

Abstract
Metastasis is the main cause of deaths related to solid cancers. Active transcriptional programmes are known to regulate the metastatic cascade but the molecular determinants of metastatic colonization remain elusive. Using an inducible piggyBac (PB) transposon mutagenesis screen, we have shown that overexpression of the transcription factor nuclear factor IB (NFIB) alone is sufficient to enhance primary mammary tumour growth and lung metastatic colonization. Mechanistically and functionally, NFIB directly increases expression of the oxidoreductase ERO1A, which enhances HIF1α‐VEGFA‐mediated angiogenesis and colonization, the last and fatal step of the metastatic cascade. NFIB is thus clinically relevant: it is preferentially expressed in the poor‐prognostic group of basal‐like breast cancers, and high expression of the NFIB/ERO1A/VEGFA pathway correlates with reduced breast cancer patient survival.
Keywords: Breast cancer, ERO1A, metastasis, NFIB, VEGFA
Subject Categories: Cancer, Signal Transduction, Vascular Biology & Angiogenesis
Transcriptional factor nuclear factor IB (NFIB) is sufficient to enhance lung metastatic colonization via enhanced angiogenesis, thus revealing a targetable network that promotes breast cancer colonization.
The paper explained.
Problem
Metastasis is the main cause of solid cancer‐related death and is a major clinical challenge in breast cancer. Although many studies have thoroughly characterized and classified breast tumours, the molecular determinants of metastatic colonization remain elusive. Their delineation and functional validation are urgently needed to improve our understanding of this currently incurable disease.
Results
We engineered a non‐metastatic cell line with the doxycycline (dox)‐inducible piggyBac (PB) transposon mutagenesis system and performed an unbiased in vivo PB‐mutagenesis genetic screen to identify drivers of metastatic colonization. We have shown that the transcription factor nuclear factor IB (NFIB) is necessary and alone sufficient for breast cancer metastasis. NFIB is upregulated in mammary tumourspheres and metastases. Transcriptional profiling of tumours and tumourspheres derived from highly metastatic cell lines and NFIB ChIP experiments showed that the lethal effect of NFIB is mediated by increased expression of the oxidoreductase ERO1A. Mechanistically and functionally, downregulation of ERO1A in NFIB‐overexpressing models decreased ROS levels and HIF1α‐VEGFA‐mediated angiogenesis, reduced metastases and prolonged overall survival of the animals. Furthermore, NFIB/ERO1A/VEGFA co‐expression correlates with the metastatic potential in triple‐negative breast cancer (TNBC) patient‐derived xenograft (PDX) models.
Impact
This study provides new molecular insights into the determinants of metastatic colonization. Our work not only highlights the power of genetic screening to identify functionally relevant metastatic networks, but also describes the mechanism by which the transcriptional factor NFIB mediates metastatic colonization. Thus, we have revealed a targetable network that influences colonization, the last and fatal step of the metastatic cascade.
Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of death in female cancer patients worldwide (Fahad Ullah, 2019). Metastasis remains the primary cause of solid‐cancer‐evoked mortality (Gupta & Massagué, 2006). The multistep process of metastasis includes local tumour cell invasion, entry into the vasculature (intravasation), exit from the circulation into the parenchyma of distant organs (extravasation), and colonization. Colonization, which results in the development of clinically manifesting metastasis and fatal disease, is dependent upon resistance of the disseminated tumour cells (DTCs) to immune and host tissue defences, survival in a foreign environment and tumour initiation capacity (Pantel & Brakenhoff, 2004; Massagué & Obenauf, 2016). However, the molecular determinants of colonization remain elusive.
Numerous oncogenic mutations and other genomic alterations co‐occur often in cancer cells and result in tumour heterogeneity, which impinges on the clinical outcome of the disease (Kennecke et al, 2010; Yates & Campbell, 2012; Koren & Bentires‐Alj, 2015). The dynamic evolution of the cancer genome is influenced by the generation of additional mutations and selective forces acting on cancer clones (Kreso & Dick, 2014). Next‐generation sequencing of DNA from clinical specimens has provided insights into breast cancer genetics and contributed to identifying potential drivers of tumour progression (Yates et al, 2015; Nik‐Zainal et al, 2016; Pereira et al, 2016; Robinson et al, 2017; Angus et al, 2019; Bertucci et al 2019; De Mattos‐Arruda et al, 2019). Mechanistic elucidation of the effects of co‐occurring genomic alterations and their functional validation is required to define the causality between these events and their contribution to specific steps of tumour progression to overt metastases.
Activating mutations of PIK3CA, which encodes for the p110α catalytic subunit of phosphoinositide 3‐kinase (PI3K), are among the most frequent alterations in human breast cancer and lead to an hyperactivated PI3K pathway signalling (Yuan & Cantley, 2008; Zhao & Vogt, 2008; Miller, 2012). We and others have shown that inducible expression of the PIK3CAH1047R mutation evokes heterogeneous mammary tumours in mice (Meyer et al, 2013; Koren & Bentires‐Alj, 2013; Koren et al, 2015), which indicates a causative effect of PIK3CA mutations in mammary tumorigenesis. In contrast, metastases were not found in mice with PIK3CA H1047R ‐derived tumours (Koren & Bentires‐Alj, 2013; Koren et al, 2015), which thus provides a model system to identify collaborating gain‐ or loss‐of‐function genomic alterations that contribute to metastasis.
Transposon insertional mutagenesis is a powerful tool in mice for discovering genes related to cancer (Ding et al, 2005; Dupuy et al, 2005; Rad et al, 2010; Dupuy, 2010). Indeed, the fact that transposons are mobile within the genome and alter gene activity in cells that express the transposase makes these systems ideal for whole‐genome screens. The piggyBac (PB) transposon was engineered to be active in mammalian cells (Ding et al, 2005) and it allows functional identification not only of oncogenes but also of tumour suppressor genes, depending on the site of insertion and the orientation of the transposon (Rad et al, 2015; Takeda et al, 2017; De La Rosa et al, 2017; Weber et al, 2019).
We performed an unbiased PB transposon insertional mutagenesis screen in cancer cells with an activating PIK3CAH1047R mutation to identify possible synergistic mechanisms of metastatic colonization. Mechanistically and functionally, we demonstrate that NFIB enhances the expression of the oxidoreductase ERO1A and VEGFA, promotes metastatic colonization and shortens overall survival of mice.
Results
Transposon mutagenesis confers metastatic potential to PIK3CA H1047R mammary cells
To identify cancer genes relevant to metastatic progression, we engineered a murine non‐metastatic mammary cancer cell line derived from PIK3CA H1047R mutant tumours (LB‐mHR1, here cited as HR1) with a doxycycline (dox)‐inducible piggyBac (PB) transposon system (HR1. PB) (Ivics et al, 2009; Rad et al, 2010) (Fig 1A). HR1 cells lacking the transposon served as control (HR1. Ctrl). To perform an unbiased in vivo PB transposon mutagenesis screen (Fig 1B), we injected HR1. PB and HR1. Ctrl orthotopically into 19 and 14 NOD/SCID mice, respectively, and monitored the animals for tumour growth and metastasis. PB transposon mutagenesis in HR1. PB cells increased tumour incidence (Fig EV1A) and promoted metastasis (Fig EV1B). Metastases were observed in half of the mice injected with HR1. PB cells of which nine lung metastases (LM)‐derived cell lines were isolated. To phenotypically characterize the LM cell lines in vivo, we injected them into mammary fat pads of NOD/SCID mice. The LM1, LM8 and LM9 lines exhibited a particularly aggressive behaviour characterized by accelerated tumour growth and metastasis (Fig EV1C–G ). These results indicate that selected transposon integrations can confer aggressive metastatic behaviour on cells.
Figure 1. Transposon mutagenesis screen identifies Nfib as a candidate metastatic inducer in breast cancer.
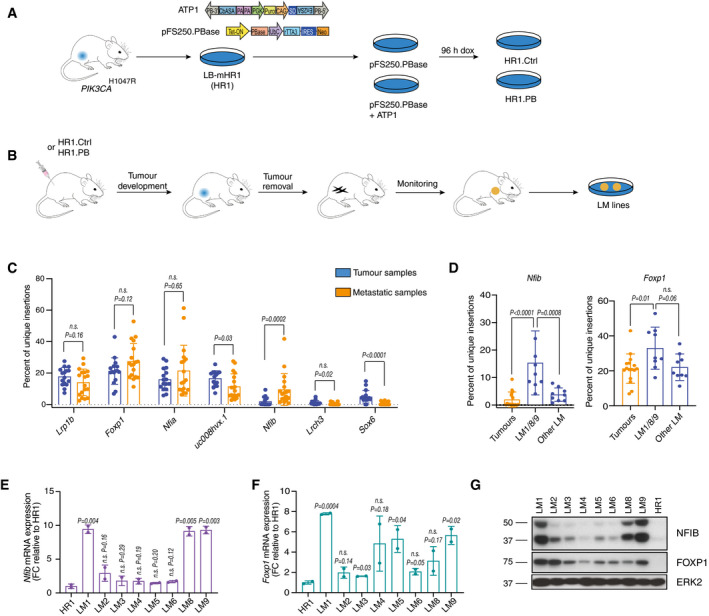
- Screen design: generation of mammary cancer cell lines LB‐mHR1 (HR1) from PIK3CA H1047R mutant transgenic animals and transfection with the piggyBac (PB) system (HR1 Ctrl and HR1. PB cells) (Ivics et al, 2009). PB transposon (ATP1‐Puro) and transposase (pFS250. PBase) plasmid design. PB‐3’/5’, piggyBac inverted terminal repeats; CβASA, Carp β‐actin splice acceptor; En2SA, Engrailed‐2 exon‐2 splice acceptor; SD, Foxf2 exon‐1 splice donor; pA, bidirectional SV40 polyadenylation signal; PGK, mouse phosphoglycerate kinase 1 promoter; Puro, puromycin resistance; CAG, cytomegalovirus enhancer and chicken beta‐actin promoter; Tet‐ON, tetracycline‐responsive element‐tight promoter; PBase, PB transposase; UbC, Ubiquitin C promoter; rTTA3, reverse tetracycline transactivator 3; IRES, internal ribosomal entry site; Neo, Neomycin/G418 selection marker. The cell pools were treated with doxycycline (1 μg/ml) for 96 h in vitro.
- In vivo screen design: generation of lung metastatic mammary cancer cell lines (LM) from HR1. PB cell lines after orthotopic injections of HR1. PB or HR1. Ctrl.
- Bar graph showing the percentage of unique insertions in the top 7 genes normalized to the total number of insertions in a given sample. Genes are annotated with a gene symbol when available otherwise with UCSC ID transcript names. Data are from all the sequenced samples (tumour samples: 16 primary tumours; metastatic samples: 3 lung micro‐metastases, 6 lung macro‐metastases and 9 LM cell lines). Means ± s.d., two‐tailed Mann–Whitney U‐test, n.s. = not significant.
- Bar graphs showing the percentage of unique insertions in Nfib and Foxp1 in tumours, LM1, LM8, LM9 and other LM cell lines normalized to the total number of insertions in a given sample. Dots represent individual samples (Tumours n = 16, LM1/8/9 n = 9, Other LM n = 9), means ± s.d., two‐tailed Mann–Whitney U‐test, n.s. = not significant.
- Bar graph representing mean Nfib mRNA expression normalized to HR1 cells. n = 2 biological replicates and n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test, FC = fold change.
- Bar graph representing mean Foxp1 mRNA expression normalized to HR1 cells. n = 2 biological replicates and n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test, FC = fold change.
- Immunoblot analysis of LM and HR1 cell lines showing NFIB and FOXP1 protein levels. ERK2 served as a loading control.
Source data are available online for this figure.
Figure EV1. In vivo unbiased transposon screen in the non‐metastatic PIK3CA H1047R mammary tumour‐derived cells allows the isolation of aggressive lung metastatic lines.
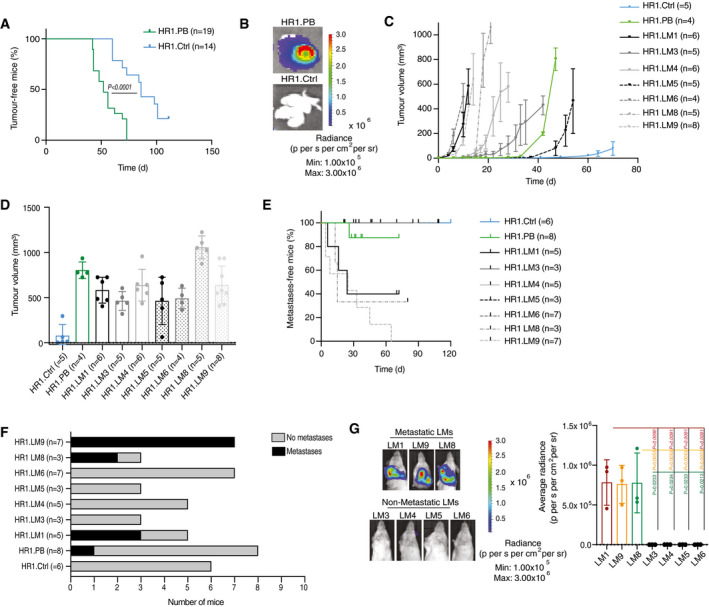
- Kaplan–Meier plot depicting tumour incidence in NOD/SCID animals inoculated in the mammary fat pad with 106 cells of HR1. Ctrl (n = 14) or HR1. PB cells (n = 19), two‐tailed log‐rank test.
