

Abstract
Introduction
Synaptic damage is a key pathology of Alzheimer's disease (AD). The mechanism underlying synaptic vulnerability in AD remains elusive.
Methods
Using a large‐scale transcriptomic dataset, we analyzed the neurogranin‐centered integrative gene network and assessed the correlation of neurogranin (NRGN) gene expression with AD pathology in post mortem brains. We studied the association of NRGN expression with Clinical Dementia Rating (CDR) and neuropathological diagnosis of AD.
Results
We find that the genes positively correlated with NRGN expression in AD are involved in synaptic transmission and cation channel pathways. NRGN expression is correlated with amyloid and tau pathology in the perirhinal cortex of post mortem brains. NRGN expression is associated with the diagnosis of AD and correlated with CDR.
Discussion
Transcriptional regulation of the gene encoding for synaptic protein is involved in selective synaptic damage in AD. Identifying the genes associated with synaptic damage pathways in AD may provide targets for intervention.
Keywords: Alzheimer's disease, amyloid, integrative gene network, neurogranin, perirhinal cortex, synapse, tau
1. INTRODUCTION
Genome‐wide transcriptome analyses of post mortem brain samples with Alzheimer's disease (AD) have provided important insights into the role of altered gene expression and gene regulatory networks in the pathogenesis of AD. 1 , 2 Brain regional vulnerability analyses revealed that temporal lobe gyri are associated with the greatest and earliest of gene expression abnormality. 1 Integrative network analysis of transcriptomic data shows that a number of coexpressed gene modules dysregulated in AD were associated with synaptic transmission and the underlying gene networks of synaptic transmission lay down a foundation to investigate specific mechanisms of dysregulated synaptic transmission and neuronal systems in general. 2 , 3
Synaptic degeneration is well recognized as a core feature of AD pathophysiology. Both post mortem and brain biopsy studies have shown a significant loss of synapses in cortices and hippocampus, which is correlated with cognitive impairment in patients with AD. 4 , 5 , 6 AD‐associated proteins, such as amyloid precursor protein and tau, are enriched in the presynaptic terminals and regulate vesicle release and structural dynamics. 7 , 8 , 9 , 10 , 11 miR‐30b, which targets molecules important for maintenance of synaptic integrity, is upregulated in the brains of AD patients. Knockdown of miR‐30b prevents amyloid beta (Aβ)‐induced synaptic loss and cognitive decline, strengthening the implication of synaptic proteins in AD pathogenesis and progression. 12
Neurogranin (Ng), a post‐synaptic protein, plays a vital role in visual–spatial learning and memory. 13 It is reported that both the gene expression and protein levels of Ng are reduced in AD brains. 14 , 15 Ng level is elevated in the cerebrospinal fluid (CSF) from patients with mild cognitive impairment (MCI) secondary to AD and AD dementia compared to those of cognitively normal controls, likely reflecting the release of Ng from damaged synapses during synaptic degeneration. 16 , 17 , 18 High levels of CSF Ng predict the longitudinal decline of memory and executive functions in patients with MCI, suggesting that altered function of Ng contributes to synaptic degeneration in AD. 19 Although extensive studies have indicated the important role of Ng in the pathogenesis of AD, the relationship between the neurogranin (NRGN) gene expression and AD pathology has not been explored.
In this study, we used a large‐scale transcriptomic dataset of the Mount Sinai National Institutes of Health NeuroBioBank (MSBB) derived from four brain regions of interest—the prefrontal cortex, the superior temporal cortex, the perirhinal cortex, and the inferior frontal cortex—obtained from post mortem brains of AD and non‐demented control subjects. We analyzed the correlation between NRGN expression and amyloid and tau pathology (plaque and tangle counts), the Clinical Dementia Rating (CDR), and the neuropathological diagnosis of AD. An integrative network analysis was performed to investigate NRGN‐associated pathways in AD.
