

Abstract
The BBSome is a protein complex consisting of BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 and BBS18 that associates with intraflagellar transport complexes and specializes in ciliary trafficking. In primary cilia, ciliary entry requires the fully assembled BBSome as well as the small GTPase, ARL6 (BBS3). Retinal photoreceptors possess specialized cilia. In light of key structural and functional differences between primary and specialized cilia, we examined the principles of BBSome recruitment to photoreceptor cilia. We performed sucrose gradient fractionation using retinal lysates of Bbs2−/−, Bbs7−/−, Bbs8−/− and Bbs3−/− mice to determine the status of BBSome assembly, then determined localization of BBSome components using immunohistochemistry. Surprisingly, we found that a subcomplex of the BBSome containing at least BBS1, BBS5, BBS8 and BBS9 is recruited to cilia in the absence of BBS2 or BBS7. In contrast, a BBSome subcomplex consisting of BBS1, BBS2, BBS5, BBS7 and BBS9 is found in Bbs8−/− retinas and is denied ciliary entry in photoreceptor cells. In addition, the BBSome remains fully assembled in Bbs3−/− retinas and can be recruited to photoreceptor cilia in the absence of BBS3. We compared phenotypic severity of their retinal degeneration phenotypes. These findings demonstrate that unlike primary cilia, photoreceptor cilia admit a partially assembled BBSome meeting specific requirements. In addition, the recruitment of the BBSome to photoreceptor cilia does not require BBS3. These findings indicate that the ciliary entry of the BBSome is subjected to cell-specific regulation, particularly in cells with highly adapted forms of cilia such as photoreceptors.
Introduction
The BBSome is a protein complex consisting of BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 and BBS18 (also known as BBIP1) (1–10). While BBS2 and BBS7 assemble into the top portion of the BBSome, BBS5, BBS8 and BBS9 form the base (11). The top and the base are joined by a corkscrew structure composed of BBS1, BBS4 and BBS18 (11). Functioning as an adaptor to intraflagellar transport (IFT) complexes specializing in ciliary transport, the BBSome recruits cargo for ciliary export and expands the cargo range of the IFT machinery (12). In other words, the BBSome participates in retrograde ciliary trafficking. In addition to retrograde trafficking, the BBSome enables the passage of cargoes through the transition zone (13), which is a physical barrier to random diffusion at the ciliary base (14). In line with its function in retrograde ciliary transport and cargo export, the absence of BBSome function results in accumulation of smoothened (15), patched 1 (15) and dopamine receptors (16) in primary cilia. However, the BBSome is also required for anterograde ciliary trafficking. In Caenorhabditis elegans, IFT-A and IFT-B particles are separated and move at different rates during anterograde IFT without the BBSome (17). Of note, the BBSome does not appear to function as a structural component that holds the IFT particles together during IFT. Rather, the BBSome is required for the assembly of the IFT-A and IFT-B particles at the ciliary base in C. elegans (17). The BBSome plays important roles in ciliary transport.
In primary cilia, components of the BBSome are localized to cilia, as well as to the basal body and centriolar satellites (18). The small GTPase, BBS3, recruits the BBSome to the basal body for loading onto IFT trains (19). The BBSome associates with the IFT trains to gain ciliary entry. The ciliary entry of the BBSome requires the complete BBSome. BBS9, which serves as a marker of at least a partially assembled BBSome, fails to localize to cilia when the levels of BBS1, BBS2, BBS4, BBS5, BBS7 or BBS8 are knocked down by RNA interference (RNAi) (20). This suggests that an incomplete BBSome and individual BBSome components cannot enter primary cilia. Besides the prerequisite of associating with IFT trains, it is not known whether there are additional mechanisms at the transition zone that screen the structural assembly of the BBSome for gaining admittance into cilia, barring ciliary entry of the BBSome when the BBSome is incomplete. In addition to the requirement of being fully assembled and loaded onto the IFT complex, the ciliary localization of the BBSome is also lost without the GTP binding of the small GTPase ARL6 (BBS3) in cultured hTERT-RPE1 cells (20, 21). In agreement with the need of BBS3 for BBSome ciliary localization, BBS1 and BBIP1 fail to localize to primary cilia when BBS3 is depleted by RNAi in RPE1 cells (22). In addition, BBS3 that contains a point mutation that abolishes the BBS3–BBS1 interaction also fails to recruit the BBSome to cilia (21). These reports support the view that BBS3 is required for BBSome recruitment to primary cilia. Although required for loading onto IFT trains and ciliary recruitment, depletion of BBS3 does not affect BBSome assembly (20, 22). These principles are established in cells possessing primary cilia.
Photoreceptors, specialized neurons of the outer retina responsible for sensitive light detection, have evolved elaborate specialized cilia termed the outer segments. These specialized outer segments, on which photoreceptors display opsin molecules for phototransduction, possess distinct structural features consisting of horizontally stacked disc membranes enclosed by the plasma membrane. The sheer size of the outer segment and its high demand for ciliary trafficking call for enhanced structural support, a need answered by the evolution of the photoreceptor-specific transition zone as an extension of the canonical transition zone (23). Photoreceptors have specialized ciliary structures and possess distinct transition zone components to meet specific demands. Many components of the BBSome, including BBS5 and BBS8, have retina-specific isoforms (24, 25), suggesting that the BBSome may have distinct features in photoreceptors. In addition, BBS3 also have an isoform that is expressed predominantly in the eye (26). It is not yet known whether functional aspects of the BBSome, for example its ciliary recruitment, differ between cells with primary cilia versus specialized cilia.
In this study, we test whether the principles for BBSome ciliary entry, which in primary cilia requires the complete BBSome and the small GTPase BBS3, hold true for the adapted, highly modified cilia of photoreceptor cells. We found the existence of a BBSome subcomplex in photoreceptor cilia in the absence of BBS2 or BBS7 and that this subcomplex can enter photoreceptor cilia. A different BBSome subcomplex is formed in the absence of BBS8, and this subcomplex is denied ciliary entry, shedding light on the prerequisite subunits for recruitment. In addition, the BBSome can be recruited to photoreceptor cilia without BBS3, in stark contrast to their primary cilia counterparts. Our findings point to the fact that highly adapted forms of cilia such as those in photoreceptor cells have evolved a set of distinct entry criteria for the BBSome, different from that in primary cilia. Ciliary entry of the BBSome is regulated in a tissue-specific manner, especially in those tissues with highly adapted forms of cilia.
Results
We performed immunohistochemistry to determine the localization of BBSome components in photoreceptor cilia. In order to clearly delineate the outer segment portion of the photoreceptor axoneme, considered equivalent to cilia, and the connecting cilia, considered equivalent to the transition zone in primary cilia, we probed retinas from postnatal day 15 (P15) mice with the anti-glycylated tubulin (TAP952) antibody as well as the anti-RP-1 antibody. RP-1 specifically decorates the outer segment portion of the photoreceptor ciliary axoneme but not the connecting cilia (27). We previously validated that this TAP952 antibody clearly demarcates the photoreceptor connecting cilia in both control and Bbs8−/− mice (28). In Bbs8−/− mice, the signal from the TAP952 antibody has minimal overlap with the signal from the anti-RP-1 antibody (28). We confirmed that this TAP952 antibody also demarcates the photoreceptor connecting cilia in both control and Bbs7−/− mice, showing minimal overlap with the signal from the anti-RP-1 antibody (Fig. 1). The RP-1 decorated, outer segment portion of the ciliary axoneme is shortened in Bbs7−/− and Bbs8−/− photoreceptors, as previously noted in another study (29). Since the anti-polyglutamylation modification antibody (GT335) is also commonly used to demarcate the photoreceptor connecting cilia, we tested the GT335 antibody in control and BBS mutant mice. The GT335 antibody clearly delineates the connecting cilia in normal photoreceptors but significantly overlaps with the signal from the anti-RP-1-antibody in Bbs7−/− photoreceptors (Fig. 1). The overlap between the signals from the GT335 antibody and the anti-RP-1 antibody suggests that the polyglutamylation mark is no longer confined to the photoreceptor transition zone in Bbs7−/− mice but is present along the entire ciliary axoneme. In contrast, the glycylation marks labeled by the TAP952 antibody clearly demarcates the connecting cilia in both control and Bbs7−/− mice. Therefore, for the rest of this study, the anti-glycylated tubulin antibody, TAP952, is used to demarcate the photoreceptor connecting cilia.
Figure 1.
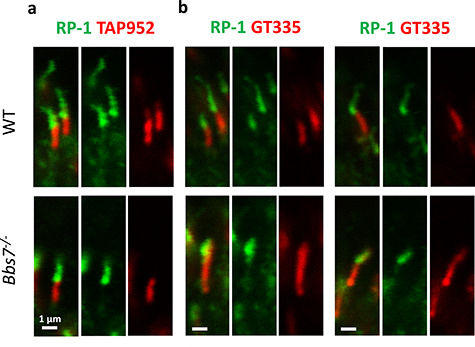
The anti-glycylated tubulin antibody (TAP952) clearly demarcates the photoreceptor connecting cilia. (a) In both wild-type and Bbs7−/− photoreceptors at P15, signal from the TAP952 antibody demarcates the connecting cilia without overlapping with the signal from the anti-RP-1 antibody marking the outer segment portion of the ciliary axoneme. (b) The GT335 antibody delineates the connecting cilia in normal photoreceptors. However, in Bbs7−/− photoreceptors, the entire ciliary axoneme, including the outer segment portion of the ciliary axoneme marked by the anti-RP-1 antibody, is polyglutamylated.
We have recently validated a mouse model in which Bbs8 is globally knocked out and shown that Bbs8−/− mice lack adequate BBSome function (28). We tested whether BBSome subunits can enter cilia without BBS8 by using sucrose gradient fractionation to determine the status of BBSome assembly in 1-month-old Bbs8−/− retinas. During sucrose gradient fractionation experiments, proteins and protein complexes in retinal lysates are subjected to a density gradient of 10–40% sucrose. Under high-speed centrifugation, they separate according to their molecular weights. In our experiments, components in the complete BBSome typically elute in fractions 12–13. Sucrose gradient fractionation shows that in the absence of BBS8, BBSome components BBS1, BBS2, BBS7, BBS9 and BBS5, whose peaks are normally found in fraction 13, are jointly shifted to fractions 10–11 which contain complexes with smaller molecular weights (Fig. 2a). This suggests that in Bbs8−/− retinas BBS1, BBS2, BBS5, BBS7 and BBS9 exist in a subcomplex (hereafter termed BBSome1·2·5·7·9). The peak fraction of IFT88, a member of the IFT-B complex, is found in fraction 14 in wild-type retinas and remains unchanged in Bbs8−/− retinas as expected (Fig. 2a). Since IFT-B complex assembly is independent from BBSome assembly, the peak fraction number of IFT88 serves as an internal control for sucrose gradient fractionation.