- Representative bioluminescence acquisition of lungs from mice injected with HR1. PB or HR1. Ctrl cells as in A.
- Graph representing the kinetics of HR1. Ctrl (n = 5), HR1. PB (n = 4), HR1. LM1 (n = 6), HR1. LM3 (n = 5), HR1. LM4 (n = 6), HR1. LM5 (n = 5), HR1. LM6 (n = 4), and HR1. LM9 (n = 8) tumour growth upon orthotopic injection of 250 × 103 cells in NOD/SCID mice, means ± s.d.
- Bar graph showing tumour volumes in mice injected with HR1. Ctrl (n = 5), HR1. PB (n = 4), HR1. LM1 (n = 6), HR1. LM3 (n = 5), HR1. LM4 (n = 6), HR1. LM5 (n = 5), HR1. LM6 (n = 4), or HR1. LM9 (n = 8), means ± s.d.
- Kaplan–Meier plot showing metastasis onset after tumour removal in mice injected with HR1. Ctrl (n = 6), HR1. PB (n = 8), HR1. LM1 (n = 5), HR1. LM3 (n = 3), HR1. LM4 (n = 5), HR1. LM5 (n = 3), HR1. LM6 (n = 7), or HR1. LM9 (n = 7).
- Bar graph showing metastatic incidence after tumour removal in mice injected with HR1. Ctrl (n = 6), HR1. PB (n = 8), HR1. LM1 (n = 5), HR1. LM3 (n = 3), HR1. LM4 (n = 5), HR1. LM5 (n = 3), HR1. LM6 (n = 7), or HR1. LM9 (n = 7).
- Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases after primary tumour removal in mice injected with metastatic or non‐metastatic LM cells. n = 3 mice, means ± s.d., two‐tailed Student’s t‐test.
Source data are available online for this figure.
Insertional PB‐mutagenesis screening identifies Nfib as a metastatic gene in mammary cancer
To investigate induced alterations that enabled cancer cells to metastasize, we mapped transposon integration sites in 16 primary tumours and 18 metastatic samples, searching for insertions enriched at metastatic sites compared with tumours. We used splinkerette PCR followed by next‐generation sequencing from both transposon arms (Friedrich et al, 2017) and identified 1,252 insertions in 1,080 genes (Dataset EV1). To evaluate the relative abundance of each integration site and minimize PCR‐induced amplification effects, we devised a diversity count measure for each integration site within each sample (percentage of unique insertions) that quantifies the number of unique sheared ends rather than all sequencing counts. To select metastatic genes, we searched for genes with more unique insertion sites (based on their diversity counts) in metastatic samples than in tumour samples (Fig 1C). Depending on the transposon integration site, insertions may lead to gene activation or inactivation. The most frequently altered genes in the primary tumours and/or metastatic samples were Lrp1b, Nfia, Foxp1, Nfib, Lrch3 and Sox6. Some of these unique insertions (e.g. Foxp1) were found in both metastatic and non‐metastatic samples at high frequency, suggesting that they are unlikely to be specifically required for metastasis. Notably, unique insertions in Nfib were particularly enriched in a subset of highly metastatic samples (LM1/8/9) compared with the other hits (Fig 1D). The same‐strand insertions upstream of both Nfib and Foxp1 transcription start sites suggest them to be candidate oncogenes. Consistent with the pattern of transposon integration, analysis of Nfib/Foxp1 mRNA expression and NFIB/FOXP1 protein abundance in the LM cell lines revealed enhanced levels of FOXP1 in all LM cell lines but selective elevation of NFIB in LM lines with high metastatic potential (Fig 1E–G). These data suggest the importance of Nfib in mammary cancer metastasis.
Depletion of Nfib delays mammary cancer growth and abrogates metastases
To assess the contribution of Nfib and Foxp1 to tumour growth and metastatic progression, we produced knockout (KO) cells for the two genes using CRISPR‐Cas 9 technology in the highly metastatic LM1 and LM9 lines and generated oligoclonal pools of cells (Appendix Fig S1A and B). Considering that NFIB has been implicated in skin stem cell maintenance (Chang et al, 2013) and tumour growth in other cancer types (Brayer et al, 2016; Semenova et al, 2016; Wu et al, 2016; Fane et al, 2017; Wu et al, 2018), we addressed its effects on mammary tumours. Nfib KO in LM1 and LM9 delayed tumour growth (Fig 2A) and markedly decreased the frequency of tumour‐initiating cells (TICs) (Fig EV2A and B). Consistently, Nfib KO also reduced tumoursphere formation (Fig EV2C and D).
Figure 2. Nfib ablation delays mammary tumour formation and abrogates metastasis.
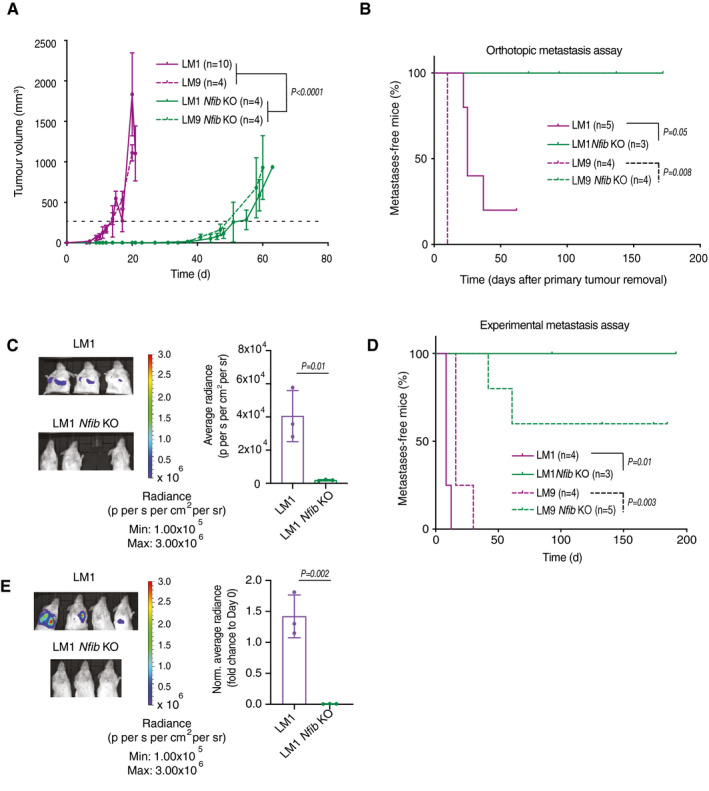
- The kinetics of LM1 (n = 10), LM9 (n = 4), LM1 Nfib KO (n = 4), and LM9 Nfib KO (n = 4) tumour growth upon orthotopic injection of 250 × 103 cells into NOD/SCID mice. Curves show means of tumour volume ± s.d., two‐way ANOVA group analysis of the times to reach 250 mm3 (dashed line).
- Kaplan–Meier plot depicting metastatic onset after tumour removal in mice injected orthotopically with LM1 (n = 5), LM9 (n = 4), LM1 Nfib KO (n = 3) or LM9 Nfib KO (n = 4) cells, two‐tailed log‐rank test.
- Nfib knockout abrogates metastasis in the orthotopic LM1 model. Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases at 25 (LM1) and 40 (KO) days after primary tumour removal; n = 3 mice, means ± s.d., two‐tailed Student’s t‐test.
- Nfib knockout impairs experimental metastases formation in the LM models. Kaplan–Meier plot showing metastatic incidence of animals inoculated i.v. with LM1 (n = 4), LM9 (n = 4), LM1 Nfib KO (n = 3), or LM9 Nfib KO (n = 5) cells, two‐tailed log‐rank test.
- Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases 16 days after i.v. injection of LM1 or LM1 Nfib KO cells, n = 3 mice, means ± s.d., two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure EV2. Nfib overexpression accelerates tumour growth.
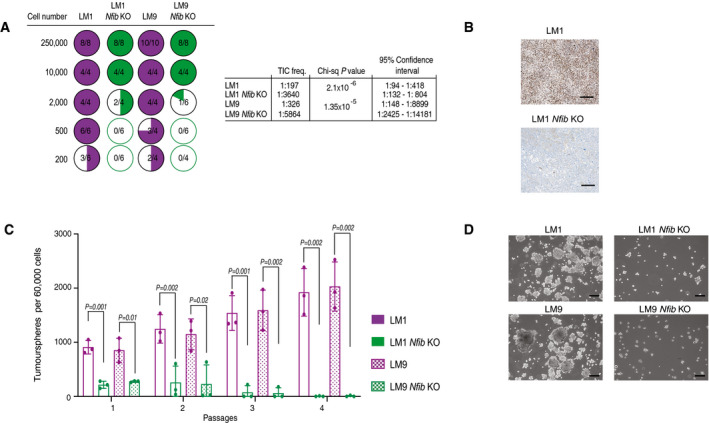
- Limiting dilution assay: (left panel) quantification of tumour initiation upon orthotopic injection of mice with LM1, LM9, LM1 Nfib KO or LM9 Nfib KO cells. (right panel) Tumour‐initiating cell (TIC) frequencies and 95% confidence intervals were calculated using ELDA (Hu & Smyth, 2009) as described in the experimental procedures.
- Representative images of immunostaining for NFIB of tumours from mice injected with LM or LM Nfib KO cancer cells. Scale bar 100 μm.
- Bar graph showing tumoursphere numbers over four passages of LM1, LM9, LM1 Nfib KO, and LM9 Nfib KO, n = 3 biological replicates and n = 3 technical replicates, means ± s.d., two‐tailed Student’s t‐test.
- Representative images of tumourspheres of LM1, LM9, LM1 Nfib KO, and LM9 Nfib KO cancer cell lines at the first passage. Scale bar 100 μm.
Source data are available online for this figure.
Deletion of Foxp1 increased mammary tumour latency (Appendix Fig S1C) and slightly decreased metastasis compared to the wild type (WT) (Appendix Fig S1D). In contrast, when LM1 and LM9 Nfib KO cells were injected orthotopically and the primary tumour removed, we observed a dramatic abrogation of lung metastasis (orthotopic metastasis assay) compared with controls (Fig 2B and C), providing a rationale to focus on the effect of Nfib in breast cancer metastasis. The data suggest that the Nfib overexpression observed in our PB screen and in metastatic lines may also enhance metastatic colonization. To test this possibility, we injected Nfib KO or control cells intravenously (i.v., experimental metastasis assay) and found that Nfib KO completely or dramatically impaired metastatic colonization in the LM1 and LM9 lines, respectively (Fig 2D and E). Finally, ablation of Nfib using CRISPR‐Cas 9 technology also increased tumour latency, decreased the number of circulating tumour cells and prolonged overall survival of BALB/c mice orthotopically injected with the highly metastatic 4T1 mammary cancer cells (Appendix Fig 2A–D). The results of these functional assays demonstrate the importance of NFIB in mammary cancer metastatic colonization.
NFIB is sufficient to induce metastasis
We next asked whether NFIB activity is alone sufficient for inducing metastasis. Using Cas9‐Activators with Synergistic Activation Mediators (SAM) (Konermann et al, 2015), we overexpressed Nfib from its endogenous promoter in the non‐metastatic parental HR1 cell line (Fig EV3A). Two different guides were designed and tested for their ability to increase Nfib expression and g4Nfib was selected for our experiments. Nfib overexpression enhanced proliferation, tumoursphere formation, migration and invasion in vitro (Appendix Fig S3A–D). Furthermore, overexpression of Nfib accelerated tumour onset (Fig EV3B), metastasis formation after orthotopic injection (Fig 3A and B) and metastatic colonization after tail‐vein injection (Fig 3C and D). Overexpression of NFIB in SUM159PT, a human TNBC cell line with low metastatic potential (Fig EV3C), similarly decreased tumour latency (Fig EV3D) and increased metastasis, both in the orthotopic (Fig EV3E and F) and the experimental metastasis assays (Fig 3E and F). Thus, NFIB appears to be sufficient to induce metastasis when overexpressed in non‐ (HR1) and low‐ (SUM159PT) metastatic mammary cancer lines.
Figure EV3. Endogenous Nfib/NFIB overexpression in HR1 and SUM159PT cell lines decreases tumour latency and enhances metastasis.
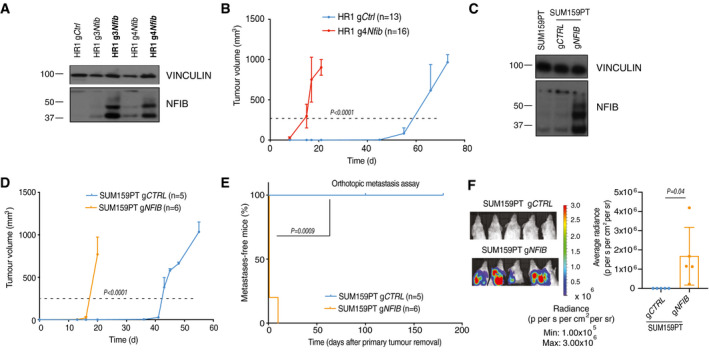
- Immunoblot showing proteins levels of NFIB in HR1 cells overexpressing Nfib via the CRISPR‐Cas 9 SAM system. In bold: cell lines after two months of culture in vitro. HR1 g4Nfib cells were selected for further studies. VINCULIN served as a loading control.
- Graph representing the kinetics of HR1 gCtrl (n = 13) and HR1 g4Nfib (n = 16) tumour growth upon orthotopic injection of 250 × 103 cells in NOD/SCID mice. Curves show means of tumour volume ± s.d., two‐tailed Student’s t‐test on the times to reach 250 mm3 (dashed line).
