

Abstract
Dystrophinopathies are allelic conditions caused by deletions, duplications and point-mutations in the DMD gene, located on the X chromosome (Xp21.2). Mutations that prematurely interrupt the dystrophin protein synthesis lead to the most severe clinical form, Duchenne muscular Dystrophy, characterized by early involvement of muscle strength. There is no known cure for dystrophinopathies. In DMD, treatment with corticosteroids have changed the natural history and the progression of the disease, prolonging ambulation, and slowing the onset of respiratory and cardiac involvement and scoliosis by several years. In the last few years, new perspectives and options are deriving from the discovery of pharmacological approaches able to restore normal, full-length dystrophin and potentially reverse the course of the disease. Read-through (RT) of nonsense mutations, thanks to its ability to bypass the premature stop codon and to act on virtually any region of the dystrophin gene, independently of the location in which the mutation resides, is one of these promising approaches. This non-systematic review shows the different steps that, passing from yeast to humans, have made it possible to use this innovative successful approach to treat serious diseases such as Duchenne muscular dystrophy.
Key words: Duchenne Muscular dystrophy, nonsense mutations, aminoglycosides, ataluren, translarna
Introduction
Duchenne muscular dystrophy (DMD) belongs to dystrophinopathies, a group of X-linked genetic degenerative disorders affecting striated muscles and caused by mutations in DMD gene 1-4.
Dystrophinopathies present with a variable spectrum of severity ranging from the exercise-induced muscle cramps and myoglobinuria, to the complete loss of muscle function, cardiomyopathy, and respiratory failure. The onset of DMD is in early childhood, characterized by a delay in motor milestones. In about 1/3 of children brain involvement with cognitive impairment and/or behavioural disorders such as ADHD (attention deficit hyperactivity disorder), autism, anxiety and obsessive-compulsive disorder can by associated 2,3.
Muscle hypertrophy is evident, especially in the calf muscles. Progression is rapid, so that by the age of 5 waddling gait and positive Gowers’ sign appear. In untreated boys, walking is lost by the age of 12 (mean 9.5 years). Following the loss of ambulation, scoliosis, respiratory failure and cardiomyopathy develop 2-5.
The incidence of DMD varies from 1:3.500 to 6.800 male births, according to the more recent estimates 6-8.
From a genetic point of view, dystrophinopathies are allelic conditions caused by deletions, duplications and point-mutations in the DMD gene, located on the X chromosome (Xp21.2). To explain how mutations in the same gene may cause the observed phenotypic variability, the rule of the “Open Reading Frame” (ORF) comes to the rescue 7,8. According to this rule, mutations that prevent the ribosome from reading the amino acid sequence correctly (“out-of-frame” or nonsense mutations) result in no functional dystrophin (see Figure 1) and produces the DMD phenotype. On the other hand, mutations that retain the reading sequence (“in frame”), generate a shortened but partly functional protein, leading to the milder BMD phenotype 7. Exceptions to this rule are observed for up to 9% of dystrophin mutations 9.
Figure 1.
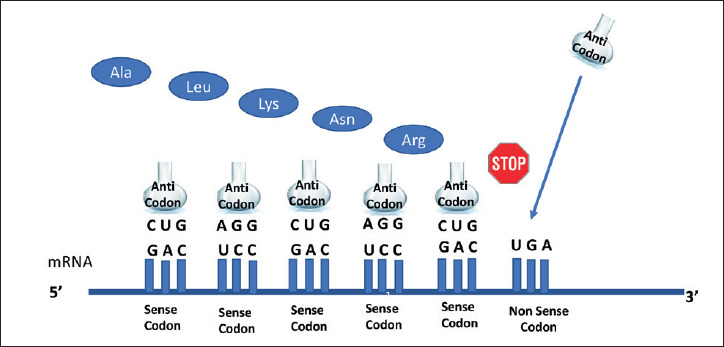
Stop of the protein synthesis, caused by a non-sense mutation (from Roy B, et al. Proc Natl Acad Sci USA 2016;113:12508-13, mod.).
There is no known cure for dystrophinopathies. In DMD, treatment with corticosteroids have changed the natural history and the progression of the disease, prolonging ambulation, and slowing the onset of respiratory and cardiac involvement and scoliosis by several years. Physiotherapy and orthotics delay the onset of joint contractures 10. Symptomatic therapy is available for cardiac and respiratory impairment 11-13. Life expectancy is shortened by cardiac and respiratory involvement but can be substantially improved with regular monitoring and pro-active management 14,15.