2. METHODS
2.1. Study population and data collection
We focused on the RNA‐sequencing data from four brain regions including Brodmann area 10 (anterior prefrontal cortex, 261 samples), Brodmann area 22 (superior temporal gyrus, 240 samples), Brodmann area 36 (perirhinal cortex, 215 samples) and Brodmann area 44 (pars opercularis, 222 samples) from 299 well‐characterized post mortem human brains in the MSBB as our discovery data set. 3 , 20 These four brain regions were found to be most vulnerable to AD based on an earlier microarray study of 19 cortical brain regions. 2 , 3 This cohort includes subjects with a complete disease spectrum of the CDR and neuropathological features of AD. All diagnostic and autopsy protocols were approved by the Mount Sinai and JJ Peters VA Medical Center Institutional Review Boards. Neuropathological diagnosis and scores of AD were established based on the diagnostic criteria of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD). 21 Quantitative measures of neuritic plaques and tau pathology were obtained in each case. 22 , 23
2.2. NGRN‐centered correlation network construction
A gene‐centered correlation network has been used to explore the potential role of that gene in a disease‐specific context. The NRGN‐centered correlation network was built as described in our previous publication. 24 , 25 Briefly, three datasets were downloaded from the Synapse data portal including Mount Sinai Brain Bank Studies BM10, 22, 36, 44 (Synapse ID: syn3157743); Harvard Brain Tissue Resource Study from prefrontal cortex (PFC), visual cortex (VC), cerebellum (CR; Synapse ID: syn3159435); and Religious Orders Study and Rush Memory and Aging Project (ROS‐MAP) Study (Synapse ID: syn3219045) from dorsolateral prefrontal cortex (DLPFC). 2 , 26 , 27 All the non‐demented control samples were excluded from the AD‐specific NRGN‐centered correlation network construction.
2.3. Statistical analysis
All of the data were analyzed using R software (version 3.6.1.) for statistical analyses. The processed gene expression data, neuropathological data, and the associated clinical information were downloaded from Synapse data portal (https://www.synapse.org, Synapse ID: syn3157743). The data were represented as mean ± standard deviation (mean ± SD) or median (interquartile range [IQR]) for the continuous variables that follow a normal distribution or not, respectively. The count variables were shown as N (%).
Non‐parametric Spearman's correlation analysis was performed to examine the correlation between NRGN expression and quantitative measures of AD pathology and CDR with Benjamini‐Hochberg (BH) correction for multiple testing. A BH‐corrected P‐value < 0.05 was considered statistically significant unless stated elsewhere.
The correlation between the NRGN expression and all other genes was calculated based on the gene expression level in each individual brain region. The gene correlation with a BH‐corrected P‐value < 0.01 was considered significant. A Fisher's exact test (FET) was applied to assess the Gene Ontology (GO) enrichment of the NGRN‐correlated genes that had consensus across all the brain regions. 25
3. RESULTS
3.1. Demographic information of the study population
A total of 299 human brains (definite AD = 131, probable AD = 42, possible AD = 39, and non‐demented control = 87) from the MSBB cohort was used for this study (Table 1). AD patients had higher global CDR scores (median, 3 for definite AD, 3 for probable AD, 1 for possible AD, and 0.5 for non‐demented control), higher mean plaque density (mean ± SD 15.1 ± 8.8 for definite AD, 6.2 ± 2.9 for probable AD, 4.5 ± 3.1 for possible AD, and 0.9 ± 1.8 for control), and higher Braak scores (median, 6 for definite AD, 4 for probable AD, 3 for possible AD, and 2 for non‐demented control). Age of death (AOD) ranged from 74 to 103 years old (mean ± SD, definite AD, 85.1 ± 9.7; probable AD, 89.3 ± 6.2; possible AD, 85.8 ± 9.1; and non‐demented control, 82.6 ± 10.5). More than 70% of the subjects in this cohort were White (87.02% for definite AD, 83.33% for probable AD, 82.05% for possible AD, and 71.26% for non‐demented control). More than 50% of the subjects were female (61.1% for definite AD, 73.8% for probable AD, 84.6% for possible AD, and 53.3% for non‐demented control). All demographic data are summarized in Table 1.