- Immunoblots showing protein levels of NFIB in SUM159PT cells overexpressing NFIB via the CRISPR‐Cas 9 SAM system. VINCULIN served as a loading control.
- Graph representing the kinetics of SUM159PT gCTRL (n = 5) and gNFIB (n = 6) tumour growth upon orthotopic injection of 250 x 103 cells into NSG mice. Curves show means of tumour volume ± s.d., two‐tailed Student’s t‐test on the times to reach 250 mm3 (dashed line).
- Kaplan–Meier plot depicting metastasis onset after tumour removal from mice injected orthotopically with SUM159PT gCTRL (n = 5) or gNFIB (n = 6), two‐tailed log‐rank test.
- Representative bioluminescence images (left panel) and quantification (right panel) of lung metastases at 30 (SUM159PT gCTRL) and 3 (SUM159PT gNFIB) days after primary tumour removal. n = 5 mice, means ± s.d., two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure 3. Nfib is sufficient to induce metastasis.
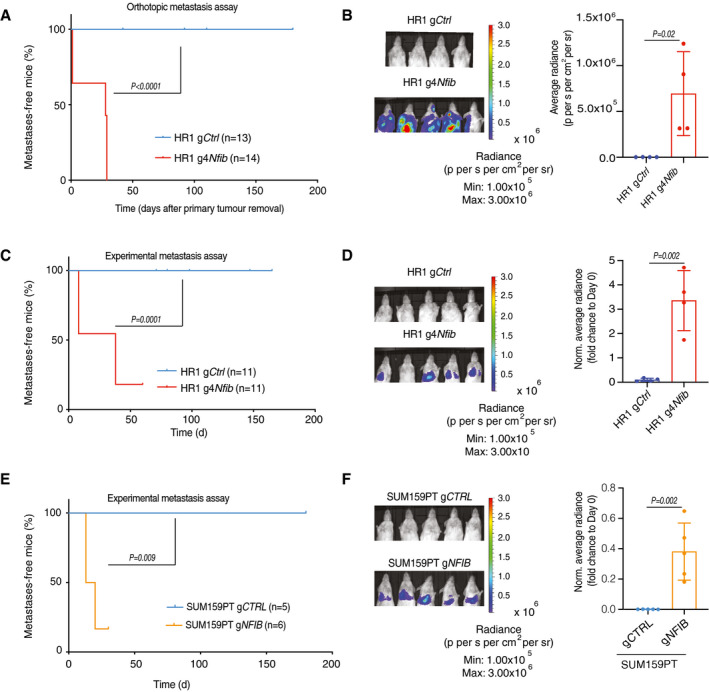
- Kaplan–Meier plot depicting metastasis onset after tumour removal in mice injected orthotopically with HR1 gCtrl (n = 13) or HR1 g4Nfib (n = 14), two‐tailed log‐rank test.
- Nfib overexpression in HR1 parental cells enhances metastasis in the orthotopic model. Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases at day 20 (HR1 gCtrl) and day 2 (HR1 g4Nfib) after primary tumour removal. n = 4 mice, means ± s.d., two‐tailed Student’s t‐test.
- Nfib overexpression in the HR1 parental cells enhances experimental metastases. Kaplan–Meier showing metastatic incidence of animals inoculated i.v. with HR1 gCtrl (n = 11) or HR1 g4Nfib (n = 11) cells, two‐tailed log‐rank test.
- Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases 16 days after i.v. injection of HR1 gCtrl or HR1 g4Nfib cells. n = 4, means ± s.d., two tailed Student’s t‐test.
- NFIB overexpression enhances experimental metastases formation in the SUM159PT model. Kaplan–Meier survival analysis of animals inoculated i.v. with SUM159PT gCTRL (n = 5) or SUM159PT gNFIB (n = 6) cells, two‐tailed log‐rank test.
- Representative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases 20 days after i.v. injection of SUM159PT gCTRL or SUM159PT gNFIB (n = 5), means ± s.d., two‐tailed Student’s t‐test.
Source data are available online for this figure.
NFIB induces metastatic colonization via increased Ero1l/ERO1A expression
To determine the molecular mechanism underlying increased metastatic colonization driven by NFIB, we sequenced total mRNA of tumourspheres and primary tumours derived from Nfib high‐expression models (LM1 and LM9) and Nfib low‐expression models (HR1, LM1 Nfib KO and LM9 Nfib KO). By comparing the 200 most differentially expressed genes with ChIP‐seq data of putative transcriptional targets of Nfib in the mammary gland (Shin et al, 2016) and epithelial‐melanocyte stem cells (Chang et al 2013; Dataset [Link], [Link], [Link]), we found endoplasmic reticulum disulphide oxidase 1 like (Ero1l) to be the single common gene (Fig 4A and B). Chromatin immunoprecipitation (ChIP) qPCR for NFIB in the LM1/LM1 NfibKO, LM9/LM9 NfibKO, 4T1 WT, 4T1 Nfib KO, HR1gCtrl and HR1g4Nfib cell lines, revealed increased NFIB binding to the promoter of Ero1l (Shin et al, 2016) (Fig 4C), indicating that ERO1L is a direct transcriptional target of NFIB. Indeed, abundance of the mouse and human ERO1L/A protein was increased in Nfib/NFIB‐overexpressing cells (Appendix Fig S4A). Consistently, Nfib KO decreased Ero1l mRNA levels (Appendix Fig S4B–D). ERO1L/ERO1A is involved in the production of hydrogen peroxidase (H2O2) (Zito, 2015) and is a poor‐prognostic factor in cancer (Takei et al, 2017; Zhou et al, 2017; Kim et al, 2018; Yang et al, 2018).
Figure 4. The NFIB‐ERO1A axis enhances lung metastatic colonization.
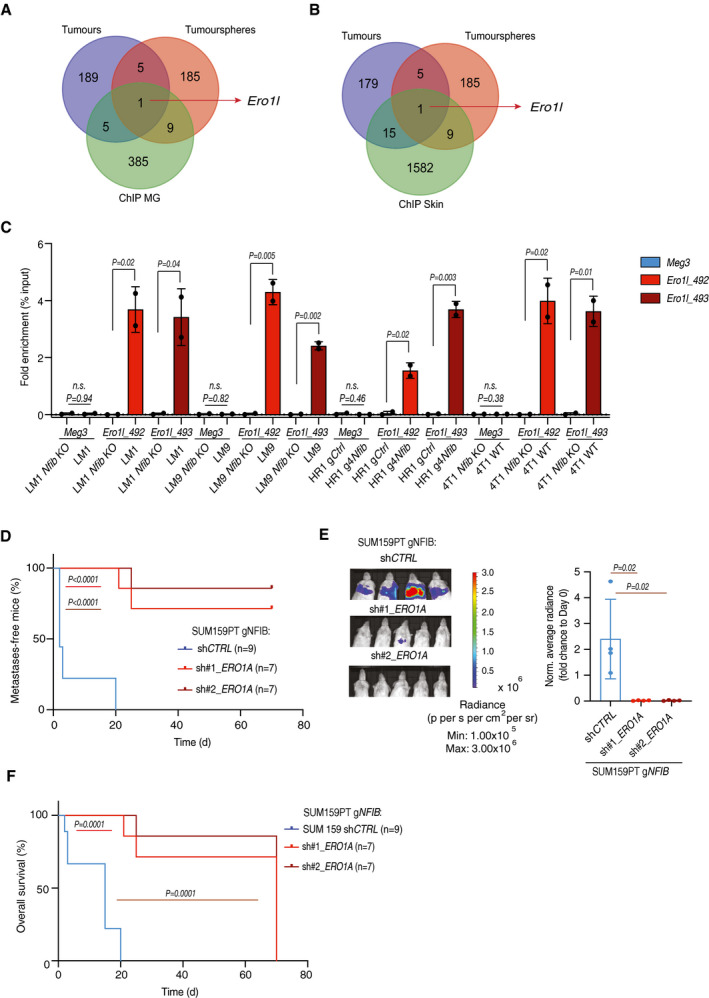
-
A, BIntersection of the top 200 differentially expressed genes (sorted by statistical significance) in tumourspheres and tumours (Nfib high‐expression vs. the respective controls) and putative transcriptional targets of Nfib determined by ChIP‐seq in mammary gland (Shin et al, 2016) (A) and epithelial‐melanocyte stem cells (Chang et al, 2013) (B) (association rule: Basal + extension: 5,000 bp upstream, 1,000 bp downstream, 10,000 bp max extension, curated regulatory domains included).
-
CChIP was performed in all murine cell lines (Nfib high‐expression and the respective Nfib low‐expression or KO controls) against Nfib followed by qPCR with primers specific for Ero1l promoter region proximal to transcription starting site (Ero1l_492 and Ero1l_493). Meg3 was used as control. Graph shows qPCR results with % input method as indicated (Nfib high‐expression vs. the respective Nfib low‐expression or KO controls). n = 2 biological replicates and n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test, n.s. = not significant.
-
DKaplan–Meier survival analysis of NSG mice inoculated i.v. with SUM159PT gNFIB shCTRL (n = 9), SUM159PT gNFIB sh1 ERO1A (n = 7), or SUM159PT gNFIB sh2 ERO1A (n = 7) cells. Two‐tailed log‐rank test.
-
ERepresentative bioluminescence images (left panel) and bar plot quantification (right panel) of lung metastases 8 (SUM159PT gNFIB shCTRL) and 20 (SUM159PT gNFIB sh1 ERO1A or SUM159PT gNFIB sh2 ERO1A) days after i.v. injection of the respective cells. n = 4, means ± s.d., two‐tailed Student’s t‐test.
-
FKaplan–Meier survival analysis of overall survival of mice injected i.v. with SUM159PT gNFIB shCTRL (n = 10), SUM159PT gNFIB sh1 ERO1A (n = 12), or SUM159PT gNFIB sh2 ERO1A (n = 13) cells, two‐tailed log‐rank test.
Source data are available online for this figure.
We next examined whether Ero1l/ERO1A mediates NFIB effects on metastatic colonization. Notably, knockdown of ERO1A in SUM159PT gNFIB, LM1 and HR1 g4Nfib, using two doxycycline‐inducible shRNA constructs (Appendix Fig S5A) reduced metastatic colonization after tail‐vein injection and prolonged survival (Fig 4D–F and Appendix Fig S5B–E). Altogether, these findings show that Ero1l/ERO1A expression is increased in Nfib/NFIB‐overexpressing models and suggest that the NFIB‐ERO1A axis is critical for metastatic colonization in breast cancer.
ERO1A induces VEGFA expression and angiogenesis via HIF1α stabilization
We found that NFIB/Nfib‐ERO1A/Ero1l overexpressing cells produce more ROS compared to the low‐expression models (Fig 5A). ROS has been shown to stabilize HIF1α (Bell et al, 2007; Yan et al, 2010), and we found an increase in nuclear HIF1α in the NFIB/Nfib‐ERO1A/Ero1l overexpressing models which was offset by ERO1A knockdown (Fig 5B). As ERO1A and HIF1α have been shown to enhance VEGFA expression (Amelino‐Camelia et al, 1998; Tanaka et al, 2016), we assessed VEGFA mRNA and protein levels in NFIB models. VEGFA increased in cells, mammary tumours and lung metastases derived from NFIB/Nfib‐overexpressing models and decreased upon ERO1A downregulation (Fig 5C and EV4A–D, Appendix Fig S6A–C). Downregulation of Nfib in HR1 g4Nfib cells decreased Vegfa and VEGFA levels (Appendix Fig S6D–F). Consistently, rescued expression of Ero1l in LM1, LM9 and 4T1 Nfib KO cell lines restored Vegfa mRNA and VEGFA protein levels and increased metastatic colonization after tail‐vein injection (Fig EV5A–E), indicating that VEGFA expression is NFIB/ERO1A dependent. Together, the data suggest that the NFIB‐ERO1A axis enhances ROS‐evoked HIF1α stabilization and VEGFA levels.
Figure 5. Ero1l/ERO1A increases Vegfa/VEGFA expression and angiogenesis.
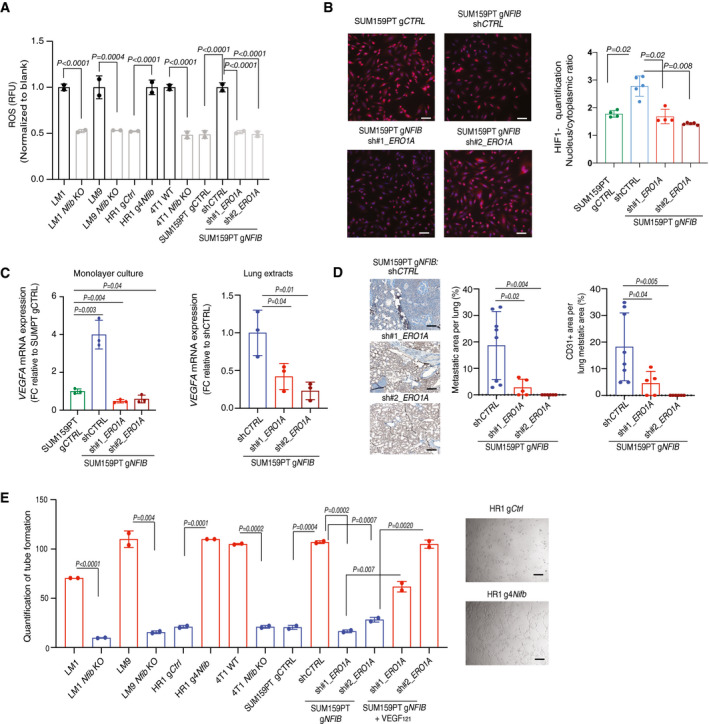
- Bar graph showing the ROS levels in the NFIB models. The fluorescence signal was normalized to the blank. n = 2 biological replicates and n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test.
