had believed to be clearly epileptic (such as the head deviation/eye deviation/tongue thrusting episodes seen in patients 1, 2, 5, and 8) proved to have no electrographic ictal correlate. It remains possible, however, that paroxysmal abnormal movements are mediated by electric discharges analogous to epilepsy but involving deeper foci that may be inaccessible by standard EEG.
Moreover, although ictal EEG was recorded for most patients, for some individuals who experienced a wide range of different event types, not every type was captured. Evaluation is further complicated by the fact that the types and frequencies of events, both epileptic and nonepileptic, often changed over time, with the frequency of particular episode types waxing and waning. A fuller understanding of the coexistence of epilepsy and paroxysmal movement disorders in patients with RRD could be achieved in the future by a prospective longitudinal study, ideally including both serial EEGs and repeated re-evaluations of the semiology of clinical events. We note that similar issues affect evaluation of paroxysmal episodes in individuals with AHC due to mutations in ATP1A3.
In all but 2 patients (patients 5 and 9, whose epilepsy started in the first months of life), developmental delay was already evident before the first seizure, suggesting a key physiologic role for RHOBTB2 in neurodevelopment. Two patients (patients 1 and 11) experienced developmental regression, either in association with exacerbation of the movement disorder (patient 1) or after an encephalopathic illness including status epilepticus (patient 11). Developmental stagnation was seen in 3 patients, again either during a period of increased abnormal movements (patient 6) or coinciding with epilepsy onset and/or worsening (patients 5, 6, and 9). Control of both seizures and paroxysmal movements may therefore be required to optimize neurodevelopmental outcome, but in view of the different therapeutic approaches required, it remains important, although challenging, to distinguish between paroxysmal movements and epileptic seizures. On the whole, control of epileptic seizures was achieved more successfully in our cohort than control of paroxysmal movements. Some agents such as carbamazepine may have been efficacious for both. Acetazolamide, used primarily for movement disorders although it does have antiepileptic activity, also showed some benefit for ataxic episodes in 2 patients (patients 4 and 6) in whom good control of epilepsy was already achieved. However, the numbers in our cohort are certainly too small to draw firm conclusions as to the most effective therapeutic approaches, and this should be a priority for future research.
RRD are complex, and our study confirms association with a number of neurologic and systemic comorbid conditions. Many patients had behavioral and/or emotional difficulties as well as gastrointestinal features and, interestingly, vasomotor symptoms (excessive flushing and/or pallor), often observed during periods of increased paroxysmal movements. These could potentially represent unexplored dysautonomic phenomena. The same may be true of the paroxysmal respiratory disturbances seen in 1 patient (patient 1).
On the basis of the findings in our cohort, there is no clear pattern of progression over time. Other than patient 11, who had a single episode of severe regression with lasting consequences, and patient 1, who experienced several episodes of regression, most patients achieved some neurodevelopmental gains over time, and neither epilepsy nor movement disorders were reported to worsen with age. However, many patients in our cohort are still very young, and an accurate longitudinal assessment of natural history would clearly be valuable to determine whether RRD are associated with progressive disease features in the longer term.
Our findings suggest that the phenotype of RRD extends beyond early infantile epileptic encephalopathy and encompasses a complex neurologic disorder characterized by prominent motor features, a wide variety of episodic phenomena, a broad spectrum of intellectual and motor disability, and, in some cases, distinctive dysautonomic or vasomotor phenomena. There seems to be a significant degree of overlap with the ATP1A3-related disease spectrum, including AHC, albeit with hemiplegic episodes emerging later in childhood, as seen in CAPOS (cerebellar ataxia, arreflexia, pes cavus, optic atrophy, and sensorineural hearing loss) and intermediate phenotypes. A registry of patients with RHOBTB2 mutations and a prospective study of their natural history would facilitate a better understanding of this disorder and identify targets for therapeutic intervention.
Acknowledgment
The authors thank the participants and their families for their involvement. M.A.K.'s research group (including D.S.) is funded by sources including the National Institute of Health Research, Rosetrees Trust, and the Jules Thorn Trust. They thank Dr. Victoria Nesbitt for her involvement in diagnosis of a patient. They also thank the Epilepsy Society for support. Part of this work was undertaken at University College London Hospitals, which received a proportion of funding from the National Institute of Health Research Biomedical Research Centres funding scheme.
