

Abstract
The fan mussel, Pinna nobilis, endemic to the Mediterranean Sea, is a critically endangered species facing mass mortality events in almost all of its populations, following the introduction of the parasite Haplosporidium pinnae. Such a unique pandemic in a marine organism, which spreads rapidly and with mortality rates reaching up to 100%, could lead to the potential extinction of the species. Only few regions, involving lagoon habitats, remain healthy throughout the entire Mediterranean Sea. This study describes the genetic structure of P. nobilis across the Gulf of Lion, including confined locations such as lagoons and ports. A total of 960 samples were collected among 16 sites distributed at 8 localities, and then genotyped using 22 microsatellite markers. Genetic diversity was high in all sites with mean allele numbers ranging between 10 and 14.6 and with observed heterozygosities (Ho) between 0.679 and 0.704. No genetic differentiation could be identified (FST ranging from 0.0018 to 0.0159) and the percentages of related individuals were low and similar among locations (from 1.6 to 6.5%). Consequently, all fan mussels, over the entire coastline surveyed, including those in the most geographically isolated areas, belong to a large genetically homogeneous population across the Gulf of Lion. Considering the ongoing mass mortality context, this result demonstrates that almost all of the genetic diversity of P. nobilis populations is still preserved even in isolated lagoons, which might represent a refuge habitat for the future of the species.
Subject terms: Population genetics, Conservation biology
Introduction
Recent extinction rates have been shown to be 100 to 1000 times their pre-human levels in taxonomically diverse groups through a wide variety of different habitat types, and they continue to accelerate1. These extinctions are mainly caused by anthropic activities such as pollution, artificialization, overexploitation, habitat loss and global change, which can lead to the isolation of populations, reduction in their size, and thus a loss of genetic diversity2. The conservation genetics concept has been pushed forward in the context of population genetics with the goal of better understanding the dynamics of genes in populations to avoid extinctions. The idea is to apply genetic methods to the conservation and restoration of biodiversity, with genetic diversity being the proxy for the level of extinction risk. Genetic diversity enhances the stability and reliability of ecosystems by providing biological insurance against environmental change3. Hence, maintaining a high level of genetic diversity is one of the main targets of conservation biology. Knowledge concerning the level and the distribution of genetic diversity within and among populations of a given species, and on the processes that ensure its maintenance are now more than ever, necessary in order to design conservation strategies.
In marine systems, coastal environments are generally viewed as discontinuous and most organisms are sessile or have very limited movement abilities. There is thus a strong interaction between habitat fragmentation of coastal environments and species life-history traits that can affect dispersal and population connectivity, as found for the pelagic early life stage which represents the main opportunity for movement4. This dispersal phase allows connectivity between distant areas and is thus a critical feature of the ecology of marine organisms and evolution, as it drives the genetic structure and composition of populations5. The degree of connectivity between populations has an impact on local population survival through replenishment processes, the maintenance of high levels of diversity and by determining their persistence and resilience in case of local disturbance6,7. Enhancing dispersal will certainly favor the long-term persistence of species, in a framework where dispersal will be affected by parameters such as habitat fragmentation, effective population size, anthropic activities and climate change8,9.
The fan mussel, Pinna nobilis (Linnaeus, 1758), is a bivalve endemic to the Mediterranean Sea, that lives half-buried in soft-bottom habitats, generally covered by seagrass meadows. Because it is associated with seagrass meadows, P. nobilis is restricted to subtidal areas, up to 30 m deep, as beyond the meadow development is limited by light intensity and water transparency. As a consequence, P. nobilis distribution is usually aggregative and fragmented. Even if this association is generally accepted, very dense aggregations of the species were recently reported in lagoons or in artificial and degraded habitats such as ports, which puts into question the species’ habitat requirements10.
P. nobilis is a successive hermaphrodite organism with asynchronous gamete maturation, thus preventing self-fertilization11. Several consecutive spawnings occur during summer, producing millions of veliger larvae. For P. nobilis, larval duration has been estimated to be around 5 to 10 days during which larvae are spread by currents12. However, there remains a significant lack of knowledge concerning the P. nobilis life cycle, as a recent study has shown that the larval stage could last up to 20 days under controlled conditions13, a much longer larval duration which would certainly enhance dispersal capabilities.
The species has been overexploited for years, for consumption or for jewelry fabrication. Over the last decades, human activities (anchoring, pollution, habitat reduction and fragmentation) have kept P. nobilis populations under high pressure which has led to population decay14. Since 1992, the fan mussel has been under strict protection, according to the European Council Directive 92/43/EEC (Annex IV) and the national laws of most Mediterranean countries. In recent years, P. nobilis abundance has increased and dense populations have been observed in anthropized habitats, such as lagoons and ports, which were previously thought to be unfavorable to their settlement as the species is generally considered as a bioindicator of the quality of coastal waters15. However, despite its endangered status, only few studies have investigated population genetics and connectivity patterns in P. nobilis16–21. Furthermore, information on the contemporary dynamics is limited, as none of the previous studies integrated the large populations recently described in lagoons.
Today, the species is facing a major ongoing pandemic that threatens its survival. Starting in October 2016, mass mortalities have been observed in fan mussel populations, probably caused by the protozoan parasite, Haplosporidium pinnae22. However, the causes of the outbreak are far from being completely understood as one recent study supported the occurrence of a multifactorial disease with non-species-specific pathogens that could have triggered mass mortalities in P. nobilis populations23 whereas another study stated that H. pinnae is the only pathological agent considered as essential for the onset of the mortality24. The very first signs of mortality were observed in southeast Spain25 and the epidemic has now spread throughout the entire Mediterranean Sea26. To date, most of the infected populations have been devastated. This is an unprecedented situation for which neither the mortality rates (around 100%) nor the speed of propagation23,26,27 have ever been recorded for a marine species, and it could lead to the potential extinction of P. nobilis. Following this critical situation, the status of the species has been reevaluated and has been recently moved to “critically endangered” on the IUCN red list28. However, a couple of populations in localities along the early infected Spanish coastline remain less affected by the parasite mortality (Alfacs Bay and Mar Menor, Spain22,26). Even if the reasons are not yet well understood, greater survival rates might be linked to salinity ranges in those areas compared to the open sea26: Alfacs Bay is subjected to high freshwater inflow of the Ebro River whereas Mar Menor is a confined coastal lagoon where salinity can reach up to 40 PSU. Even if observations are limited, lagoons are now a priority habitat in Spanish conservation plans, as large populations have recently been described in this habitat type throughout the Mediterranean Sea.
While accounting for about 250 km of coastline in the Mediterranean Sea, the Gulf of Lion is characterized by numerous lagoons with various hydromorphological characteristics29. Recent surveys in these habitats revealed the presence of P. nobilis in high densities, even much higher than those observed in the usual seagrass meadows (T. Morage, pers. com.). Recent monitoring data demonstrated that almost 90% of the population of P. nobilis is found in the lagoons of Thau and Salses-Leucate along the Gulf of Lion10 and, to date, these areas are still free from parasite contamination. Considering the current pandemic situation for P. nobilis, these lagoons may represent a key habitat in a conservation strategy and may provide hope for the persistence of the species. In the meantime, as these lagoons are unstable environments, facing variations in nutrient concentrations or fresh water inflows29, the long-term survival of the species may be uncertain. Lastly, these lagoons are rendering the distribution of P. nobilis highly fragmented and isolated in small patches, and require better knowledge about the population structure and dispersal patterns of the species. Here, we used highly polymorphic markers to (i) assess the genetic diversity of fan mussel aggregations in the different types of habitats, (ii) estimate whether the habitat fragmentation tends to create genetic differentiation and spatial genetic structuring between aggregations and (iii) assess the regional patterns of dispersal throughout the Gulf of Lion. This work is the first genetic study of P. nobilis populations in the highly fragmented environment of the Gulf of Lion where lagoon habitats were also considered, before the spread of the pandemic along the French Mediterranean coast.