- Left panel: representative images of HIF1α immunofluorescence. HIF1α was stained with Alexa‐633 (red) and nucleus with DAPI (blue). Scale bar, 100 μm. Right panel: bar graph showing quantification of HIF1α nuclear/cytoplasmatic ratio. n = 5 biological replicates, means ± s.d., two‐tailed Mann–Whitney U‐test.
- Left panel: Bar graph representing mean VEGFA mRNA expression in SUM159PT gCTRL and SUM159PT gNFIB breast cancer cell lines upon downregulation of ERO1A with two independent shRNAs (sh1 and sh2). Right panel: Bar graph representing mean VEGFA mRNA expression in lung extracts from animals inoculated i.v. with SUM159PT gNFIB shCTRL, SUM159PT sh1 ERO1A, or SUM159PT sh2 ERO1A cells. In both graphs n = 3 biological replicates and n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test, FC = fold change.
- Left panel: Representative images of CD31‐positive endothelial structures in lungs. Scale bar, 1 mm. Right panel: Bar graphs showing lung metastases quantified as percentage of metastatic area per lung area and quantification of CD31 staining in the metastatic area. Means ± s.d., n = 8 shCTRL, n = 5 per sh1 ERO1A, and n = 6 sh2 ERO1A, two‐tailed Student’s t‐test.
- Left panel: Bar graph showing average of master segments counted after incubation of HUVEC cells with conditioned medium from LM1, HR1, 4T1 (Nfib high‐expression and the respective Nfib low‐expression or KO controls) and SUM159PT gNFIB shCTRL, SUM159PT gNFIB sh1 ERO1A, SUM159PT gNFIB sh2 ERO1A cells. Human VEGF121 (0.4 ng/μl) was added to SUM159PT gNFIB sh1 and sh2 ERO1A cells, n = 2 technical replicates, means ± s.d., two‐tailed Student’s t‐test. Right panel: Representative images showing tube formation four hours after addition of conditioned medium from HR1 gCtrl or g4Nfib. Scale bar, 100 μm.
Source data are available online for this figure.
Figure EV4. The NFIB‐ERO1A‐VEGFA axis promotes lung metastatic colonization through angiogenesis.
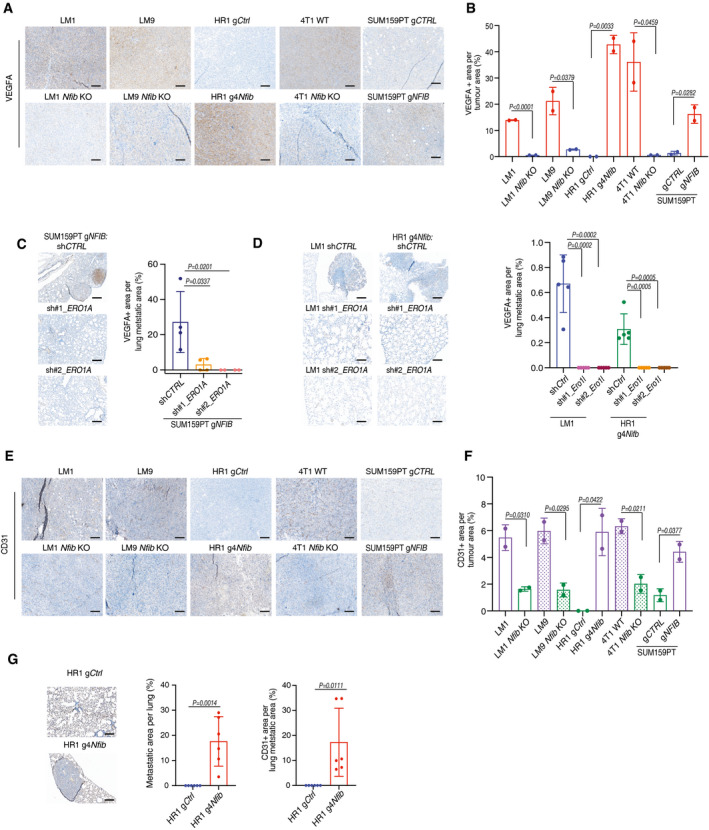
- Representative images of VEGFA‐stained tumours. Scale bar, 1 mm.
- Bar graph showing quantification of VEGFA staining in the tumours. Means ± s.d., n = 2, two‐tailed Student’s t‐test.
- Left panel: Representative images of VEGFA‐positive lung metastases. Scale bar, 1 mm. Right panel: Bar graph showing quantification of VEGFA. Means ± s.d., n = 4 SUM159PT gNFIB shCTRL, n = 4 sh1 ERO1A, and n = 4 sh2 ERO1A, two‐tailed Student’s t‐test.
- Left panel: Representative images of VEGFA‐positive lung metastases. Scale bar, 1 mm. Right panel: Bar graph showing quantification of VEGFA. Means ± s.d., n = 5 HR1g4Nfib and LM1 shCTRL, n = 5 sh1 ERO1A, and n = 5 sh2 ERO1A, two‐tailed Student’s t‐test.
- Representative image of CD31‐positive endothelial structures in tumours. Scale bar, 1 mm.
- Bar graph showing quantification of CD31 staining in tumours. Means ± s.d., n = 2, two‐tailed Student’s t‐test.
- Left panel: Representative images of CD31‐positive endothelial structures in lung metastases. Scale bar, 1 mm. Right panel: Bar graphs showing quantification of metastases as percentage of metastatic area per lung and quantification of CD31 staining in the metastatic area. Means ± s.d., n = 6 HR1 gCtrl and n = 6 HR1 g4Nfib, two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure EV5. Ero1l overexpression in Nfib KO cells restores Vegfa expression and metastasis.
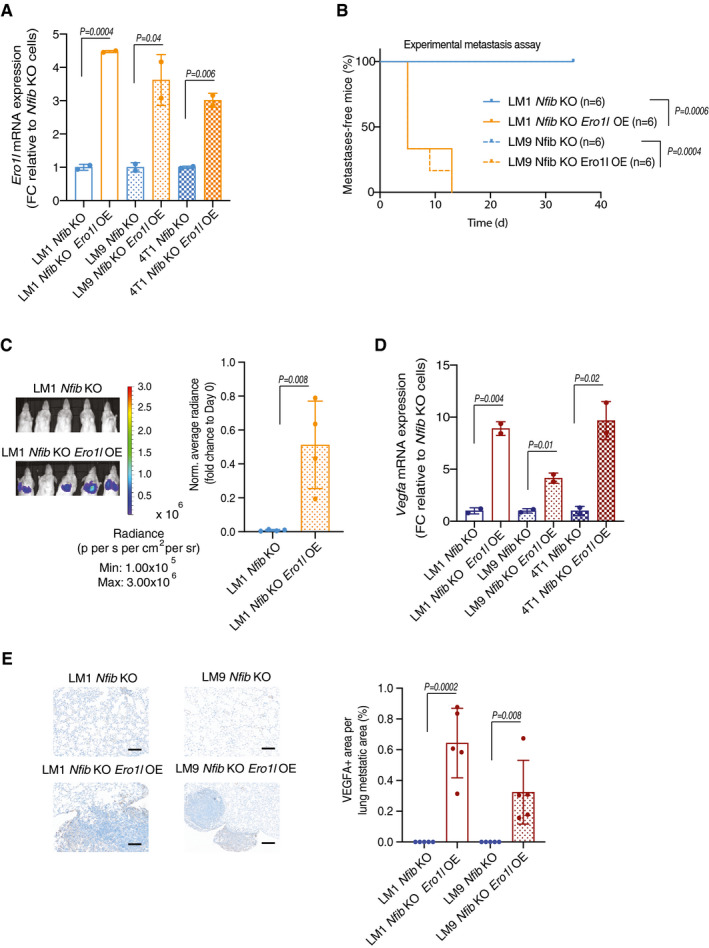
- Bar graph representing mean Ero1l mRNA expression in the LM1, LM9, 4T1 WT with respective Nfib KO cell lines and Ero1l overexpression (OE) as indicated. Means ± s.d., n = 2 biological replicates and n = 2 technical replicates, two‐tailed Student’s t‐test, FC = fold change.
- Kaplan–Meier survival analysis of NOD/SCID mice inoculated i.v. with LM1 Nfib KO (n = 6), LM1 Nfib KO Ero1l OE (n = 6), LM9 Nfib KO (n = 6) or LM9 Nfib KO Ero1l OE (n = 6) cells. Two‐tailed log‐rank test.
- Representative bioluminescence images (left panel) and quantification (right panel) of lung metastases 5 days after i.v. injection of LM1 Nfib KO (n = 4) or LM1 Nfib KO Ero1l OE (n = 4) cells, means ± s.d., two‐tailed Student’s t‐test.
- Bar graph representing mean Vegfa mRNA expression in the LM1, LM9, 4T1 WT with respective Nfib KO cell lines and Ero1l overexpression. Means ± s.d., n = 2 biological replicates and n = 2 technical replicates, two‐tailed Student’s t‐test, FC = fold change.
- Left panel: Representative images of VEGFA‐positive lung metastases. Scale bar, 1 mm. Right panel: Bar graph showing quantification of VEGFA staining in the metastatic area. Means ± s.d., n = 5 LM1/LM9 Nfib KO and LM1/LM9 Nfib KO Ero1l OE, two‐tailed Student’s t‐test.
Source data are available online for this figure.
We assessed angiogenesis by endothelial‐specific CD31 staining in sections of mouse mammary tumours and metastases from NFIB overexpression and KO models and observed higher and lower frequency of vascular structures than controls, respectively (Fig EV4E‐G). CD31‐positive blood vessels were also decreased in lung metastases upon ERO1A knockdown in SUM159PT gNFIB cells (Fig 5D and Fig EV4G). In vitro, conditioned medium from NFIB/Nfib/Ero1l/ERO1A high‐expression cell lines increased endothelial tube formation compared with low‐expression models in the matrigel‐based tube formation assay. Addition of human recombinant VEGF121 to the conditioned medium from ERO1A low‐expression models enhanced endothelial tube formation (Fig 5E). Taken together, these data indicate that NFIB is a breast cancer metastasis transcriptional regulator which, through increased expression of ERO1A and VEGFA, promotes angiogenesis at the metastatic site. This creates a permissive microenvironment for metastatic colonization.
High NFIB‐ERO1A‐VEGFA expression is associated with poor prognosis in TNBC patients
NFIB is expressed preferentially in basal‐like breast cancers and is a potential prognostic factor in TNBC human breast cancer, as observed in previous reports (Moon et al, 2011; Liu et al, 2019). NFIB overexpression discriminated between metastatic versus non‐metastatic TNBC breast cancer patient‐derived xenograft (PDX) models (Fig 6A; Appendix Fig S7A) and NFIB, ERO1A and VEGFA were co‐overexpressed in these samples (Fig 6A and B). Next, analysis of NFIB mRNA expression levels from METABRIC (Fig 6C) (Curtis et al, 2012) and TCGA (Koboldt et al, 2012) (Appendix Fig S7B) confirmed that NFIB expression is elevated in basal‐like breast cancer, according to PAM50 classification, and in iC10 by integrated cluster classification (Dawson et al, 2013; Mukherjee et al 2018). Increased NFIB gene copy numbers were observed in TNBC (Appendix Fig S7C and D). Notably, we found that high expression of NFIB and the combined signature of ERO1A and NFIB in patients with breast cancer of the basal‐like subtype were predictive of decreased distant metastasis‐free survival and/or overall survival (Fig 6D and E; Appendix Fig S7E and F).
Figure 6. NFIB‐ERO1A‐VEGFA overexpression is associated with aggressive tumours.
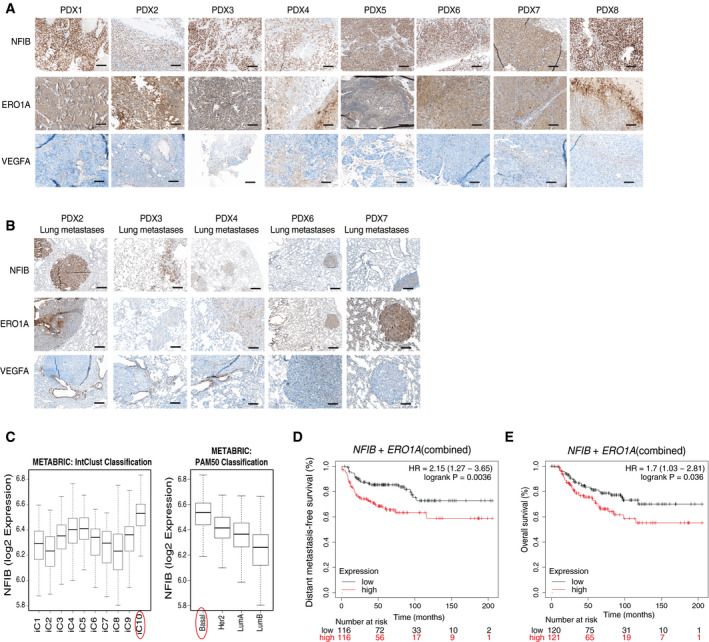
-
ARepresentative images of NFIB, ERO1A, VEGFA‐positive TNBC PDX primary tumours. Scale bar, 1 mm.
