

Abstract
We report a family carrying a previously described truncating mutation, NM_001267550.2(TTN):c.107889del p.(Lys35963Asnfs*9) in exon 364, and a novel truncating mutation, NM_001267550.1:c.100704C > A p.(Tyr33568*) in exon 358 in the titin gene. The c.107889del mutation, which was maternally transmitted, has been previously described in patients from the Iberian Peninsula. The mother was of Peruvian descent suggesting a potential European ancestral origin of this mutation. In this family, a daughter, who is a compound heterozygote carrying both these mutations, developed a peripartum cardiomyopathy during her second pregnancy. Subsequently, she was diagnosed with a myopathy following electromyography testing and a muscle biopsy which showed fiber type disproportion. Her brother, who carries only the paternally inherited c.100704C > A mutation, developed a cardiomyopathy following a suspected viral illness. Their father, who transmitted this mutation, has no evidence of a cardiomyopathy. We hypothesize that the c.100704C > A mutation confers susceptibility to the development of cardiomyopathy which may be brought on by cardiovascular stress. Our study of this family expands the genotype and phenotype spectrum of disorders that can be associated with mutations in the titin gene.
Key words: TTN gene truncating mutations, cardiomyopathy, susceptibility mutation
Introduction
The titin gene, TTN, is the largest known polypeptide in humans and has multiple functions that include participating in myogenesis and contributing to the elasticity of muscle tissues. There are a variety of clinical and pathological phenotypes that have been described ranging from early onset myopathy with fatal cardiomyopathy, congenital centronuclear myopathy to adult onset muscular dystrophy. The pattern of inheritance can also vary from autosomal recessive to autosomal dominant 1. We present a family with siblings suffering from cardiomyopathy, one of whom also developed a myopathy.
Case history
The index patient is a 37-year-old woman who was admitted for delivery of her second pregnancy and was diagnosed with toxemia of pregnancy. Her first pregnancy had been uneventful. As part of the investigations for the toxemia, an echocardiogram was performed and revealed a left ventricular ejection fraction of 30-35% (normal range 55-70%) with impaired relaxation of left ventricular diastolic filling. She was treated for a global cardiomyopathy and delivered by cesarean section. At age 39 years, the ejection fraction had improved to 45% and she continued to follow up with a cardiologist. Over the next four years it was noted that she had reduced exercise tolerance and for several years she was having difficulty climbing stairs. These issues were attributed to the cardiomyopathy. At age 43 years she developed an episode of plantar fasciitis and was evaluated by a physical therapist who noted weakness. She was sent for an initial neurological consultation and subsequently referred for a neuromuscular evaluation. Her neurological review of systems revealed no complaints of difficulty chewing, swallowing, speaking, double vision or sensory loss. She had complaints of imbalance and difficulties climbing stairs and arising from a deep seated chair. Physical examination revealed normal vital signs and neurological examination showed no abnormalities of the mental status, cranial nerve examination, cerebellar system and sensation assessing light touch, pin prick, vibration and proprioception. Testing her power revealed normal strength in her arms evaluating the biceps, triceps, internal rotation, external rotation, wrist flexion and extension. She had no scapular winging. In her right leg, she had weakness with Medical Research Council (MRC) scale of 3/5 testing hip flexion, extension and abduction, knee extension and foot dorsiflexion. On the left, power in these muscles was diffusely MRC Grade 4/5. She had normoactive reflexes in her arms and reduced reflexes in her legs. Her plantar reflexes were flexor and she had a “waddling” gait.
Follow up with the cardiologist at age 44 years showed an ejection fraction of 50% and a negative nuclear stress test.
A family history revealed that her two children do not suffer from a neuromuscular disorder. She has 5 siblings who are all healthy with no complaints or ongoing diagnosis of a neurological or cardiac disorder. However, a brother, age 53 years, was diagnosed with non-ischemic cardiomyopathy at age 43 years when he presented with symptoms of congestive heart failure following a prodrome of a presumed viral infection. Investigations revealed a left ventricular ejection fraction of 10%. Currently he is well controlled with medical management and has an ejection fraction of 55%. He has no symptoms of muscle weakness. Her mother, who was the product of a consanguineous marriage between first cousins died at age 46 years from an unclear cause. Her father, age 84 years, has no neuromuscular complaints and no history or clinical evidence of a cardiomyopathy. Both of her parents are from Peru and are not related to each other.