In the last few years, new perspectives and options are deriving from the discovery of pharmacological approaches able to restore normal, full-length dystrophin and potentially reverse the course of the disease. Read-through (RT) of nonsense mutations, thanks to its ability to bypass the premature stop codon and to act on virtually any region of the dystrophin gene, independently of the location in which the mutation resides, is one of these promising approaches.
Brief history of read-through approach for stop mutations
The history of readthrough of premature stop mutations in eukaryotes began in 1979, when 2 papers describing suppression of these mutations by aminoglycosides were published 16,17. Several of this class of antibiotics were tested for relative capacity to read through premature stop mutations 18,19. These observations led to testing of gentamicin, initially in a cystic fibrosis cell line 20, then in the mdx mouse, an animal model for Duchenne muscular dystrophy that fortuitously harbors a UAA premature stop mutation in exon 23 of DMD gene 21. This mdx proof-of-concept study demonstrated an about 20% of normal expression of muscle fibre dystrophin in vitro and in vivo, and showed that dystrophin was properly localized to the sarcolemma. These muscle fibres showed an increased resistance to eccentric contraction injury, while a decrease in blood creatine kinase levels suggested a reduced muscle cell fragility 19.
On the basis of these findings, human trials of intravenous gentamicin were undertaken on patients with Duchenne/Becker muscular dystrophies in which readthrough strategies may be applicable in about 13% of patients 20,21. With this strategy, small administered molecule drugs producing a conformational change in the mRNA, can allow the ribosome to insert an amino acid at a UGA, UAG, or UAA premature stop codon site during translation 16.
Drugs inducing suppression of these nonsense mutations increase the readthrough of the premature stop signal, and the production of full-length protein. The minimal quantity of full-length dystrophin required to achieve normal muscle function is not known, but a 30% of normal seems to be sufficient to avoid muscular dystrophy in humans. Lesser amounts of dystrophin may ameliorate symptoms or temper disease progression 22.Two small pilot studies on gentamicin administration in nonsense mutations DMD (nmDMD) patients appeared in the years 2001-2003. The initial study was performed by Wagner et al. 23. In this trial, 4 patients (2 DMD and 2 BMD) aged 7-16, were daily administered gentamicin intravenously, at a dosage of 7.5 mg/kg, for 2 weeks. Over this short period of drug exposure, drug activity, as assessed by muscle dystrophin expression on biopsies, and muscle strength, was not detected. No renal or ototoxicity was observed.
In 2003, Politano et al. 24 reported the results of intravenous gentamicin administration in 4 DMD subjects, 3 ambulant and 1 wheelchair-bound, aged between 4 and 9. They used a gentamicin regimen comprising 2 six-day courses of therapy at a dosage of 6.0 and 7.5 mg/kg respectively, separated by an intervening period of 7 weeks. They demonstrated an increase in dystrophin expression in 3 of 4 subjects in end-of treatment biopsies. The best results were obtained in the younger patient. They also shown a re-expression of sarcoglycan complex consistent with the restore of the sarcolemmal integrity. No renal or ototoxicity was identified.
These reports aroused the attention of many researchers on this new treatment option for patients with nonsense mutations 25-27.
In 2010, Malik et al. 28 published the results of a more extensive study on 34 DMD patients, who were subdivided in four cohorts. Two of them received 14-day gentamicin (7.5 mg/kg/day): Cohort 1, had ten stop codon patients (nmDMD) and Cohort 2, eight frameshift controls. Two additional nmDMD cohorts were treated, at the same dosage for 6 months: Cohort 3 (n = 12) weekly, and Cohort 4 (n = 4) twice weekly. Pre- and post-treatment biopsies were assessed for dystrophin levels, as were clinical outcomes. After 6 months of gentamicin, dystrophin levels significantly increased with the highest levels to 15% of normal (1 in Cohort 3, and 2 in Cohort 4), accompanied by reduced serum CK levels. Stabilization of strength and a slight increase in forced vital capacity (FVC) were also observed. All subjects were carefully monitored for adverse events and no persistent findings of nephrotoxicity or ototoxicity were reported.