-
BRepresentative images of NFIB, ERO1A, VEGFA‐positive lung metastases from TNBC PDX. Scale bar, 1 mm.
-
CHigher NFIB expression in iC10 and basal breast cancer subtype compared with the other subtypes using intClust (Dawson et al, 2013) and PAM50 classifications in the METABRIC cohort (n = 1,980 across the 10 Intclust, iC). Boxplot represents NFIB expression values comprised between the 1st and the 3rd quartiles with the central band representing median expression value, and whiskers indicating the farthest data points that are still within the distance of 1.5 times the interquartile range (IQR).
-
D, EDistant metastasis‐free survival (D, n = 232) and overall survival (E, n = 241) plots generated using the Kaplan–Meier plotter based on the mean expression using the signal intensity of the NFIB (213029_at) combined with the ERO1A (218498_s_at) probes. The cut‐off was automatically set to split patients into two groups (median), high and low. The plots were generated using the signal intensity of the different probes in Affymetrix microarray gene expression data from TNBC patients of The Cancer Genome Atlas. Number of patients (n) and P values (two tailed log‐rank test) are presented in the panels.
Source data are available online for this figure.
Discussion
Metastasis, the final, lethal hallmark of cancer, remains a major burden for breast cancer patients but there are numerous mechanisms that may prevent circulating cancer cells from colonizing distant organs. Only when these are circumvented and metastases develop is the prognosis of patients dismal (Massagué & Obenauf, 2016). Several interacting oncogenic pathways contribute to metastasis, but the exact molecular mechanisms of colonization are not fully understood. A detailed understanding of these molecular mechanisms is urgently needed.
Previous studies showed that NFIB governs epithelial‐melanocyte stem cell behaviour and facilitates melanoma cell migration and invasion (Chang et al, 2013; Fane et al, 2017). NFIB has also been implicated in models of small‐cell lung carcinoma (SCLC) and breast cancer (Moon et al, 2011; Denny et al, 2016; Semenova et al, 2016; Campbell et al, 2018; Liu et al, 2019), and NFIB or ERO1A has been associated with different tumour types (Becker‐Santos et al, 2017; Zhou et al, 2017; Yang et al, 2018). Furthermore, in accordance with our results, ERO1A has been shown to enhance VEGFA expression in a TNBC human cell line (Tanaka et al, 2016). NFIB was shown to enhance TNBC cell survival and progression by suppressing CDKN1A (Liu et al, 2019), and to confer oestrogen independency in oestrogen receptor‐positive breast cancer models (Campbell et al, 2018).
Here, we provide new insights into the effects of the NFIB‐ERO1A‐VEGFA axis on breast cancer progression to metastasis. Via an unbiased in vivo PB functional genetic screen, we have shown that NFIB induces mammary cancer metastatic progression and colonization. We have collected functional and mechanistic evidence that NFIB is a mammary cancer metastatic transcriptional regulator. First, depletion of NFIB delayed mammary tumour growth and abrogated metastases in three different cell lines when using both the orthotopic and experimental metastasis assays. Second, Nfib KO reduced tumoursphere formation and the frequency of tumour‐initiating cells. Third, endogenous Nfib/NFIB overexpression was sufficient to evoke metastasis in non‐metastatic murine and human mammary cancer lines in both the orthotopic and experimental metastasis assays. It also decreased overall survival of the animals.
Mechanistically, we have shown that ERO1A is a direct target of NFIB and a critical effector in evoking metastatic colonization. ERO1A enhanced ROS levels, causing HIF1α stabilization, increased VEGFA levels and angiogenesis, which resulted in a permissive microenvironment for colonization. These recorded effects of the NFIB‐ERO1A‐VEGFA axis on breast cancer progression to metastasis identify a targetable network for cancer therapy.
Materials and Methods
Cell lines
LB‐mHR1 is a mouse mammary cancer cell line derived from a transgenic mouse expressing PIK3CAH1047R under the CAG promoter (Meyer et al 2011). In brief, a mammary tumour was dissociated into single cells through mechanical and enzymatic digestion. The cells were sorted using GFP and established in DMEM supplemented with 10% FBS. To generate HR1. PB cells, LB‐mHR1 cells were engineered with the PB system (Ivics et al, 2009; Rad et al, 2010) by transduction with the pFS250‐PBase vector (HR1. Ctrl) followed by transfection with an ATP1 construct. To induce transposon mobilization and generate pools of mutagenized cells, HR1. PB cells were treated with dox for up to 96 h in vitro. LM (1 to 9) cell lines are lung‐derived tumour cell lines isolated from NOD/SCID mice bearing HR1. PB‐derived mammary tumours. In brief, lungs were dissociated into single cells and cultured until tumour cell colonies became apparent. For increased purity, tumour cells were sorted for GFP. HEK293T and 4T1 cell lines were purchased from the ATCC and cultured according to the ATCC protocol. LB‐mHR1, 4T1, LM1‐9 and HEK293T cells lines were cultured in DMEM supplemented with 10% FCS. SUM159PT cells were kindly provided by Dr. Charlotte Kupperwasser (Boston, USA). SUM159PT cells were cultured in Ham’s F12 with 5% foetal calf serum (Gibco, Invitrogen), 5 µg/ml bovine insulin (Sigma), 1 μg/ml hydrocortisone (Sigma) and 1× penicillin/streptomycin (Gibco, Invitrogen). SUM159PT cell line identity was confirmed and routinely tested using short tandem repeat (STR) sequencing. HUVEC cells were cultured in endothelial growth medium 2 (EGM2 basal medium, Promocell) completed with endothelial growth supplements according to manufacturer’s instructions and 1% penicillin/streptomycin. HUVEC were used at passage 5–6. All cell lines were tested routinely for mycoplasma contamination.
PDX models
The PDXs used in this study were previously described (Derose et al, 2011; Gao et al, 2015) and their metastatic potential examined in lungs by haematoxylin and eosin staining, and expression of ER, PR and HER2.
Animal experiments
All in vivo experiments were performed in accordance with the Swiss animal welfare ordinance and approved by the cantonal veterinary office Basel‐Stadt. Female NOD/SCID, NSG, and BALB/c mice were maintained in the Friedrich Miescher Institute for Biomedical Research and the Department of Biomedicine animal facilities in accordance with Swiss guidelines on animal experimentation. BALB/c, NOD/SCID and NSG mice originally are from the Janvier Labs and from in‐house colonies. Mice were maintained in a sterile controlled environment (a gradual light–dark cycle with light from 7:00 to 17:00, 21–25°C, 45–65% humidity). For orthotopic engraftment of cancer cell lines in the limiting dilution assay, 250 × 103, 10 × 103, 2 × 103, 500, or 200 LM cells were suspended in 50 μl Matrigel:PBS (1:1) and injected into the fourth mouse mammary gland of six‐ to eight‐week‐old female NOD/SCID mice. The frequencies of tumour‐initiating cells in the different conditions were calculated and compared statistically using the Extreme Limiting Dilution Analysis (ELDA) online tool (Hu & Smyth, 2009). For the PB in vivo screen, HR1. Ctrl and HR1. PB cells (1 × 106) were resuspended in 50 μl Matrigel:PBS (1:1) and injected into the mammary fat pads of four‐ to eight‐week‐old female NOD/SCID mice. For orthotopic engraftment of cancer cell lines, LB‐mHR1 or LM cells (250 × 103 cells) were resuspended in 50 μl Matrigel:PBS (1:1) and injected into the mammary fat pads of four‐ to eight‐week‐old female NOD/SCID mice. SUM159PT or 4T1 (250 × 103 cells) cells were injected into the mammary fat pads of four‐ to eight‐week‐old female NSG or BALB/c mice, respectively. Tumours were measured with Vernier callipers and volume calculated as 0.5 × ([large diameter] × [smallest diameter]2). Tumours were resected before they reached 1,500 mm3 and mice were monitored regularly for signs of metastatic outgrowth and distress. For survival studies, animals were sacrificed when tumours reached 1,500 mm3 or when they showed any signs of distress (e.g. breathing disorders, weight loss or immobility). All orthotropic experimental procedures (tumour resection and tumour cell implantation) were undertaken on anaesthetized mice by a single investigator according to protocols approved by the cantonal veterinary office Basel‐Stadt. Experimental metastasis assays were performed by injecting 100 × 103 cells suspended in 100 μl of PBS into tail veins (i.v.). After intravenous injection of LM, LB‐mHR1 or SUM159PT cells, in vivo bioluminescence imaging was performed to confirm injection and to monitor metastatic outgrowth. For bioluminescent imaging, mice were injected i.p. with 100 μl of D‐luciferin (15 mg/ml, Biosynth L8220). Mice were anesthetized with isoflurane (2% in 1 l/min oxygen) and bioluminescence imaging performed using an IVIS Lumina XR instrument (Caliper LifeSciences) upon injection of luciferin. Acquisition times ranged from 3 to 10 min.
Splinkerette PCR for the amplification of transposon integration sites
For PB sequencing, we adapted the splinkerette PCR protocol described previously (Friedrich et al, 2017). Tumour samples (16 primary tumours) and 18 metastatic samples (3 lung metastases, 6 lung macro‐metastases and 9 LM cell lines) were sequenced. Genomic DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen) and sheared with a Covaris sonicator to a fragment length of 250 bp. After end repair and A‐tailing, purified DNA fragments were ligated to a splinkerette adaptor (obtained after annealing of top 5′‐gttcccatggtactactcatataatacgactcactataggtgacagcgagcgct‐3′ and bottom 5′‐gcgctcgctgtcacctatagtgagtcgtattataatttttttttcaaaaaaa‐3′). Transposon‐containing fragments were enriched by 18 cycles of transposon‐specific PCR1 for the 5′ transposon ends in a unique library (5′‐gacggattcgcgctatttagaaagagag‐3′ for the 5′ arm of PB, and common splinkerette primer 5′‐gttcccatggtactactcata‐3′). Bar coding of individual samples and completion of Illumina adaptor sequences were achieved by an additional 12 cycles (primary tumours) and 18 cycles (metastatic samples) of transposon‐specific PCR and a custom array of 35 unique bar‐coding primers. For the 5′ arm, we used 5′‐aatgatacggcgaccaccgagatctacacatgcgtcaattttacgcagactatc‐ 3′ and for the splinkerette side, we used 5′‐caagcagaagacggcatacgagatcggtXXXXXXXXtaatacgactcactatagg‐3′ primers. The Xs represent the bar code of eight nucleotides. After magnetic bead purification (Beckman Ampure XP), libraries were assembled in two pools and sequenced on an HiSeq Desktop Sequencer (Illumina): HiSeq 2500 and MiSeq, Rapid Run, Paired‐End, 2 × 100 bp, with 15% PhiX. A 7‐pM aliquot was loaded on to the instrument.
Mapping of insertion sequences to the mouse genome and identification of integration sites
Pre‐processing and alignment
Paired‐End reads were first pre‐processed by removing the expected transposon‐derived sequence (5’‐TAGGGTTAA‐3’) from the beginning of the first read (read pairs with non‐matching first reads were discarded; typically around 1%) using the preprocessReads function from QuasR (version 1.12.0). Read pairs were then aligned to the mouse genome (BSgenome. Mmusculus. UCSC.mm10) using the QuasR qAlign function and parameters “‐m 1 ‐‐best ‐‐strata ‐‐maxins 1000”; only uniquely mapping pairs with up to 1,000 bp between‐pair distance were reported. Mapping rates were recorded and non‐mapped read pairs were further aligned against the non‐mobilized transposon sequence to estimate the probable fraction of read pairs that originate from non‐mobilized copies of the transposon. For each aligned read pair, the piggyBac insertion coordinate was identified as the coordinate of the first (most 5’) base of the first read.
Integration site identification and quantification
For each unique integration site, the number of distinct supporting alignments (distinct read pairs), and the genomic sequence from the four base pairs on the same strand as the first read directly upstream of the insertion site were recorded. Only integration sites with the expected upstream TTAA sequence were used for the downstream analysis.
Association of integration sites with genes
Coordinates of known genes (exons/introns, 5’‐untranslated region [UTR], coding sequence [CDS], 3’UTR) were obtained from the TxDb. Mmusculus. UCSC.mm10.knownGene—Bioconductor package (version 3.2.2). Promoter regions were defined as regions 2,000 bp upstream of known transcript start sites. Integration sites were matched against these genomic regions to identify overlaps on any strand and orientation, selecting the first overlap in the case of multiple overlaps. Integration sites were classified hierarchically as follows: sites without overlaps to any transcript were labelled as promoter sites (in the case of an overlap with a promoter region) or intergenic sites. All other sites were labelled with the first region type that they overlapped, in the following order: 5’UTR, CDS, 3’UTR, intron or ncRNA (defined as an overlap with a transcript without annotated CDS). Sites were further labelled according to their orientation with respect to the associated gene (same or opposite). Finally, sites were grouped according to the gene they overlapped (including promoter sites) or, for intergenic sites, according to the pair of flanking genes.
Selection of the enriched unique integration sites
The diversity has been calculated for each insertion, i.e., the number of distinct alignment pairs starting at the insertion coordinate (Chapeau et al, 2017). For each gene, the diversity was summed to give the number of unique insertions in a gene. Finally, this value was divided by the total number of unique insertions for a given sample. In order to find putative metastatic genes, we looked at genes with more unique insertions in the metastatic than in the tumour samples.
