

Abstract
Background
Activation of pancreatic stellate cells (PSCs) is a key cause of chronic pancreatitis (CP), while inhibition of transforming growth factor-β (TGF-β) signaling renders PSCs inactive. Inhibitory Smads (I-Smads) impede TGF-β intracellular signaling and may provide a way to alleviate CP. Thus, we aimed to investigate the molecular mechanism of I-Smads in CP animals and freshly-isolated PSCs.
Methods
Sixteen male C57BL/6 mice were randomly divided into two groups; a control group (treated with saline) and a CP group (treated with caerulein) for 6 weeks. Masson’s staining was performed to identify fibrosis, and immunohistochemistry (IHC) was performed to measure the levels of Smad6 between the two groups. An improved method derived from internal digestion was used to isolate PSCs from male Sprague Dawley rats. Quantitative real-time polymerase chain reaction (qRT-PCR) and immunofluorescence staining were used to measure the messenger ribonucleic acid (mRNA) and protein levels of alpha-smooth muscle actin (α-SMA). Plasmids of I-Smads or SB431542 were transfected into freshly-isolated PSCs, and relative mRNA levels of marker genes were quantified by qRT-PCR. The two-tailed Student’s t-test was performed to assess significance.
Results
The Smad6 protein level was significantly higher in the pancreas tissue of CP mice compared to the control group. A large number of PSCs were isolated from rat pancreas using an improved isolating method and were confirmed by quiescent and active PSC markers including cluster differentiation antigen 133 (CD133), perilipin 2 (Plin2), α-SMA, Desmin, and collagen 1 (Col1). The mRNA levels of both Smad6 and Smad7 were down-regulated during freshly-isolated PSC activation. Over-expression of both Smad6 and Smad7 in freshly-isolated PSC reduced the mRNA level of α-SMA, glial fibrillary acidic protein (GFAP), Desmin, Col1, Col3, and fibronectin 1 (Fn1) significantly. SB431542 reduced the mRNA level of α-SMA, Col1, Col3, and Fn1 significantly in freshly-isolated PSCs.
Conclusions
This study demonstrated that CP promoted the expression of I-Smads, which suppressed the activation of freshly-isolated PSCs via a negative feedback loop.
Keywords: I-Smads, freshly isolated pancreatic stellate cells, chronic pancreatitis, negative feedback
Introduction
Pancreatic stellate cells (PSCs) play important roles in pancreatic diseases, such as chronic pancreatitis (CP). Quiescent PSCs contain a burden of vitamin A droplets, acting as a kind of vitamin A storage cell (1,2). Under pathological conditions, quiescent PSCs become activated, lose their vitamin A droplets, and develop a myofibroblast-like cell type. With the activation of PSC, its properties of proliferation and migration activity increases, releasing a large number of cytokines, such as transforming growth factor-β (TGF-β1) and tumor necrosis factor alpha (TNF-α), and secreting extracellular matrix (ECM), which ultimately leads to pancreatic fibrosis (3).
It has been demonstrated that TGF-β signaling is involved in PSC activation progress (4,5). Quiescent PSCs become activated upon TGF-β stimulation, and activated PSCs release cytokines, including TGF-β1, to further activate themselves. The positive feedback loop of TGF-β signaling makes it difficult to deactivate PSCs, thus making the alleviation of CP particularly challenging.
The inhibitory Smads (I-Smads), which comprise Smad6 and Smad7, inhibit TGF-β intracellular signaling via interaction with type I receptors and receptor-regulated Smads (R-Smads) (6). I-Smads also belong to the Smad family containing carboxy-terminal Mad Homology 2 (MH2) domains. Hama et al. have shown that adenovirus-mediated Smad7 over-expression inhibits TGF-β1-induced nuclear translocation of Smad3 (an R-Smad) and Smad4 (known as a common-partner Smad or co-Smad) in PSCs. The over-expression of Smad7 enhances PSC proliferation (7). Hepatic stellate cells (HSCs) are located in the liver and share similar characteristics with PSCs. It has been shown that the over-expression of Smad7 in HSCs suppresses expression of alpha-smooth muscle actin (α-SMA), a stellate cell activation marker, and reduces the synthesis of ECM proteins such as collagen. Smad7 expression blocks the TGF-β signal by inhibiting Smad2/3 phosphorylation (8). Bian et al. have further demonstrated that 5-aza-2-deoxycytidine (5-azadC), a deoxyribonucleic acid (DNA) methylation inhibitor, prevents the phosphorylation of Smad2 and Smad3 by up-regulation of Smad7 expression (9).