Figure 2.
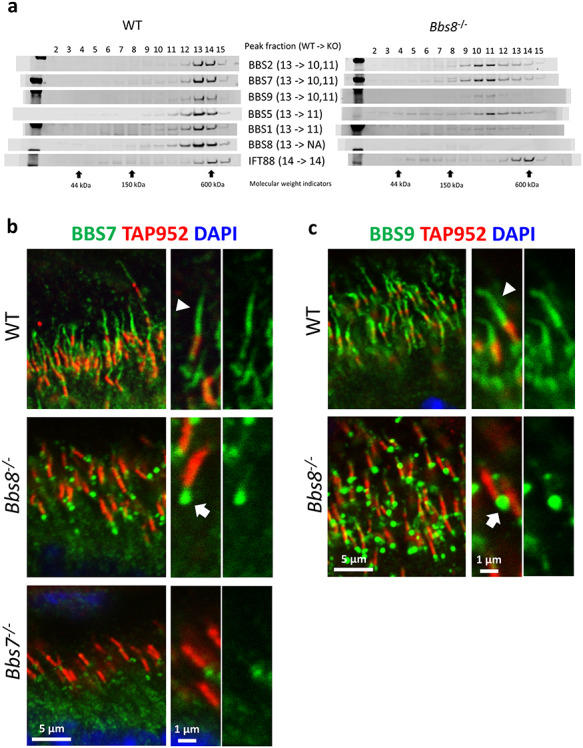
BBSome subcomplex consisting of BBS1, BBS2, BBS5, BBS7 and BBS9 (BBSome1·2·5·7·9) is denied ciliary entry in the absence of BBS8 in Bbs8−/− retinal photoreceptors. (a) Sucrose gradient fractionation shows that the peak fractions of BBS1, BBS2, BBS5, BBS7 and BBS9 are shifted from fraction 13 to fraction 10–11 in retinas of Bbs8−/− mice. (b) BBS7, a member of the BBSome1·2·5·7·9 subcomplex, is denied ciliary entry and accumulates at the base of the connecting cilia in retinal photoreceptors of Bbs8−/− mice (arrow). (c) BBS9, a member of the BBSome1·2·5·7·9 subcomplex, is also denied ciliary entry in retinal photoreceptors of Bbs8−/− mice (arrow).
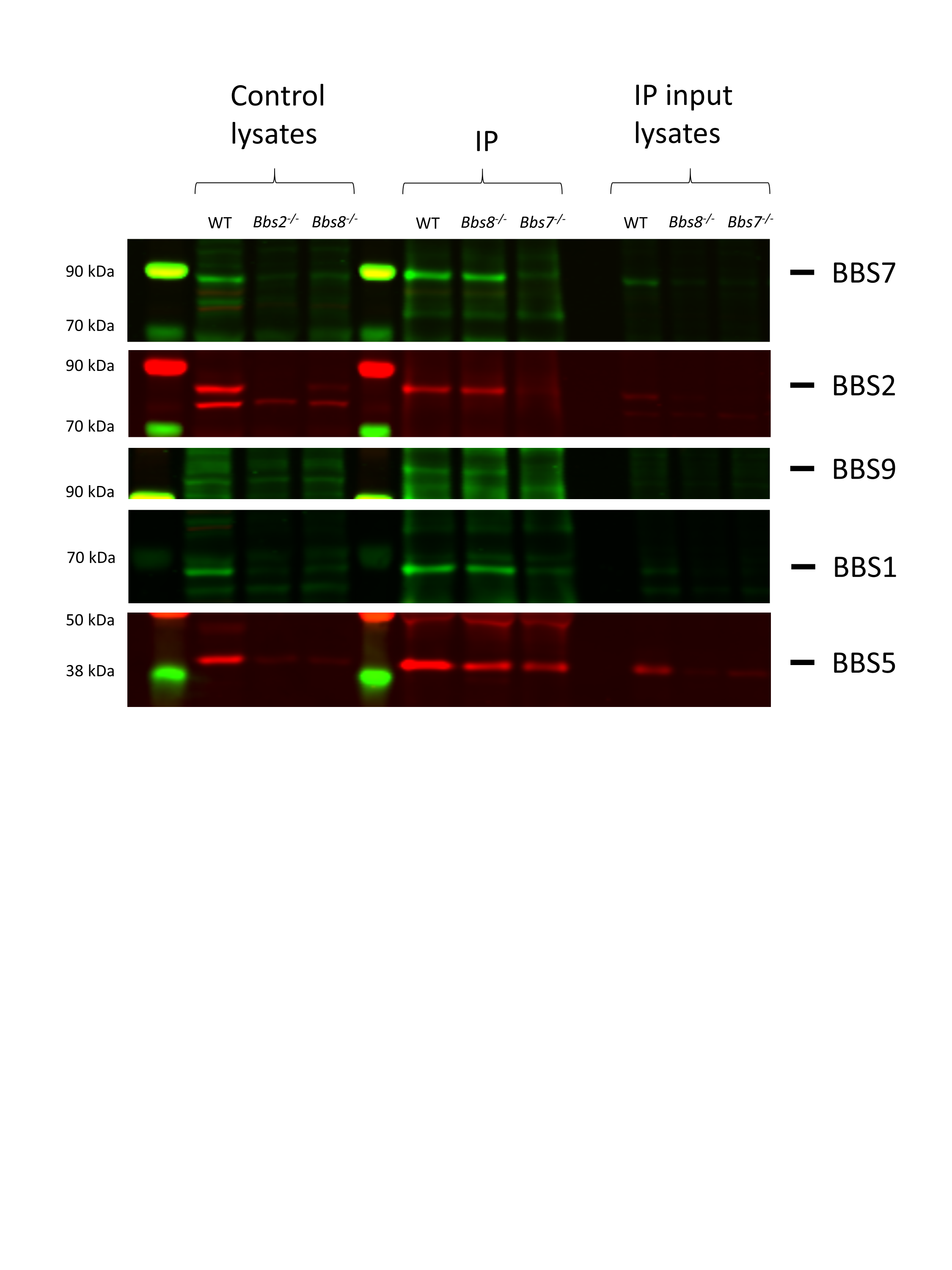
To verify that BBS1, BBS2, BBS5, BBS7 and BBS9 exist in a subcomplex in Bbs8−/− retinas, immunoprecipitation (IP) was performed using the anti-BBS7 antibody to pull down proteins associated with BBS7. Retinas from 1-month-old wild-type and Bbs8−/− mice were disrupted and the retinal lysates were incubated with magnetic beads coupled to the anti-BBS7 antibody. Retinal lysates prepared from a Bbs7−/− mouse were used as the negative control for the IP experiment. The immunoblot was then probed with antibodies against BBS1, BBS2, BBS5, BBS7 and BBS9 proteins. A robust BBS7 signal is observed in the IP samples from wild-type retinas as well as from Bbs8−/− retinas but not in the IP sample from Bbs7−/− retinas, indicating that BBS7 is successfully pulled down in control and in Bbs8−/− mice (Supplementary Material, Fig. S1). A robust signal from BBS2 is also observed in the IP samples from wild-type retinas as well as from Bbs8−/− retinas but not in the IP sample from Bbs7−/− retinas, indicating that BBS2 and BBS7 interact in both wild-type and Bbs8−/− retinas. Similarly, BBS1, BBS5 and BBS9 are detected in IP samples of both wild-type and Bbs8−/− retinas (Supplementary Material, Fig. S1). This result indicates that these proteins interact and exist in a subcomplex in Bbs8−/− retinas. It is noted that BBS5 is weakly detected even in the IP sample prepared from Bbs7−/− retinas, suggesting that this anti-BBS7 antibody may cross-react with BBS5. However, even if this is the case, it does not change the conclusion of the IP experiment. BBS1, BBS2, BBS5, BBS7 and BBS9 exist in a subcomplex in Bbs8−/− retinas.
To determine whether the BBSome1·2·5·7·9 subcomplex can enter cilia, we performed immunohistochemistry to visualize BBS7 and BBS9 in Bbs8−/− retinas. In P15 wild-type retinas, BBS7 localizes to the outer segment portion of the photoreceptor ciliary axoneme (Fig. 2b, arrowhead), as well as within the connecting cilia marked by the TAP952 antibody (Fig. 2b; Supplementary Material, Fig. S2). This localization pattern is absent in photoreceptors of Bbs7−/− littermates, negative controls exposed to identical experimental conditions (Fig. 2b; Supplementary Material, Fig. S2). Similarly, BBS5 has been observed in the photoreceptor ciliary axoneme, including the outer segment portion of the axoneme and the connecting cilia (24). BBS8 localization to the photoreceptor axoneme was also demonstrated elsewhere (30), supporting the observation patterns we noted here. A gap is often observed between the BBS7 signal in the outer segment portion of the ciliary axoneme and that in the connecting cilia (Fig. 2b; Supplementary Material, Fig. S2).
To determine whether the BBSome subcomplex detected in retinas of Bbs8−/− mice can enter photoreceptor cilia, we probed Bbs8−/− retinas with the anti-BBS7 and anti-BBS9 antibodies, respectively. We have shown that components of this BBSome1·2·5·7·9 subcomplex interact. In Bbs8−/− retinas, BBS7 and BBS9, components of the BBSome1·2·5·7·9 subcomplex, both accumulate at the bottom of the connecting cilia (Fig. 2b and c, arrows; Supplementary Material, Figs S3 and S4), suggesting that this subcomplex cannot gain entry into photoreceptor cilia.