Chromatin immunoprecipitation assay (ChIP)
Cells growing as monolayer cultures were fixed in 1% formaldehyde (SIGMA) for 10 min, quenched by addition of 0.125 M glycine (SIGMA), washed twice with 10 ml of PBS and 3.5 ml of SDS buffer (NaCl 100 mM, Tris–Cl pH8 50 mM, 5 mM EDTA pH 8.0, NaN3 0,02%, SDS 0,5%) including inhibitors (Complete Mini, ROCHE) were added. Lysed cells were incubated in IP buffer (100 mM Tris at pH 8.6, NaCl 100 mM, 0.5% SDS, 5% Triton X‐100, and 5 mM EDTA with proteinase inhibitor) and the chromatin disrupted by sonication using a Diagenode Bioruptor sonicator UCD‐300 to obtain fragments of 200–500 bp (15 cycles with 30 s of ON and 30 s OFF). For each ChIP, suitable amount of fragmented and pre‐cleared chromatin was diluted and incubated with specific antibodies overnight. Antibodies used were normal rabbit IgG (Diagenode, C15410206) as a control and anti‐NFIB (Sigma, HPA003956‐100UL; 2 µl of 1 µg/µl dilution). Immunoprecipitated complexes were recovered on Dynabead Protein A beads (Thermo Fisher) and, after extensive washes, DNA was recovered by reverse crosslinking and purification using QIAquick PCR purification kit (Qiagen). For ChIP‐quantitative PCR (qPCR), real‐time PCRs were performed using Fast SYBR green master mix reagent (Applied Biosystems). Primer sequences used for ChIP‐qPCR experiments were designed using Cistrome Data Browser (http://cistrome.org/db/), Integrated Viewer Genome software (Robinson et al, 2011) and data from GSE74826 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74826; (Shin et al, 2016): Ero1l Peak 18492 FW 5’‐GAGACTGCAGAGGGACAAGA‐3’, REV 5’‐GCGCTCAGTTGAAACTCTGT‐3’; Ero1l Peak 18493 FW 5’‐AAAGACGCGGTCCTTCC‐3’, REV 5’‐AAGGCTTAGGCAGCCAGA‐3’; Meg3_TSS FW 5’‐AAACAACGCTCTCCTTTCCTAAG‐3’, REV 5’‐AAATAACCCCAACTGGTGATTG‐3’.
Genome editing by CRISPR‐Cas9
A single sgRNA that produced a frameshift mutation in first and second exons was designed using the Zhang’s lab online CRISPR (http://crispr.mit.edu) and cloned into a modified PX330 (Addgene plasmid 42230) in which the puromycin cassette was replaced by red fluorescent protein (RFP; provided by the group of M. Bühler at the Friedrich Miescher Institute, Basel). The sgRNA sequences selected for Nfib (based on the lowest number of predicted off‐targets and highest predicted efficiency) were, guide 1: 5’‐CACCGCTCCGGGAAAGTGCGTTTTA‐3’ and 5’‐AAACTAAAACGCACTTTCCCGGAGC‐3’; guide 2: 5’‐CACCGTAGGCAATTGCACGGACGTG‐3’ and 5’‐AAACCACGTCCGTGCAATTGCCTAC−3’. The sgRNA sequences selected for Foxp1 were, guide1: 5’‐CACCGCTTCGTGACACTCGGTCCAA‐3’ and 5’‐AAACTTGGACCGAGTGTCACGAAGC‐3’; guide 2: 5’‐CACCGTAGTAAGTGGTTGCCACCGC‐3’ and 5’‐AAACGCGGTGGCAACCACTTACTAC‐3’. The sgRNA vectors were transduced into LM and 4T1 cells and the cells sorted for RFP positivity into 96‐well plates. Single cell clones were expanded and screened by immunoblotting. Different numbers of clones were pooled in equal proportions to minimize undesired off‐target and clonal effects (LM1 KO = 5, LM9 KO = 2, 4T1 WT = 2, 4T1 KO = 5). For human NFIB overexpression, we used SAM‐engineered Cas9 activation complexes (Konermann et al, 2015). LB‐mHR1 and SUM159PT cells were lentivirus infected in order to express the full murine and human NFIB from an endogenous promoter and maintained in culture for two months. sgRNA sequences were designed using Zhang’s lab online CRISPR (http://sam.genome‐engineering.org) and cloned into the lentiviral vector Addgene; plasmid 61427. Lenti‐sgRNA‐MS2 was digested with BsmbI and purified in a gel. The two annealed primers were ligated using the golden gate reaction. The sgRNA sequences selected for mouse Nfib were, guide 2: 5’‐CACCGCAGGAGGAGGAGGAGTAAAG‐3’ and 5’‐AAACCTTTACTCCTCCTCCTCCTGC‐3’; guide 3: 5’‐CACCGTGGGGGAGGCGCGCGGGAGG‐3’ and 5’‐AAACCCTCCCGCGCGCCTCCCCCAC‐3’; guide 4: 5’‐CACCGTGTGGAGAGGCTGGTGCAAA‐3’ and 5’‐AAACTTTGCACCAGCCTCTCCACAC‐3’. The sgRNA sequences selected for human NFIB were, guide 1: 5’‐CACCGAGCTGAGCCATCCATTCCTC‐3’ and 5’‐AAACGAGGAATGGATGGCTCAGCTC‐3’; guide 2: 5’‐CACCGACTAGGCTTGCAGTAAACGC‐3’ and 5’‐AAACGCGTTTACTGCAAGCCTAGTC‐3’; guide 3: 5’‐CACCGGAAGAGACTTGTCAGTATA‐3’ and 5’‐AAACTATACTGACAAGTCTCTCCC‐3’.
Lentiviral infections
The ATP1 vector was described previously (Rad et al, 2010). The transposon has PB inverted terminal repeats and can, therefore, be mobilized with the transposon system. ITRs were cloned into pBlueScript and the following genetic elements introduced between the ITRs of the transposon: Carp β‐actin splice acceptor (CβASA), En2SA splice acceptor from exon 2 of the mouse Engrailed‐2 gene, Lun‐SD from exon 1 of the mouse Foxf2 gene, and two bidirectional SV40 polyAs. Promoter elements carried by the transposons were unique to individual transposons: CAG (CMV immediate early enhancer and chicken beta‐actin gene promoter) for ATP1. The piggyBac transposase was cloned out of the Super PiggyBac Transposase expression (System Biosciences) by PCR (primers 5’ AGCTAGCACCGGTCGGAATTGTACCCAATTCGTTAAG 3’ and 5’ AGAATTCTTAATTAATTCTGGCGGCCGTTACG 3’) and cloned into a doxycycline‐inducible vector pFS250 (a kind gift from Novartis). For human ERO1A knockdown, we used two shRNA constructs (V2THS_85712 and V2THS_85710: Dharmacon pTRIPZ). Non‐targeting shRNAs (pTRIPZ) were used as control. For mouse Ero1l knockdown, we used two shRNA constructs (MSH100691‐LVRU6MH‐a and MSH100691‐LVRU6MH‐b: GeneCopoeia), non‐targeting shRNA (CSHCTR001‐1‐LVRU6MH) was used as control. For mouse Ero1l overexpression we used a pLenti‐C‐Myc‐DDK‐P2A‐Puro (Origene, MR207416L3). A dual green fluorescent protein‐luciferase 2 reporter (eGFP‐Luc2; Liu et al, 2010) vector was used for in vivo bioluminescence imaging experiments. Lentivirus batches were produced using PEI transfection on 293T cells as previously described (Britschgi et al, 2017). The titre of each lentivirus batch was determined in 4T1, LM, FMI‐LB‐mHR1 and SUM159PT cells. Cells were infected overnight in the presence of polybrene (8 µg/ml). p250. PBase selection was performed with 500 µg/ml Neomicin G418 (InvivoGen) and applied 48 h after infection. ATP1 selection was performed with 2.5 µg/ml puromycin (Sigma) and applied 48 h after transfection. The SAM‐engineered Cas9 activation complex (Konermann et al, 2015) consists of three lentiviral vectors: Lenti MS2‐P65‐HSF1_Hygro (Addgene plasmid 61426), Lenti‐sgRNA‐MS2_Zeo (Addgene; plasmid 61427), and Lenti dCAS‐VP64_Blast (Addgene 61425). Cells were infected first with a 1:1 mass ratio of Lenti MS2‐P65‐HSF1 and Lenti dCAS‐VP64 at a MOI of 10 viral particles per cell; selection was carried out in 1 µg/ml Blasticidin (Gibco) and 500 µg/ml Hygromicin (InvivoGen) after a 24‐h transduction. Selection in 500 µg/ml Zeomicin (Invitrogen) was also applied after a 24‐h infection with Lenti‐sgRNA‐MS2_Zeo. Cells were cultured for two months in vitro before performing experiments.
Transient gene silencing
The siRNA IDs were as follows: siNfib (SI00178451, SI02668085 and SI02733983), (s4823 and s4825), non‐targeting controls (D‐001810‐10‐05) were ordered as onTarget‐plus SMART pools (Dharmacon). Transfections of siRNAs were performed according to the manufacturer’s guidelines (DharmaFect 1, Dharmacon).
Fluorescence‐activated cell sorting
For FACS, cell lines were detached using trypsin‐EDTA, resuspended in growth medium and counted. Cells were passed through a 40‐μm strainer (Falcon) and resuspended in PBS with 1% FCS. DAPI (0.2%, Invitrogen) was added (1:250) 2 min before cell sorting. Single cells were gated on the basis of their forward and side‐scatter profiles and pulse‐width was used to exclude doublets. Dead cells (DAPI bright) were gated out and RFP and GFP bright cells were selected. FACS was carried out with a BD FACSAria III (Becton Dickinson) using a 70‐μm nozzle for SUM159PT, 4T1, HR1 and LM cell lines.
Immunoblotting
Cells were lysed in RIPA buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 1x protease inhibitor cocktail (Complete Mini, Roche), 0.2 mM sodium orthovanadate, 20 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride. Extracted proteins were measured using a DC protein assay kit (Bio‐Rad #5000112) and their concentrations adjusted. Whole‐cell lysates (30–50 μg) were subjected to 8% SDS–PAGE, transferred to PVDF membranes (Immobilon‐P, Millipore) and blocked for 1 h at room temperature with 5% milk in PBS–0.1% Tween 20. Membranes were then incubated overnight with antibodies as indicated and exposed to secondary HRP‐coupled anti‐mouse or ‐rabbit antibody at 1:5,000–10,000 for 1 h at room temperature. For each of the blots presented, the results represent at least three independent experiments. The following antibodies were used: anti‐NFIB (Sigma HPA003956‐100UL, 1:1,000), anti‐Vinculin (Abcam ab18058, 1:500), anti‐ERK2 (Santa Cruz sc‐1647, 1:2,000), anti‐FOXP1 (Cell Signaling 4402, 1:1,000) and anti‐ERO1L (Abnova H0003001‐M01 Clone 4G3, 1:500).
ELISA
Murine and human VEGFA protein levels were assessed using the mVEGFA Duo‐set (R&D System, DY493‐05) and hVEGFA Duo‐set (R&D System, DY293B‐05), respectively. Reagent Diluent Concentrate 2 (R&D Systems, DY995) was used. VEGFA ELISAs from R&D Systems were used according to the manufacturer’s protocol.
RNA isolation and qPCR
Total RNA was extracted using a Qiagen, RNeasy Plus Mini kit (cat. number 74136) according to the manufacture’s protocol. Total RNA (1 µg) was transcribed using the iScript cDNA synthesis kit (Bio‐Rad, cat. number 170‐8891). PCR and fluorescence detection were performed using the StepOnePlus Sequence Detection System or the 7500 ABI Fast Cycler (Applied Biosystems) according to the manufactures’ protocols in a reaction of 20 μl containing 1× TaqMan Universal PCR Master Mix (Applied Biosystems) and 25–50 ng cDNA. Primetime qPCT IDT assays (Integrated DNA technologies) were used for quantification of Nfib (Mm. PT.58.16634012), Gapdh (Mm. PT.39a.1), Foxp1 (Mm. PT.58.2734907), Ero1l (Mm. PT.58.6905093), Vegfa (Mm. PT.58.1400306), NFIB (Hs. PT.58.40046929), ERO1A (Hs. PT.58.2554238), VEGFA (Hs. PT.58.1149801) and HPRT1 (Hs. PT.58.45621572). All measurements were performed in technical duplicates and the arithmetic mean of the Ct values used for calculations: target gene mean C t values were normalized to the respective housekeeping genes (Hprt1 or Gapdh), mean C t values (internal reference gene, C t), and then to the experimental control. The values obtained were 2−ΔΔ C t expressed as fold changes in regulation compared with the experimental control using the 2−ΔΔ C t method of relative quantification.
Tumoursphere cultures
LM and HR1 cells were plated in ultra‐low attachment plates (Corning) for six days at 10,000 cells per mL in DMEM:F12 supplemented with 1x B27 (Gibco, Invitrogen), 20 ng/ml human or mouse EGF (PeproTech), 20 ng/ml basic FGF (PeproTech) and 1× penicillin/streptomycin (Gibco, Invitrogen; Hilsenbeck et al, 2008). Primary tumourspheres were dissociated with 0.05% trypsin and replated at the same density for six further days for secondary tumoursphere assessment; additional rounds of culture were performed. All measurements were performed in technical triplicates.