Pancreatic fibrosis is an important feature of CP. PSC activation promotes fibrosis progress by secreting cytokines and ECM proteins (10), and thus, PSCs are an important target for antifibrotic therapies. The continuous activation of TGF-β signaling is a key basis for the activation of PSCs, resulting in CP’s development. If TGF-β signaling can be down-regulated, it will provide important supports for the clinical relief of CP. Fortunately, I-Smads, which are endogenous TGF-β inhibitors, can hinder TGF-β signaling activity by directly binding to R-Smads, a potential target for CP's remission and treatment. Therefore, exploring the expression level and function of I-Smads in the CP process may provide a new strategy for the clinical treatment of CP in the future.
SB431542 is a well-tested chemical that specifically inhibits type I receptor, also known as activin receptor-like kinase 5 (ALK5) (11). In renal epithelial carcinoma A498 cells, SB431542 inhibits Smad3 phosphorylation, TGF-β1-induced nuclear Smad3 localization, as well as collagen 1 (Col1) and fibronectin 1 (Fn1) messenger ribonucleic acid (mRNA) expression (12). In pancreatic cancer cell PANC-1, SB431542 inhibits TGF-β regulated epithelial to mesenchymal transition (EMT) (13). However, the function of SB431542 has not been well investigated during PSC activation, especially in freshly-isolated PSCs.
Rodents, including mice and rats, are good animal models for studying human diseases' related mechanisms at the histological and molecular levels. Injection of caerulein in mice was a widely used procedure for inducing histological CP (14). On the other hand, since the rat’s pancreas is larger, more freshly-isolated PSCs can be obtained from rats than from mice.
Herein, we aimed to investigate the molecular mechanism of I-Smads in CP animals and freshly-isolated PSCs. We constructed a CP animal model and found that CP promoted Smad6 expression in pancreatic tissues. We modified the method of rat PSC isolation and harvested as many cells as possible for experimentation. We over-expressed I-Smads or SB431542 in freshly-isolated PSCs and found that both I-Smads and SB431542 can inactive PSCs during its early activation progress. Our data suggested that negative feedback of TGF-β signaling by I-Smads might provide a novel treatment strategy for CP.
We present the following article following the ARRIVE (Animal Research: Reporting of In Vivo Experiments) reporting checklist (available at http://dx.doi.org/10.21037/atm-20-4282).
Methods
Animal model
Experiments were performed under a project license (NO.: 20200103017) granted by the Experimental Animals Ethics Committee of Southeast University, in compliance with national and Southeast University guidelines for the care and use of animals.
After adaptation for 7 days under a 12-hour light/dark cycle at 21–25 °C, male C57BL/6 mice (aged 6–8 weeks, 18–22 g, purchased from Animal Laboratory of Nanjing Medical University, Nanjing, China) were randomly divided into a CP group and a normal control group (n=8 mice per group), which were treated with 50 µg/kg caerulein (Bachem AG, Bubendorf, Switzerland) or saline, respectively. After 6 weeks of treatment, the control and CP mice were sacrificed with 4% halothane anesthesia followed by carbon dioxide (CO2). The mice’s pancreases were then collected, fixed with paraformaldehyde (PFA), and embedded in paraffin.