We then turn our attention to Bbs2−/− retinas. Sucrose gradient fractionation of retinal lysates from 1-month-old mice shows that in the absence of BBS2, peak fractions of BBS1, 5, 8 and 9 are jointly shifted from fraction 13 to fraction 11, indicating the presence of a subcomplex BBSome1·5·8·9 (Fig. 3a). In contrast, the peak fraction of BBS7 has shifted from fraction 13 to fraction 7, indicating that BBS7 is not a part of this subcomplex (Fig. 3a). To determine whether subcomplex BBSome1·5·8·9 can gain ciliary entry, we performed immunohistochemistry to visualize BBS8 and BBS9, components of the subcomplex, in Bbs2−/− retinas. In wild-type photoreceptors, BBS8 localizes to the photoreceptor ciliary axoneme (Fig. 3b, arrowhead; Supplementary Material, Fig. S5), similar to the pattern observed for BBS7. BBS8 is readily observed in the outer segment portion of the ciliary axoneme, and to a lesser extent, within the connecting cilia (Fig. 3b; Supplementary Material, Fig. S5). These patterns are absent in photoreceptors of Bbs8−/− mice, which serves as negative controls subjected to the same experimental procedures (Fig. 3b; Supplementary Material, Fig. S5). BBS9 also localizes to the outer segment portion of the ciliary axoneme in wild-type retinas (Fig. 3c, arrowhead). Surprisingly, in the photoreceptors of Bbs2−/− mice, BBS8 still localizes to the outer segment portion of the photoreceptor ciliary axoneme (Fig. 3b, arrow; Supplementary Material, Fig. S6). BBS9, another member of the BBSome1·5·8·9 subcomplex found in Bbs2−/− retinas, also localizes to photoreceptor ciliary axonemes in the absence of BBS2 (Fig. 3c, arrow; Supplementary Material, Fig. S4). These data show that the subcomplex of the BBSome consisting of at least BBS1, BBS5, BBS8 and BBS9 identified in Bbs2−/− retinas can gain ciliary entry in photoreceptor cells.
Figure 3.
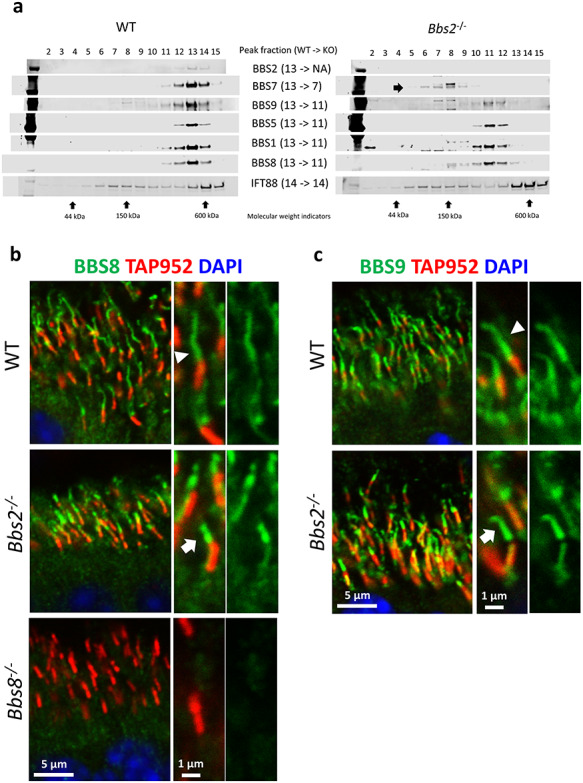
BBSome subcomplex consisting of BBS1, BBS5, BBS8 and BBS9 is recruited to photoreceptor cilia in the absence of BBS2 in Bbs2−/− retinal photoreceptors. (a) Sucrose gradient fractionation shows that the peak fractions of BBS1, BBS5, BBS8 and BBS9 are shifted from fraction 13 to fraction 11 in retinas of Bbs2−/− mice. (b) BBS8, a member of this subcomplex, localizes to photoreceptor ciliary axoneme in Bbs2−/− mice (arrow). (c) BBS9, a member of this subcomplex, is also observed in photoreceptor ciliary axoneme in Bbs2−/− mice (arrow).
In Bbs7−/− retinas, the BBSome1·5·8·9 subcomplex is also observed. When retinas of 1-month-old Bbs7−/− mice are subject to sucrose gradient fractionation, peak fractions of BBS1, BBS5, BBS8 and BBS9 are jointly shifted from fraction 12 to fractions 10 and 11 containing complexes with molecular weights smaller than the complete BBSome (Fig. 4a). In eluted fractions of Bbs7−/− retinas, BBS2 is barely detectable, consistent with the notion that the stability of BBS2 depends on BBS7 (6). To discern whether the BBSome1·5·8·9 subcomplex, which can gain ciliary entry in Bbs2−/− retinas can also gain ciliary entry in the absence of BBS7, we probed the localization of BBS8 and BBS9 in the photoreceptor cells of Bbs7−/− mice. Immunohistochemical data show that in the retinas of Bbs7−/− mice, both BBS8 and BBS9 are observed in the outer segment portion of the photoreceptor ciliary axoneme (Fig. 4b and c, arrows; Supplementary Material, Figs S6 and S4), suggesting that this subcomplex is recruited to photoreceptor cilia. Together, these data indicate that similar to Bbs2−/− retinas, a subcomplex of the BBSome consisting of at least BBS1, BBS5, BBS8 and BBS9 exists in Bbs7−/− retinas, and this subcomplex can be recruited to photoreceptor cilia in the absence of BBS7. Therefore, in certain cases, a partially assembled BBSome is admitted to photoreceptor cilia. However, the data suggest that specific subunits are needed to gain photoreceptor ciliary entry.
Figure 4.
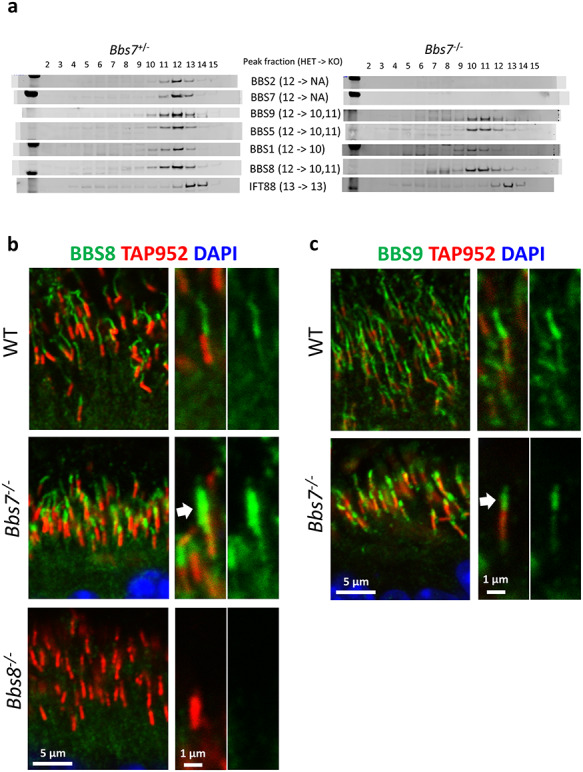
The BBSome subcomplex (BBSome1·5·8·9) consisting of BBS1, BBS5, BBS8 and BBS9 is found in photoreceptor cilia in the absence of BBS7 in Bbs7−/− retinal photoreceptors. (a) Sucrose gradient fractionation shows that the peak fractions of BBS1, BBS5, BBS8 and BBS9 are shifted from fraction 12 to fraction 10–11 in retinas of Bbs7−/− mice. (b) BBS8, a member of this BBSome1·5·8·9 subcomplex, localizes to photoreceptor ciliary axoneme in Bbs7−/− mice (arrow). (c) BBS9, a member of this BBSome1·5·8·9 subcomplex, is also observed in photoreceptor ciliary axoneme in Bbs7−/− mice (arrow).
The BBSome1·5·8·9 subcomplex found in both Bbs2−/− and Bbs7−/− retinas can gain entry to photoreceptor cilia, whereas the BBSome1·2·5·7·9 subcomplex found in Bbs8−/− retinas is denied photoreceptor ciliary entry. This distinction shows that the ciliary entry mechanism of the BBSome in photoreceptor cilia remains selective, and this process likely depends upon certain components of the BBSome. Inferring from the data, BBS8, or another unidentified member of this BBSome1·5·8·9 subcomplex, could be indispensable for the ciliary entry of the BBSome in photoreceptor cells. Alternatively, the presence of BBS2 and/or BBS7 in a partially assembled BBSome complex could exclude the ciliary entry of the BBSome1·2·5·7·9 subcomplex. It is not known whether the requirement for specific subunits is due to direct interaction or due to the structural surface features maintained by the incorporation of BBS8 into the BBSome. Together, we found that in contrast to primary cilia, where only the completely assembled BBSome can gain ciliary entry, a partially assembled BBSome subcomplex can gain entry to photoreceptor cilia if specific criteria are met. This photoreceptor ciliary entry requires the presence of specific BBSome components, which can involve BBS8.
BBS3 is a small GTPase required for the ciliary localization of BBSome components in cultured RPE1 cells (20, 21). We tested whether the recruitment of the BBSome to the specialized cilia of photoreceptor cells requires BBS3 similar to in primary cilia. The fractionation of 1-month-old Bbs3−/− retinal lysates by sucrose gradient shows that in the retinas of Bbs3−/− mice, the BBSome is assembled, as the peak fraction number remains unchanged in control versus Bbs3−/− mice (Fig. 5a). Next, we probed the localization of BBS7, BBS8 and BBS9 in the photoreceptors of Bbs3−/− mice to determine whether the BBSome can be recruited to photoreceptor cilia without BBS3. Unexpectedly, all these three BBSome components are readily observed in the photoreceptor ciliary axoneme of Bbs3−/− photoreceptors (Fig. 5b-d, arrows; Supplementary Material, Figs S3, S6 and S4), indicating that the BBSome can gain photoreceptor ciliary entry in the absence of BBS3. In addition, the aberrant escape of the polyglutamylation marks to the entire photoreceptor ciliary axoneme observed in photoreceptors of Bbs7−/− mice is not observed in Bbs3−/− photoreceptors (Supplementary Material, Fig. S7). Together, these data demonstrate that unlike in primary cilia, where ciliary recruitment of the BBSome requires BBS3, the BBSome is recruited to cilia without BBS3 in the specialized cilia of photoreceptor cells.
Figure 5.
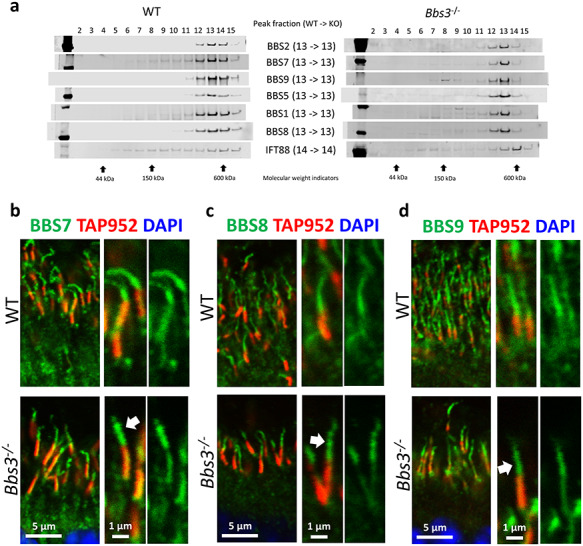
The BBSome localizes to photoreceptor cilia in the absence of BBS3 in Bbs3−/− retinal photoreceptors. (a) Sucrose gradient fractionation shows that the peak fractions of BBS1, BBS2, BBS5, BBS7, BBS8 and BBS9 are unchanged in retinas of Bbs3−/− mice. (b) BBS7 localizes to photoreceptor ciliary axonemes in retinas of Bbs3−/− mice (arrow). (c) BBS8 localizes to photoreceptor ciliary axonemes in Bbs3−/− mice (arrow). (d) BBS9 is also observed in the photoreceptor ciliary axoneme in Bbs3−/− mice (arrow).