Routine serum chemistries including a cell count and differential, comprehensive metabolic panel, aldolase and inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein were normal. Her creatine phosphokinase was 976 U/L (nl-24-173 U/L) and after discontinuing fenofibrate came down to 477 U/L. Magnetic resonance imaging (MRI) of the pelvis with and without contrast showed atrophy of the right piriformis and gluteus maximus muscles with no enhancement. This was interpreted as showing evidence suggestive of a neurogenic process. An MRI of the lumbosacral spine showed no significant abnormalities.
She had an electromyography (EMG) done which showed normal motor nerve conduction parameters testing bilateral ulnar, median, peroneal and tibial nerves. Evoked sensory nerve action potentials were normal testing the ulnar, median, radial, sural and superficial peroneal nerves. A concentric needle EMG showed no spontaneous activity in any muscle sampled of her arms or legs. Low amplitude polyphasic motor unit potentials and a full interference of submaximal effort was noted in the proximal muscles of her arms. These abnormalities were noted to a greater degree in analyzing the muscles of her legs.
Muscle biopsies of the left deltoid and tibialis anterior were obtained, and show moderate myopathic changes (Fig. 1). H&E stained frozen section (1A) show an increase in variation of fiber size, with several smaller/hypotrophic fibers, and an increase in the internally and centrally placed nuclei. NADH-TR (1B) illustrate several ring and lobulated fibers. ATPase Ph-4.3 (1C) in which the type 1 myofibers show darker staining, and ATPase Ph-9.4 (1D) where the type 2 myofibers show darker staining. Both stains show that the type 1 myofibers are small/hypotrophic and are more numerous, comprising more than 60% of the myofibers, compared with type 2 fibers. Based upon these non-specific pathological changes, the differential diagnosis includes fiber type disproportion, limb-girdle muscular dystrophy, myotonic dystrophy and centronuclear myopathy.
Figure 1.
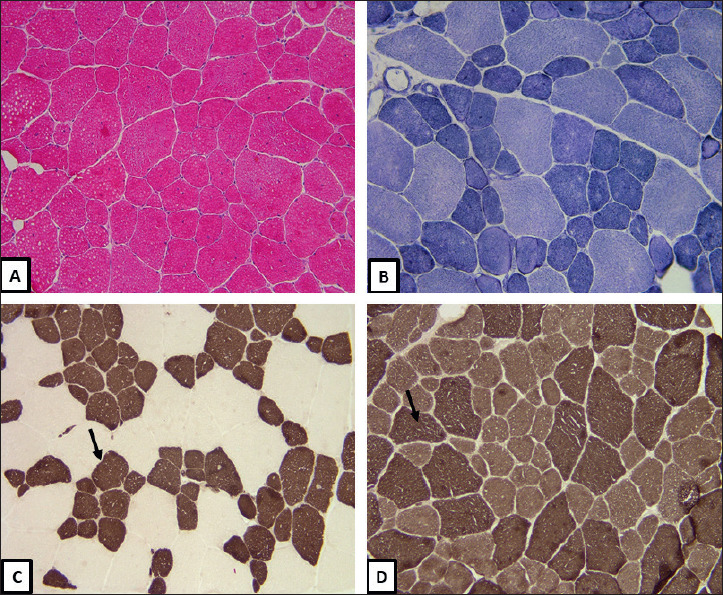
(A) H&E, Frozen section 200X, show an increase in variation of fiber size and internally and centrally placed nuclei. (B) NADH-TR, Frozen section 200X, ring fibers. (C) ATPase Ph 4.3, Frozen section 200X, type 1 myofibers (arrow) are hypotrophic and more numerous (> 60%). (D) ATPase Ph 9.4, Frozen section 200X type 2 myofibers (arrow) are of normal size and fewer in number.
Genetic analysis
The patient underwent commercial genetic analysis by performing whole exome sequencing of eighty genes known to cause genetic myopathies. This analysis included negative test results of myotonic disorders DM1 and DM2. All testing was done with the patient and family members following local IRB approved policies and procedures.
In the index patient, the results showed a c.617G>A variant (rs373108373) in exon 4 of the TRPV4 gene resulting in an R206H protein alteration. This variant is reported in the ExAC database in a heterozygous state in three individuals (https://gnomad.broadinstitute.org/variant/12-110240891-C-A?dataset=gnomad_r2_1) 2.
This R206H change involves a conservative amino acid substitution which is not likely to impact secondary protein structure. Our protein modeling analysis with PolyPhen 3 suggests that this is a benign variant. In addition, a c.409G > A variant was detected in exon 4 of the LAMA2 gene. This A137T variant (rs368349321) is predicted to be deleterious to the protein by SIFT 4, PolyPhen and Mutation taster analysis 5. Although it has not been reported in the ClinVar database, pathogenic variants in LAMA2 gene typically cause an autosomal recessive disorder and a second variant was not detected by sequencing and deletion/duplication analysis. It is unlikely that this is a disease producing variant in this patient.