Although these promising results, the need for regular intravenous administration, and safety laboratory parameters discouraged the clinical practical application. Furthermore, multiple forms of gentamicin were identified, with significant variation in their potential to promote dystrophin expression 25. Hence, the need to study new pharmacological molecules with the same pharmacological characteristics.
Ataluren (PTC124)
Ataluren (formerly known as PTC124) is a small molecule (Fig. 2) developed by PTC Therapeutics as an orally bioavailable product able to bypass nonsense mutations (Fig. 3) and avoid potential renal- and ototoxicity of aminoglycosides. It was originally developed by means of an optimized high-throughput screening campaign. A dose dependent readthrough of all three nonsense codons (UGA, UAG, UAA) was observed, with the highest readthrough at UGA, followed by UAG and then UAA 29. PTC124 soon proved to be a more potent nonsense-suppressing agent than gentamicin 30-32.
Figure 2.
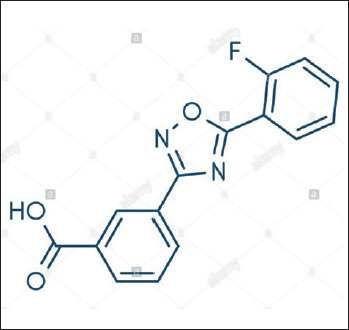
Ataluren chemical structure.
Figure 3.
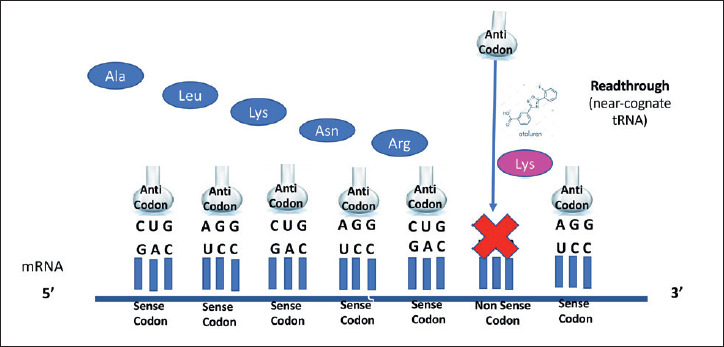
Ataluren’s mechanism of action (from Roy B, et al. Proc Natl Acad Sci U S A 2016;113:12508-12513, mod.).
When administered in mdx mice, treatment with PTC124 restored dystrophin production in all skeletal muscles examined, including the diaphragm, and cardiac muscle. The dystrophin levels were found to be 20-25% those of control mouse muscles, and partially restored force generation and resistance against eccentric exercise were observed suggesting that PTC124 was able to reduce muscle fragility 33. PTC-treated mdx mice also exhibited increased levels of sarcoglycans, consistent with stabilization of the dystrophin-associated proteins. The rescue of skeletal muscle was seen within 2 to 8 weeks of exposure. Readthrough by PTC124 was selective and specific to disease-causing premature termination, without evidence of changes in the ribosomal readthrough of normal stop codons. These encouraging results led to the initiation of studies in humans 34-39.
Hirawat et al. 34 assessed safety and tolerability of PTC124 in a Phase 1 study enrolling 62 healthy adult volunteers and concluded that the drug was well tolerated, except for mild headaches, dizziness, and gastrointestinal discomfort at high dose.
Finkel et al. 36, in a Phase 2a open-label, sequential dose-ranging trial recruited 38 boys with nonsense mutation DMD, subdivided in three cohorts according to the administered dosage of Ataluren: 16 mg/kg/day (6 patients); 40 mg/kg/day (20 patients); and 80 mg/kg/day (12 patients). Treatment duration was 28 days. Change in full-length dystrophin expression, as assessed by immunostaining in pre- and post-treatment muscle biopsy specimens, was the primary endpoint. They found that dystrophin expression was increased in 61% of subjects post-treatment, associated neither with nonsense mutation type nor exon location. Ataluren was generally well tolerated, supporting the evaluation of ataluren 40 mg/kg/day and 80 mg/kg/day in a Phase 2b, double-blind, long-term study, having 6-minute walk distance (6MWD) as primary endpoint. This phase IIb double-blind, placebo-controlled, dose-ranging, efficacy and safety study failed to demonstrate improvement in the 6MWT.