Isolation and culture of PSCs
Rat PSCs were isolated as previously described, with some modification (1). Briefly, the pancreases from male Sprague Dawley rats (200–250 g) were digested by injecting pre-warmed 0.08% collagenase V (Sigma, USA, C9263) in Gey’s balanced salt solution (GBSS) into the common bile duct. The cell suspension was centrifuged in a 13.1% Nycodenz (Axis-Shield, Norway, 1002424) gradient with 0.3% bovine serum albumin (BSA) at 1,400 g for 20 minutes. PSCs were separated into a fuzzy band just above the Nycodenz solution interface and 0.3% BSA. PSCs were harvested and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Hyclone, USA, SH30023.01) containing 10% fetal bovine serum, 2.5 mM glutamine, 15 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) Buffer, and antibiotics (penicillin 100 units/mL and streptomycin 100 µg/mL).
Plasmids, inhibitor, and cell transfection
Plasmids of Homo Smad6-Flag and Mus Smad7-Flag were kindly provided by Prof. Yeguang Chen, Tsinghua University, China. The plasmid of Green Fluorescent Protein (GFP) was kindly provided by Prof. Qinghua Tao, Tsinghua University, China. 1 µg of Smad6 or Smad7 was used for plasmid transfection using Lipofectamine 2000 (Thermo, USA, 11668019), while GFP was used as a negative control. Cells were cultured to indicated time points before harvesting for RNA isolation.
SB431542 was purchased from Selleck (USA). The storage concentration of this inhibitor was 10 mM in dimethyl sulfoxide (DMSO). 1 or 5 µM of SB431542 was used for the inhibiting experiments. Cells were cultured to indicated time points before harvesting for RNA isolation.
RNA isolation, reverse transcription, and real-time PCR
PSCs were cultured in 6-well dishes before harvesting. Total RNA was extracted using TRIzol reagent (Invitrogen, USA, 15596026) and was subjected to reverse transcription using HiScript II 1st Strand complementary DNA (cDNA) Synthesis Kit (Vazyme, China, R212). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the ABI (USA) QuantStudio 6 platform according to the manufacturer’s instructions. All qPCR primers were optimized, as previously described (15) and are listed in Table S1. β-Actin was used as an internal reference. One well of 6-well dishes was used as the unit of analysis for each dataset. The two-tailed Student’s t-test was performed to assess significance.
Masson’s staining
Masson’s staining was described previously (16). Briefly, pancreatic tissue was fixed in 4% PFA for 24 h and dehydrated via a graduated ethanol series followed by embedding in paraffin blocks. Pancreas sections (5 µm) were dewaxed in xylene and subjected to Masson’s staining.
Immunofluorescence (IF) and immunohistochemistry (IHC) staining
Briefly, PSCs were fixed with ice-cold PFA and incubated with antibody against α-SMA (Sigma, USA, A5228) overnight at 4 °C, followed by Cy3–conjugated Goat Anti-Mouse immunoglobulin G (IgG, Proteintech, China, SA00009-1). Stained cells were visualized on a Zeiss Axio Observer microscope (Germany). 4',6-Diamidino-2-phenylindole (DAPI) was used to stain the nucleus.
The fixed pancreatic tissues were cut into sections followed by IHC protocol as previously described (16). Sections were incubated with mouse monoclonal Smad6 (Sigma, USA, SAB1412513, 1:100 dilution).
Statistical analysis
Data in this study were described as mean ± standard deviation (SD), and the differences between groups with normal distribution were calculated by the Student’s t-test using SPSS 22.0 (IBM, USA). The criterion of significance was set at P<0.05.
Results
Chronic pancreatitis promoted Smad6 expression
To examine the expression level of I-Smads in CP tissues, we established a model of CP in mice. There were no differences in the health status of the animals between the two groups. Masson’s staining was used to confirm the model (Figure 1A,B). All mice in the CP group (8/8) showed pancreatic fibrosis phenotype,, while all animals in the control group (8/8) did not. Since an antibody of Smad7 was unavailable, we tested Smad6 in the CP model and normal tissues. The IHC staining data showed that the protein level of Smad6 was significantly higher in the pancreas tissue of CP mice compared to those in the control group (Figure 1C,D).
Figure 1.

Chronic pancreatitis promoted Smad6 expression. (A,B) Masson’s staining was performed in (A) normal pancreas tissue and (B) CP pancreas tissue. (C,D) IHC of Smad6 was performed in (C) normal pancreas tissue and (D) CP pancreas tissue (magnification, ×400).