Glossary
- AHC
alternating hemiplegia of childhood
- CAPOS
cerebellar ataxia, arreflexia, pes cavus, optic atrophy, and sensorineural hearing loss
- RRD
RHOBTB2-related disorders
Appendix. Authors
Study Funding
No specific funding of sponsorship was received for this work. M.A. Kurian and D. Steel are funded by a National Institute of Health Research Professorship, and M.A.K.'s research group also benefits from funding from the Sir Jules Thorn Trust and Rosetrees Trust. All research at UCL Great Ormond Street Institute of Child Health is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Lopes F, Barbosa M, Ameur A, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet 2016;53:190–199. [DOI] [PubMed] [Google Scholar]
- 2.Straub J, Konrad EDH, Grüner J, et al. Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in drosophila. Am J Hum Genet 2018;102:44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belal H, Nakashima M, Matsumoto H. De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum Mutat 2018;39:1070–1075. [DOI] [PubMed] [Google Scholar]
- 4.IHS Classification ICHD-3. London: International Headache Society; 2019. Available at: ichd-3.org/appendix/a1-migraine/a1-6-episodic-syndromes-that-may-be-associated-with-migraine/a1-6-5-alternating-hemiplegia-of-childhood/. Accessed September 2019. [Google Scholar]
- 5.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522–530. [DOI] [PubMed] [Google Scholar]
- 7.ClinicalTrials.gov. Use of Betashot in Children and Adults With Epilepsy (study in progress). Available at: clinicaltrials.gov/ct2/show/NCT02825745. Accessed October 2019.
- 8.UK Brain Expression Consortium (UKBEC). Available at:braineac.org. Accessed November 2019. [Google Scholar]
- 9.Heinzen EL, Swoboda J, Hitomi Y, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 2012;44:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosewich H, Sweney MT, DeBrosse S, et al. Research conference summary from the 2014 International Task Force on ATP1A3-related disorders. Neurol Genet 2017;3:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Patient 2 as a toddler is sitting in his high chair. While he is laughing, choreiform dyskinesia and rhythmic tremulous movements involving all 4 limbs, together with some dystonic posturing of the feet, is seen.Download Supplementary Video 1 (8.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_1
Patient 1 as a baby with severe generalized hyperkinesia and chorea involving the trunk (leading to truncal instability) and all limbs. He is seen to arch to the right side and also backward.Download Supplementary Video 1 (10MB, mp4) via http://dx.doi.org/10.1212/011543_Video_2
Patient 10 at 18 years of age. Subtle dysmetria (double-tapping), hyperkinesia (possibly including low-amplitude myoclonic jerks) of the extremities, and orofacial dyskinesia are evident.Download Supplementary Video 1 (8MB, mp4) via http://dx.doi.org/10.1212/011543_Video_3
Patient 1, 1 year of age, is seen playing on the floor. Note dystonic posturing of the feet and intermittent eye deviation.Download Supplementary Video 1 (8.4MB, mp4) via http://dx.doi.org/10.1212/011543_Video_4
Patient 2 as a baby is seen after a bath. Parents report that changes of temperature such as bathing often trigger these episodes. Asymmetrical posturing of all 4 limbs and with dystonic tremor are present.Download Supplementary Video 1 (6.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_5
Patient 3, 37 years of age, is seen walking, with asymmetrical dystonic posturing of the upper limbs. Note also his broad-based, slightly asymmetrical and unsteady gait pattern.Download Supplementary Video 1 (2.6MB, mp4) via http://dx.doi.org/10.1212/011543_Video_6
Patient 1 is seen as a toddler experiencing an episode of generalized weakness. He is quiet, still, and drooling (with tenting of the mouth) but is still alert and responsive to touch. Two brief episodes of upward eye deviation are seen.Download Supplementary Video 1 (7.7MB, mp4) via http://dx.doi.org/10.1212/011543_Video_7
Patient 2 as a baby experiences an episode of generalized weakness. He is noticeably pale, floppy, and weak, with tenting of the mouth, but remains alert. At the end of the video, as he begins to recover some movement, and his color starts to improve.Download Supplementary Video 1 (7.5MB, mp4) via http://dx.doi.org/10.1212/011543_Video_8
Data Availability Statement
The data relevant to this article are published within the text and tables.