However, further detailed analysis of the Phase IIb data suggested the possibility of a bell-shaped dose-response with clinical benefit to DMD boys receiving low dose, and in high-dose patients in whom serum levels of PTC124 were low 37,38.
In 2014, Bushby et al. 39 published the results of a randomized, double-blind, placebo-controlled study enrolling 174 DMD patients ≥ 5 years with nm-dystrophinopathy who received orally, 3 times daily, ataluren 40 mg/kg/day (n = 57); ataluren 80 mg/kg/day (n = 60); or placebo (n = 57) for 48 weeks. The primary endpoint was change in 6-Minute Walk Distance (6MWD) at Week 48. Ataluren was generally well tolerated. Unexpectedly, both the primary and secondary endpoints (timed function tests) favoured ataluren versus placebo, at the lower dosage.
As a consequence, the European Medicines Agency (EMA) reviewed ataluren for the treatment of ambulant patients aged 5 and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene 40-44.
In 2017, Mc Donald et al. 45 published the results of a major international multicentre, randomised, double-blind, placebo-controlled, phase 3 trial carried out on Ataluren, including 54 sites in 18 countries located in North America, Europe, the Asia-Pacific region, and Latin America. Two-hundred-thirty boys aged 7-16 years, with nonsense mutation DMD and a baseline 6MWD of ≥ 150 m and 80% or less of the predicted normal value for age and height, were enrolled and randomly assigned (1:1) between March 26, 2013 and Aug 26, 2014. Randomisation was stratified by age (< 9 vs ≥ 9 years), duration of previous corticosteroid use (6-12 months vs ≥ 12 months), and baseline 6MWD (< 350 m vs ≥ 350 m). The primary endpoint was change in 6MWD from baseline to week 48. The results showed that changes in 6MWD did not differ significantly between patients in the ataluren group and those in the placebo group. However, a significant effect of ataluren in a subgroup of patients with a baseline 6MWD between 300 m and 400 m was observed. Baseline 6MWD values within this range were in fact associated with a more predictable rate of decline over 1 year. The preliminary patient registry data also indicated a longer ambulatory period in ataluren-treated patients compared with published natural history 42. Because the phase 2 clinical trial results were not satisfactory in terms of meeting the primary endpoint, ataluren was first rejected for approval by the European Medicines Agency (EMA) 43. However, as the conclusions drawn from further analysis suggested ataluren to be effective in terms of slowing down the disease course, EMA has subsequently granted ataluren a conditional marketing authorization in 2014 43,44. Ataluren is indicated for the treatment of Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene, in ambulatory patients aged 2 years and older in the European Member States and Iceland, Liechtenstein, Norway, Great Britain, Northern Ireland, Kazakhstan, Israel, and Republic of Korea, and aged 5 years and older in Chile, Brazil, and Ukraine (under special state registration). The presence of a nonsense mutation in the dystrophin gene should be determined by genetic testing (Ref Translarna 43 Summary of Product Characteristics (SmPC) for respective countries) whereas ataluren is still an investigational drug in the United States 44.
In 2018, Ebrahimi-Fakhari et al. 46 reported their experience in 4 non-ambulatory nmDMD patients. Routine investigations included cardiac function, pulmonary function tests and muscle strength. They compared changes in left ventricular fractional shorting (LVFS), FVC and BMI from two defined time periods (18-26-month period prior to and after Ataluren start). There were no adverse clinical effects or relevant abnormalities in routine laboratory values. They conclude that Ataluren appears to mildly ameliorate the clinical course in their patients with a good safety profile.
In the same year, Ruggiero et al. 47, describing how the best results at 1 year were seen in the younger of the 3 DMD patients, aged 5,8 and 10 and treated with Ataluren, suggested earlier initiation of therapy. D’Ambrosio et al. 48, broadening the spectrum of patients potentially benefiting from this type of treatment, reported the promising results obtained in a 24-year-old nmDMD symptomatic carrier who took ataluren for a period of 9 months.
Several papers were published in the last 3 years stressing that a new era is emerging for the treatment of DMD patients with nm mutations 49-59.