Materials and methods
Sampling
As P. nobilis is an endangered species, we received necessary permissions for sampling from the DREAL (Direction Régionale de l’Environnement, de l’Aménagement et du Logement) of Occitanie (prefectural order n°2018-s-24). All Methods were carried out in accordance with relevant guidelines and regulations. Samples were collected from all sites where aggregations of P. nobilis were observed following a year of extensive and systematic searching in all habitats along the coast of the Gulf of Lion, from the Spanish border to the Rhône estuary. A total of 960 tissue samples were collected from 16 sites along the coast during the summer of 2018 (see Fig. 1 and Table 1 for collection information). We assume that these 16 sites provide a fairly complete sampling of the majority of the main aggregations of P. nobilis in the area. Sampled sites were segregated by location, type of habitat (sea, lagoon, port or lagoon linked to the sea via a port) or population density for further genetic analyses. Sites were structured in 8 localities which refer to the geomorphological entities where they were sampled (Table 1). For each site, SCUBA divers biopsied ~ 1 cm3 of mantle tissue on random individuals, without moving the individual. This method was shown to be non-lethal after an initial test and survey (T. Morage, pers. com). As a preventive measure, the smallest individuals were not sampled to avoid lethal biopsies. Each tissue sample was stored in absolute ethanol at room temperature.
Figure 1.

Location of 16 sampled sites for Pinna nobilis in the Gulf of Lion, north-western Mediterranean Sea. This map was created using R 4.0.3 (https://www.r-project.org/) using coastline and surface water data provided by https://www.data.gouv.fr/fr/.
Table 1.
Collection information for the 16 sampling sites in the Gulf of Lion (north-western Mediterranean Sea) where the 960 Pinna nobilis individuals were collected for this study.
| Geomorphological entity | Site | Type of environment | Number of individuals sampled | Population density (/100 m2) | Estimated abundance | Percentage of abundance sampled (%) | GPS | |
|---|---|---|---|---|---|---|---|---|
| Longitude | Latitude | |||||||
| Côte des Albères | Peyrefite | Sea | 157 | 1.25 | 17,000 | 1 | 3.158431 | 42.460506 |
| Port-Vendres | 19 | 0.15 | 3.113699 | 42.518787 | ||||
| Leucate | Leucate Chenal | Port + Lagoon | 50 | 4.58 | 140,000 | 0.2 | 3.02771 | 42.808951 |
| Leucate Sud | 53 | 14.5 | 3.02322 | 42.81098 | ||||
| Leucate Port | 105 | 70.83 | 3.03474 | 42.86486 | ||||
| Leucate Nord | 54 | 7.72 | 3.043567 | 42.884103 | ||||
| Ayrolle | Ayrolle | Lagoon | 51 | 0.63 | 2500 | 2 | 3.07408 | 43.06163 |
| Cap d'Agde | Cap d'Agde | Sea | 116 | 4.83 | 200 | 58 | 3.49488 | 43.27064 |
| Port Ambonne | Port Ambonne | Port | 35 | 1.22 | 50 | 70 | 3.5272 | 43.29226 |
| Thau | Thau Sud | Port + Lagoon | 47 | 7.29 | 90,000 | 0.2 | 3.544564 | 43.328782 |
| Lido | 54 | 1.42 | 3.638553 | 43.394586 | ||||
| Mèze | 24 | 1.59 | 3.609889 | 43.423402 | ||||
| Eaux Blanches | 34 | 2.64 | 3.678688 | 43.41625 | ||||
| Port de Sète | 61 | 4.03 | 3.701066 | 43.408866 | ||||
| Frontignan | Frontignan | Port | 49 | 5.97 | 150 | 32.7 | 3.776224 | 43.434929 |
| Port-St-Louis | Port-St-Louis | Sea | 51 | 1.74 | 4000 | 1.3 | 4.88147 | 43.37038 |
Size estimation
The size of each individual sampled was calculated to identify different size groups that will correspond to fan mussels that settled over successive years and which could therefore represent different clouds of larvae. Because P. nobilis lives half-buried in the sediment, it is impossible to directly measure the total length of the shell and thus the size of each sampled individual had to be estimated from three morphometric measurements: the maximum width, the minimum width and the unburied length, as described in Garcia-March et al.30. The following equation, described in De Gaulejac31, is used to determine the total length:
where Ht is the total length; H the unburied length; l the minimum width and L the maximum width.
The size estimates were used to further differentiate size groups and to investigate genetic structure in relation to size structure in order to search for genetic heterogeneity of recruitment.
Molecular analyses
Extraction of genomic DNA from the 960 samples was performed using the QIAcube HT robot following the manufacturer’s instructions (Qiagen, Hilden, Germany). All samples were genotyped using 30 microsatellite markers from Peyran et al.32 and González-Wangüemert et al.33.
PCRs were performed following the same method as described in Peyran et al.32. Microsatellite markers were amplified using Type-it Microsatellite PCR kit (Qiagen, Hilden, Germany) in a final volume of 12.5 µL including 4 µL Type-it Multiplex PCR Master Mix (2X), 6 µL RNase-free water, 1 µL of each primer (2 µM forward and reverse primers diluted in TE pH 8 buffer) and 1.5 µL of DNA template with a concentration of around 50 ng/µL. PCR programs consisted of an initial denaturing step of 5 min at 95 °C, followed by 40 cycles of 30 s at 95 °C, 1 min 30 s at annealing temperature (53–63 °C, depending on the locus32,33), 30 s at 72 °C and a final elongation step at 60 °C for 30 min. Loci were combined in multiplex panels according to their allele size and primer annealing temperature. Individuals were genotyped by assessing the allele size. Forward primers were labelled with fluorescent dyes (PET, NED, VIC, 6-FAM, Applied Biosystems) and PCR products were sent to a private company, GenoScreen (Lille, France), for fragment genotyping where they were visualized using an Applied Biosystems 3730 Sequencer. GeneScan 500 LIZ (Applied Biosystems) was used for accurate sizing. Allele sizes were scored and checked manually using GENEMAPPER v.5 (Applied Biosystems). Samples presenting ambiguous peak profiles were re-amplified, genotyped and re-scored and all peak profiles that were still unclear were treated as missing data.
Data analysis
As mollusks often present amplification troubles during PCR, the quality of the microsatellite markers was investigated using MICROCHECKER v 2.2.334 to search for the presence of null alleles, scoring errors and large allele dropout. On the original panel of 30 markers, eight loci presented evidence of null allele high frequencies and were removed from the study to avoid any bias in further analyses. The final set of markers contained 22 polymorphic loci (Supplementary Table S1).