Freshly-isolated PSCs from Sprague Dawley rats
To investigate the function of I-Smads and SB431542 in the early stage of PSC activation, we freshly isolated rat PSCs using the density gradient centrifugation method. These PSCs were cultured in a medium of DMEM plus 10% FBS, which caused them to activate automatically. The PSCs remained quiescent with a stellate, angular appearance on day 1 after isolation (Figure 2A). During PSC activation, they progressively developed a myofibroblast-like phenotype (Figure 2B,C,D,E,F). In order to verify the status of freshly-isolated PSCs, we carried out qRT-PCR for the relative mRNA expression level of α-SMA, which is a well-known PSC activation marker (Figure 2G). We found that there was almost no α-SMA mRNA expression on day 1. The α-SMA expression level reached its relatively high level on day 5 and maintained this level through day 10. We further investigated the protein level of α-SMA during PSC activation. Immunofluorescence staining data confirmed that the α-SMA protein level increased gradually from day 1 to day 7 (Figure 2H,I,J,K,L,M,N,O,P). According to mRNA or protein level of α-SMA, these results indicate that PSCs were fully activated after at least the fifth day.
Figure 2.

Freshly-isolated rat pancreatic stellate cells. (A,B,C,D,E,F) Freshly-isolated PSCs were cultured and monitored on the indicated days. (G) Relative mRNA expression level of α-SMA was measured by comparing with β-actin during freshly-isolated PSC activation. (H,I,J,K,L,M,N,O,P) Immunofluorescence staining of α-SMA was carried out on days 1, 3, and 7. DAPI was used as a nuclear marker (magnification, ×400).
I-Smads were down-regulated during activation of freshly-isolated PSCs
In addition to α-SMA, we also investigated the mRNA levels of other important markers during the activation of freshly-isolated PSCs. Firstly, we found that Desmin, an intermediate filament marker involved in myogenic differentiation (17), was at a low level on day 1 and peaked on day 7 (Figure 3A). This data confirmed that Desmin was not a quiescent PSC marker (18). Furthermore, collagen 1 (Col1) expression, an ECM marker, increased gradually as expected (Figure 3B). We then investigated perilipin 2 (Plin2, also known as adipose differentiation-related protein or ADRP), which is associated with intracellular neutral lipid storage droplets and controlled cellular lipolytic activity (19). Plin2 was highly expressed on day 1, and reduced gradually during PSC activation (Figure 3C). The expression pattern of Plin2 was consistent with the phenomenon of PSCs losing their vitamin A droplets. Kordes et al. have shown that the cluster differentiation antigen 133 (CD133) positive hepatic stellate cell was a progenitor cell (20), and thus, we tested whether CD133 was expressed in PSCs. Our data showed that CD133 was highly expressed in day-1 PSCs and then reduced dramatically. After day 7, there was no expression of CD133 (Figure 3D). We then examined the expression patterns of Smad6 and Smad7 in freshly-isolated PSCs. We found that both I-Smads had a relatively high expression level on day 1, which reduced by approximately half on day 3 (Figure 3E,F). However, the level did not decrease further during PSC activation. These results suggest that both of these I-Smads might have important roles in maintaining the quiescent status of PSCs.
Figure 3.