TABLE 1.
Demographic information of the study population
| NC (87) | Definite AD (131) | Probable AD (42) | Possible AD (39) | |
|---|---|---|---|---|
|
Age of death Mean ± SD |
82.6 ± 10.5 | 85.1 ± 9.7 | 89.3 ± 6.2 | 85.79 ± 9.1 |
| Sex: Male (%) | 39 (44.82%) | 51 (38.93%) | 11 (26.19%) | 6 (15.38%) |
|
CDR Median (IQR) |
0.5 (0,1) | 3 (2,5) | 3 (2,3) | 1(0.5,2.5) |
|
Mean plaque density Mean ± SD |
0.9 ± 1.8 | 15.1 ± 8.8 | 6.2 ± 2.9 | 4.5 ± 3.1 |
|
Braak score Median (IQR) |
2 (1,3) | 6 (5,6) | 4 (3,6) | 3(2,4) |
|
Ethnicity White (%) |
62 (71.26%) | 114 (87.02%) | 35 (83.33%) | 32 (82.05%) |
Abbreviations: Definite AD, definite Alzheimer's disease; IQR, interquartile range; NC, non‐demented control; Possible AD, possible Alzheimer's disease; Probable AD, probable Alzheimer's disease; SD, standard deviation.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts. Cerebrospinal fluid (CSF) neurogranin (Ng) was associated with CSF tau protein in patients with Alzheimer's disease (AD) and mild cognitive impairment. The relationship between neurogranin gene (NRGN) expression and AD pathology has not been reported. The NRGN‐centered correlation network has not been explored.
Interpretation: Association of NRGN expression with AD pathology in the perirhinal cortex suggests that transcriptional regulation of the genes encoding for synaptic protein is involved in selective synaptic damage in AD. Dysfunction of synaptic transmission pathways might be associated with synaptic degeneration in AD.
Future directions: The article proposes a framework for studying the molecular mechanism underlying regional vulnerability to AD. The additional studies include: (a) the NRGN expression in apolipoprotein E (APOE) ε4 carriers compared to non‐APOE ε4 carriers in different brain regions and (b) the role of potassium channels associated with Ng in synaptic degeneration of AD.
3.2. Neuronal function relevance of NRGN expression in AD
To analyze the genes related to the NRGN gene expression in AD, we constructed an NRGN‐centered gene correlation network and explored the functional implications of these NRGN‐correlated genes in the AD context. Table 2 shows the functional annotation of the relevance to neurogranin‐correlated/co‐expressed genes in AD. Synaptic transmission and cation channel pathways are the top significantly enriched functional pathways. The enriched gene category includes transmission of nerve impulse, synaptic transmission, and potassium channel pathways. In addition, the expression of gene category of sister chromatid cohesion is reduced. Figure 1 shows the integrative regulatory network of NRGN depicted according to Spearman's correlation, with all 263 gene correlates/co‐expresses with NRGN (BH correction P < 0.01).
TABLE 2.