A preliminary genetic analysis was performed to confirm that some sites within the same area could be agglomerated into localities. As no genetic differentiation was identified (Supplementary Table S2), samples were grouped into 8 localities considered as units for further analyses.
Genetic diversity was investigated using GenAlex 6.50335, for each locality and between size groups within locality through mean allele numbers (Na), number of private alleles (Ap), and expected (He) and observed (Ho) heterozygosities. As sample size ranged from 35 to 262 across localities, standardized allelic richness (Ar) and standardized private allelic richness (Apr) were estimated using ADZE software36, based on a standardized sample size of 35 (smallest sample size). Hardy–Weinberg exact test was also performed for each loci using GENEPOP software37. The p-value of the exact test of Hardy–Weinberg was estimated based on a Markov Chain algorithm (dememorization = 10,000, number of batches = 1000 and iterations per batch = 10,000). Values for inbreeding coefficient (FIS) were calculated for all loci and for each localities using the method of Weir and Cockerham38, implemented in Genetix 39 and significance of values was established by permutations (1000 permutations per population). Differentiation index (FST) was calculated between locality pairs and between size groups by using the Robertson and Hill estimator for FST40 corrected by Raufaste and Bonhomme41, RH’, implemented in Genetix, as this estimator is unbiased and shows a lower variance when values of FST are low (< 0.05). The Bonferroni sequential correction for multiple tests was then applied to correct the p-values42.
An exploration for any genetic structuring was also performed with a MultiDimensional Scaling (MDS), using the stats package implemented in R software v. 1.1.45343 and based on the matrix of Nei’s genetic distances between localities which was calculated using GenAlex. The molecular genetic variance was analyzed between and within localities using the AMOVA framework in Arlequin suite v. 3.544.
Genetic structure was also explored using a model-based clustering method implemented in STRUCTURE 2.3.445. An admixture model with no prior information about sampling location was used with a burn-in period of 50 000 iterations, followed by a 100,000 Monte Carlo Markov Chain (MCMC) replicates for K = 1 to K = 8 clusters and 10 iterations for each K. The choice of the most likely number of clusters (K) was made by calculating an ad hoc statistic (ΔK) using Structure Harvester online46 and based on the rate of change in the log probability of data between successive K as described by Evanno et al.47. Several authors pointed out that uneven subpopulations sampling may bias STRUCTURE results and lead to overestimate the number of clusters48. StructureSelector49, a web based software, was thus also used to estimate the number of clusters (K) based on four estimators, MedMedK, MedMeaK, MaxMedK and MaxMeaK, developed by Puechmaille48 and that were shown to be less sensitive to uneven sampling.
A Mantel test was performed to test the isolation by distance hypothesis, based on geographic (distances with straight lines or those which followed the coastline were both tested) and Nei’s genetic distances between locations. The analysis was conducted using the R package ape50.
Lastly, the genetic relatedness was calculated between all pairs of individuals by the rxy coefficient51 using the R package Demerelate52 to investigate family structure within localities and within sites. The relatedness coefficient indicates the proportion of shared alleles between two individuals. Thus, with a sufficient number of highly polymorphic loci, a rxy > 0.25 between two individuals assumes that they have at least one parent in common. The percentage of individuals sharing at least one parent (Prxy) was calculated within each locality (i.e. the percentage of relations with rxy > 0.25) and for each size group that could be identified in some localities. This percentage gives a proxy of the admixture in each locality as relatedness indexes are calculated between individuals of different size classes. Because the percentage of individuals biopsied in relation to the estimated abundance is different among localities and very low in some cases (Table 1), the effects of sampling effort on the final results needed to be estimated. We thus modeled Prxy depending on the number of individuals in a given sample, using a repeated random sub-sampling method, similar to rarefaction curves. For each locality, new datasets of 10 individuals were created by randomly selecting individuals in the existent database. Other datasets were then created by successively adding individuals that were randomly drawn (without replacement) in the existing database (3 individuals for localities where sample size was small, or 5 individuals for localities where sample size was high). For each locality, we thus had new datasets with 10, 13, 16, etc. or 10, 15, 20, etc. individuals until approaching the sample size. The procedure was repeated five times to increase the number of new datasets. Relatedness coefficients were calculated between all pairs of individuals within each of these new datasets and Prxy was estimated. A non-linear regression was then performed to model the relation between the number of individuals sampled and Prxy in each locality in order to estimate the threshold of sampling effort where adding more individuals does not increase the percentage of related individuals found. The non-linear regression was also used to extrapolate the maximum Prxy in localities where sample size was too small.
Ethical approval
The sampling was non-lethal and approved by the DREAL (Direction Régionale de l’Environnement, de l’Aménagement et du Logement) of Occitanie (prefectural order n°2018-s-24).
Results
Genetic diversity
13 out of 22 markers (see Table S1) as well as 5 out of 8 localities (Table 1) showed low but significant FIS values, indicating a significant heterozygote deficiency and departure from Hardy–Weinberg equilibrium. The analyses revealed a high level of genetic diversity as the mean allele number per locality (Na) ranged from 10 to 14.6 (Table 2). Standardized allelic richness (Ar) was very similar between localities as it ranged from 8.1 to 8.5. The number of private alleles (Ap) was higher in Leucate and Thau, which are the two most isolated localities. However, the standardized private allelic richness (Apr) ranged from 0.26 to 0.37 and is thus very similar within localities. The observed (Ho) and expected (He) heterozygosities ranged from 0.679 to 0.704 and from 0.705 to 0.726, respectively. Overall, the number of alleles and the heterozygosity values were very homogeneous among localities and there was no decrease in diversity in isolated lagoon areas.
Table 2.
Summary statistics of genetic diversity indices of Pinna nobilis in each locality.
| Locality | N | Na | Ap | Ar | Apr | Ho | He | FIS |
|---|---|---|---|---|---|---|---|---|
| Côte des Albères | 176 | 13.5 | 5 | 8.5 | 0.37 | 0.679 | 0.707 | 0.02903*** |
| Leucate | 262 | 14.6 | 20 | 8.3 | 0.26 | 0.685 | 0.707 | 0.04461*** |
| Ayrolle | 51 | 10.9 | 1 | 8.5 | 0.28 | 0.684 | 0.705 | 0.05279*** |
| Cap d'Agde | 116 | 12.6 | 6 | 8.5 | 0.35 | 0.688 | 0.711 | 0.01862* |
| Port Ambonne | 35 | 10.0 | 1 | 8.4 | 0.29 | 0.691 | 0.705 | 0.009 |
| Thau | 220 | 14.3 | 14 | 8.3 | 0.27 | 0.704 | 0.726 | 0.03524*** |
| Frontignan | 49 | 10.5 | 1 | 8.1 | 0.26 | 0.701 | 0.707 | –0.009 |
| Port-St-Louis | 51 | 10.9 | 3 | 8.2 | 0.29 | 0.703 | 0.717 | 0.007 |
Significant values of FIS are indicated with *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001.
N number of individuals sampled, Na mean number of alleles, Ap number of private alleles, Ar standardized allelic richness, Apr standardized private allele richness, Ho observed heterozygosity, He expected heterozygosity and FIS inbreeding coefficient.