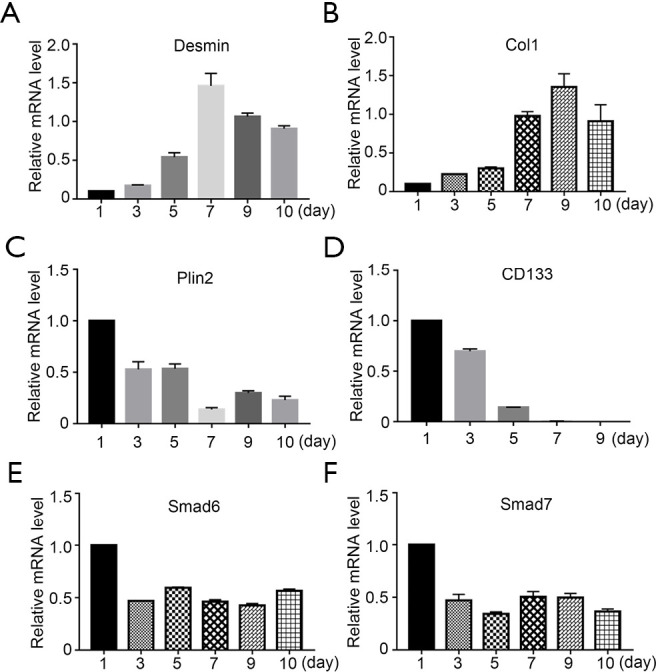
The mRNA levels of I-Smads and other markers during activation of freshly-isolated PSCs. The relative mRNA expression level of (A) Desmin, (B) Col1, (C) Plin2, (D) CD133, (E) Smad6, and (F) Smad7 was measured by comparing with β-actin during activation of freshly-isolated PSCs.
I-Smads and inhibitor suppressed the activation of freshly-isolated PSCs
The above results suggest that over-expression of I-Smads might act to suppress the activation of freshly-isolated PSCs. According to the data from Figure 1, we chose PSCs separated by only 5 days for subsequent experiments. We investigated whether over-expression of either Smad6 or Smad7 could suppress PSC activation markers α-SMA, GFAP, and Desmin. We found that α-SMA and Desmin were inhibited by both Smad6 and Smad7 after 24 h of over-expression, while GFAP was not inhibited (Figure 3A,B,C, left panels). After 48 h of over-expression, all three markers were inhibited by the I-Smads (Figure 4A,B,C, right panels). We then tested the ECM synthesis function of PSCs. After 48 h of over-expression, Smad6 suppressed the expression of Col1, Col3, and Fn1, while Smad7 suppressed Col3 and Fn1, but not Col1 (Figure 4D,E,F). Lastly, we found that the quiescent PSCs marker, CD133, was not changed significantly by either Smad6 or Smad7 after 48 h of over-expression (Figure 4G). These results demonstrated that both I-Smads could inhibit both the activation of freshly-isolated PSCs and the ECM synthesis function of PSCs.
Figure 4.

I-Smads suppressed the activation of freshly-isolated PSCs. The relative mRNA expression level of (A) α-SMA, (B) GFAP, (C) Desmin, (D) Col1, (E) Col3, (F) Fn1, and (G) CD133 was measured after 24 h and/or 48 h under the condition of over-expression of both Smad6 and Smad7 during activation of freshly-isolated PSCs. GFP was a negative control. β-Actin acted as an internal reference. The two-tailed Student’s t-test was performed to assess significance. ns, not significant; *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001.
The results above have proved the function of endogenous inhibitors of TGF-β signaling. We then tested SB431542, which is a chemical that inhibits the TGF-β type I receptor. Different doses of SB431542 were used for inhibiting the activation of freshly-isolated PSCs and ECM synthesis. Both 1 and 5 µM had significant inhibitory effects on PSC activation at different time points (Figure S1A). As expected, we also found that both doses suppressed the expression of the ECM markers Col1, Col3, and Fn1 at 12 or 24 h after SB431542 stimulation (Figure S1B,C,D). These results demonstrate that SB431542 inhibited both the activation of freshly-isolated PSCs and the ECM synthesis function of PSCs.
Discussion
During CP progression, TGF-β signaling is active, and numerous targets of TGF-β signaling are induced (4). It has been demonstrated that both Smad6 and Smad7 are the target genes of TGF-β signaling (21). This study provided evidence that Smad6 was highly expressed in the CP animal model (Figure 1C,D). This result confirmed that Smad6 was a target of TGF-β signaling and that TGF-β could induce the expression of Smad6 through R-Smads-mediated transcriptional activation (21).