Matrigel‐based tube formation assay
50 μl of growth factor reduced matrigel phenol red‐free (Corning) were spread evenly in a 96‐well plate, which was put in the incubator (37°C) for 30 min to allow matrigel to solidify. Then HUVEC cells (10–15,000 cells per well) were seeded on the matrigel in a total volume of 200 μl of conditioned medium from LM, 4T1, HR1 and SUM159PT cells. Recombinant VEGF121 (PrepoTech 100‐20A, 0.4 ng/μl) was added to the conditioned medium. After 4 h, cells were imaged using a ZEISS Axio Vert. A1 Inverted Microscope and finally fixed with 4% PFA. HUVEC network and number of master segments were quantified using the Angiogenesis Analyzer tool from ImageJ (Fiji) (Carpentier et al, 2020).
Transwell migration and invasion assay
Migration assay was performed using transwell chambers 8 μm pore size (Corning) accordingly to the manufacturer’s protocol. Cells were starved for 16 h, and 15,000 cells were seeded on the insert in 500 μl medium containing 0.5% FBS, and 750 μl of full growth medium including 10% FBS was added to the well. Cells were allowed to migrate towards the FBS gradient for 24 h before the inserts were washed with PBS and remaining cells on the upper surface of the membrane were removed using cotton swabs. The cells that had migrated though the membrane were fixed with 4% PFA and stained with 0.2% Crystal violet. The whole membranes were then imaged using a ZEISS Axio Vert. A1 Inverted Microscope.
For the invasion assay, 1,000 cells were seeded in a 96‐well plate on top of solidified growth factor reduced matrigel (Corning) in 200 μl assay medium (DMEM medium supplemented with 2% matrigel and 2% FBS). After 2 h at 37°C, 300 μl of DMEM medium with 10% matrigel and 2% FBS were added and the medium was replaced with 400 μl of assay medium every three days until the end of the experiment (10/12 days total). After two weeks, the number of invasive structures was counted.
Sulphorhodamine‐B (SRB) cell proliferation assay
1,000 cells per well were seeded in five 96‐well plates and fixed after two‐three hours or every consecutive day until day five in 100 μl of cold 3.3% trichloroacetic acid and incubated at 4°C for 1 h or O/N. The plates were washed four times with slow‐running tap water and air‐dried at RT. 100 μl of 0.057% sulphorhodamine B (SRB) dye were added to each well and plates were left at RT for 30 min, then rinsed four times with 1% (vol/vol) acetic acid and air‐dried at RT. For optical density (OD) measurement, 200 μl of 10 mM Tris base solution (pH 10.5) were added and the OD was read using the Synergy H1 microplate reader (at 510 nm; BioTek).
Circulating tumour cell quantification
Circulating tumour cells (CTCs) were isolated from the peripheral blood of animals bearing tumours just before tumour resection. Peripheral blood was plated in DMEM medium supplemented with 10% FCS and colonies counted on day 10 of culture. The number of CTCs was expressed as the total number of colonies in the dish divided by the volume of blood taken.
RNA sequencing and analysis
Tumour RNA preparation and sequencing
Sorted GFP‐positive cells from tumours were collected and total RNA was extracted using a Qiagen, RNeasy Plus Mini kit (cat. number 74136) according to the manufacture’s protocol. RNA integrity was measured on an Agilent 2100 Bioanalyzer using RNA Pico reagents (Agilent Technologies). The library was prepared using an Illumina TruSeq stranded mRNA‐seq preparation kit (according to recommendations from the manufacturer). Library quality was measured on an Agilent 2100 Bioanalyzer for product size and concentration. Single‐end libraries were sequenced with an Illumina HiSeq 2500 (50‐nt read length).
Tumoursphere RNA preparation and sequencing
Cells were dissociated with 0.05% trypsin, and total RNA extracted using a Qiagen, RNeasy Plus Mini kit (cat. number 74136) according to the manufacture’s protocol. RNA integrity was measured on an Agilent 2100 Bioanalyzer using RNA Pico reagents (Agilent Technologies). The library was prepared using an Illumina TruSeq stranded mRNA‐seq preparation kit (according to recommendations from the manufacturer). Library quality was measured on an Agilent 2100 Bioanalyzer for product size and concentration. Single‐end libraries were sequenced by a NextSeq 500 (76‐nt read length).
Analysis
Reads were aligned to the mouse genome (UCSC version mm10) with STAR (Dobin et al, 2013). The output was sorted and indexed with samtools. Stand‐specific coverage tracks per sample were generated by tiling the genome in 20‐bp windows and counting 5' end of reads per window using the function bamCount from the Bioconductor package bamsignals (https://bioconductor.org/packages/release/bioc/html/bamsignals.html). These window counts were exported in bigWig format using the Bioconductor package rtracklayer (https://bioconductor.org/packages/release/bioc/html/rtracklayer.html). The qCount function of QuasR (https://bioconductor.org/packages/release/bioc/html/QuasR.html) was used to count the number of reads (5' ends) overlapping the exons of each gene, assuming an exon union model. Differential gene expression analysis was performed using the limma‐voom framework (Carriço et al, 2011).
Cellular ROS quantification
To quantify ROS levels, we used the cellular reactive oxygen species detection assay kit (Deep Red fluorescence; Abcam, #ab186029). 20,000 cells were plated in a 96‐well black wall/clear bottomed microplate and incubated ON. The day after, ROS Deep Read working solution was added for 30–60 min in the dark at RT. Wells with no dye solution were used as control. The deep red fluorescence was read using the Synergy H1 microplate reader (at excitation 650 nm, emission 675 nm; BioTek).
Immunohistochemistry
All xenograft tissues were fixed in formalin fix (Shandon, Thermo Fisher) for 24 h at 4°C. Samples were then dehydrated, embedded in paraffin, sectioned (3–4 µm) and processed for haematoxylin/eosin staining and for immunohistochemistry. All immunohistochemistry experiments were performed using a Ventana DiscoveryXT instrument (Roche Diagnostics) following the Research IHC DAB Map XT procedure. For NFIB staining, slides were pretreated with CC1 for 90 min. NFIB primary antibody (1:250, Sigma‐Aldrich, HPA003956) was incubated for 44 min, followed by secondary antibody incubation and detection. The RUO Discovery Universal procedure was used for CD31, VEGFA and ERO1A staining, with a CC1 pre‐treatment (40 min) and incubation with a rat anti‐CD31 (SZ31 dianova, 1:50), or with rabbit anti‐VEGFA (GeneTex GTX61510, 1:100), or with rabbit anti‐ERO1A (GeneTex, GTX112589 1:1,000) antibodies for 1 h at 37°C, respectively. Next, a polymer Immpress goat anti‐rat for CD31 (Vector Lab, MP‐7444) or Universal Immuno‐enzyme Polymer (UIP) anti‐rabbit (Nichirei, 414142F) for VEGFA and ERO1A was applied for 28 min at 37°C, and the peroxidase reaction revealed with the Discovery ChromoMap DAB kit (Ventana, Roche diagnostics). Counter staining was performed with haematoxylin II and bluing reagent (Ventana, Roche diagnostics). For PDXs, the overall procedure was the same but the signal was amplified using the amplification kit (Ventana, Roche diagnostics, 760‐080). Two images of two to eight lung metastases, two images for tumours and PDX of each condition were captured and quantified manually or with ImageJ (Fiji).
CD31, NFIB, ERO1A and VEGFA quantification
The images were analysed using Fiji open source software (Schindelin et al, 2009). First, the lung area for each tissue section was measured by manual selection. Second, for each metastasis in each tissue section, a region of interest (ROI) was selected manually. Each ROI was then subjected to background subtraction (rolling ball with a 50‐px radius) followed by colour deconvolution (Ruifrok & Johnston, 2001) using [H DAB] vectors. The resulting colour channel (2) corresponding to the DAB stain was blurred (Gaussian Blur with a sigma of 5) and was then thresholded (using the value for all images [222]) to measure the CD31‐positive areas. For NFIB, ERO1A and VEGFA IHC images, stained tissues areas were selected manually and the pixels quantified by colour deconvolution ([H DAB] vectors) and thresholding the colour channel (2) (using a value of [170] for all images). Scripts for these semi‐automatic analyses are available upon request.
Immunofluorescence
12 mm glass slides were coated with 200 μl of 0.1 mg/ml Poly‐L‐Lysine (SIGMA) for two hours at 37°C, washed twice with PBS, and 10,000 cells seeded per well. The day after, slides were washed once with ice‐cold PBS and fixed with 4% PFA 4% for 15 min and washed three times 5 min with ice‐cold PBS, then permeabilized with Triton 0.3% PBS for 15 min at RT, and washed three times 5 min with ice‐cold PBS. Then cells were blocked with 2.5 % normal goat serum (NGS) in PBS for one hour at RT, incubated with the primary antibody in 2.5 % NGS in PBS ON at 4°C or for two hours at RT on rocking table. Next, cells were washed three times 5 min with ice‐cold PBS and incubated with the secondary antibody 2.5 % NGS in PBS for 2 h at RT on rocking table in the dark. After three times 5‐min washing in PBS, the nuclei were stained with DAPI (2 mg/ml; Invitrogen) for 10 min at RT, after two more washes for 5 min with PBS, the slides were dried and mounted with one drop Prolong Gold antifade mounting medium (Invitrogen), and 24 h later the slides were analysed or stored at 4°C. The following antibodies were used: anti‐rabbit HIF1a (Abcam ab2185, 1:1,000) and anti‐rabbit Alexa 633 (Thermo Fisher, 1:1,000).
Microscopy image acquisition
For immunohistochemistry sections, images were captured using a Nikon Ni‐e upright microscope coupled to a PRIOR slide loader. Acquisition was performed with a 4× AIR objective with a DS‐Fi3 camera using NIS software. Quantification of the images was performed using Fiji. Phase‐contrast imaging used an inverted Zeiss Axioscope (10×, NA = 0.25 and 5×, NA = 0.15) equipped with an Axiocam 503 mono 60N‐C camera (pixel size 4.54 μm) and images were acquired using the Zen lite software. For immunofluorescence, images of stained sections were captured using Nikon T2 microscope, with a Photometrics Prime 95B camera using NIS software.
Data analysis from publicly available datasets
Mutation, segmented DNA copy number and mRNA expression data of breast tumours were from METABRIC (Curtis et al, 2012; Pereira et al, 2016) and TCGA Research Network (Koboldt et al, 2012) from cBioPortal (Cerami et al, 2012; Gao et al, 2013). We renamed the segmented DNA copy number data levels (produced using GISTIC 2.0) from −2, −1, 0, 1, 2 to "Deep Deletion", "Deletion", "Diploid", "Gain", and "High‐level Amplification", respectively. Relapse‐free survival, distant metastasis‐free survival and overall survival of NFIB and ERO1A were generated using the 2017 version of KMplotter (Györffy et al 2010) (http://kmplot.com/analysis/index.php?p=service&cancer=breast). Venn diagrams were produced using Bioinformatics & Evolutionary Genomics http://bioinformatics.psb.ugent.be/webtools/Venn/.
Statistical data analysis
The standard laboratory practice randomization procedure was used for cell‐line groups and animals of the same age and sex. The investigators were not blinded to allocation during experiments and outcome assessment. The number of mice was calculated by power analysis using data from small pilot experiments. Values represent the means ± s.d. unless stated otherwise. P values were determined using unpaired two‐tailed t‐tests and statistical significance set at P = 0.05. The variance was similar between the groups compared. Biological replicates correspond to different cell lines and tumour material. Technical replicates are tests or assays run on the same sample multiple times. The means of technical replicates, if available, were used for analysis and visualization. Biological replicates are tests or assays run on different samples and were used for statistical analysis and for reporting the number of experimental entities. Data were tested for normal distribution, Student’s t‐tests and two‐way ANOVA (if normally distributed) or nonparametric Mann–Whitney U‐test or Wilcoxon tests were applied unless stated otherwise. Kaplan–Meier plots were generated using the survival calculation tool from GraphPad Prism and significance was calculated using the two‐tailed log‐rank test at P < 0.05. For the analysis of tumour growth, we extracted for each individual sample the time to reach a tumour volume of 250 mm3 as follows: a linear model was fit to the log of the tumour volume as a function of time using all volumes greater than zero and least squares fitting in R (lm function). The resulting fit typically had an R‐squared of 0.7 or greater. The estimated times were then used to compare conditions, either in a two‐way ANOVA or Student’s t‐test, as indicated.
Author contributions
Study conception and manuscript writing: FZ, PMR, MB‐A; Design and experiments, data analysis and result interpretation: FZ, PMR Computational analysis of RNA‐seq, data analysis and result interpretation: P‐AM; Intersection between RNA‐seq data and Nfib putative targets, data analysis and result interpretation: CJ; Computational analysis of RNA‐seq and PB‐seq experiments, data analysis and result interpretation: AS, MBS; Performing and optimizing immunohistochemical staining, data analysis and result interpretation: M‐MC; PB library preparation and quality control for PB‐seq: TE; Algorithm for immunohistochemistry and quantification: LS; LB‐mHR1 cell line: LB; Experiments for the generation of HR1‐PB cell lines: JPC; PBase and ATP1 vectors and conceptual development of the project: RR; Conceptual development of the project: MRJ; Endothelial cells and reagents and conceptual development of the project: AR and AB; Reading and approval of the final manuscript: All authors.
Conflict of interest
P. M. R., L. B., J. P. C. and M. R. J. are employees of Novartis Pharma AG, M. B.‐A. owns equities in, and receives laboratory support and compensation from Novartis, and serves as consultant for Basilea. The other authors declare that they have no conflict of interest.
For more information
Kaplan–Meier plotter: http://kmplot.com/analysis/.
cBioportal: https://www.cbioportal.org/.