Genetic differentiation
All of the pairwise FST values were low (ranging from 0.0018 to 0.0159) and only two were significant after Bonferroni sequential correction: between Thau and Leucate and between Thau and Cap d’Agde (Table 3). The multivariate analysis (MDS) based on Nei’s genetic distance (Fig. 2) did not reveal any clear partitioning of the localities. All localities were grouped together with the exception of Port-St-Louis and Frontignan which were distant from all other localities, a result inconsistent with the FST values described above. However, as the MDS is a graphical representation, these distances between Port-St-Louis, Frontignan and other localities may not be significant. The AMOVA analyses showed that 99% of the total variation was supported by the variability among individuals (Supplementary Table S3a,b). The model-based clustering method analysis for population structure identified 4 clusters using Evanno’s method and 2 clusters using Puechmaille’s method (Supplementary Fig. S1). However, the analysis did not clearly assign each individual to a distinct cluster and there was a considerable level of admixture that did not allow clusters to be identified in relation to spatial distribution of individuals in localities. The isolation-by-distance test did not reveal any significant correlation between the genetic and geographic distances when distances were calculated using straight lines (Mantel test; Z = 45,468.91; p-value = 0.3863) but was significant with distances following the coastline (Mantel test; Z = 64,534.25; p-values = 0.0209). Overall, none of the analyses revealed a clear and consistent genetic differentiation between sampled localities that can be linked to the geographic localization or habitat type.
Table 3.
FST values of pairwise comparisons (Robertson and Hill estimator for FST, 1984 corrected by Raufaste & Bonhomme, 2000) between the 8 localities where Pinna nobilis specimens were collected.
| FST | Leucate | Ayrolle | Cap d'Agde | Port Ambonne | Thau | Frontignan | Port St Louis |
|---|---|---|---|---|---|---|---|
| Côte des Albères | 0.00181 | 0.00919 | 0.00338 | 0.00634 | 0.00225 | 0.0085 | 0.00773 |
| Leucate | 0.00443 | 0.00407 | 0.01594 | 0.01269* | 0.00853 | 0.0099 | |
| Ayrolle | 0.00397 | 0.00495 | 0.00624 | 0.00771 | 0.00308 | ||
| Cap d'Agde | 0.01354 | 0.01952* | 0.00866 | 0.00577 | |||
| Port Ambonne | 0.01181 | 0.00551 | 0.00383 | ||||
| Thau | 0.01332 | 0.00846 | |||||
| Frontignan | 0.00463 |
*Indicates significant values after Bonferroni sequential correction.
Figure 2.

Multidimensional Scaling (MDS) based on Nei’s genetic distances among the 8 localities where Pinna nobilis individuals were collected.
Relatedness
The non-linear regression between Prxy and sampling effort (Supplementary Fig. S2) showed that all the curves quickly reached an asymptote with a sampling effort of approximately 50 individuals per sample, which is about the minimum sample size we have when considering localities, except in Port Ambonne (35 individuals). Prxy was then estimated following the nonlinear regressions obtained for each locality and for all individuals mixed (Fig. 3). Prxy was similar among localities (ranging from 2.8 to 4.7%) except for Frontignan (1.6%) and Port Ambonne (6.5%). Similar results were found when calculating Prxy within sites (Supplementary Table S4). Then, when considering all individuals within a single sample Prxy was similar to the mean percentage in each locality (3.1%).
Figure 3.

Percentage (Prxy) of Pinna nobilis individuals sharing at least one common parent (i.e. percentage of relations with rxy > 0.25) calculated for each locality and for all sampled individuals mixed. Based on Queller and Goodnight (1989) rxy index for relatedness.
Size classes
The mean sizes ranged between 30 and 45 cm except in Thau and Ayrolle where there was a larger number of small individuals (Fig. 4). Two clear size classes were segregated in Thau, Port-St-Louis and Ayrolle (Fig. 4). In the three localities, the genetic diversity was similar between size groups (Table 4) and there was no genetic difference between the two size classes in each locality as all FST values were non-significant (Table 4). Then, Prxy in each size class were not different from each other and were also similar to Prxy values when size classes were grouped as a single sample (Table 4). Only in Port-St-Louis, smaller individuals showed a very low Prxy (< 1%).
Figure 4.
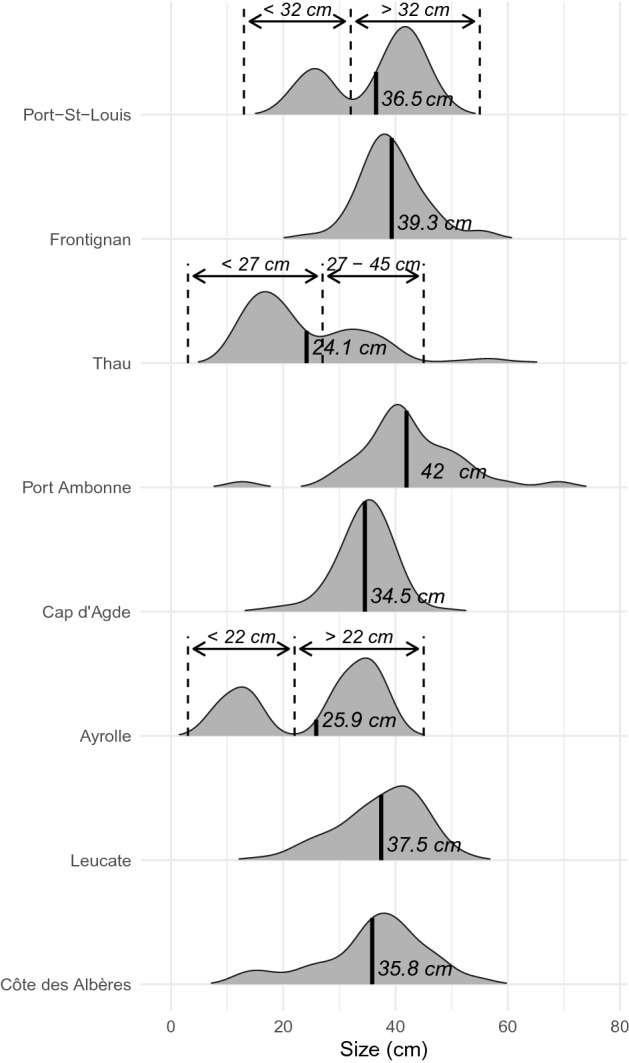
Ridgeline plot showing the size density distribution of Pinna nobilis individuals sampled in each locality. Mean sizes in each locality are represented by solid lines. Arrows represent the range of the size groups identified in Port-St-Louis, Thau and Ayrolle.
Table 4.
Summary statistics for the two size classes in Ayrolle, Thau and Port-St-Louis.
| Size groups | N | Na | Ho | He | FIS | FST | Prxy (%) |
|---|---|---|---|---|---|---|---|
| Ayrolle | 3.51 | ||||||
| 0–22 cm | 18 | 8.9 | 0.684 | 0.694 | 0.04265* | 0.01601 | 4.57 |
| 22–50 cm | 32 | 9.2 | 0.677 | 0.697 | 0.06160*** | 3.02 | |
| Thau | 3.24 | ||||||
| 0–27 cm | 114 | 12.8 | 0.694 | 0.713 | 0.03117*** | 0.00368 | 2.73 |
| 27–45 cm | 55 | 10.8 | 0.701 | 0.710 | 0.02229* | 3.30 | |
| Port-St-Louis | 3.60 | ||||||
| 0–32 cm | 16 | 7.9 | 0.699 | 0.683 | 0.00941 | 0.01703 | 0.83 |
| 32–50 cm | 35 | 9.8 | 0.701 | 0.689 | 0.00620 | 4.03 | |
Significant values of FIS are indicated with *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001.