I-Smads have been implicated in the down-regulation of TGF-β signaling (6). In this study, we provided evidence that both I-Smads were highly expressed in freshly-isolated PSCs and down-regulated during the activation of freshly-isolated PSCs. The over-expression of I-Smads inhibited the activation of freshly-isolated PSCs by measuring PSC activation markers, including α-SMA, GFAP, and Desmin, and also suppressed the ECM synthesis function of PSCs by measuring markers Col1, Col3, and Fn1. SB431542, a known TGF-β type I receptor inhibitor, is involved in TGF-β signaling inhibition within many cell types (22-24). This study demonstrated that SB431542 suppressed both the activation of freshly-isolated PSCs and PSC ECM synthesis. Our findings provide a novel insight into the progression of freshly-isolated PSC activation.
Additionally, we observed that the protein level of Smad6, an endogenous TGF-β signaling inhibitor, increased significantly in the pancreatic tissue of CP mice. These results suggested that PSCs themselves might alleviate the progression of CP by synthesizing TGF-β inhibitors. However, this negative feedback of TGF-β signaling seems insufficient to alleviate CP. If I-Smads of PSCs could be induced more in vivo, this would likely block TGF-β signaling activation, thereby alleviating CP. Therefore, establishing an in vivo I-Smads induction system will provide support for future translational medicine research.
We investigated the mRNA expression levels of several genes during the activation of freshly-isolated PSCs, and found that Smad6 and Smad7 were highly expressed 1-day after PSC isolation, following by down-regulation on day 3 (Figure 3E,F). These results implied that Smad6 and Smad7 might participate in maintaining the quiescent state by inhibiting TGF-β signaling.
Both Smad6 and Smad7 are inhibitors of the TGF-β superfamily, but there are still some differences between these two proteins (6). Smad6 generally mediates Bone Morphogenetic Protein (BMP) signals, whereas Smad7 mediates TGF-β signaling (25-27). Although both of these I-Smads contain MH2 domains, which are required for interactions with type I receptors, Hanyu et al. demonstrated that the MH2 domain of Smad7 is not sufficient for maximum inhibition of TGF-β signaling (28). Our data showed that Smad6, but not Smad7, suppressed Col1 expression 48 h after the over-expression of plasmids, although both of them suppressed Col3 and Fn1 expression at the same time point (Figure 4D,E,F). These results reflected the differences between the two I-Smads, and BMP signaling should be investigated carefully during activation of freshly-isolated PSCs.
PSCs are the key target of pancreatic diseases, such as CP and pancreatic cancer. However, at present, scientists have not yet found a unique marker that distinguishes PSCs from other cell types. For example, GFAP was used as an active PSC marker; however, some other types of cells also express GFAP, such as astrocytes from the nervous system. In this study, we found that CD133 and Plin2 could act as quiescent PSCs markers (Figure 3C,D). CD133, a stem/progenitor cell marker, was highly expressed in PSCs (Figure 3D) and in HSCs (20), meaning that PSCs possess stem/progenitor cell properties. Unfortunately, inhibition of TGF-β signaling did not transform active PSCs into quiescent ones based on the mRNA level of CD133 (Figure 4G). These results suggest that inhibition of TGF-β signaling is not sufficient to transfer active PSCs into quiescent ones.
Early prevention is important for controlling and alleviating CP mainly because PSCs are in the early stage of activation. Fully activated PSCs are very difficult to inactivate during experiments. Therefore in this study, we focused primarily on freshly-isolated PSCs, which are easier to revert to an inactive state. Further study should investigate the sufficient and necessary conditions required to inactivate PSCs, including signaling pathways, transcription factors, or chemicals.
There were some limitations in this study that should be noted. Firstly, due to the limitations of experimental conditions, we only used animal models and did not obtain patients' pathological sections with CP. We need further to verify our findings in human samples in future studies. Secondly, we observed that I-Smads could inhibit PSC activation-related genes’ expression level at the cellular level. However, we did not construct transgenic mice that specifically and continuously activated I-Smads in vivo. Therefore, we cannot observe the remission effect of I-Smads’ over-expression in mice with CP at the animal level. Thirdly, SB431542, a known inhibitor, also showed good inhibitory activity on TGF-β signaling in PSCs. However, the inhibitory effect should be compared between the inhibitor and I-Smads both in vivo and in vitro. Lastly, our results need to be validated in other CP models, such as pancreatic duct ligation.