Bioinf. & Evol. Gen: http://bioinformatics.psb.ugent.be/webtools/Venn/.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank members of the Bentires‐Alj laboratory for advice and discussions. The authors are grateful for the support of the Genomics Core Facility at FMI and the BSSE‐ETHZ, to the Animal facilities of the University of Basel and FMI. We thank S. Bichet (FMI) for help with immunohistochemistry, G. Galli for discussion and advice, S. Smallwood and S. Dessus‐Babus for optimizing and running the PB‐seq experiments. We thank A. L. Welm (University of Utah) for the PDX models. We thank C. Kuperwasser and S. Ethier for the SUM159 cells. This research and also the publication fees were funded by the Swiss National Science Foundation (#310030_184673 20). Research in the Bentires‐Alj laboratory is supported by the Swiss Initiative for Systems Biology—SystemsX, the European Research Council (ERC advanced grant 694033 STEM‐BCPC), Novartis, the Krebsliga Beider Basel, the Swiss Cancer League, the Swiss Personalized Health Network (Swiss Personalized Oncology driver project), and the Department of Surgery of the University Hospital Basel.
EMBO Mol Med (2021) 13: e13162.
Data availability
Tumour mRNA‐seq data (GEO accession GSE144392): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144392.
Tumoursphere mRNA‐seq data (GEO accession GSE144393): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144393.
PiggyBac screen genomic GEO accession (GSE144898): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144898.
References
- Amelino‐Camelia G, Ellis J, Mavromatos NE, Nanopoulos DV, Sarkar S (1998) Tests of quantum gravity from observations of γ‐ray bursts. Nature 393: 763–765 [Google Scholar]
- Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, Nik‐Zainal S, Steenbruggen TG, Tjan‐Heijnen VCG, Labots M et al (2019) The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 51: 1450–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker‐Santos DD, Lonergan KM, Gronostajski RM, Lam WL (2017) Nuclear factor I/B: a master regulator of cell differentiation with paradoxical roles in cancer. EBioMedicine 22: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS (2007) Mitochondrial reactive oxygen species trigger hypoxia‐inducible factor‐dependent extension of the replicative life span during hypoxia. Mol Cell Biol 27: 5737–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, Soria JC, Dien AT, Adnani Y, Kamal M et al (2019) Genomic characterization of metastatic breast cancers. Nature 569: 560–564 [DOI] [PubMed] [Google Scholar]
- Brayer KJ, Frerich CA, Kang H, Ness SA (2016) Recurrent fusions in MYB and MYBL1 define a common, transcription factor‐driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov 6: 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi A, Duss S, Kim S, Couto JP, Brinkhaus H, Koren S, De Silva D, Mertz KD, Kaup D, Varga Z et al (2017) The Hippo kinases LATS1 and 2 control human breast cell fate via crosstalk with ERα. Nature 541: 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TM, Castro MAA, de Oliveira KG, Ponder BAJ, Meyer KB (2018) Era binding by transcription factors NFIB and YBX1 enables FGFR2 signaling to modulate estrogen responsiveness in breast cancer. Cancer Res 78: 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier G, Berndt S, Ferratge S, Rasband W, Cuendet M, Uzan G, Albanese P (2020) Angiogenesis analyzer for ImageJ — A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci Rep 10: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriço C, Costa JM, Santos TC (2011) Descrição da larva de neocordulia machadoi santos, costa & carriço, 2010 (Odonata: Corduliidae) para o Brasil. Biota Neotrop 11: 71–73 [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Pasolli HA, Giannopoulou EG, Guasch G, Gronostajski RM, Elemento O, Fuchs E (2013) NFIB is a governor of epithelial–melanocyte stem cell behaviour in a shared niche. Nature 495: 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapeau EA, Gembarska A, Durand EY, Mandon E, Estadieu C, Romanet V, Wiesmann M, Tiedt R, Lehar J, de Weck A et al (2017) Resistance mechanisms to TP53‐MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc Natl Acad Sci 114: 3151–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SJ, Rueda OM, Aparicio S, Caldas C (2013) A new genome‐driven integrated classification of breast cancer and its implications. EMBO J 32: 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Rosa J, Weber J, Friedrich MJ, Li Y, Rad L, Ponstingl H, Liang Q, De Quirós SB, Noorani I, Metzakopian E et al (2017) A single‐copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN‐cooperating tumor suppressor genes. Nat Genet 49: 730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mattos‐Arruda L, Sammut SJ, Ross EM, Bashford‐Rogers R, Greenstein E, Markus H, Morganella S, Teng Y, Maruvka Y, Pereira B et al (2019) The genomic and immune landscapes of lethal metastatic breast cancer. Cell Rep 27: 2690–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny SK, Yang D, Chuang CH, Brady JJ, Lim JSS, Grüner BM, Chiou SH, Schep AN, Baral J, Hamard C et al (2016) Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 166: 328–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E et al (2011) Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 17: 1514–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T (2005) Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122: 473–483 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy AJ (2010) Transposon‐based screens for cancer gene discovery in mouse models. Semin Cancer Biol 20: 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA (2005) Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436: 221–226 [DOI] [PubMed] [Google Scholar]
- Fahad Ullah M (2019) Breast cancer: current perspectives on the disease status. Adv Exp Med Biol 1152: 51–64 [DOI] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Hollingsworth DEJ, Simmons JL, Spoerri L, Oh TG, Chauhan J, Chin T, Harris L, Harvey TJ et al (2017) NFIB mediates brn2 driven melanoma cell migration and invasion through regulation of EZH2 and MITF. EBioMedicine 16: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich MJ, Rad L, Bronner IF, Strong A, Wang W, Weber J, Mayho M, Ponstingl H, Engleitner T, Grove C et al (2017) Genome‐wide transposon screening and quantitative insertion site sequencing for cancer gene discovery in mice. Nat Protocols 12(2): 289–309 [DOI] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y et al (2015) High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med 21: 1318–1325 [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massagué J (2006) Cancer metastasis: building a framework. Cell 127: 679–695 [DOI] [PubMed] [Google Scholar]
- Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123: 725–731. [DOI] [PubMed] [Google Scholar]
- Hilsenbeck S, Michalowska AM, Medina D, Green JE, Tsimelzon A, Behbod F, Zhang M, Kittrell F, Landis MD, Rosen JM et al (2008) Identification of tumor‐initiating cells in a p53‐null mouse model of breast cancer. Cancer Res 68: 4674–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK (2009) ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347: 70–78 [DOI] [PubMed] [Google Scholar]
- Ivics Z, Li MA, Mátés L, Boeke JD, Nagy A, Bradley A, Izsvák Z (2009) Transposon‐mediated genome manipulation in vertebrates. Nat Methods 6: 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, Nielsen TO, Gelmon K (2010) Metastatic behavior of breast cancer subtypes. J Clin Oncol 28: 3271–3277 [DOI] [PubMed] [Google Scholar]
- Kim KM, An AR, Park HS, Jang KY, Moon WS, Kang MJ, Lee YC, Ku JH, Chung MJ (2018) Combined expression of protein disulfide isomerase and endoplasmic reticulum oxidoreductin 1‐α is a poor prognostic marker for non‐small cell lung cancer. Oncol Lett 16: 5753–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki‐Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER et al (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H et al (2015) Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 517: 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Bentires‐Alj M (2013) Mouse models of PIK3CA mutations: One mutation initiates heterogeneous mammary tumors. FEBS J 280: 2758–2765 [DOI] [PubMed] [Google Scholar]
- Koren S, Bentires‐Alj M (2015) Breast tumor heterogeneity: source of fitness, hurdle for therapy. Mol Cell 60(4): 537–546. [DOI] [PubMed] [Google Scholar]
- Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O et al (2015) PIK3CAH1047R induces multipotency and multi‐lineage mammary tumours. Nature 525: 114–118 [DOI] [PubMed] [Google Scholar]
- Kreso A, Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14: 275–291 [DOI] [PubMed] [Google Scholar]
- Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, Wen S, Chang YF, Bachmann MH, Shimono Y et al (2010) Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci USA 107: 18115–18120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RZ, Vo TM, Jain S, Choi WS, Garcia E, Monckton EA, Mackey JR, Godbout R (2019) NFIB promotes cell survival by directly suppressing p21 transcription in TP53‐mutated triple‐negative breast cancer. J Pathol 247: 186–198 [DOI] [PubMed] [Google Scholar]
- Massagué J, Obenauf AC (2016) Metastatic colonization by circulating tumour cells. Nature 529: 298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DS, Brinkhaus H, Müller U, Müller M, Cardiff RD, Bentires‐Alj M (2011) Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res 71: 4344–4351 [DOI] [PubMed] [Google Scholar]
- Meyer DS, Koren S, Leroy C, Brinkhaus H, Müller U, Klebba I, Müller M, Cardiff RD, Bentires‐Alj M (2013) Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. Oncogenesis 2(9): e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TW (2012) Initiating breast cancer by PIK3CA mutation. Breast Cancer Res 14: 2010–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HG, Hwang KT, Kim JA, Kim HS, Lee MJ, Jung EM, Ko E, Han W, Noh DY (2011) NFIB is a potential target for estrogen receptor‐negative breast cancers. Mol Oncol 5: 538–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Russell R, Chin S‐F, Liu B, Rueda OM, Ali HR, Turashvili G, Mahler‐Araujo B, Ellis IO, Aparicio S et al (2018) Associations between genomic stratification of breast cancer and centrally reviewed tumour pathology in the METABRIC cohort. NPJ Breast Cancer 4: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC et al (2016) Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature 534: 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel K, Brakenhoff RH (2004) Dissecting the metastatic cascade. Nat Rev Cancer 4: 448–456 [DOI] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HKM, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ et al (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7: 11479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, Campos LS, Yusa K, Banerjee R, Li MA et al (2010) PiggyBac transposon mutagenesis: A tool for cancer gene discovery in mice. Science 330: 1104–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad R, Rad L, Wang W, Strong A, Ponstingl H, Bronner IF, Mayho M, Steiger K, Weber J, Hieber M et al (2015) A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat Genet 47: 47–56 [DOI] [PubMed] [Google Scholar]
- Robinson DR, Wu Y‐M, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar‐Sinha C, Raymond V et al (2017) Integrative clinical genomics of metastatic cancer. Nature 548: 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruifrok AC, Johnston DA (2001) Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23: 291–299 [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carrera I, Frise E, Verena K, Mark L, Tobias P, Stephan P, Curtis R, Stephan S, Benjamin S et al (2009) Fiji – an Open platform for biological image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova EA, Kwon M‐C, Monkhorst K, Song J‐Y, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF et al (2016) Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep 16: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HY, Willi M, Yoo KH, Zeng X, Wang C, Metser G, Hennighausen L (2016) Hierarchy within the mammary STAT5‐driven Wap super‐enhancer. Nat Genet 48: 904–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Wei Z, Koso H, Rust AG, Yew CCK, Mann MB, Ward JM, Adams DJ, Copeland NG, Jenkins NA et al (2017) A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet 47: 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Yoneda A, Sakai‐sawada K, Kosaka M, Minomi K (2017) Hypoxia‐inducible ERO1 α promotes cancer progression through modulation of integrin‐ β 1 modification and signalling in HCT116 colorectal cancer cells. Sci Rep 7: 9389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V (2016) Cancer‐associated oxidoreductase ERO1‐ a drives the production of VEGF via oxidative protein folding and regulating the mRNA level. Br J Cancer 114: 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, de la Rosa J, Grove CS, Schick M, Rad L, Baranov O, Strong A, Pfaus A, Friedrich MJ, Engleitner T et al (2019) PiggyBac transposon tools for recessive screening identify B‐cell lymphoma drivers in mice. Nat Commun 10: 1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Zhu X, Liu W, Ruan T, Wan W, Tao K (2018) NFIB promotes cell growth, aggressiveness, metastasis and EMT of gastric cancer through the Akt/Stat3 signaling pathway. Oncol Rep 40: 1565–1573 [DOI] [PubMed] [Google Scholar]
- Wu N, Jia D, Ibrahim AH, Bachurski CJ, Gronostajski RM, MacPherson D (2016) NFIB overexpression cooperates with Rb/p53 deletion to promote small cell lung cancer. Oncotarget 7: 57514–57524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Colandrea VJ, Hale JJ (2010) Prolyl hydroxylase domain‐containing protein inhibitors as stabilizers of hypoxia‐inducible factor: small molecule‐based therapeutics for anemia. Expert Opin Ther Pat 20: 1219–1245 [DOI] [PubMed] [Google Scholar]
- Yang S, Yang C, Yu F, Ding W, Hu Y, Cheng F, Zhang F, Guan B, Wang X, Lu L et al (2018) Endoplasmic reticulum resident oxidase ERO1‐Lalpha promotes hepatocellular carcinoma metastasis and angiogenesis through the S1PR1/STAT3/VEGF‐A pathway. Cell Death Dis 9: 1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Campbell PJ (2012) Evolution of the cancer genome. Nat Rev Genet 13: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H et al (2015) Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 21: 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK (2008) Class I PI3K in oncogenic cellular transformation. Oncogene 27: 5486–5496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Wang G, Gao S, Chen YE, Jin C, Wang Z (2017) Expression of ERO1L in gastric cancer and its association with patient prognosis. Experimental and Therapeutic Medicine 14: 2298–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito E (2015) ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic Biol Med 83: 299–304 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
Tumour mRNA‐seq data (GEO accession GSE144392): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144392.
Tumoursphere mRNA‐seq data (GEO accession GSE144393): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144393.
PiggyBac screen genomic GEO accession (GSE144898): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144898.