Functional annotation of relevance to neurogranin‐correlated/co‐expressed gene in Alzheimer's disease
| GeneSet | Size | System | Gene category | Overlap | GeneSet | Popoverlap | Pop | FET_P | Corr_P | Fold_Enrich |
|---|---|---|---|---|---|---|---|---|---|---|
| Positive | 226 | Bio Pro | Transmission of nerve impulse | 30 | 201 | 440 | 13178 | 4.82E‐12 | 4.80E‐08 | 4.470149254 |
| Positive | 226 | Bio Pro | Synaptic transmission | 27 | 201 | 415 | 13178 | 1.80E‐10 | 1.80E‐06 | 4.265491818 |
| Positive | 226 | Bio Pro | Cell–cell signaling | 35 | 201 | 906 | 13178 | 2.90E‐07 | 0.0029 | 2.532755648 |
| Positive | 226 | Mol Fun | Potassium channel activity | 12 | 201 | 133 | 13178 | 8.72E‐07 | 0.0086 | 5.915385479 |
| Positive | 226 | Cell Com | Vol‐gated potassium channel complex | 10 | 201 | 88 | 13178 | 8.75E‐07 | 0.0087 | 7.450248756 |
| Positive | 226 | Bio Pro | Potassium ion transport | 13 | 201 | 174 | 13178 | 2.60E‐06 | 0.026 | 4.89832447 |
| Positive | 226 | Bio Pro | Neuron development | 14 | 201 | 205 | 13178 | 3.08E‐06 | 0.031 | 4.477417789 |
| Negative | 37 | Bio Pro | Sister chromatid cohesion | 3 | 33 | 12 | 13178 | 3.10E‐06 | 0.031 | 99.83333333 |
| Positive | 226 | Bio Pro | Neuron differentiation | 17 | 201 | 299 | 13178 | 3.31E‐06 | 0.033 | 3.7276161 |
| Positive | 226 | Mol Fun | Vol‐gated potassium channel activity | 10 | 201 | 104 | 13178 | 4.09E‐06 | 0.041 | 6.30405664 |
| Positive | 226 | Mol Fun | Cation channel activity | 16 | 201 | 273 | 13178 | 4.41E‐06 | 0.044 | 3.82472619 |
Abbreviations: FET_P, Fisher exact test P value; Corr_P, BH‐corrected P value.
FIGURE 1.
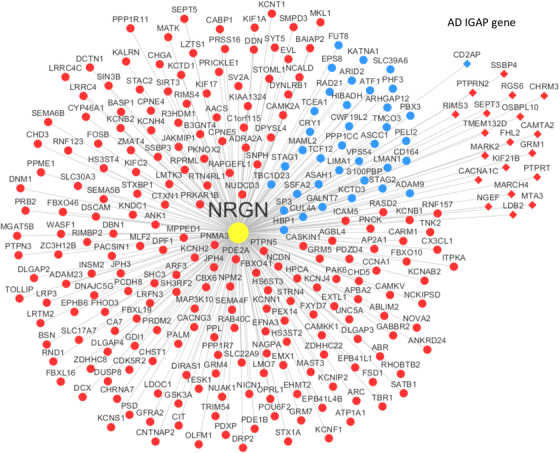
Neurogranin (NRGN)‐centered gene co‐expression network. The network includes the 263 genes significantly correlated with NRGN by Spearman correlation analysis (Benjamini‐Hochberg correction P < 0.01). Nodes in red indicate the genes positively correlated with NRGN while those in blue represent genes positively correlated with NRGN. The nodes in the diamond shape are the Alzheimer's disease risk genes identified by the International Genomics of Alzheimer's Project (IGAP)
3.3. Correlation analyses between the NRGN expression and amyloid and tau pathology
A correlation of the NRGN expression with the quantitative amyloid pathology (plaque mean) in four brain regions is seen in Figure 2. The NRGN expression is negatively correlated with amyloid plaque density only in the perirhinal cortex but not in the superior temporal gyrus, anterior prefrontal gyrus, and pars opercularis. The lower NRGN expression correlates with higher density of amyloid plaque in the perirhinal cortex (Rho = –0.25, BH‐corrected P = 0.001106). Figure 3 shows a correlation study of the NRGN expression with quantitative neurofibrillary neuropathology score in four brain regions. Among four brain regions, the NRGN expression correlates with neurofibrillary neuropathology only in the perirhinal cortex (Rho = –0.26, BH‐corrected P = 0.001106). Lower NRGN expression correlates with higher neurofibrillary neuropathology in the perirhinal cortex of AD brains.
FIGURE 2.