Since animal experiments' results were a better reflection of the situation in vivo, the number of related animal experiments and the number of animals used cannot be reduced.
In summary, we demonstrated that CP promotes the expression of Smad6. We also showed that both I-Smads suppressed the activation of freshly-isolated PSCs through inhibition of TGF-β signaling and its targets. Taken together, I-Smads and TGF-β signaling are involved in a negative feedback loop in CP. These experimental results offer a new perspective to clarify the role of the TGF-β signaling pathway in CP. Simultaneously, the significant effect of I-Smads and inhibitors also provides new targets for drug screening and clinical treatment.
Supplementary
The article’s supplementary files as
Acknowledgments
We thank Prof. Yeguang Chen and Prof. Qinghua Tao from Tsinghua University for the plasmid gifts.
Funding: This study was supported by the National Natural Science Foundation of China (NSFC) [grant number 81800571 (HL)]; the Basic Scientific Research Foundation of Southeast University [grant number 2242018K40162 (HL)]; the Talent Project of Dual Creative Ph.D. from Jiangsu Province [grant number 1190000047 (HL)]; the National Key Laboratory of Bioelectronics and Zhongda Hospital Cross Innovation Research Foundation [grant number 2018yy-jccx009 (HL)]; and the Horizontal Scientific Research Project of Zhongda Hospital [grant number 2018010015 (HL)].
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Experiments were performed under a project license (NO.: 20200103017) granted by the Experimental Animals Ethics Committee of Southeast University, in compliance with national and Southeast University guidelines for the care and use of animals.
Reporting Checklist: The authors have completed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) checklist. Available at http://dx.doi.org/10.21037/atm-20-4282
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-4282
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-4282). The authors have no conflicts of interest to declare.
(English Language Editor: A. Kassem)
References
- 1.Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998;115:421-32. 10.1016/S0016-5085(98)70209-4 [DOI] [PubMed] [Google Scholar]
- 2.Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43:128-33. 10.1136/gut.43.1.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneider E, Schmid-Kotsas A, Zhao J, et al. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am J Physiol Cell Physiol 2001;281:C532-43. 10.1152/ajpcell.2001.281.2.C532 [DOI] [PubMed] [Google Scholar]
- 4.Apte MV, Haber PS, Darby SJ, et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 1999;44:534-41. 10.1136/gut.44.4.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelmann R, Ruf D, Wagner M, et al. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-beta1 transgenic mouse model. Am J Physiol Gastrointest Liver Physiol 2001;280:G164-72. 10.1152/ajpgi.2001.280.1.G164 [DOI] [PubMed] [Google Scholar]
- 6.Miyazawa K, Miyazono K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol 2017;9:a022095. 10.1101/cshperspect.a022095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hama K, Ohnishi H, Aoki H, et al. Angiotensin II promotes the proliferation of activated pancreatic stellate cells by Smad7 induction through a protein kinase C pathway. Biochem Biophys Res Commun 2006;340:742-50. 10.1016/j.bbrc.2005.12.069 [DOI] [PubMed] [Google Scholar]
- 8.Dooley S, Hamzavi J, Breitkopf K, et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003;125:178-91. 10.1016/S0016-5085(03)00666-8 [DOI] [PubMed] [Google Scholar]
- 9.Bian EB, Huang C, Wang H, et al. Repression of Smad7 mediated by DNMT1 determines hepatic stellate cell activation and liver fibrosis in rats. Toxicol Lett 2014;224:175-85. 10.1016/j.toxlet.2013.10.038 [DOI] [PubMed] [Google Scholar]