We then determined whether the presence of a partial BBSome within photoreceptor cilia (such as in Bbs2−/− and Bbs7−/− mice) or a complete BBSome without BBS3 function in photoreceptor cilia (such as in Bbs3−/− mice) confer benefits in regard to the retinal degeneration phenotype and alter phenotypic severity. We visualized retinal histology in 4-month-old BBS mice that were crossed onto the 129 background for at least 7 generations to minimize the background effect (Fig. 6a). The thicknesses of their outer nuclear layers were measured and compared using one-way ANOVA followed by post hoc Tukey’s test (Fig. 6b). The thicknesses of the outer nuclear layers in Bbs2−/− mice and Bbs7−/− mice are not significantly different from each other (Bbs2−/− mice: 27.94 ± 2.47 μm, n = 3; Bbs7−/− mice: 33.51 ± 0.42 μm, n = 4; P = 0.28). This is consistent with the finding that BBSome subcomplexes in Bbs2−/− and Bbs7−/− retinas have a similar composition. In Bbs8−/− mice, in which the partial BBSome complex is denied ciliary entry and accumulates underneath photoreceptor cilia, retinal degeneration is more severe than that in both Bbs7−/− mice (Bbs8−/− mice: 19.17 ± 0.85 μm, n = 4; Bbs7−/− mice: 33.51 ± 0.42 μm, n = 4; P = 0.0085) and Bbs2−/− mice, although the difference between Bbs8−/− and Bbs2−/− does not reach statistical significance (Fig. 6b). We then examined outer nuclear layer thickness in Bbs3−/− mice, which have completely assembled BBSomes that are able to gain entry into the photoreceptor cilia. Surprisingly, retinal degeneration in Bbs3−/− mice (Bbs3−/− mice: 6.56 ± 0.09 μm, n = 3) is more severe than that in Bbs2−/− (P < 0.0001), Bbs7−/−(P < 0.0001) or Bbs8−/− mice (P < 0.0001). This result indicates that the presence of a complete BBSome complex in Bbs3−/− mice, even if it is able to gain entry into cilia without BBS3, lacks adequate function and does not delay retinal degeneration in Bbs3−/− mice (Fig. 6). We also compared the severity of retinal degeneration in Bbs4−/− mice with the abovementioned BBS mouse models. The BBSome is largely assembled in the absence of BBS4, at least in cell culture, since BBS4 is among the last subunits incorporated into the BBSome (6). Retinal degeneration in Bbs4−/− mice is faster than that in Bbs8−/− mice (Bbs4−/− mice: 11.60 ± 0.22, n = 3; Bbs8−/− mice: 19.17 ± 0.85 μm, n = 4; P = 0.005) and not statistically different from that in Bbs3−/− mice (Bbs4−/− mice: 11.60 ± 0.22, n = 3; Bbs3−/− mice: 6.56 ± 0.09 μm, n = 3; P = 0.44) (Fig. 6). This supports the notion that the presence of nearly complete, BBS4-defieicnt BBSomes does not confer an advantage in terms of photoreceptor survival. The full list of P-values is listed in Supplementary Material, Table S1.
Figure 6.
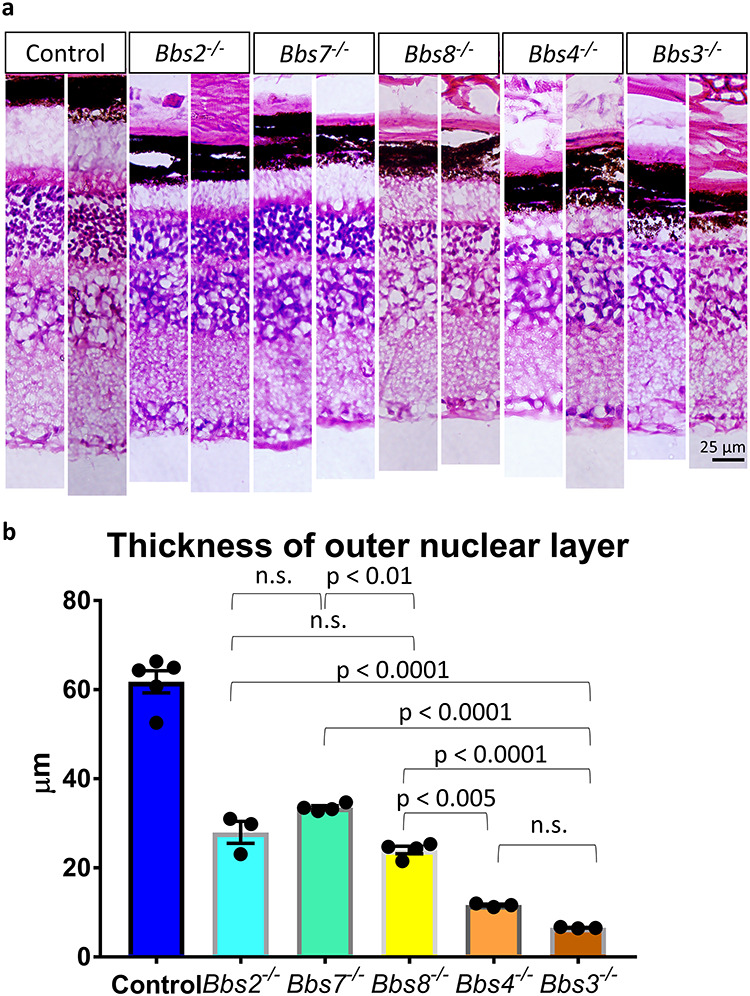
Retinal degeneration is not prevented by the presence of a partial BBSome nor a complete BBSome without BBS3 function within photoreceptor cilia. (a) Retinal histology of 4-month-old Bbs2−/−, Bbs7−/−, Bbs8−/−, Bbs4−/− and Bbs3−/− mice and their control littermates was visualized by light microscopy, and the thicknesses of their outer nuclear layers were quantified (b). Retinal histology of two animals per genotype is shown. Retinal degeneration in Bbs8−/− mice, in which BBSome1·2·5·7·9 is denied photoreceptor ciliary entry, is more severe than that in Bbs7−/− mice, in which a partial BBSome1·5·8·9 is in photoreceptor cilia. Retinal degeneration in Bbs3−/− mice, which possess a completely assembled BBSome in photoreceptor cilia, is more severe than that in Bbs2−/−, Bbs7−/− or Bbs8−/− mice, which possess partial BBSomes. Each data point represents the thickness of the outer nuclear layer of a single animal. The error bars represent the standard error of the mean.
We then determined whether the presence of partial BBSomes within photoreceptor cilia (such as in Bbs2−/− and Bbs7−/− mice) or a complete BBSome without BBS3 function in photoreceptor cilia (in Bbs3−/− mice) can traffic membrane cargo proteins and prevents the mislocalization of putative BBSome cargo syntaxin-3 (STX3) to the photoreceptor outer segment. The localization of STX3 was visualized by immunohistochemistry in P15–16 retinas, and the photoreceptor outer segment was visualized by the anti-rhodopsin (RHO) antibody. STX3 mislocalizes to the photoreceptor outer segment in Bbs2−/−, Bbs7−/−, Bbs8−/−, Bbs4−/− and Bbs3−/− mice (Supplementary Material, Fig. S8), supporting the notion that a partial BBSome in photoreceptor cilia or a complete BBSome in cilia but without BBS3 results in membrane cargo mislocalization due to the lack of adequate BBSome function.
Since we did not observe a notable difference in regard to mislocalization of STX3 among BBS mouse models, we explored other early phenotypes in BBS mice that may be relevant to photoreceptor survival. We previously found that the connecting cilia are abnormally elongated in Bbs8−/− photoreceptors as early as P15–16 (28). It is not clear whether this phenotype is present in other BBS mouse models or whether differences in phenotypic severity among BBS models can be detected at this early stage. Therefore, we measured the length of photoreceptor connecting cilia in P15–16 Bbs2−/−, Bbs7−/− and Bbs3−/− mice in addition to Bbs8−/− mice to determine whether these mutant mouse models have abnormal connecting cilia length. To measure connecting cilia length, connecting cilia were visualized using the TAP952 antibody and 30 connecting cilia were randomly selected and measured in each animal. The experiment was repeated in a different set of animals. The measurements from these two experiments were combined for interpretation, resulting in 60 connecting cilia measurements per genotype. The results confirm that connecting cilia in Bbs8−/− mice are abnormally elongated (wild type: 1.49 ± 0.030 μm; Bbs8−/−: 1.83 ± 0.029 μm; P < 0.0001; Supplementary Material, Fig. S9). Interestingly, compared with Bbs8−/− mice, this connecting cilia phenotype is milder in both Bbs7−/− mice (Bbs7−/−: 1.59 ± 0.029 μm; P < 0.0001) and in Bbs2−/− mice (Bbs2−/−: 1.72 ± 0.023 μm; P = 0.053), and more severe in Bbs3−/− mice (Bbs3−/−: 1.98 ± 0.030 μm; P < 0.005), mirroring the phenotypic severity noted in retinal degeneration (Supplementary Material, Fig. S9). The full list of P-values is shown in Supplementary Material, Table S2. The connecting cilia phenotype and the retinal degeneration phenotype have a Pearson’s correlation coefficient of −0.94, where a Pearson’s correlation coefficient of −1 would indicate a perfect negative correlation. In other words, these BBS mouse models possess various degrees of connecting cilia abnormality at a young age highly correlated with their rates of photoreceptor degeneration.