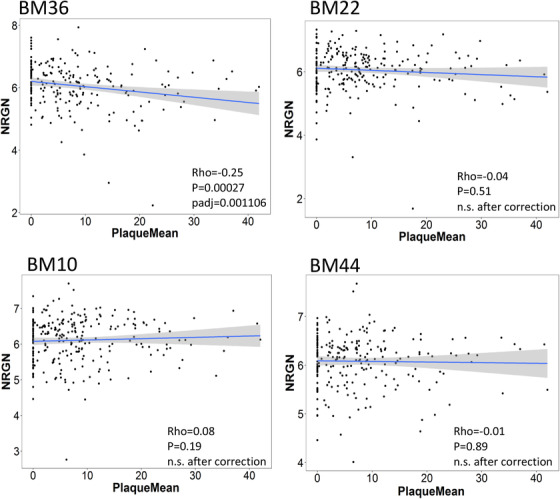
Correlations between amyloid plaque load and neurogranin (NRGN) mRNA expression in four brain regions investigated in this study. The significant correlation between NRGN mRNA expression and plaque load was shown in the perirhinal cortex of donor brains. Abbreviations: BA 10, Brodmann area 10, the prefrontal cortex; BA 22, Brodmann area 22, the superior temporal gyrus, BA 36, Brodmann area 36, the perirhinal cortex; and BA 44, Brodmann area 44, the pars opercularis of the inferior frontal gyrus. P‐value < 0.05 was considered statistically significant
FIGURE 3.
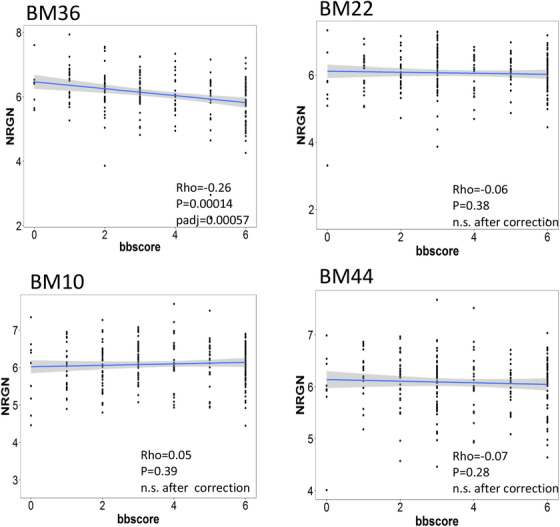
Correlations between tau pathology and neurogranin (NRGN) mRNA expression in four brain regions investigated in this study. The significant correlation between NRGN mRNA expression and plaque load was shown in the perirhinal cortex of donor brains. Abbreviations: BA 10, Brodmann area 10, the prefrontal cortex; BA 22, Brodmann area 22, the superior temporal gyrus, BA 36, Brodmann area 36, the perirhinal cortex; and BA 44, Brodmann area 44, the pars opercularis of the inferior frontal gyrus. BB score, Braak score; P‐value < 0.05 was considered statistically significant
3.4. A correlation analysis between the NRGN expression and cognitive and functional scores
Figure 4a shows the NRGN expression correlates with the functional score of clinical dementia (CDR score). Lower NRGN expression correlates with higher CDR scores in AD (Rho = –0.28, P = 3.1e‐05, Padj = 1.3e‐04). The relationship between the NRGN expression and neuropathological diagnosis of AD is investigated. Among four diagnostic categories, the lower NRGN expression is observed more in the brains of definite AD, followed by probable AD, possible AD, and non‐demented control (Figure 4b).
FIGURE 4.