Among these, some deserve to be reported: 1) the paper by Landfeldt et al. 52 who published a targeted mini-review of the literature from 1995 to 2018, which included cohort studies, guidelines, randomised clinical trials, clinical commentaries and reviews. The review covered the pathophysiology, epidemiology, and burden of nmDMD and the clinical programme for ataluren. Based on the current evidence, and their experiences, they recommend patients with nmDMD should be given ataluren as soon as possible after diagnosis; 2) that of Muntoni et al. 53 who presented patient demographics and characteristics of nmDMD patients treated with ataluren, included in the STRIDE registry. STRIDE (Strategic Targeting of Registries and International Database of Excellence) is a multicentre registry providing real-world evidence regarding ataluren use in patients with nmDMD in clinical practice. Patients should be followed up from enrolment for ≥ 5 years or until study withdrawal. As of 9 July 2018, 213 DMD boys have been enrolled from 11 countries. Mean (standard deviation) ages at first symptoms and at study treatment start were 2.7 (1.7) years old and 9.8 (3.7), respectively. Mean (standard deviation) ataluren exposure was 639.0 (362.9) days. Six patients withdrew. STRIDE represents the first drug registry for patients with DMD and the largest real-world registry of patients with nonsense mutations; 3) the paper by Mercuri et al. 57 who examined the safety and effectiveness of ataluren, comparing the results from the STRIDE Registry and CINRG DMD Natural History. A propensity score matching was performed to identify STRIDE and CINRG DNHS patients who were comparable in established disease progression predictors (registry cut-off date, 9 July 2018). The results confirmed those of previous clinical trials, showing a series of benefits in nmDMD patients treated with ataluren + Standard of Care (SoC), compared with DMD patients receiving SoC only. Patients receiving ataluren plus standard of care (SoC) in STRIDE had a significantly delayed age at loss of ambulation, worsening of performance in timed function tests and worsening of pulmonary function versus patients receiving SoC alone in the CINRG DNHS (all p ≤ 0.0386), over a mean treatment period of 632 days. Ataluren plus SoC was also associated with delayed deterioration of cardiac function versus SoC alone, although this did not reach statistical significance likely due to the short period of observation. No correlation was observed between DMD genotype (type and location of nonsense mutation) and disease progression or treatment benefits; 4) the paper by Campbell et al. 58 who combined data from the two completed randomized controlled trials (ClinicalTrials.gov: NCT00592553; NCT01826487) of ataluren in nmDMD and examined the intent-to-treat (ITT) populations and two patient subgroups (baseline 6-min walk distance [6MWD] ≥ 300-< 400 or < 400 m). The meta-analyses evaluated 6MWD change from baseline to week 48. The Authors found statistically significant differences in 6MWD change with ataluren versus placebo across all three meta-analyses, supporting previous evidence for ataluren in slowing disease progression versus placebo in patients with nmDMD over 48 weeks. Treatment benefit was most evident in patients with a baseline 6MWD ≥ 300-< 400 m (the ambulatory transition phase).
Conclusions
DMD is a genetic degenerative disorder affecting muscles, caused by mutations in the dystrophin gene, the biggest gene described in humans. They can be deletions of one or more exons in prevalence (65-75%), or duplications (10-15%). The remaining are due to nonsense mutations that prematurely stop the protein synthesis. DMD boys present with motor delay and usually loss ambulation before the age of 12. Death is due to respiratory or heart failure. Treatment consists in steroid administration and cardiological and respiratory support. Gene therapy is under consideration.
Ataluren is a treatment for patients with nmDMD, designed to promote ribosomal readthrough of an in-frame premature stop codon and thereby enable the production of full-length dystrophin protein, which has the non-negligible advantage of being orally administered. All the scientific papers published so far have shown that ataluren is a drug capable of slowing the evolution of the disease in patients with nmDMD. Respiratory and cardiac parameters also appear to be positively influenced by treatment with ataluren, though they have not reached statistical significance, probably due to the short period of observation. Early diagnosis and treatment appear to be a key point. The expansion of treatment to female symptomatic carriers of the disease seems a viable option.
Figures and tables
Acknowledgments
Pacini Editore Srl received unconditional support by PTC Italy Medical Affairs for Medical Writing.