In summary, we found that in contrast to primary cilia, which requires a completely assembled BBSome for ciliary entry, the BBSome subcomplex BBSome1·5·8·9 in both Bbs2−/− and Bbs7−/− retinas can enter photoreceptor cilia. On the other hand, the BBSome subcomplex BBSome1·2·5·7·9 is denied ciliary entry, demonstrating that the entry mechanism is selective and depends on composition of the complex. We may deduce that BBS8, or another undetected member in this subcomplex, is critical for gaining ciliary entry. Alternatively, BBS2 or BBS7 could inhibit the ciliary entry of an incompletely assembled BBSome complex. In addition, unlike the requirement for BBS3 for BBSome entry in primary cilia, the complete BBSome is found within photoreceptor cilia in Bbs3−/− mice. BBS3 is not required for BBSome entry in the specialized cilia of photoreceptor cells. We then examined whether the existence of a partial BBSome or a complete BBSome without BBS3 in photoreceptors modifies the retinal degeneration phenotype. Interestingly, BBS mouse models with nearly completely assembled (such as Bbs4−/−) or completely assembled BBSomes without BBS3 (such as Bbs3−/−) tend to have more severe retinal degeneration. A summary schematic depicting BBSome subcomplexes and their relative localization in photoreceptors is shown in Figure 7. The rendering of the BBSome and its subcomplexes is derived from the 3D structure of the mammalian BBSome (31).
Figure 7.
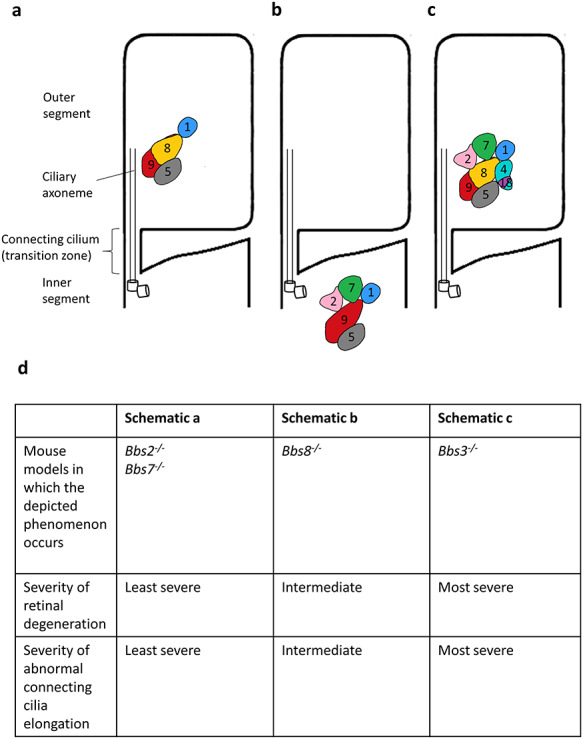
Summary schematic of the BBSome subcomplexes and their localization in photoreceptor cilia in BBS mouse models. (a) The BBSome1·5·8·9 subcomplex localizes to photoreceptor cilia in Bbs2−/− and Bbs7−/− mice. These mice have comparatively mild retinal degeneration. (b) The BBSome1·2·5·7·9 subcomplex is denied ciliary entry in photoreceptor cells in Bbs8−/− mice. These mice have more severe retinal degeneration compared with that in Bbs2−/− and Bbs7−/− mice. (c) The entire BBSome is present in photoreceptor cilia in Bbs3−/− mice. These mice have the most severe retinal degeneration. These observations are listed in (d).
Discussion
Primary cilia do not grant entry to a partially assembled BBSome. It is not known whether this is due to the loss of association between the IFT trains and partially assembled BBSomes or whether there are additional mechanisms at the transition zone that discriminate between completely assembled or partially assembled BBSomes. We examined whether the recruitment of the BBSome to photoreceptor cilia follows the principles observed in primary cilia, which admit only the completely assembled BBSome. We found in the absence of BBS2 or BBS7, the BBSome1·5·8·9 subcomplex exists and is capable of entering photoreceptor cilia. This observation is supported by an independent study reporting that BBS5 localization is not severely altered in the photoreceptors of Bbs2−/−, Bbs7−/− and Bbs4−/− mice (32). However, ciliary entry is not indiscriminate. Specific subunits, or perhaps structural conformations or charge distribution afforded by the incorporation of these subunits, appear to be required. In the absence of BBS8, for example, the BBSome1·2·5·7·9 subcomplex is found, but this subcomplex is denied ciliary entry and accumulates at the bottom of the photoreceptor cilia. The existence of this subcomplex is confirmed by IP. From these results, it could be inferred that BBS8, or an unidentified member of the subcomplex, is key to gaining ciliary entry. It is also admissible that BBS2 and/or BBS7, when the BBSome is incomplete, prevents the ciliary entry of the partial BBSome. The gating mechanism that discriminates between these two subcomplexes, granting entry to one but denying entry to the other, is not fully understood.
The BBSome associates with the IFT trains to enter cilia. It has been found that the association between the BBSome and IFT-B (anterograde IFT) complex is mediated by the interface between IFT38, BBS1, BBS2 and BBS9 (33). It has been reported that BBS1 interacts with IFT144, a member of the IFT-A (retrograde IFT) complex when co-expressed (17). Since the BBSome1·2·5·7·9 subcomplex containing the above-mentioned subunits is denied ciliary entry, it is therefore unlikely that failure to mount onto the IFT trains is the main cause behind the denial of ciliary access. It is more likely that other mechanisms such as gating are at play. However, we cannot exclude the possibility that the difference in shape and conformation of the BBSome1·2·5·7·9 subcomplex renders it unable to mount onto the IFT trains despite the presence of the BBS1, BBS2 and BBS9 interface.
It is also not clear how a BBSome subcomplex is assembled in the absence of BBS2 or BBS7. The BBS/CCT complex consists of BBS6, BBS10, BBS12, BBS7 and CCT family chaperonin proteins CCT1, CCT2, CCT3, CCT4, CCT5 and CCT8 (34). BBS7 and also BBS2 show robust interactions with the CCT chaperonin complex (34). BBS6, BBS10 and BBS12 likely act as substrate-binding subunits that mediate the interaction between BBS7 and the CCT chaperonins, which regulate protein folding. When individual BBSome subunits are overexpressed, both BBS2 and BBS7 can robustly pull down CCT/TRiC proteins in 293 T cells (6). BBS6 is found to directly interact with BBS2 in Co-IP experiments in transfected cells, consistent with the idea that chaperonin-like BBS proteins mediate the interaction between BBS2 and the CCT complex (34). Even though association of BBS2 and BBS7 with BBS9 is disrupted in BBS6-, BBS10- or BBS12-depleted cells, association between BBS9 and BBS1 or BBS9 and BBS4 were barely or only slightly affected (34). This suggests that although BBS2 and BBS7 are required for the assembly of the complete BBSome, a subcomplex of the BBSome could be formed in their absence. How this subcomplex is assembled is unknown. BBS12 is found to interact with BBS1, BBS2, BBS4, BBS7 and BBS9 in co-IP experiments of transfected cells (34). Whether or not BBS12 or another unknown protein plays a role in mediating a partially assembled BBSome remains unknown. The BBSome cannot fully assemble in the absence of BBS6 in both eyes and testis of Bbs6−/− mice (34). Interestingly, in eyes of Bbs6−/− mice, the peak fraction of BBS1 is shifted to the same fraction as the peak fraction of BBS8, colocalizing in the fraction containing complexes with molecular weight ranging from 150 to 200 kDa (34). These data support the potential existence of a subcomplex containing BBS1 and BBS8 in the absence of BBS6, which interacts with BBS2 and likely mediates the interaction of BBS2 with the CCT complex (34).
In addition to the admittance of a BBSome subcomplex to photoreceptor cilia under specific criteria, we found that the localization of the BBSome to photoreceptor cilia does not require BBS3, which is required for recruitment of the BBSome to primary cilia. It is not clear whether the ability of the BBSome to enter photoreceptor cilia without BBS3 is due to distinct features of the photoreceptor transition zone or distinct features of the BBSome itself.
We examined whether the presence of the BBSome1·5·8·9 subcomplex in photoreceptor cilia in Bbs2−/− and Bbs7−/− mice, or the presence of the entire BBSome in cilia in Bbs3−/− mice, delay retinal degeneration. We performed this comparison in 4-month-old BBS mice on the 129 background. Interestingly, at this age, the least severe retinal degeneration is observed in Bbs2−/− and Bbs7−/− mice. Bbs8−/− mice, in whose photoreceptors the partial BBSome is denied ciliary entry, have more severe degeneration than both Bbs2−/− and Bbs7−/− mice, even though the difference between Bbs2−/− and Bbs8−/− mice is not statistically significant. However, degeneration in Bbs8−/− mice is not nearly as severe as in Bbs3−/− mice, which have complete BBSomes in their photoreceptor cilia but lack adequate function due to the lack of BBS3. It is contrary to expectation that the severity of the phenotype is not alleviated but aggravated in Bbs3−/− mice. We also evaluated the severity of retinal degeneration in Bbs4−/− mice. The BBSome is fully assembled except the absence of BBS4 (6). The severity of retinal degeneration in Bbs4−/− mice is closest to and not statistically different from that in Bbs3−/− mice. It has been reported that BBS5 localization to photoreceptor cilia is not severely altered in Bbs4−/− mice (32), suggesting that BBS4-deficient BBSomes may be in photoreceptor cilia in Bbs4−/− mice. Based on these results, having a near-complete BBSome such as in Bbs4−/− mice does not confer a survival advantage to photoreceptor cells. Having a completely assembled BBSome but without BBS3 function also does not delay retinal degeneration. In all BBS mutant models examined, STX3 mislocalizes to the outer segment, suggesting that the critical function of the BBSome in trafficking membrane protein cargo is missing.
Bbs5−/− mice have reportedly much slower retinal degeneration compared with Bbs8−/− mice, even though both BBS5 and BBS8 are members of the BBSome complex (35). Interestingly, BBSome assembly is largely unaffected in the absence of BBS5, at least in cells (20). It is not known whether a BBSome subcomplex is recruited to photoreceptor cilia without BBS5 and retains limited functions. The assembly and localization of BBSome components in the photoreceptors of Bbs5−/− mice merits further investigation.
BBS2 has two calcium-binding loops and mutations in the first calcium-binding loop in the β-propeller domain cause BBS, indicating that the function of the BBSome requires the calcium binding of BBS2 (36). This is congruent with the finding that even though the subcomplex containing at least BBS1, BBS5, BBS8 and BBS9 (BBSome1·5·8·9) can enter photoreceptor cilia without BBS2, it does not possess the ability to regulate membrane cargo and does not prevent retinal degeneration in Bbs2−/− mice. In bovine retinal BBSomes, ARL6/BBS3 interacts with the BBSome through BBS1 and BBS7 (31). BBS3 mediates the interaction between the BBSome with membranes and regulates the recruitment of membrane cargo (22). This is consistent with the fact that completely assembled BBSome in the photoreceptor cilia of Bbs3−/− mice fails to regulate membrane cargo localization such as STX3. In other words, we found that neither the partially assembled BBSome subcomplex that we identified, nor the completely assembled BBSome without BBS3 can successfully regulate membrane cargo localization even if they can gain ciliary entry. However, we cannot completely rule out the possibility that BBSome subcomplexes or fully assembled BBSomes without BBS3 perform other limited functions. The cause of varying phenotypic severity in retinal degeneration remains unclear.
