

SUMMARY
Transcription is epigenetically regulated by the orchestrated function of chromatin binding proteins that tightly control the expression of master transcription factors, effectors and supportive housekeeping genes required for establishing and propagating the normal and malignant cell state. Rapid advances in chemical biology and functional genomics have facilitated exploration of targeting epigenetic proteins, yielding effective strategies to target transcription while reducing toxicities to untransformed cells. Here, we review recent developments in conventional active site and allosteric inhibitors, peptidomimetics, and novel PROTAC technology that have deepened our understanding of transcriptional processes and led to promising preclinical compounds for therapeutic translation, particularly in cancer.
Keywords: Transcription, Epigenetics, Inhibitor, Degrader, Chromatin, PROTAC, peptidomimetic
eTOC blurb
New methods for targeting epigenetic regulatory proteins have led to rapid improvements in the mechanistic understanding of cancer biology and provided novel approaches for cancer therapies. This review summarizes these new developments with a focus on regulation of transcription, and discusses fundamental challenges and future directions for cancer chemical epigenetics.
INTRODUCTION
Many types of cancer cells display selective addiction to transcriptional processes that play critical roles in driving their malignant behavior. This process is tightly controlled by networks of epigenetic proteins and is required for the maintenance of normal cell growth, induction of differentiation, and in disease states, initiation, maintenance and propagation of the aberrant cell state. The process of transcription is epigenetically regulated by the orchestrated function of chromatin binding proteins, resulting in accessible DNA and active transcription. This process results in the highly regulated expression of master transcription factors (TFs), effector and supportive housekeeping genes that are required for establishing and propagating cell state. Critically, these processes are dysregulated in neoplastic states by a variety of events, including feedback transcriptional amplification by MYC family TFs (Bradner et al., 2017; Brien et al., 2019). Genome-wide surveys indicate that transcription is globally dysregulated in a wide-array of cancer states, (Grobner et al., 2018; Ma et al., 2018) nominating this process as an important area for therapeutic exploration in cancer.
During the process of transcription, cell-type and context-specific DNA-binding master TFs, coactivators, co-repressors and RNA-binding proteins are necessary to guide the RNA polymerase complex to accessible DNA. Direct targeting of transcription has been possible since the identification of the direct effects of α-amanitin on RNA polymerase (Lindell et al., 1970), however, this compound displays significant toxicities that have limited its use clinically. Other inhibitors with this function, such as triptolide, flavopiridol, and the newer compounds THZ531 and KL1, display improved toxicity profiles, and have in some cases advanced to clinical trials (Table S1). Further, successful compounds directly targeting TFs have been reported. One such example is the small molecule nutlin, which binds to MDM2, displacing p53 from the MDM2 binding site. This indirectly stabilizes p53 by preventing its MDM2-mediated polyubiquitination and proteasomal degradation. Other compounds that similarly target TFs directly by binding to protein-protein interaction interfaces, include stapled peptides targeting p53-MDM2/4 and molecules that stabilize or preclude binding to partner proteins for the TFs MYB, c-MYC, and STAT3 (Table S1). In each case, targeting the TF requires a deep understanding of its interaction domains with binding partners and cognate DNA binding motifs, and is therefore by definition low-throughput. This makes it exceedingly difficult to potently and specifically target individual transcription factors in a high-throughput manner.
As a result of these issues, interest has accrued in the targeting of proteins and cofactors involved in the recruitment of TFs. These targets, including the mammalian SWI/SNF nucleosome remodeling complex, histone writers, readers and erasers, are involved in the epigenetic programming of cells by establishing sites for transcription, termed the “histone code.” Like dysregulation of transcription, the function of these complexes is similarly dysregulated in neoplastic cell states (Grobner et al., 2018; Ma et al., 2018). These complexes contain enzymes, and therefore are susceptible to inhibitor strategies using active site binding, in addition to allosteric approaches. By targeting these enzymes specifically, generalized and pleiotropic effects on cell growth, survival and apoptosis are realized, mediated by general disruption of transcriptional processes. A key area of concern for inhibitors targeting epigenomic regulators is toxicity due to their propensity for extensive alteration of transcriptional processes genome-wide. Despite this concern, inhibitors of histone methyltransferase, histone deacetylase and lysine demethylases have demonstrated an appropriate therapeutic window, sufficient for clinical translation.
In parallel with the development of potent and specific inhibitors, advances in chemical biology have resulted in a novel strategy, termed proteolysis-targeted chimera (PROTAC) based degradation (Burslem and Crews, 2020). A PROTAC is a small molecule that binds to both a target protein of interest and an E3 ubiquitin ligase-containing complex receptor, such as CRBN, VHL, or MDM2, resulting in these two protein components being brought physically close together, with subsequent polyubiquitination of the target protein (Fig 1). This leads to proteasomal degradation of the target protein, that may be sufficient to cause near complete target protein loss. Exciting advances using this technology have resulted in the generation of multiple compounds targeting epigenetic regulatory proteins including, among others, the bromodomain and extraterminal domain-containing (BET) proteins. The early BET degrader, dBET1, which utilized N-butyl-2-hydroxyacetamide to link the BET-inhibitor JQ1 with a thalidomide tail, induced highly selective cereblon (CRBN)-dependent BET protein degradation and delayed leukemia progression in mice (Winter et al., 2015). Other BET degraders including ARV825 and MZ1 were simultaneously developed with dBET1 (Table S1). In some cases, these compounds may distinguish between closely related proteins, such as the SJF⍺ and SJFδ compounds that display selectivity for distinct p38 MAP kinase isoforms (Smith et al., 2019). The full potential of PROTACs, enzyme inhibitors and compounds that disrupt protein-protein interaction surfaces to broadly dysregulate epigenetic targets in cancer treatment have yet to be realized, though PROTACs do represent an area of exciting development and a major focus of enquiry for many groups. Exciting advances using this technology have resulted in compounds targeting the Androgen Receptor (AR) in AR+ prostatic carcinoma, and the estrogen receptor (ER) in ER+ breast cancer moving into clinical trials. Examples such indicating the therapeutic potential of PROTACs have fueled increasing interest in PROTAC design and resulted in a variety of new molecules targeting epigenetic and signaling regulators (Burslem and Crews, 2020). Thus, this chemical biology approach represents a novel area of exciting development and a major focus of enquiry for groups interested in targeting transcriptional processes.