References
- 1.Emery AEH. The muscular dystrophies. Lancet 2002;359:687-695. https://doi.org/10.1016/S0140-6736(02)07815-7 10.1016/S0140-6736(02)07815-7 [DOI] [PubMed] [Google Scholar]
- 2.Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet 2019;394:2025-2038. https://doi.org/10.1016/S0140-6736(19)32910-1 10.1016/S0140-6736(19)32910-1. [DOI] [PubMed] [Google Scholar]
- 3.Brandsema JF, Darras BT. Dystrophinopathies. Semin Neurol 2015;35:369-384. https://doi.org/10.1055/s-0035-1558982 10.1055/s-0035-1558982 [DOI] [PubMed] [Google Scholar]
- 4.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919-928. https://doi.org/10.1016/0092-8674(87)90579-4 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- 5.Koeks Z, Bladen CL, Salgado D, et al. Clinical outcomes in duchenne muscular dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis 2017;4:293-306. https://doi.org/10.3233/JND-170280 10.3233/JND-170280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prior TW. Genetic analysis of the Duchenne muscular dystrophy gene. Arch Pathol Lab Med 1991;115:984-990. PMID: 1898245 [PubMed] [Google Scholar]
- 7.Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90-5. https://doi.org/10.1016/0888-7543(88)90113-9 10.1016/0888-7543(88)90113-9 [DOI] [PubMed] [Google Scholar]
- 8.Hoffman EP. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol 2020;39:179-186. https://doi.org/10.36185/2532-1900-020 10.36185/2532-1900-020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34(2):135-144. https://doi.org/10.1002/mus.20586 10.1002/mus.20586 [DOI] [PubMed] [Google Scholar]
- 10.McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 2018;391:451-461. https://doi.org/10.1016/S0140-6736(17)32160-8 10.1016/S0140-6736(17)32160-8 [DOI] [PubMed] [Google Scholar]
- 11.Feingold B, Mahle WT, Auerbach S, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 2017;136:e200-e231. https://doi.org/10.1161/CIR.0000000000000526 10.1161/CIR.0000000000000526 [DOI] [PubMed] [Google Scholar]
- 12.Nigro G, Comi LI, Politano L, et al. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990;26:271-277. https://doi.org/10.1016/0167-5273(90)90082-g 10.1016/0167-5273(90)90082-g [DOI] [PubMed] [Google Scholar]
- 13.Nigro G, Comi LI, Politano L, et al. Cardiomyopathies associated with muscular dystrophies. Engel & Franzini-Armstrong. Myology. McGRAW-HILL; 2004;45, pp. 1239-1256. [Google Scholar]
- 14.Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol 2012;31:121-125. PMID: 23097603 [PMC free article] [PubMed] [Google Scholar]
- 15.San Martín PP, Solis FF, Cavada Ch G. Survival of patients with Duchenne muscular dystrophy. Rev Chil Pediatr 2018;89:477-483. https://doi.org/10.4067/S0370-41062018005000704 10.4067/S0370-41062018005000704 [DOI] [PubMed] [Google Scholar]
- 16.Palmer E, Wilhelm JM, Sherman F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature 1979;277:148-150. https://doi.org/10.1038/277148a0 10.1038/277148a0 [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Ursic D, Davies J. Phenotypic suppression and misreading in Saccharomyces cerevisae. Nature 1979;277:146-148. https://doi.org/10.1038/277146a0 10.1038/277146a0 [DOI] [PubMed] [Google Scholar]
- 18.Kopelowitz J, Hompe C, Goldman R, et al. Influence of codon context in UGA suppression and readthrough. J Mol Biol 1992;225:261-265. https://doi.org/10.1016/0022-2836(92)90920-f 10.1016/0022-2836(92)90920-f [DOI] [PubMed] [Google Scholar]
- 19.Engelberg-Kulka H, Schoulaker-Schwarz R. Stop is not the end: physiological implications of translational readthrough. J Theor Biol 1988;131:477-485. https://doi.org/10.1016/s0022-5193(88)80042-0 10.1016/s0022-5193(88)80042-0 [DOI] [PubMed] [Google Scholar]
- 20.Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 1997;3:1280-1284. https://doi.org/10.1038/nm1197-1280 10.1038/nm1197-1280 [DOI] [PubMed] [Google Scholar]
- 21.Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clinical Invest 1999;104:375-381. https://doi.org/10.1172/JCI7866 10.1172/JCI7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dent KM, Dunn DM, von Niederhausern AC, et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet 2005;134:295-298. https://doi.org/10.1002/ajmg.a.30617 10.1002/ajmg.a.30617 [DOI] [PubMed] [Google Scholar]