Two variants were observed in the TTN gene (NM_001267550), a previously reported pathogenic variant in the ClinVar database, rs281864930, c.107889del in the last exon (364/364), resulting in frameshift and premature truncation of protein (Lys35963Asnfs*9) (National Center for Biotechnology Information. ClinVar; [VCV000038439.5], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000038439.5). This variant is located in the portion of the TTN gene encoding the M-band region of the protein. A second variant, c.100704C > A in exon 358 (358/364) of TTN gene results in a nonsense variant Tyr33568*. This is a novel variant not reported in either the 1000 Genome 6 or gnomAD (ExAC) databases 2 and is predicted to cause loss of normal protein function through protein truncation or nonsense-mediated decay. This variant is located in that portion of the TTN gene which encodes the A-band of the titin protein and, overall, our analysis suggest that it is pathogenic.
Her father agreed to directed testing for the TTN gene and results show he is a carrier of the c.100704C > A variant. This indicates that the mutations in the index case are in trans confirming that she is a compound heterozygote inheriting the c.100704C > A mutation from the father and the c.107889del mutation from her mother. Her brother with the history of cardiomyopathy, also underwent directed testing which showed that he inherited the c.100704C > A paternal allele (Fig. 2).
Figure 2.
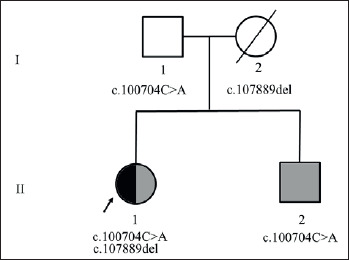
Pedigree showing the inheritance of the titin variants in this family. Dark fill indicates muscle phenotype and grey fill indicates cardiac phenotype. Index patient (arrow) inherited two variants one from each parent and shows both muscle and cardiac phenotype.
Discussion
The first description of a titinopathy resulting from mutations in the titin gene was published in 2002 by Hackman et al. 7. This was a study of Finnish patients with tibial muscular dystrophy and there was evidence for a founder effect in this population. In a follow up study in 2008 8 by this same group, the first report of the c.107889del is described in two unrelated Spanish families and designated as g.29337delA. This report described adult patients developing muscular dystrophy in an autosomal dominant fashion. In 2013, this mutation was again described but following an autosomal recessive pattern of inheritance in a 7 year old boy who initially developed symptoms at age 3 9. The ethnic origin of this patient’s mother who carried the c.107889del mutation is not indicated. Interestingly, in follow up study of the patients involved in the prior publication by Hackman et al. 8, a second mutation is described confirming an autosomal recessive mode of transmission 10. Evila et al. published an additional paper in 2016 describing another Spanish family carrying this mutation 11. In 2020, Saverese et al. reported six individuals with this mutation one of whom is from France and the remaining five are from Spain 12. Our genetic analysis demonstrates the presence of this c.107889del mutation in the TTN gene in our family which is of Peruvian descent, a population that has an admixture of genes from Spain. This strongly suggests a founder effect for the c.107889del mutation in this admixed population that may have originated in Spain. Alternatively, this mutation could be the result of a separate mutational event at this site. The second mutation, c.100704C > A in exon 358 of TTN gene is novel and following the ACMG/AMP guidelines 13 the variant is pathogenic (or probably pathogenic). These results, in combination with the neurological examination, EMG testing and muscle biopsy provide strong evidence that the index patient has a titinopathy.
Our patient can be compared to others carrying biallelic protein truncating variants described in recent report by Savarese et al. 1. In this report, analysis of five patients indicates onset in the thirties with distal lower limb weakness and walking difficulties in three of them. Our patient shows proximal and distal lower extremity weakness which was described in the two of the patients with biallelic truncating mutations but unlike our patient, their onset was in infancy and childhood. The presence of the c.107889del causes the production of a ‘quasi-normal’ protein, resulting in an adult-onset myopathy. The muscle biopsy of our patient demonstrates fiber type disproportion. This is a common pathologic feature of not only those patients with biallelic truncation mutations but also those carrying missense and monoallelic truncating mutations 1.