When we examined the length of photoreceptor connecting cilia in P15–16 BBS mice, we found that although BBS mice all have abnormal connecting cilia elongation, phenotypic differences already exist at this early age when degeneration has just begun. Interestingly, mouse models with the greatest connecting cilia elongation at a young age are the models with the fastest retinal degeneration, and these two phenotypes have a strong negative correlation. The cause of abnormal connecting cilia lengthening is unknown but likely pertains to the loss of some functional roles of the BBSome and/or BBS3 at the photoreceptor transition zone. This abnormality appears to be partially mitigated by the ability of the BBSome1·5·8·9 subcomplex to cross the transition zone into cilia, as the connecting cilia phenotype is the mildest in Bbs7−/− and Bbs2−/− mice. The crossing of the BBSome without BBS3, on the other hand, does not seem to alleviate it, as the most severe phenotype is found in Bbs3−/− mice. It is important to note that correlation does not equal causality. Therefore, whether connecting cilia defects contribute to the variability observed in retinal degeneration is unknown.
Together, we show that knockout mouse models of BBS have genuine biological differences in phenotypic severity that are not due to their genetic backgrounds. We propose that photoreceptor degeneration is caused by defects in more than one biological processes, some of which require the intact, fully assembled BBSome, while others can be mitigated by partial BBSomes. Our data suggest that membrane cargo transport requires the intact BBSome and BBS3, since membrane cargo STX3 mislocalizes in all BBS mice even in those with partial complexes. In other processes, however, it is possible that partial BBSomes may retain limited functions (such as at the transition zone) and the mitigation of such defects by partial complexes and/or BBS3 causes differences in phenotypic severity among BBS mutant mice. We postulate that this is the reason why BBS mice share classical BBS phenotypes such as retinal degeneration but their severity varies. Although speculative, this is an intriguing hypothesis that merits investigation.
Retinal degeneration is not the only BBS phenotype where phenotypic variations have been observed among BBS knockout mouse models, even though these mouse models all have Bardet-Biedl syndrome (16, 28, 35, 37–39). For example, knockout mouse models of the BBSome demonstrate differential sensitivity to the effects of leptin on renal sympathetic nerve activity (SNA). Bbs2−/− mice have normal baseline renal SNA and normal blood pressure in the presence of high leptin levels, because they are fully resistant to the effects of leptin. In contrast, Bbs4−/− and Bbs6−/− mice have elevated baseline renal SNA and elevated blood pressure (40). Bbs2−/− mice are resistant to the effect of leptin on renal SNA and are spared from developing hypertension, whereas Bbs4−/− and Bbs6−/− mice are sensitive to the high leptin levels resulting in elevated blood pressure. The difference in renal phenotypes could be explained by the existence of a BBSome subcomplex in the absence of BBS4 (the BBS4-deficient BBSome). This subcomplex, retaining residual BBSome function, could potentially regulate receptor localization that enables the detection of elevated leptin levels, thereby contributing to increased SNA and hypertension in Bbs4−/− mice. Even though only a completely assembled BBSome can gain entry to primary cilia, the leptin receptor localizes to the cellular plasma membrane (41). It is not clear whether partial BBSomes may retain residual functions in regard to these cargos. The explanation of the phenotypic difference between Bbs2−/− and Bbs4−/− mice remains speculative. We also cannot rule out the possibility of non-BBSome functions of individual BBS gene products.
Our findings suggest that BBS8 could be a candidate for mediating the ciliary entry of the BBSome in photoreceptors. Previously, different apparent molecular weights of the BBSome between eye and testis tissues have been reported (34), pointing to tissue-specific differences in BBSome composition. In addition, BBS5 and BBS8 have retina-specific isoforms, offering further support for the existence of tissue-specific BBSomes. For example, BBS8 has a retina-specific isoform, which encodes an additional 10 amino acids from a previously unknown BBS8 exon (25). Mutations in this exon cause non-syndromic retinal degeneration. BBS5, a component of the BBSome, also has a retina-specific splice variant (24). In this study, we found that while primary cilia deny entry to an incomplete BBSome, photoreceptor cilia admit a subcomplex of the BBSome meeting certain criteria. It is possible that the retinal-specific Bbs8 exon is required for ciliary recruitment of the BBSome in photoreceptor cells. We postulate that cell-specific function and therefore cargo range of the BBSome can be enabled by cell- or tissue-specific isoforms of certain BBSome components. It is possible that these isoforms, as a part of the modified BBSome, enable the BBSome to perform photoreceptor-specific functions in their specialized cilia. Together, these data indicate that the ciliary entry of the BBSome, and potentially its functions, is subjected to cell-specific regulation.
In this study, we have observed that the polyglutamylation marks that characterize the photoreceptor connecting cilia aberrantly expands to the entire axoneme in BBSome mutant mice, but this is not the case in Bbs3−/− mice, pointing to the fact that certain functions pertaining to the role of the BBSome in ciliary structure may be preserved in Bbs3 mutants. There is an intact BBSome in Bbs3−/− mice. It is possible that such an intact BBSome, despite the loss of its function to transport membrane cargos due to the lack of BBS3, can nevertheless perform other functions. Such possibilities merit further investigation. However, these limited functions of the BBSome in Bbs3−/− mice did not result in a survival advantage in photoreceptor cells.
The glutamylation of ciliary subdomains is the result of a careful balancing act by glutamylases and deglutamylases (42). The knockdown of deglutamylase ccp5 in zebrafish increases ciliary tubulin glutamylation and causes ciliopathy phenotypes (43). Interestingly, targeted deglutamylation of cilia reduces the entry of IFT into cilia and preferentially slows down anterograde IFT, indicating that axonemal glutamylation affects ciliary transport (44). In addition, Hedgehog signaling and the agonist-induced accumulation of signaling molecules in cilia are hampered by the targeted deglutamylation of the ciliary axoneme (44). Together, these studies suggest that alterations in ciliary glutamylation could affect ciliary transport and signaling. Alterations of glutamylation observed in BBS photoreceptor cilia could be caused by mislocalization of glutamylases or deglutamylases that normally maintains the boundaries of these ciliary subdomains by regulating posttranslational modifications.
Together, this study demonstrates that a subcomplex of the BBSome containing at least BBS1, BBS5, BBS8 and BBS9 (BBSome1·5·8·9) enters photoreceptor cilia in the absence of BBS2 or BBS7, in contrast to primary cilia. A different subcomplex (BBSome1·2·5·7·9) identified in the absence of BBS8 is unable to gain ciliary entry in photoreceptor cells. BBS8 could be important for gaining ciliary entry of the BBSome in photoreceptors. In addition, the recruitment of the BBSome to cilia in photoreceptors does not require the small GTPase BBS3. However, the existence of complete BBSomes but without the function of BBS3 does not confer a survival advantage to photoreceptors. The findings of this study point to cell- or tissue-specific recruitment criteria of the BBSome to cilia especially in cells with adapted forms of cilia such as photoreceptor cells. The determination of specific BBSome subunits required for the ciliary entry of the BBSome may shed light on novel mechanisms at the ciliary transition zone that regulate the admittance of the BBSome to cilia.
Materials and Methods
Ethics statement and mouse models
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were handled according to approved Institutional Animal Care and Use Committee (IACUC) protocol #8072147 at the University of Iowa. Animals were housed according to IACUC recommendations. Euthanasia was performed by either carbon dioxide inhalation followed by cervical dislocation or ketamine/xylazine-induced anesthesia followed by transcardiac perfusion. Humane endpoints were strictly observed, and every effort was made to minimize suffering. The validation of mouse models Bbs2−/−, Bbs7−/−, Bbs8−/−, Bbs4−/− Bbs3−/− were published elsewhere (16, 28, 38, 39, 45). Validation of the specific genotype of each mouse used in the study was performed as previously published. For comparison of retinal degeneration, mice were bred onto a 129 background for at least 7 generations.
Histology
Retinal histology was performed as previously described (28, 46). Briefly, animals were anesthetized by intraperitoneal ketamine and xylazine mixture, and transcardial perfusion was performed using 10% formalin (approximately 4% formaldehyde) at 2.5 ml/min for a total volume of 1.25 ml/g body weight. Eyes were enucleated, and a small puncture was created through the lens using a 26 G syringe. Eyes were then embedded in Tissue-Tek O.C.T. compound and frozen in a 2-methybutane bath chilled with liquid nitrogen. Eyes were sectioned using a Cryostat microtome at thickness of 10 μm and stored at −80°C for further use. Sections were processed with hematoxylin and eosin histology and visualized under a light microscope at 40× magnification. Retinal images near the optic nerve were acquired. Four images per animal were randomly selected and thicknesses of retinal layers were quantified in these four images using ImageJ. Thicknesses of retinal layers from these four images were then averaged to yield the values for that animal. Comparison of the thicknesses of the outer nuclear layers between different models is performed by one-way ANOVA followed by post hoc Tukey’s test.
Immunohistochemistry
We have previously demonstrated that the expression of BBS proteins in the eye peaks around P15, likely due to the requirement of BBS proteins for outer segment morphogenesis (28). Therefore, animals aged between P15–16 were used for immunohistochemistry. At this age, there is minimal loss of photoreceptor cells in mutant mouse models of the BBSome (28, 46). The antibody against BBS7 was purchased from Proteintech (#18961-1-AP) and used at 1:200 dilution. The antibody against BBS8 was purchased from Sigma-Aldrich (#HPA003310) and used at 1:200 dilution. The antibody against glycylated tubulin (TAP952) was purchased from EMD Millipore (#MABS277) and used at 1:200 dilution. The antibody against polyglutamylation (GT335), a marker for the connecting cilia, was purchased from Adipogen (AG-20B-0020) and used at 1:1000 dilution.