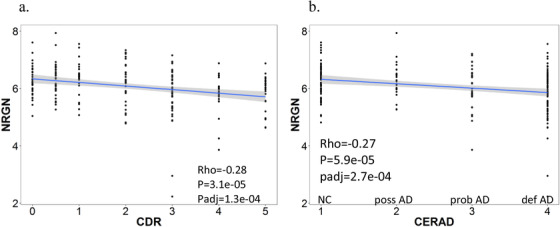
Correlation of neurogranin (NRGN) mRNA expression with functional score and diagnosis of AD. a, Lower NRGN mRNA expression corresponds to higher CDR scores. b, Lower NRGN mRNA expression is associated with more definite pathological diagnosis of AD. AD, Alzheimer's disease; CDR, Clinical Dementia Rating; CERAD, Consortium to Establish a Registry for Alzheimer's Disease
4. DISCUSSION
To our knowledge, this investigation represents the first integrative network analysis of a large‐scale transcriptomic dataset of the post mortem brains of AD in which NRGN is an area of focus. We find that NRGN expression in AD is positively associated with synaptic transmission, cation channels, neuron development, and neuron differentiation genes. Although the previous integrative network analysis has shown that synaptic transmission genes SNAP91(synaptosome associated protein 91), BSN (Bassoon presynaptic cytomatrix protein), and GLS (glutaminase) are highly enriched in late‐onset AD‐associated modules, 2 our study has extended the previous finding by demonstrating that NRGN is also involved in synaptic transmission in AD. Our study suggests that the regulation of synaptic function at gene expression level in AD plays an important role in AD pathogenesis.
Regarding the association of NRGN expression with AD pathology, we demonstrated that lower NRGN expression was correlated with higher amyloid and tau pathology in the BA 36 (perirhinal cortex [PRC]). Previous studies have shown that amyloid plaques are associated with local synaptic loss in mouse models and human brains. 28 Aβ oligomers are reported to be toxic to synapses and inhibit long‐term potential in vivo and ex vivo. 29 , 30 , 31 With respect to tau protein, it is found to be translocated to the somatodendritic compartment aberrantly and contributes to synaptic and neuronal dysfunction in AD. 32 , 33 In this study, the negative correlation between NRGN expression and amyloid and tau pathology indicates that the pathological accumulation of amyloid and tau protein may downregulate the NRGN expression in vulnerable brain regions of AD, contributing to synaptic degeneration in AD. Conversely, certain brain regions with low NRGN expression may increase vulnerability to the AD pathology.
The relationship between NRGN expression and AD is further demonstrated by the association of NRGN expression with the post mortem diagnostic certainty of AD. We show that the low level of NRGN expression is mostly associated with definite pathological diagnosis of AD, followed by probable AD, possible AD, and non‐demented controls. This finding supports a previous report that the increased CSF Ng concentration is specific to the diagnosis of AD. 34 In addition, we show that lower NRGN expression correlates with higher CDR scores in this cohort. This result is consistent with previous findings that loss of synapses is well correlated with cognitive and functional impairment in patients with AD. 4 , 6 , 35 , 36
For many years, the brain's selective vulnerability to AD has been extensively investigated. 37 The mechanism underlying region‐specific vulnerability to AD is poorly understood. The studies on post mortem AD brains show that AD specifically affects brain regions in a predictable pattern. 37 Extracellular senile plaques (SPs) formed by aggregated Aβ peptides spread from neocortical regions to the allocortex, diencephalon, striatum, brainstem nuclei, and finally to the cerebellum. In contrast, intracellular neurofibrillary tangles (NFTs) composed of hyper‐phosphorylated tau protein evolve from entorhinal cortex and hippocampus to locus coeruleus, basal forebrain, and finally the neocortex.
In this study, a correlation between the NRGN expression and amyloid and tau pathology is significant only in the PRC (Brodmann area 36) among four brain regions we examined. The finding is consistent with a previous study. 38 It is reported that the PRC has very high density of NFTs and moderate loading of SPs in a post mortem brain of the patient with preclinical dementia. 38 Taken together, it is suggested that the PRC is one of the critical sites for NRGN expression in association with both amyloid and tau pathology in AD. The PRC is a part of medial temporal lobe and localized in the parahippocampal region. The studies show that PRC (Brodmann area 35 and 36) is involved in coarse and detail‐orientated cortical hippocampal networks. 39 It is suggested that the damage of the perirhinal cortex may affect hippocampal function in a disease process such as AD.