- 23.Howard MT, Shirts BH, Petros LM, et al. Sequence specificity of aminoglycoside-induced stop codon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol 2000;48:164-169. PMID: 10939566 [PubMed] [Google Scholar]
- 24.Neri M, Torelli S, Brown S, et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord 2007;17:913-8. https://doi.org/10.1016/j.nmd.2007.07.005 10.1016/j.nmd.2007.07.005 [DOI] [PubMed] [Google Scholar]
- 25.Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 2001;49:706-711. PMID: 11409421 [PubMed] [Google Scholar]
- 26.Politano L, Nigro G, Nigro V, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol 2003;22:15-21. PMID: 11409421 [PubMed] [Google Scholar]
- 27.Karpati G, Lochmuller H. When running a stop sign may be a good thing. Ann Neurol 2001;49:693-694. PMID: 11409418 [PubMed] [Google Scholar]
- 28.Aurino S, Nigro V. Readthrough strategies for stop codons in Duchenne muscular dystrophy. Acta Myol 2006;25:5-12. PMID: 17039975 [PubMed] [Google Scholar]
- 29.Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. Bio Drugs 2009;23:165-174. https://doi.org/10.2165/00063030-200923030-00003 10.2165/00063030-200923030-00003 [DOI] [PubMed] [Google Scholar]
- 30.Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 2010;67:771-780. https://doi.org/10.1002/ana.22024 10.1002/ana.22024 [DOI] [PubMed] [Google Scholar]
- 31.Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447:87-91. https://doi.org/10.1038/nature05756 10.1038/nature05756 [DOI] [PubMed] [Google Scholar]
- 32.Hamed SA. Drug evaluation: PTC-124 – a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs 2006;9:783-789. PMID: 17096300 [PubMed] [Google Scholar]
- 33.No Authors Listed. Molecule of the month. Ataluren. Drug News Perspect 2010;23:135. PMID: 20369078 [PubMed] [Google Scholar]
- 34.European Medicines Agency. Translarna™ (ataluren) summary of product characteristics, 2019. (https://www.ema.europa.eu/en/documents/product-information/translarna-epar-product-information_en.pdf).
- 35.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, et al. Development of novel aminoglycosidem (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem 2009;52:2836-2845. https://doi.org/10.1021/jm801640k 10.1021/jm801640k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirawat S, Welch EM, Elfring GL, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol 2007;47:430-444. https://doi.org/10.1177/0091270006297140 10.1177/0091270006297140 [DOI] [PubMed] [Google Scholar]
- 37.Finkel RS. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124). J Child Neurol 2010;25:1158-1164. https://doi.org/10.1177/0883073810371129 10.1177/0883073810371129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkel RS, Flanigan KM, Wong B, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 2013;8:e81302. https://doi.org/10.1371/journal.pone.0081302 10.1371/journal.pone.0081302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan NJ. Ataluren: first global approval. Drugs 2014;74:1709-1714. https://doi.org/10.1007/s40265-014-0287-4 10.1007/s40265-014-0287-4 [DOI] [PubMed] [Google Scholar]
- 40.Mullard A. EMA reconsiders ‘read-through’ drug against Duchenne muscular dystrophy following appeal. Nat Biotechnol 2014;32:706. https://doi.org/10.1038/nbt0814-706 10.1038/nbt0814-706 [DOI] [PubMed] [Google Scholar]
- 41.Bushby K, Finkel R, Wong B, et al. PTC124-GD-007-DMD Study Group . Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014;50:477-487. https://doi.org/10.1002/mus.24332 10.1002/mus.24332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namgoong JH, Bertoni C. Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy. Degener Neurol Neuromuscul Dis 2016;6:37-48. https://doi.org/10.2147/DNND.S71808 10.2147/DNND.S71808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas M, Vlcek V, Balabanov P, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord 2015;25:5-13. https://doi.org/10.1016/j.nmd.2014.11.011 10.1016/j.nmd.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 44. [(accessed on 11 November 2018)]; Available online: http://ir.ptcbio.com/news-releases/news-release-details/ptc-therapeutics-announces-initial-data-patient-registry.
- 45. [(accessed on 11 November 2018)]; Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/translarna.