N the number of individuals in the size classes, Na the mean number of alleles, Ho the observed heterozygosity, He the expected heterozygosity, FIS the inbreeding coefficient, FST values of pairwise comparisons between size classes and Prxy the percentage of individuals sharing at least one common parent (i.e. percentage of relations with rxy > 0.25) within the two size groups and considering the two size groups mixed (in bold).
Discussion
The genetic structure of P. nobilis across the Gulf of Lion, based on the analysis of 960 fan mussels sampled at 8 localities, with different environmental conditions, appeared homogeneous throughout the area including samples isolated in enclosed lagoons and harbors. High genetic diversity was highlighted in all locations sampled, no significant genetic differentiation was identified and the percentages of related individuals were very limited and similar between all locations. In a given site, genetic parameters were also very homogeneous between size classes. These results suggest that all fan mussels belong to a large, homogeneous and highly connected population at the scale of the Gulf of Lion, which has direct implications for genetics-based conservation strategies hereinafter discussed.
Several loci showed evidence of heterozygote deficiency, which is a common phenomenon in mollusks53,54. It could be caused by different factors such as null alleles, inbreeding, Wahlund effect or selection. The possible reasons for heterozygote deficiency in P. nobilis were further detailed in a previous study32. All the loci that showed evidence of null alleles were removed from the dataset to avoid bias in the results.
Genetic diversity and relatedness
Genetic diversity was similar among the different localities, except in Thau, Leucate and for the Côte des Albères which showed higher values because the number of private alleles was higher. Such higher genetic diversity likely reflects the larger sample sizes from these three localities (i.e. increasing rare private alleles in these localities) as allelic richness was similar among localities when standardizing sampling effort. The levels of diversity and heterozygosity of P. nobilis are similar to those found in other bivalve species: Cerastoderma glaucum55, Ostrea edulis56 and Crepidula fornicata57. Genetic diversity values found here were slightly higher compared to what was found in other P. nobilis populations in the Baleares19 or along the Spanish coast20 but this is also likely due to a lower sampling effort in their studies. This high level of diversity in all sampled localities supports the hypothesis of a large effective population size that maintains high polymorphism, even in the presence of fragmented and isolated habitats58. Such hypothesis of large effective population is quite common in marine sessile benthic species that have external reproduction as they synchronize the release of their gametes to ensure a higher probability of external fecundation. This is the case for several bivalves such as the scallop Pecten fumatus, where synchronized spawning events are triggered by high density and abundance of conspecifics59 and thus annual recruitments involve a large number of spawners. This phenomenon was also observed in other bivalves such as the date mussel60 and in other sessile organisms such as sea urchins61, brittle stars62 and other invertebrates63. In the case of P. nobilis, the reproduction patterns are quite complex and still poorly understood. For example, Trigos et al.13 reported the occurrence of internal fertilization with females that maintain eggs in their body cavity whereas external fertilization was largely admitted as the main mode of reproduction64,65. Then, while fan mussels are described as a successive hermaphrodite, 38% of individuals in controlled conditions and 20% in wild population in Alfacs Bay, Spain, appeared simultaneously male and female, leading to self-fertilization13,66. Such high rates of self-fertilization would decrease genetic admixture and thus genetic diversity, and could induce higher rates of inbreeding, particularly in enclosed areas such as Thau or Leucate. However, Prado et al.66 add caution to their findings by saying that unfavorable ecological situations may have the potential to trigger the development of simultaneous hermaphrodites in bivalves populations. This phenomenon has not been observed in the populations of the Cabrera National Park Marine Protected Area in the Balearic Islands67 which also shows evidence of large admixture19. Considering the high level of genetic diversity and low inbreeding coefficients that were observed in all localities in the present study, such high rates of self-fertilization are unlikely to occur here and the hypothesis of synchronized spawning events involving a large number of individuals is more relevant with the genetic data gathered thus far.
The percentage of related individuals was not larger in a given locality than considering all individuals mixed in a single sample, even for the most isolated localities such as lagoons. The two extreme values (Frontignan: 1.6% and Port Ambonne: 6.5%) were found in the smallest harbors with the smallest abundances. Low abundances increase the effects of the heterogeneity of larval supply, during recruitment, in both directions, thus leading randomly to more or less related individuals. Overall, only a small proportion of individuals showed relatedness values > 0.25 (mean of 3.3%) emphasizing evidence of large effective population size and substantial admixture during reproductive events. This result also correlates with similarity of the diversity indices among localities. Moreover, in the three sites where two size classes could be identified, individuals did not show more relatedness within a given size group. Only few studies investigated relatedness in wild marine species and to date, none of them have studied relatedness in the Gulf of Lion and within lagoons. However, Costantini et al.68 studied the relatedness of red coral individuals and found that 25.98% of pairwise comparisons involved related individuals which is much higher than what we are reporting for P. nobilis.
Both diversity indices and relatedness estimates demonstrate that even if patchy and isolated, aggregations of P. nobilis originate from large effective population sizes. This is mainly linked to the reproductive mechanism that consists of the synchronic release of gametes by all individuals to ensure success in the fecundation and an increase in admixture. In this framework, the question of connectivity between aggregations remains, since the large population size in lagoons could maintain high genetic diversity indexes while, at the same time, this high diversity in small isolated harbors could indicate a supply from outside populations.
Regional patterns of dispersal
None of the analyses performed in this study showed genetic differentiation among localities when considering genetic diversity levels, allele frequencies or relatedness index (i.e. combinations of alleles). All fan mussel aggregations belong to a large homogeneous, highly connected, population across the entire Gulf of Lion. Even small aggregations located in isolated areas (lagoons or ports) were not genetically segregated. The absence of spatial genetic differentiation is often interpreted as a signal for significant connectivity but this is primarily an equilibrium between genetic drift and migration. In the context of large effective population size, the genetic drift will be limited and only a small amount of migration is required to maintain genetic homogeneity. Considering the isolated aggregations of P. nobilis located in lagoons, the potential for a high self-recruitment rate remains likely as these lagoons have limited water inflow with the open sea which could limit the exportation of pelagic larvae. Then, as we are dealing with a species characterized by large effective population size, in relation to its reproductive mode, only a small amount of larval supply from other localities should be necessary to maintain low FST values, limited relatedness and the overall genetic homogeneity. In the meantime, due to their properties, larvae from all localities, even enclosed lagoons, have the potential to be exported to the open sea and spread to all localities, drifted by currents, which would also maintain the overall genetic homogeneity. Both scenarios would explain the low proportion of related individuals, as recruitment is the result of a large reproductive event, involving a large number of spawners, and relatives will not necessarily remain together until settlement (i.e. no kin aggregation).