For immunohistochemistry of BBS proteins, animals were euthanized with CO2 asphyxiation followed by cervical dislocation without transcardial perfusion. Eyes were enucleated, a small puncture was created through the lens with a 26 G syringe and eyes were embedded in Tissue-Tek O.C.T. medium without fixation. These blocks were frozen in a 2-methylbutane bath chilled with liquid nitrogen. Sections were prepared at 10 μm thickness and stored at −80 °C until use. Slides were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at 4°C prior to permeabilization and primary antibody incubation. Sections were permeabilized with 0.3% Triton X-100 in PBS for 15 min at room temperature. Sections were then blocked in a blocking buffer containing 5% bovine serum albumin, 5% normal goat serum and 0.05% Triton X-100 in PBS for 1 h at room temperature and incubated with primary antibodies at 4°C overnight. The next day, slides were washed three times using PBS followed by secondary antibody incubation at room temperature for 1 h. After another round of washing using PBS, slides were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). Confocal images were taken using the Leica SP8 STED microscope at the Central Microscopy Research Facility at the University of Iowa at 40× magnification with 8× digital magnification. All images displayed in a single panel were (1) derived from the same experiment and (2) the fluorescence of the target BBS protein, including the negative control samples, was captured with identical imaging settings and exposure times to facilitate the ability to directly compare this BBS protein across different BBS mutant models. Negative controls—retinas derived from knockout animals of the target BBS protein—were included in every imaging experiment except for BBS9. Of note, retinal sections prepared from eyes fixed by the transcardial perfusion of the animal with paraformaldehyde did not produce a meaningful staining pattern for BBS7 nor BBS8, indicating that these antibodies cannot recognize the antigen when it is masked by extensive fixation.
For immunohistochemistry of syntaxin-3 (STX3) and rhodopsin (RHO), sections were prepared from animals that were transcardially perfused using 10% formalin as previously described (46). For primary antibody incubation, anti-STX3 antibody was purchased from Proteintech (#155561-1-AP) and used at 1:1000 dilution. The anti-RHO antibody (1D4) was purchased from Santa Cruz Biotechnology (#sc-57 432) and used at 1:500 dilution.
Sucrose gradient
For preparing a 10–40% sucrose gradient, 10 and 40% sucrose solutions were prepared in PBS with 0.04% Triton-X 100. In 4 ml ultracentrifuge tubes, 1.8 ml of 40% sucrose solution was added, followed by the careful addition of 1.8 ml of 10% sucrose solution. The tubes were carefully maintained in an incline for 3 h at room temperature to allow the gradient to form. Dissected retinas from 1-month-old control and BBS mutant mice were homogenized, respectively, with a glass homogenizer using 220 μl of homogenization buffer consisting of PBS with 0.5% Triton-X 100 and protease inhibitors. After homogenization, lysates were kept on ice for 10 min and then centrifuged for 10 min at 10 000 × g at 4°C. Then, 200 μl of the supernatant was carefully loaded onto the chilled ultracentrifuge tubes containing 10–40% sucrose gradient while minimizing disturbance to the interface between solutions. The samples were subject to ultracentrifugation at 32 000 rpm for 18 h at 4°C using a TH-660 rotor. Twenty fractions, each containing 190 μl of eluent, were collected. Fraction 20 (located at the bottom of the tube) contains the portion of the sample with the largest molecular weights, whereas fraction 1 (located at the top of the tube) contains the portion of the sample with the smallest molecular weights. These samples were frozen at −80°C until further use.
Fractions 2 to 15 were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis western blotting using 4–12% Bis-Tris denaturing gels. Twenty-two microliters of each fraction were mixed with the loading dye and reducing agent following manufacturer’s instructions and heated at 95°C for 5 min prior to loading onto the gels. The proteins in the gels were transferred onto nitrocellulose membranes. The membranes were blocked with the LICOR Intercept (PBS) blocking buffer for 1 h at room temperature with gentle rotation, and the proteins of interest on the membranes were probed with primary antibodies prepared in the ODYSSEY blocking buffer containing 0.2% Tween-20 overnight at 4°C with gentle rotation. The antibodies used for western blotting are listed in Table 1. The next day, the membranes underwent three rounds of washing with TBST (TBS containing 0.1% Tween-20) and the primary antibodies were detected with LICOR secondary antibody conjugates used at 1:20 000 dilutions. The membranes were incubated with the secondary antibodies for 1 h at room temperature with gentle rotation. After another four rounds of washing with TBST, the fluorescence signal of the target proteins of interest was detected using the ODYSSEY CLx imaging system.
Table 1.
Antibodies and their dilutions used for western blotting
| Antibody (clone) | Source | Product number | Dilution |
|---|---|---|---|
| BBS1 | Bethyl Laboratories | Custom made | 1:200 |
| BBS2 (A-12) | Santa Cruz | Sc-365 355 | 1:150 |
| BBS5 (B-11) | Santa Cruz | Sc-515 331 | 1:500 |
| BBS7 | Proteintech | 18 961-1-AP | 1:500 |
| BBS8 (E-2) | Santa Cruz | Sc-271 009 | 1:500 |
| BBS9 | Sigma-Aldrich | HPA021289 | 1:500 |
| IFT88 | Proteintech | 13 967 | 1:1000 |
Immunoprecipitation
IP was performed with Dynabeads Protein G from Thermo Fischer Scientific (#10003D) following manufacturer’s instructions. The lysis buffer contains 150 mm of KCl, 2 mm of MgCl2 and ethylene glycol-bis(β-aminoethyl ether)-N,N, ,
, -tetraacetic acid (EGTA), 50 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10% glycerol, 0.5% NP-40 and protease inhibitors. Five micrograms of the anti-BBS7 antibody from Proteintech (#18961-1-AP) were coupled to 50 μl of the Dynabeads suspension following manufacturer’s instructions. For IP, retinas from a 1-month-old mouse were disrupted in 1 ml of lysis buffer, kept on ice for 10 min, and centrifuged at 10 000 × g for 10 min at 4°C. After removing 15 μl of the retinal lysates for input, retinal lysates were incubated with the antibody–bead complex overnight at 4°C with rotation. The following day, the antibody–bead–protein complexes were washed and collected following manufacturer’s instructions, and the antibody–protein complexes were dissociated from the magnetic beads by heating the sample with LDS buffer at 70°C for 10 min. IP samples were subject to western blotting as described above, and the fluorescence signal of the target proteins of interest was detected using the ODYSSEY CLx imaging system. Whole eye lysates from Bbs2−/− and Bbs8−/− mice were included in the western blot as negative controls to facilitate the identification of proteins of interest.
-tetraacetic acid (EGTA), 50 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10% glycerol, 0.5% NP-40 and protease inhibitors. Five micrograms of the anti-BBS7 antibody from Proteintech (#18961-1-AP) were coupled to 50 μl of the Dynabeads suspension following manufacturer’s instructions. For IP, retinas from a 1-month-old mouse were disrupted in 1 ml of lysis buffer, kept on ice for 10 min, and centrifuged at 10 000 × g for 10 min at 4°C. After removing 15 μl of the retinal lysates for input, retinal lysates were incubated with the antibody–bead complex overnight at 4°C with rotation. The following day, the antibody–bead–protein complexes were washed and collected following manufacturer’s instructions, and the antibody–protein complexes were dissociated from the magnetic beads by heating the sample with LDS buffer at 70°C for 10 min. IP samples were subject to western blotting as described above, and the fluorescence signal of the target proteins of interest was detected using the ODYSSEY CLx imaging system. Whole eye lysates from Bbs2−/− and Bbs8−/− mice were included in the western blot as negative controls to facilitate the identification of proteins of interest.
Quantification of connecting cilia length
In P15–16 wild type, Bbs2−/−, Bbs7−/−, Bbs8−/− and Bbs3−/− mice, the photoreceptor connecting cilia were marked using the TAP952 antibody. For each animal, 30 connecting cilia were randomly selected and their lengths were measured using ImageJ. This experiment was repeated in a different set of animals, and connecting cilia length measurements from these two experiments were combined, resulting in 60 connecting cilia measurements per genotype. Connecting cilia lengths among wild-type and mutant mice were compared using one-way ANOVA followed by post hoc Tukey’s test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors gratefully acknowledge Dr Qihong Zhang for helpful discussions. The authors gratefully acknowledge Charles C. Searby for management of laboratory resources. The authors gratefully acknowledge the contributions of Dr Darryl Nishimura.
Conflict of Interest. The authors declare that they have no competing interests.
Contributor Information
Ying Hsu, Department of Pediatrics, Division of Medical Genetics and Genomics, University of Iowa Carver College of Medicine, Iowa City, IA 52242, USA.
Seongjin Seo, Department of Ophthalmology and Visual Sciences, University of Iowa, Iowa City, IA 52242, USA.
Val C Sheffield, Department of Pediatrics, Division of Medical Genetics and Genomics, University of Iowa Carver College of Medicine, Iowa City, IA 52242, USA.
Ethics Approval and Consent to Participate
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were handled according to approved Institutional Animal Care and Use Committee (IACUC) protocol #8072147 at the University of Iowa. Animals were housed according to IACUC recommendations.
Authors’ Contributions
Y.H. performed the sucrose gradient fractionation experiments, the immunohistochemistry experiments, IP, analyzed the data and wrote the original draft. S.S. analyzed the data, discussed hypotheses, performed revisions of the manuscript and figures, in addition to providing invaluable technical advice. V.C.S. analyzed the data, discussed hypotheses, participated in the design of experiments, co-wrote the manuscript draft as well as performed revisions of the manuscript and figures.
Funding
National Institutes of Health (R01 EY011298 and R01 EY017168 to V.C.S., R01 EY022616 and R21 EY027431 to S.S.); Roy J. Carver Charitable Trust (V.C.S.); University of Iowa core facilities funded by National Institutes of Health (P30 EY025580 to V.C.S.).