- 46. [(accessed on 11 November 2018)]; Available online: http://www.ptcbio.com/en/pipeline/clinical-trials-ataluren. [Google Scholar]
- 47.McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:1489-1498. https://doi.org/10.1016/S0140-6736(17)31611-2 10.1016/S0140-6736(17)31611-2 [DOI] [PubMed] [Google Scholar]
- 48.Ebrahimi-Fakhari D, Dillmann U, Flotats-Bastardas M, et al. Off-label use of ataluren in four non-ambulatory patients with nonsense mutation Duchenne muscular dystrophy: effects on cardiac and pulmonary function and muscle strength. Front Pediatr 2018;6:316. https://doi.org/10.3389/fped.2018.00316 10.3389/fped.2018.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruggiero L, Iodice R, Esposito M, et al. One-year follow up of three Italian patients with Duchenne muscular dystrophy treated with ataluren: is earlier better? Ther Adv Neurol Disord 2018;11:1756286418809588. https://doi.org/10.1177/1756286418809588 10.1177/1756286418809588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Ambrosio P, Orsini C, Nigro V, et al. Therapeutic approach with Ataluren in Duchenne symptomatic carriers with nonsense mutations in dystrophin gene. Results of a 9-month follow-up in a case report. Acta Myol 2018;37:272-274. eCollection 2018 Dec. PMID: 30944907 [PMC free article] [PubMed] [Google Scholar]
- 51.Shimizu-Motohashi Y, Komaki H, Motohashi N, et al. Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J Pers Med 2019;9:1. https://doi.org/10.3390/jpm9010001 10.3390/jpm9010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ristl R, Urach S, Rosenkranz G, et al. Methods for the analysis of multiple endpoints in small populations: a review. J Biopharm Stat 2019;29:1-29. https://doi.org/10.1080/10543406.2018.1489402 10.1080/10543406.2018.1489402 [DOI] [PubMed] [Google Scholar]
- 53.Korinthenberg R. A new era in the management of Duchenne muscular dystrophy. Dev Med Child Neurol 2019;61:292-297. https://doi.org/10.1111/dmcn.14129 10.1111/dmcn.14129 [DOI] [PubMed] [Google Scholar]
- 54.Landfeldt E, Sejersen T, Tulinius M. A mini-review and implementation model for using ataluren to treat nonsense mutation Duchenne muscular dystrophy. Acta Paediatr 2019;108:224-230. https://doi.org/10.1111/apa.14568 10.1111/apa.14568. [DOI] [PubMed] [Google Scholar]
- 55.Muntoni F, Desguerre I, Guglieri M, et al. Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. J Comp Eff Res 2019;8:1187-1200. https://doi.org/10.2217/cer-2019-0086 10.2217/cer-2019-0086 [DOI] [PubMed] [Google Scholar]
- 56.Li D, McDonald CM, Elfring GL, et al. Assessment of treatment effect with multiple outcomes in 2 clinical trials of patients with Duchenne muscular dystrophy. JAMA Network Open 2020;3:e1921306. https://doi.org/10.1001/jamanetworkopen.2019.21306 10.1001/jamanetworkopen.2019.21306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pascual-Morena C, Cavero-Redondo I, Álvarez-Bueno C, et al. Restorative treatments of dystrophin expression in Duchenne muscular dystrophy: a systematic review. Ann Clin Transl Neurol 2020;7:1738-1752. https://doi.org/10.1002/acn3.51149 10.1002/acn3.51149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernert G, Hahn A, Köhler C, et al. Expert recommendation: treatment of nonambulatory patients with Duchenne muscular dystrophy. Nervenarzt 2020;November 19. https://doi.org/10.1007/s00115-020-01019-3 10.1007/s00115-020-01019-3 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mercuri E, Muntoni F, Osorio AN, et al. Safety and effectiveness of ataluren: comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J Comp Eff Res 2020;9:341-360. https://doi.org/10.2217/cer-2019-0171 10.2217/cer-2019-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell C, Barohn RJ, Bertini E, et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. Comp Eff Res 2020;9:973-984. https://doi.org/10.2217/cer-2020-0095 10.2217/cer-2020-0095 [DOI] [PubMed] [Google Scholar]
- 61.Sheikh O, Yokota T. Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin Investig Drugs 2021;30:167-176. https://doi.org/10.1080/13543784.2021.1868434 10.1080/13543784.2021.1868434 [DOI] [PubMed] [Google Scholar]