In summary, this study reports that NRGN expression is selectively correlated with amyloid and tau pathology in the perirhinal cortex among four brain regions we examined. The lower level of the NRGN expression in post mortem brains of AD is associated with poorer pre‐mortem functional score in these patients with AD. Although highly statistically significant after correction for multiple comparisons, the modest magnitude of effect suggests that other covariates in addition to NRGN expression might relate to amyloid and tau pathology in the perirhinal cortex. Alternatively, the modest magnitude of effect might reflect the inherent heterogeneity of the patients with AD. An integrative network analysis reveals that the positively correlated genes with NRGN in AD are involved in synaptic transmission and cation channel pathways. Understanding of synaptic functional pathways associated with synaptic damage in AD may provide additional avenues to prioritize therapeutic targets for intervention.
CONFLICTS OF INTEREST
Henrik Zetterberg has served on scientific advisory boards for Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies, and CogRx; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, and Biogen; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). Kaj Blennow has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, Biogen, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). MMc has served on a scientific advisory board for Novartis Pharmaceuticals in the area of multiple sclerosis research.
The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP‐AD) Target Discovery Program and other National Institute on Aging (NIA)‐supported programs to enable open‐science practices and accelerate translational learning. Data are available for general research use according to the requirements for data access and data attribution that can be found at: https://adknowledgeportal.synapse.org/#/DataAccess/Instructions.
ACKNOWLEDGMENTS
Xiaoyan Sun thanks Mss. Jacqueline Allee Smith for her generous support of Alzheimer's disease research. Henrik Zetterberg is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C and #ADSF‐21‐831377‐C), the Olav Thon Foundation, the Erling‐Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019‐0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. Kaj Blennow is supported by the Swedish Research Council (#2017‐00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement (#ALFGBG‐715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236). Bin Zhang, Qian Wang, and Zhenyu Yue were supported by the NIH/NIA grant U01AG046170. Bin Zhang was also supported by RF1AG054014, RF1AG057440, R01AG057907, U01AG052411, and R01AG068030.
Sun X, Wang Q, Blennow K, et al. Association of neurogranin gene expression with Alzheimer's disease pathology in the perirhinal cortex. Alzheimer's Dement. 2021;7:e12162. 10.1002/trc2.12162
Xiaoyan Sun, Qian Wang, and Bin Zhang contributed equally to this study.
DATA AVAILABILITY STATEMENT
The Mount Sinai Brain Bank (MSBB) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3159438) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The Religious Orders Study and Rush Memory and Aging Project (ROS‐MAP) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3219045) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The Harvard Brain Tissue Resource Center (HBTRC) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3159435) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org).