References
- 1. Mykytyn, K. and Sheffield, V.C. (2004) Establishing a connection between cilia and Bardet-Biedl syndrome. Trends Mol. Med., 10, 106–109. [DOI] [PubMed] [Google Scholar]
- 2. Nishimura, D.Y., Searby, C.C., Carmi, R., Elbedour, K., Van Maldergem, L., Fulton, A.B., Lam, B.L., Powell, B.R., Swiderski, R.E. and Bugge, K.E. (2001) Positional cloning of a novel gene on chromosome 16q causing Bardet–Biedl syndrome (BBS2). Hum. Mol. Genet., 10, 865–874. [DOI] [PubMed] [Google Scholar]
- 3. Mykytyn, K., Braun, T., Carmi, R., Haider, N.B., Searby, C.C., Shastri, M., Beck, G., Wright, A.F., Iannaccone, A. and Elbedour, K. (2001) Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat. Genet., 28, 188–191. [DOI] [PubMed] [Google Scholar]
- 4. Nishimura, D.Y., Swiderski, R.E., Searby, C.C., Berg, E.M., Ferguson, A.L., Hennekam, R., Merin, S., Weleber, R.G., Biesecker, L.G. and Stone, E.M. (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet., 77, 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nachury, M.V., Loktev, A.V., Zhang, Q., Westlake, C.J., Peranen, J., Merdes, A., Slusarski, D.C., Scheller, R.H., Bazan, J.F., Sheffield, V.C. and Jackson, P.K. (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell, 129, 1201–1213. [DOI] [PubMed] [Google Scholar]
- 6. Zhang, Q.H., Yu, D.H., Seo, S.J., Stone, E.M. and Sheffield, V.C. (2012) Intrinsic protein-protein interaction-mediated and Chaperonin-assisted sequential assembly of stable Bardet-Biedl syndrome protein complex, the BBSome. J. Biol. Chem., 287, 20625–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheidecker, S., Etard, C., Pierce, N.W., Geoffroy, V., Schaefer, E., Muller, J., Chennen, K., Flori, E., Pelletier, V., Poch, O.et al. (2014) Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J. Med. Genet., 51, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Badano, J.L., Ansley, S.J., Leitch, C.C., Lewis, R.A., Lupski, J.R. and Katsanis, N. (2003) Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am. J. Hum. Genet., 72, 650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katsanis, N., Lewis, R.A., Stockton, D.W., Mai, P.M.T., Baird, L., Beales, P.L., Leppert, M. and Lupski, J.R. (1999) Delineation of the critical interval of Bardet-Biedl syndrome 1 (BBS1) to a small region of 11q13, through linkage and haplotype analysis of 91 pedigrees. Am. J. Hum. Genet., 65, 1672–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mykytyn, K., Nishimura, D.Y., Searby, C.C., Shastri, M., Yen, H.J., Beck, J.S., Braun, T., Streb, L.M., Cornier, A.S., Cox, G.F.et al. (2002) Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet., 31, 435–438. [DOI] [PubMed] [Google Scholar]
- 11. Chou, H.T., Apelt, L., Farrell, D.P., White, S.R., Woodsmith, J., Svetlov, V., Goldstein, J.S., Nager, A.R., Li, Z.X., Muller, J.et al. (2019) The molecular architecture of native BBSome obtained by an integrated structural approach. Structure, 27, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu, P.W. and Lechtreck, K.F. (2018) The Bardet-Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc. Natl. Acad. Sci. U. S. A., 115, E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ye, F., Nager, A.R. and Nachury, M.V. (2018) BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol., 217, 1847–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chih, B., Liu, P., Chinn, Y., Chalouni, C., Komuves, L.G., Hass, P.E., Sandoval, W. and Peterson, A.S. (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol., 14, 61–72. [DOI] [PubMed] [Google Scholar]
- 15. Zhang, Q., Seo, S., Bugge, K., Stone, E.M. and Sheffield, V.C. (2012) BBS proteins interact genetically with the IFT pathway to influence SHH related phenotypes. Hum. Mol. Genet. 21, 1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, Q.H., Nishimura, D., Vogel, T., Shao, J.Q., Swiderski, R., Yin, T., Searby, C., Carter, C.S., Kim, G., Bugge, K., Stone, E.M. and Sheffield, V.C. (2013) BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci., 126, 2372–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei, Q., Zhang, Y.X., Li, Y.J., Zhang, Q., Ling, K. and Hu, J.H. (2012) The BBSome controls IFT assembly and turnaround in cilia. Nat. Cell Biol., 14, 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chamling, X., Seo, S., Searby, C.C., Kim, G., Slusarski, D.C. and Sheffield, V.C. (2014) The Centriolar satellite protein AZI1 interacts with BBS4 and regulates Ciliary trafficking of the BBSome. PLoS Genet., 10, e1004083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xue, B., Liu, Y.X., Dong, B., Wingfield, J.L., Wu, M.F., Sun, J., Lechtreck, K.F. and Fan, Z.C. (2020) Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc. Natl. Acad. Sci. U. S. A., 117, 2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seo, S., Zhang, Q., Bugge, K., Breslow, D.K. and Searby, C.C. (2011) A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and smoothened. PLoS Genet., 7, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mourao, A., Nager, A.R., Nachury, M.V. and Lorentzen, E. (2014) Structural basis for membrane targeting of the BBSome by ARL6. Nat. Struct. Mol. Biol., 21, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin, H., White, S.R., Shida, T., Schulz, S., Aguiar, M., Gygi, S.P., Bazan, J.F. and Nachury, M.V. (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell, 141, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dharmat, R., Eblimit, A., Robichaux, M.A., Zhang, Z.X., Nguyens, T.M.T., Jung, S.Y., He, F., Jain, A., Li, Y.M., Qin, J.et al. (2018) SPATA7 maintains a novel photoreceptor-specific zone in the distal connecting cilium. J. Cell Biol., 217, 2851–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolch, S.N., Dugger, D.R., Chong, T., McDowell, J.H. and Smith, W.C. (2016) A splice variant of Bardet-Biedl syndrome 5 (BBS5) protein that is selectively expressed in retina. PLoS One, 11, e0148773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Riazuddin, S.A., Iqbal, M., Wang, Y., Masuda, T., Chen, Y., Bowne, S., Sullivan, L.S., Waseem, N.H., Bhattacharya, S., Daiger, S.P.et al. (2010) A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet., 86, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pretorius, P.R., Baye, L.M., Nishimura, D.Y., Searby, C.C., Bugge, K., Yang, B.L., Mullins, R.F., Stone, E.M., Sheffield, V.C. and Slusarski, D.C. (2010) Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet., 6, e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu, Q., Zuo, J. and Pierce, E.A. (2004) The retinitis pigmentosa 1 protein is a photoreceptor microtubule- associated protein. J. Neurosci., 24, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsu, Y., Garrison, J.E., Kim, G., Schmitz, A.R., Searby, C.C., Zhang, Q., Datta, P., Nishimura, D.Y., Seo, S. and Sheffield, V.C. (2017) BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet., 13, e1007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dilan, T.L., Singh, R.K., Saravanan, T., Moye, A., Goldberg, A.F.X., Stoilov, P. and Ramamurthy, V. (2018) Bardet-Biedl syndrome-8 (BBS8) protein is crucial for the development of outer segments in photoreceptor neurons. Hum. Mol. Genet., 27, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Datta, P., Hendrickson, B., Brendalen, S., Ruffcorn, A. and Seo, S.J. (2019) The myosin-tail homology domain of centrosomal protein 290 is essential for protein confinement between the inner and outer segments in photoreceptors. J. Biol. Chem., 294, 19119–19136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh, S.K., Gui, M., Koh, F., Yip, M.C. and Brown, A. (2020) Structure and activation mechanism of the BBSome membrane protein trafficking complex. elife, 9, e53322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robichaux, M.A., Potter, V.L., Zhang, Z.X., He, F., Liu, J., Schmid, M.F. and Wensel, T.G. (2019) Defining the layers of a sensory cilium with STORM and cryoelectron nanoscopy. Proc. Natl. Acad. Sci. U. S. A., 116, 23562–23572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nozaki, S., Francisco, R., Araya, C., Katoh, Y. and Nakayama, K. (2019) Requirement of IFT-B-BBSome complex interaction in export of GPR161 from cilia. Biol. Open, 8, bio043786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seo, S., Baye, L.M., Schulz, N.P., Beck, J.S., Zhang, Q., Slusarski, D.C. and Sheffield, V.C. (2010) BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. U. S. A., 107, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kretschmer, V., Patnaik, S.R., Kretschmer, F., Chawda, M.M., Hernandez-Hernandez, V. and May-Simera, H.L. (2019) Progressive characterization of visual phenotype in Bardet-Biedl syndrome mutant mice. Invest. Ophthalmol. Vis. Sci., 60, 1132–1143. [DOI] [PubMed] [Google Scholar]
- 36. Patel, N., Aldahmesh, M.A., Alkuraya, H., Anazi, S., Alsharif, H., Khan, A.O., Sunker, A., Al-Mohsen, S., Abboud, E.B., Nowilaty, S.R.et al. (2016) Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med., 18, 554–562. [DOI] [PubMed] [Google Scholar]
- 37. Carter, C.S., Vogel, T.W., Zhang, Q.H., Seo, S., Swiderski, R.E., Moninger, T.O., Cassell, M.D., Thedens, D.R., Keppler-Noreuil, K.M., Nopoulos, P.et al. (2012) Abnormal development of NG2(+)PDGFR-alpha(+) neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat. Med., 18, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nishimura, D.Y., Fath, M., Mullins, R.F., Searby, C., Andrews, M., Davis, R., Andorf, J.L., Mykytyn, K., Swiderski, R.E., Yang, B.L.et al. (2004) Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. U. S. A., 101, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mykytyn, K., Mullins, R.F., Andrews, M., Chiang, A.P., Swiderski, R.E., Yang, B.L., Braun, T., Casavant, T., Stone, E.M. and Sheffield, V.C. (2004) Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc. Natl. Acad. Sci. U. S. A., 101, 8664–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rahmouni, K., Fath, M.A., Seo, S., Thedens, D.R., Berry, C.J., Weiss, R., Nishimura, D.Y. and Sheffield, V.C. (2008) Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J. Clin. Invest., 118, 1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo, D.F., Cui, H.X., Zhang, Q.H., Morgan, D.A., Thedens, D.R., Nishimura, D., Grobe, J.L., Sheffield, V.C. and Rahmouni, K. (2016) The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet., 12, e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. He, K., Ling, K. and Hu, J. (2020) The emerging role of tubulin posttranslational modifications in cilia and ciliopathies. Biophysics Reports, 6, 89–104. [Google Scholar]
- 43. Pathak, N., Austin-Tse, C.A., Liu, Y., Vasilyev, A. and Drummond, I.A. (2014) Cytoplasmic carboxypeptidase 5 regulates tubulin glutamylation and zebrafish cilia formation and function. Mol. Biol. Cell, 25, 1836–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hong, S.R., Wang, C.L., Huang, Y.S., Chang, Y.C., Chang, Y.C., Pusapati, G.V., Lin, C.Y., Hsu, N., Cheng, H.C., Chiang, Y.C.et al. (2018) Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and hedgehog signaling. Nat. Commun., 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang, Q.H., Nishimura, D., Seo, S., Vogel, T., Morgan, D.A., Searby, C., Bugge, K., Stone, E.M., Rahmouni, K. and Sheffield, V.C. (2011) Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc. Natl. Acad. Sci. U. S. A., 108, 20678–20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hsu, Y., Garrison, J.E., Seo, S. and Sheffield, V.C. (2020) The absence of BBSome function decreases synaptogenesis and causes ectopic synapse formation in the retina. Sci. Rep., 10, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.