- 10.Jaster R. Molecular regulation of pancreatic stellate cell function. Mol Cancer 2004;3:26. 10.1186/1476-4598-3-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callahan JF, Burgess JL, Fornwald JA, et al. Identification of novel inhibitors of the transforming growth factor beta1 (TGF-beta1) type 1 receptor (ALK5). J Med Chem 2002;45:999-1001. 10.1021/jm010493y [DOI] [PubMed] [Google Scholar]
- 12.Laping NJ, Grygielko E, Mathur A, et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol 2002;62:58-64. 10.1124/mol.62.1.58 [DOI] [PubMed] [Google Scholar]
- 13.Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005;7:509-21. 10.1593/neo.04640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo BM, Oh TY, Kim YB, et al. Novel antioxidant ameliorates the fibrosis and inflammation of cerulein-induced chronic pancreatitis in a mouse model. Pancreatology 2005;5:165-76. 10.1159/000085268 [DOI] [PubMed] [Google Scholar]
- 15.Lin H, Zhu X, Chen G, et al. KDM3A-mediated demethylation of histone H3 lysine 9 facilitates the chromatin binding of Neurog2 during neurogenesis. Development 2017;144:3674-85. 10.1242/dev.144113 [DOI] [PubMed] [Google Scholar]
- 16.Xiao W, Jiang W, Shen J, et al. Retinoic Acid Ameliorates Pancreatic Fibrosis and Inhibits the Activation of Pancreatic Stellate Cells in Mice with Experimental Chronic Pancreatitis via Suppressing the Wnt/beta-Catenin Signaling Pathway. PLoS One 2015;10:e0141462. 10.1371/journal.pone.0141462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mato E, Lucas M, Petriz J, et al. Identification of a pancreatic stellate cell population with properties of progenitor cells: new role for stellate cells in the pancreas. Biochem J 2009;421:181-91. 10.1042/BJ20081466 [DOI] [PubMed] [Google Scholar]
- 18.Nielsen MFB, Mortensen MB, Detlefsen S. Identification of markers for quiescent pancreatic stellate cells in the normal human pancreas. Histochem Cell Biol 2017;148:359-80. 10.1007/s00418-017-1581-5 [DOI] [PubMed] [Google Scholar]
- 19.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology 2008;149:942-9. 10.1210/en.2007-1713 [DOI] [PubMed] [Google Scholar]
- 20.Kordes C, Sawitza I, Muller-Marbach A, et al. CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun 2007;352:410-7. 10.1016/j.bbrc.2006.11.029 [DOI] [PubMed] [Google Scholar]
- 21.Afrakhte M, Moren A, Jossan S, et al. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem Biophys Res Commun 1998;249:505-11. 10.1006/bbrc.1998.9170 [DOI] [PubMed] [Google Scholar]
- 22.Schade D, Lanier M, Willems E, et al. Synthesis and SAR of b-annulated 1,4-dihydropyridines define cardiomyogenic compounds as novel inhibitors of TGFbeta signaling. J Med Chem 2012;55:9946-57. 10.1021/jm301144g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng C, Fang Y, Tong W, et al. Synthesis and biological evaluation of novel tetrahydro-β-carboline derivatives as antitumor growth and metastasis agents through inhibiting the transforming growth factor-β signaling pathway. J Med Chem 2014;57:600-12. 10.1021/jm401117t [DOI] [PubMed] [Google Scholar]
- 24.Roth GJ, Heckel A, Brandl T, et al. Design, synthesis, and evaluation of indolinones as inhibitors of the transforming growth factor beta receptor I (TGFbetaRI). J Med Chem 2010;53:7287-95. 10.1021/jm100812a [DOI] [PubMed] [Google Scholar]
- 25.Imamura T, Takase M, Nishihara A, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature 1997;389:622-6. 10.1038/39355 [DOI] [PubMed] [Google Scholar]
- 26.Jung SM, Lee JH, Park J, et al. Smad6 inhibits non-canonical TGF-beta1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat Commun 2013;4:2562. 10.1038/ncomms3562 [DOI] [PubMed] [Google Scholar]
- 27.Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997;389:631-5. 10.1038/39369 [DOI] [PubMed] [Google Scholar]
- 28.Hanyu A, Ishidou Y, Ebisawa T, et al. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J Cell Biol 2001;155:1017-27. 10.1083/jcb.200106023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as