REFERENCES
- 1. Wang M, Roussos P, McKenzie A, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer's disease. Genome Med. 2016;8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late‐onset Alzheimer's disease. Cell. 2013;153:707‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang M, Li A, Sekiya M, et al. Transformative network modeling of multi‐omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer's disease. Neuron. 2021;109:257‐272.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457‐464. [DOI] [PubMed] [Google Scholar]
- 5. Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer's disease. Ann Neurol. 1993;33:190‐199. [DOI] [PubMed] [Google Scholar]
- 6. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572‐580. [DOI] [PubMed] [Google Scholar]
- 7. Laßek M, Weingarten J, Wegner M, et al. APP is a context‐sensitive regulator of the hippocampal presynaptic active zone. PLoS Comput Biol. 2016;12:e1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212‐7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spires TL, Orne JD, SantaCruz K, et al. Region‐specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voelzmann A, Okenve‐Ramos P, Qu Y, et al. Tau and spectraplakins promote synapse formation and maintenance through Jun kinase and neuronal trafficking. eLife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee SH, Kang J, Ho A, Watanabe H, Bolshakov VY, Shen J. APP family regulates neuronal excitability and synaptic plasticity but not neuronal survival. Neuron. 2020;108:676‐690.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song Y, Hu M, Zhang J, Teng ZQ, Chen C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA‐30b in Alzheimer's disease. EBioMedicine. 2019;39:409‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pak JH, Huang FL, Li J, et al. Involvement of neurogranin in the modulation of calcium/calmodulin‐dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci U S A. 2000;97:11232‐11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer's disease. Int Psychogeriatr. 1998;10:11‐23. [DOI] [PubMed] [Google Scholar]
- 15. George AJ, Gordon L, Beissbarth T, et al. A serial analysis of gene expression profile of the Alzheimer's disease Tg2576 mouse model. Neurotox Res. 2010;17:360‐379. [DOI] [PubMed] [Google Scholar]
- 16. Kester MI, Teunissen CE, Crimmins DL, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015;72:1275‐1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 2015;11:1180‐1190. [DOI] [PubMed] [Google Scholar]
- 18. Sun X, Dong C, Levin B, et al. APOE epsilon4 carriers may undergo synaptic damage conferring risk of Alzheimer's disease. Alzheimers Dement. 2016;12:1159‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Headley A, De Leon‐Benedetti A, Dong C, et al. Neurogranin as a predictor of memory and executive function decline in MCI patients. Neurology. 2018;90:e887‐e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang M, Beckmann ND, Roussos P, et al. The Mount Sinai cohort of large‐scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data. 2018;5:180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479‐486. [DOI] [PubMed] [Google Scholar]
- 22. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 23. Haroutunian V, Perl DP, Purohit DP, et al. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185‐1191. [DOI] [PubMed] [Google Scholar]
- 24. Litvinchuk A, Wan YW, Swartzlander DB, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer's disease. Neuron. 2018;100:1337‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q, Zhang Y, Wang M, et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson's disease. Nat Commun. 2019;10:5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bennett DA, Schneider JA, Buchman AS, Mendes de Leon C, Bienias JL, Wilson RS. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology. 2005;25:163‐175. [DOI] [PubMed] [Google Scholar]
- 28. Colom‐Cadena M, Spires‐Jones T, Zetterberg H, et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer's disease. Alzheimers Res Ther. 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koffie RM, Hashimoto T, Tai HC, et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid‐beta. Brain. 2012;135:2155‐2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448‐6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo. Nature. 2002;416:535‐539. [DOI] [PubMed] [Google Scholar]
- 32. Tracy TE, Gan L. Tau‐mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr Opin Neurobiol. 2018;51:134‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid {beta}‐induced impairment of hippocampal long‐term potentiation. J Neurosci. 2011;31:1688‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wellington H, Paterson RW, Portelius E, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86:829‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer's disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm (Vienna). 1996;103:603‐618. [DOI] [PubMed] [Google Scholar]
- 36. de Wilde MC, Overk CR, Sijben JW, Masliah E. Meta‐analysis of synaptic pathology in Alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016;12:633‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, Montine TJ. The basis of cellular and regional vulnerability in Alzheimer's disease. Acta Neuropathol. 2019;138:729‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer's disease cases. Acta Neuropathol. 1992;85:23‐30. [DOI] [PubMed] [Google Scholar]
- 39. Burke SN, Gaynor LS, Barnes CA, et al. Shared functions of perirhinal and parahippocampal cortices: implications for cognitive aging. Trends Neurosci. 2018;41:349‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Mount Sinai Brain Bank (MSBB) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3159438) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The Religious Orders Study and Rush Memory and Aging Project (ROS‐MAP) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3219045) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The Harvard Brain Tissue Resource Center (HBTRC) study data are available at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage?Study=syn3159435) via the AD Knowledge Portal (https://adknowledgeportal.synapse.org).