The absence of genetic differentiation is common in mollusks, particularly at this spatial scale, as species are known to have large effective population sizes and long larval durations (for example in Concholepas concholepas69, Crepidula fornicata57 and Pecten maximus70). Population genetics studies were performed for P. nobilis in Balearic Islands and along the Spanish coast and authors reported no, or very weak, genetic differentiation between sites19,20. Populations were situated in open sea locations and authors associated the lack of genetic structure to a high connectivity in their study area. Simulations performed by Wesselmann et al.20, highlight the high dispersal potential of fan mussels during the pelagic larval state. In the present study, considering the size of the study area and the dispersal potential of larvae, the hypothesis of high connectivity could thus be consistent, at least between the most exposed locations, and even between the most distant ones such as Côte des Albères and Port-St-Louis.
Sea-lagoon genetic connectivity is poorly documented in the Gulf of Lion. To our knowledge, fine-scale genetic structure among lagoons in this area was investigated, in bivalve species, only for Ruditapes decussatus71 and Mytilus galloprovincialis72 and, similarly to the present study for P. nobilis, no genetic differentiation could be identified. Similar results were also found for the sand goby Pomatoschistus minutus73. These species are known to have large population sizes. We could thus be in the same situation as that of P. nobilis, where it is difficult to differentiate both scenarios of high connectivity vs reproductive events involving large number of spawners and low migration which maintains the genetic homogeneity.
Other studies on P. nobilis showed isolated populations in Greece16 and in the Venetian lagoon18, where geographic configuration and environmental conditions are similar to those in the lagoons in the Gulf of Lion. Genetic differentiations can thus occur in isolated areas, when a population is segregated by geography or by marine currents, which create natural barriers and prevent populations from exchanging larvae16. Given the particular geographic configuration of the Gulf of Lion’s coast, compartmentalization of populations could be expected, especially for lagoons. Finally, Pérez-Ruzafa et al.74 reported that connectivity between the sea and lagoon must be much lower than expected, by simulating larval dispersal of several species with different life history traits. This could thus be a clue to support the hypothesis of high self-recruitment rates in lagoons.
Implication for conservation
Results from the present study on P. nobilis demonstrate genetic homogeneity across localities, wherever they are (lagoons, harbors, open sea) that implies some connectivity among these localities that maintain genetic diversity. The question concerning the contribution of this connectivity within the context of repopulating localities remains, as we could oppose two scenarios involving a dominance of migration vs one with a negligible migration and dominance of self-recruitment from large local populations. In the case of large lagoons such as Thau and Leucate, it is likely that self-recruitment dominates the local recruitment since all size classes and more regular settlement patterns are present. Conversely, in the case of small harbors, repopulation is highly dependent on migration that is typically more variable, leading to more variability in size classes.
P. nobilis is a sedentary endemic species with a highly fragmented distribution. This study highlights a large genetically homogenous population with high genetic diversity across the entire coast of the Gulf of Lion. This high genetic diversity may involve a large number of spawners and thus the population may have a large effective size. Maintaining a high level of genetic diversity in populations is one of the fundamentals of genetic conservation as it preserve the species’ abilities to adapt to environmental changes and increase resilience75. Considering the results of the present study and regarding the pandemic occurring throughout the Mediterranean Sea, preserving fan mussels in a single locality, for example, might allow for the entire genetic variability of fan mussels in the Gulf of Lion to be preserved. Then, as migration between localities, even small, might exist, it could help to repopulate other areas. In a context of developing a species rescue plan, conserving a relatively small number of individuals in aquariums, under controlled conditions to protect them from being infested by the parasite, might be enough to provide a backup of almost the entire genetic diversity of the species.
This study also showed the wide diversity of habitat types that the fan mussels can colonize. The epidemic, caused by the parasite H. pinnae and leading to mass mortality, has now spread throughout almost the entire Mediterranean Sea and has already reached the coast of the Gulf of Lion where all of the infected populations have been devastated. To date, only individuals settled in lagoons remain less or unaffected and the survival of the species is likely to rest on them, as there is no longer dense population in the open sea. The present study thus has important implications for conservation as it highlights the opportunity to find a habitat that is suitable for fan mussels but which is unfavorable to the development of the parasite. Lagoons, thanks to their particular physico-chemicals conditions, might thus represent a refuge habitat for fan mussels, which could be the key for the persistence of the species in the wild.
Supplementary Information
Acknowledgements
This research was implemented thanks to the financial support of the Region Occitanie, the General Council of Pyrénées-Orientales and the French Ministry for Ecological Transition. C. Peyran was supported by a PhD scholarship granted by Sorbonne University. We are grateful for the support of the DREAL (Direction Régionale de l’Environnement, de l’Aménagement et du Logement) Occitanie and all of the ports involved in the study: the ports of Sète, Port-la-Nouvelle, Port-Vendres and Frontignan. We also thank the staff from the Cerbère‐Banyuls Natural Marine Reserve and the Agathoise Coast Marine Protected Area for their collaboration and permission to work within their protected areas.
Author contributions
S.P. designed the study, T.M., E.N.C., G.I. and C.P. did the fieldwork, C.P. performed genetic analyses and drafted the paper, E.B. and S.P. contributed to data interpretation and reviewed the paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-87493-4.
References
- 1.Ceballos G, et al. Accelerated modern human–induced species losses: entering the sixth mass extinction. Sci. Adv. 2015;1:e1400253. doi: 10.1126/sciadv.1400253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baillie, J. E. ., Hilton-Taylor, C. & Stuart, S. N. 2004 IUCN Red List of Threatened Species. A Global Species Assessment. (2004).
- 3.Hughes AR, Stachowicz JJ. Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. Proc. Natl. Acad. Sci. 2004;101:8998–9002. doi: 10.1073/pnas.0402642101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowen RK. Scaling of connectivity in marine populations. Science. 2006;311:522–527. doi: 10.1126/science.1122039. [DOI] [PubMed] [Google Scholar]
- 5.Ronce O. How does it feel to be like a rolling stone? Ten questions about dispersal evolution. Annu. Rev. Ecol. Evol. Syst. 2007;38:231–253. doi: 10.1146/annurev.ecolsys.38.091206.095611. [DOI] [Google Scholar]
- 6.Cowen RK, Sponaugle S. Larval dispersal and marine population connectivity. Ann. Rev. Mar. Sci. 2009;1:443–466. doi: 10.1146/annurev.marine.010908.163757. [DOI] [PubMed] [Google Scholar]
- 7.White JW, et al. Connectivity, dispersal, and recruitment. Oceanography. 2019;32:50–59. doi: 10.5670/oceanog.2019.310. [DOI] [Google Scholar]
- 8.Munday PL, et al. Climate change and coral reef connectivity. Coral Reefs. 2009;28:379–395. doi: 10.1007/s00338-008-0461-9. [DOI] [Google Scholar]
- 9.Saunders MI, et al. Human impacts on connectivity in marine and freshwater ecosystems assessed using graph theory: A review. Mar. Freshw. Res. 2016;67:277. doi: 10.1071/MF14358. [DOI] [Google Scholar]
- 10.Peyran, C., Morage, T., Nebot-Colomer, E., Iwankow, G. & Planes, S. Unexpected residual habitats raise hope for the survival of the over the edge of extinction fan mussel, Pinna nobilis, along the Occitan coast (north-western Mediterranean Sea) (2020).