The role of truncating mutations in the etiology of cardiomyopathy was initially reported in 2012 based upon analysis of 312 patients with dilated cardiomyopathy 14. More recently, a large population study supports the role of truncating mutations in patients affecting heart function 15. In four patients with biallelic truncating mutations described by Savarese et al., the presence of a dilated cardiomyopathy was noted in one and some abnormality of cardiac function in two others 1. One patient had no evidence of cardiac involvement indicating the cardiac abnormalities are an inconsistent feature of those with titinopathy. In our family, it is interesting to note that the index patient and her brother carrying the c.100704C > A truncating mutation both developed a cardiomyopathy. Their father, in his eighties, who passed this mutation to both siblings remains without evidence of a cardiomyopathy. It appears that this mutation may represent a susceptibility gene for the development of cardiomyopathy. It is important to note that the index patient did not develop the cardiomyopathy until the hemodynamic stress brought on by her second pregnancy. In her brother, it is likely that the viral illness the patient suffered contributed to the development of his cardiomyopathy. There is experimental evidence of studies in rats carrying a single truncating TTN mutations on the impact upon heart physiology during cardiac stress 15. These experiments provide support that these mutations contribute to a susceptibility to the development of cardiomyopathy which manifests after cardiac stress. In this family, the c.100704C > A mutation encodes the A-band region of the titin protein. It has been reported that mutations associated with dilated cardiomyopathy were overrepresented in the titin A-band and were absent from the Z-disc and M-band regions of titin 14. The authors concluded that TTN truncating mutations are the most common known genetic cause of dilated cardiomyopathy, occurring in approximately 25% of familial cardiomyopathy cases and 18% of sporadic cases 15,16. We hypothesize that in this family, the c.100704C > A represents a susceptibility variant that requires the presence of an additional cardiac stressor for the development of cardiomyopathy.
Our genetic analysis of this family strongly suggests the possibility of a founder effect of the c.107889del mutation originating in the Iberian Peninsula and spreading to South America. In addition, our study reports a novel truncating mutation, c.100704C > A, that expands the spectrum of mutations that can cause a titinopathy. Furthermore, we provide evidence that this mutation may confer carrier susceptibility to the development of dilated cardiomyopathy. Finally, our report confirms that in individuals with a titan related cardiomyopathy, the clinician must monitor such patients for the development of myopathic symptoms prompting further genetic analysis for a second TTN mutation.
Figures and tables
References
- 1.Savarese M, Maggi L, Vihola A, et al. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol 2018;75:557-565. https://doi.org/10.1001/jamaneurol.2017.4899 10.1001/jamaneurol.2017.4899 Erratum in: JAMA Neurol 2018;75:1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434-443. https://doi.org/10.1038/s41586-020-2308-7 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248-249. https://doi.org/10.1038/nmeth0410-248 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073-1081. https://doi.org/10.1038/nprot.2009.86 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 5.Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361-362. https://doi.org/10.1038/nmeth.2890 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 6.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68-74. https://doi.org/10.1038/nature15393 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002;71:492-500. https://doi.org/10.1086/342380 10.1086/342380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 2008;18:922-928. https://doi.org/10.1016/j.nmd.2008.07.010 10.1016/j.nmd.2008.07.010 [DOI] [PubMed] [Google Scholar]
- 9.Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 2013;81:1205-1214. https://doi.org/10.1212/WNL.0b013e3182a6ca62 10.1212/WNL.0b013e3182a6ca62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evilä A, Vihola A, Sarparanta J, et al. Atypical phenotypes in titinopathies explained by second titin mutations. Ann Neurol 2014;75:230-240. https://doi.org/10.1002/ana.24102 10.1002/ana.24102 [DOI] [PubMed] [Google Scholar]
- 11.Evilä A, Palmio J, Vihola A, et al. Targeted next-generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol Neurobiol 2017;54:7212-7223. https://doi.org/10.1007/s12035-016-0242-3 10.1007/s12035-016-0242-3 [DOI] [PubMed] [Google Scholar]
- 12.Savarese M, Vihola A, Oates EC, et al. Genotype-phenotype correlations in recessive titinopathies. Genet Med 2020;22:2029-2040 (2020). https://doi.org/10.1038/s41436-020-0914-2 10.1038/s41436-020-0914-2 [DOI] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-423. https://doi.org/10.1038/gim.2015.30 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619-628. https://doi.org/10.1056/NEJMoa1110186 10.1056/NEJMoa1110186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 2017;49:46-53. https://doi.org/10.1038/ng.3719 10.1038/ng.3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270ra6. https://doi.org/10.1126/scitranslmed.3010134 10.1126/scitranslmed.3010134 [DOI] [PMC free article] [PubMed] [Google Scholar]