- 11.De Gaulejac B. Mise en évidence de l’hermaphrodisme successif à maturation asynchrone de Pinna nobilis. Biol. Pathol. Anim. 1995;1:99–103. [Google Scholar]
- 12.Butler A, Vicente N, de Gaulejac B. Ecology of the pterioid bivalves Pinna bicolor Gmelin and Pinna nobilis L. Mar. Life. 1993;3:37–45. [Google Scholar]
- 13.Trigos S, Vicente N, Prado P, Espinós FJ. Adult spawning and early larval development of the endangered bivalve Pinna nobilis. Aquaculture. 2018;483:102–110. doi: 10.1016/j.aquaculture.2017.10.015. [DOI] [Google Scholar]
- 14.Öndes F, Kaiser MJ, Güçlüsoy H. Human impacts on the endangered fan mussel, Pinna nobilis. Aquat. Conserv. Mar. Freshw. Ecosyst. 2020;30:31–41. doi: 10.1002/aqc.3237. [DOI] [Google Scholar]
- 15.IOPR. Premier séminaire international sur la grande nacre de Méditerranée : Pinna nobilis. Mém. Inst. Océanogr. Paul Ricard 134 (2003).
- 16.Katsares V, Tsiora A, Galinou-Mitsoudi S, Imsiridou A. Genetic structure of the endangered species Pinna nobilis (Mollusca: Bivalvia) inferred from mtDNA sequences. Biologia. 2008;63:412–417. doi: 10.2478/s11756-008-0061-8. [DOI] [Google Scholar]
- 17.Rabaoui L, et al. Genetic variation among populations of the endangered fan mussel Pinna nobilis (Mollusca: Bivalvia) along the Tunisian coastline. Hydrobiologia. 2011;678:99–111. doi: 10.1007/s10750-011-0827-9. [DOI] [Google Scholar]
- 18.Sanna D, et al. Mitochondrial DNA reveals genetic structuring of Pinna nobilis across the mediterranean sea. PLoS ONE. 2013;8:e67372. doi: 10.1371/journal.pone.0067372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.González-Wangüemert M, et al. Gene pool and connectivity patterns of Pinna nobilis in the Balearic Islands (Spain, Western Mediterranean Sea): Implications for its conservation through restocking. Aquat. Conserv. Mar. Freshw. Ecosyst. 2019;29:175–188. doi: 10.1002/aqc.2976. [DOI] [Google Scholar]
- 20.Wesselmann M, et al. Genetic and oceanographic tools reveal high population connectivity and diversity in the endangered pen shell Pinna nobilis. Sci. Rep. 2018;8:4770. doi: 10.1038/s41598-018-23004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanna D, et al. New mitochondrial and nuclear primers for the Mediterranean marine bivalve Pinna nobilis. Mediterr. Mar. Sci. 2014;15:416. doi: 10.12681/mms.459. [DOI] [Google Scholar]
- 22.Catanese G, et al. Haplosporidium pinnae sp. nov., a haplosporidan parasite associated with mass mortalities of the fan mussel, Pinna nobilis, in the Western Mediterranean Sea. J. Invertebr. Pathol. 2018;157:9–24. doi: 10.1016/j.jip.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Scarpa F, et al. Multiple non-species-specific pathogens possibly triggered the mass mortality in Pinna nobilis. Life. 2020;10:238. doi: 10.3390/life10100238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grau, A. et al. Wide-geographic and long-term analysis of the role of pathogens in the decline of Pinna nobilis to critically endangered species. (2021).
- 25.Vázquez-Luis M, et al. Pinna nobilis: A mass mortality event in Western Mediterranean Sea. Front. Mar. Sci. 2017;4:1–6. doi: 10.3389/fmars.2017.00220. [DOI] [Google Scholar]
- 26.Cabanellas-Reboredo M, et al. Tracking a mass mortality outbreak of pen shell Pinna nobilis populations: A collaborative effort of scientists and citizens. Sci. Rep. 2019;9:13355. doi: 10.1038/s41598-019-49808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-March JR, et al. Can we save a marine species affected by a highly infective, highly lethal, waterborne disease from extinction? Biol. Conserv. 2020;243:108498. doi: 10.1016/j.biocon.2020.108498. [DOI] [Google Scholar]
- 28.Kersting, D. et al. Pinna nobilis. The IUCN Red List of Threatened Species 2019. (2019). 10.2305/IUCN.UK.2019-3.RLTS.T160075998A160081499.en
- 29.Ifremer. Réseau de Suivi Lagunaire du Languedoc-Roussillon. (2014).
- 30.García-March JR, García-Carrascosa AM, Pena ÁL. In situ measurement of Pinna nobilis shells for age and growth studies: A new device. Mar. Ecol. 2002;23:207–217. doi: 10.1046/j.1439-0485.2002.02781.x. [DOI] [Google Scholar]
- 31.De Gaulejac, B. Etude écophysiologique du mollusque bivalve méditerranéen Pinna nobilis L. reproduction; croissance; respiration. (1993).
- 32.Peyran C, Planes S, Tolou N, Iwankow G, Boissin E. Development of 26 highly polymorphic microsatellite markers for the highly endangered fan mussel Pinna nobilis and cross-species amplification. Mol. Biol. Rep. 2020 doi: 10.1007/s11033-020-05338-1. [DOI] [PubMed] [Google Scholar]
- 33.González-Wangüemert M, et al. Highly polymorphic microsatellite markers for the Mediterranean endemic fan mussel Pinna nobilis. Mediterr. Mar. Sci. 2014;16:31. doi: 10.12681/mms.949. [DOI] [Google Scholar]
- 34.Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 2004;4:535–538. doi: 10.1111/j.1471-8286.2004.00684.x. [DOI] [Google Scholar]
- 35.Peakall R, Smouse PE. GenAlEx 65: Genetic analysis in Excel. Population genetic software for teaching and research: An update. Bioinformatics. 2012;28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szpiech ZA, Jakobsson M, Rosenberg NA. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics. 2008;24:2498–2504. doi: 10.1093/bioinformatics/btn478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rousset F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 38.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 39.Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N. & Bonhomme, F. GENETIX 4.05, Population genetics software for Windows TM. Université de Montpellier II (2004).
- 40.Robertson A, Hill WG. Deviations from Hardy–Weinberg proportions: Sampling variances and use in estimation of inbreeding coefficients. Genetics. 1984;107:703–718. doi: 10.1093/genetics/107.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raufaste N, Bonhomme F. Properties of bias and variance of two multiallelic estimators of FST. Theor. Popul. Biol. 2000;57:285–296. doi: 10.1006/tpbi.2000.1457. [DOI] [PubMed] [Google Scholar]
- 42.Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- 43.R Core Team. R: A Language and Environment for Statistical Computing. (2018).
- 44.Excoffier L, Lischer HEL. Arlequin suite ver 35: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 45.Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. (2000). [DOI] [PMC free article] [PubMed]
- 46.Earl DA, VonHoldt BM. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 47.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 48.Puechmaille SJ. The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 2016;16:608–627. doi: 10.1111/1755-0998.12512. [DOI] [PubMed] [Google Scholar]
- 49.Li Y-L, Liu J-X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018;18:176–177. doi: 10.1111/1755-0998.12719. [DOI] [PubMed] [Google Scholar]
- 50.Paradis E, Schliep K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 51.Queller DC, Goodnight KF. Estimating relatedness using genetic markers. Evolution. 1989;43:258. doi: 10.1111/j.1558-5646.1989.tb04226.x. [DOI] [PubMed] [Google Scholar]
- 52.Kraemer P, Gerlach G. Demerelate: Calculating interindividual relatedness for kinship analysis based on codominant diploid genetic markers using R. Mol. Ecol. Resour. 2017;17:1371–1377. doi: 10.1111/1755-0998.12666. [DOI] [PubMed] [Google Scholar]
- 53.Hare MP, Karl SA, Avise JC. Anonymous nuclear DNA markers in the American oyster and their implications for the heterozygote deficiency phenomenon in marine bivalves. Mol. Biol. Evol. 1996;13:334–345. doi: 10.1093/oxfordjournals.molbev.a025593. [DOI] [PubMed] [Google Scholar]
- 54.Giantsis IA, Mucci N, Randi E, Abatzopoulos TJ, Apostolidis AP. Microsatellite variation of mussels (Mytilus galloprovincialis) in central and eastern Mediterranean: Genetic panmixia in the Aegean and the Ionian Seas. J. Mar. Biol. Assoc. UK. 2014;94:797–809. doi: 10.1017/S0025315414000174. [DOI] [Google Scholar]
- 55.Tarnowska K, Chenuil A, Nikula R, Féral J, Wolowicz M. Complex genetic population structure of the bivalve Cerastoderma glaucum in a highly fragmented lagoon habitat. Mar. Ecol. Prog. Ser. 2010;406:173–184. doi: 10.3354/meps08549. [DOI] [Google Scholar]
- 56.Šegvić-Bubić T, et al. Translocation and aquaculture impact on genetic diversity and composition of wild self-sustainable Ostrea edulis populations in the Adriatic sea. Front. Mar. Sci. 2020;7:1–13. doi: 10.3389/fmars.2020.00084. [DOI] [Google Scholar]
- 57.Dupont L, Ellien C, Viard F. Limits to gene flow in the slipper limpet Crepidula fornicata as revealed by microsatellite data and a larval dispersal model. Mar. Ecol. Prog. Ser. 2007;349:125–138. doi: 10.3354/meps07098. [DOI] [Google Scholar]
- 58.Ellegren H, Ellegren N. Determinants of genetic diversity. Nat. Publ. Gr. 2016;17:422–433. doi: 10.1038/nrg.2016.58. [DOI] [PubMed] [Google Scholar]
- 59.Mendo T, Moltschaniwskyj N, Lyle JM, Tracey SR, Semmens JM. Role of density in aggregation patterns and synchronization of spawning in the hermaphroditic scallop Pecten fumatus. Mar. Biol. 2014;161:2857–2868. doi: 10.1007/s00227-014-2551-2. [DOI] [Google Scholar]
- 60.Žuljević A, Despalatović M, Cvitković I, Morton B, Antolić B. Mass spawning by the date mussel Lithophaga lithophaga. Sci. Rep. 2018;8:10781. doi: 10.1038/s41598-018-28826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lamare MD, Stewart BG. Mass spawning by the sea urchin Evechinus chloroticus (Echinodermata: Echinoidea) in a New Zealand fiord. Mar. Biol. 1998;132:135–140. doi: 10.1007/s002270050379. [DOI] [Google Scholar]
- 62.Soong K, Chang D, Chao S. Presence of spawn-inducing pheromones in two brittle stars (Echinodermata: Ophiuroidea) Mar. Ecol. Prog. Ser. 2005;292:195–201. doi: 10.3354/meps292195. [DOI] [Google Scholar]
- 63.Watson G, Bentley M, Gaudron S, Hardege J. The role of chemical signals in the spawning induction of polychaete worms and other marine invertebrates. J. Exp. Mar. Biol. Ecol. 2003;294:169–187. doi: 10.1016/S0022-0981(03)00264-8. [DOI] [Google Scholar]
- 64.Gaulejac BD, Henry M, Vicente N. An ultrastructural study of gametogenesis of the marine bivalve Pinna nobilis (Linnaeus 1758) II, Spermatogenesis. J. Molluscan Stud. 1995;61:393–403. doi: 10.1093/mollus/61.3.393. [DOI] [Google Scholar]
- 65.Cabanellas-Reboredo M, et al. Recruitment of Pinna nobilis (Mollusca: Bivalvia) on artificial structures. Mar. Biodivers. Rec. 2009;2:e126. doi: 10.1017/S1755267209001274. [DOI] [Google Scholar]
- 66.Prado P, et al. Breeding, planktonic and settlement factors shape recruitment patterns of one of the last remaining major population of Pinna nobilis within Spanish waters. Hydrobiologia. 2020;847:771–786. doi: 10.1007/s10750-019-04137-5. [DOI] [Google Scholar]
- 67.Deudero S, et al. Reproductive investment of the pen shell Pinna nobilis Linnaeus, 1758 in Cabrera National Park (Spain) Mediterr. Mar. Sci. 2017;18:271. doi: 10.12681/mms.1645. [DOI] [Google Scholar]
- 68.Costantini F, Rugiu L, Cerrano C, Abbiati M. Living upside down: Patterns of red coral settlement in a cave. Mediterr. Mar. Sci. 2018 doi: 10.7717/peerj.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cárdenas L, Castilla JC, Viard F. Hierarchical analysis of the population genetic structure in Concholepas concholepas, a marine mollusk with a long-lived dispersive larva. Mar. Ecol. 2016;37:359–369. doi: 10.1111/maec.12286. [DOI] [Google Scholar]
- 70.Morvezen R, et al. Genetic structure of a commercially exploited bivalve, the great scallop Pecten maximus, along the European coasts. Conserv. Genet. 2016;17:57–67. doi: 10.1007/s10592-015-0760-y. [DOI] [Google Scholar]
- 71.Borsa P, Jarne P, Belkhir K, Bonhomme F. Genetic structure of the palourde 103. Genet. Evol. Aquat. Org. 1994;103:1–12. [Google Scholar]
- 72.Skalamera J, Renaud F, Raymond M, de Meeûs T. No evidence for genetic differentiation of the mussel Mytilus galloprovincialis between lagoons and the seaside. Mar. Ecol. Prog. Ser. 1999;178:251–258. doi: 10.3354/meps178251. [DOI] [Google Scholar]
- 73.Boissin E, Hoareau TB, Berrebi P. Effects of current and historic habitat fragmentation on the genetic structure of the sand goby Pomatoschistus minutus (Osteichthys, Gobiidae) Biol. J. Linn. Soc. 2011;102:175–198. doi: 10.1111/j.1095-8312.2010.01565.x. [DOI] [Google Scholar]
- 74.Pérez-Ruzafa A, et al. Connectivity between coastal lagoons and sea: Asymmetrical effects on assemblages’ and populations’ structure. Estuar. Coast. Shelf Sci. 2019;216:171–186. doi: 10.1016/j.ecss.2018.02.031. [DOI] [Google Scholar]
- 75.Frankham R. Quantitative genetics in conservation biology. Genet. Res. 1999;74:237–244. doi: 10.1017/S001667239900405X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.