Figure 1. PROTACs bridge a target protein of interest and an E3 ligase complex to induce selective degradation.
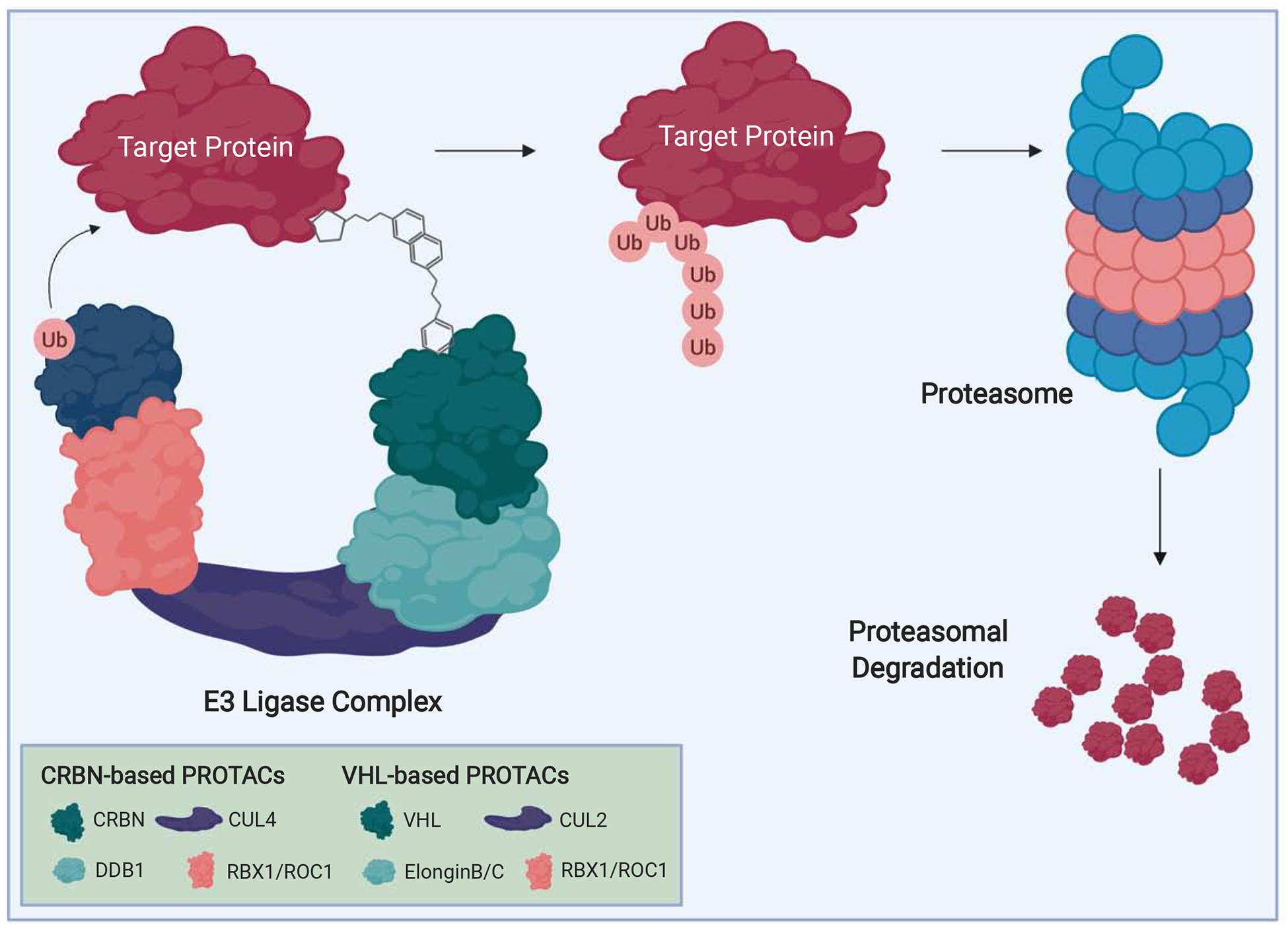
By the PROTAC strategy, a chemical structure bridging a target protein of interest (red) and the substrate receptor of an E3 ubiquitin ligase containing complex (green) results in E2-dependent ubiquitination of the target protein. This leads to recruitment of the target protein to the proteasome and proteasomal degradation. Differences between CRBN and VHL-based PROTAC complexes are demonstrated.
Targeting Epigenetic Proteins
Indirect strategies to target proteins that manipulate transcription offer unique opportunities to establish a comprehensive understanding of the transcriptional landscape. Specific post-translational modifications (PTMs) of histone proteins are highly associated with induction and repression of transcription. These modifications, catalyzed by epigenetic writer and eraser proteins and recognized by epigenetic reader proteins, may be manipulated through enzymatic inhibition or degradation (Fig 2). For clarity, this review will focus on epigenetic regulation of mRNA transcription, though these epigenetic proteins display far-ranging activities on both transcription, epigenetic regulation and indeed, other cellular processes including DNA repair and replication.
Figure 2. Epigenetic proteins drive alterations that can be targeted using chemical inhibitors and degraders.
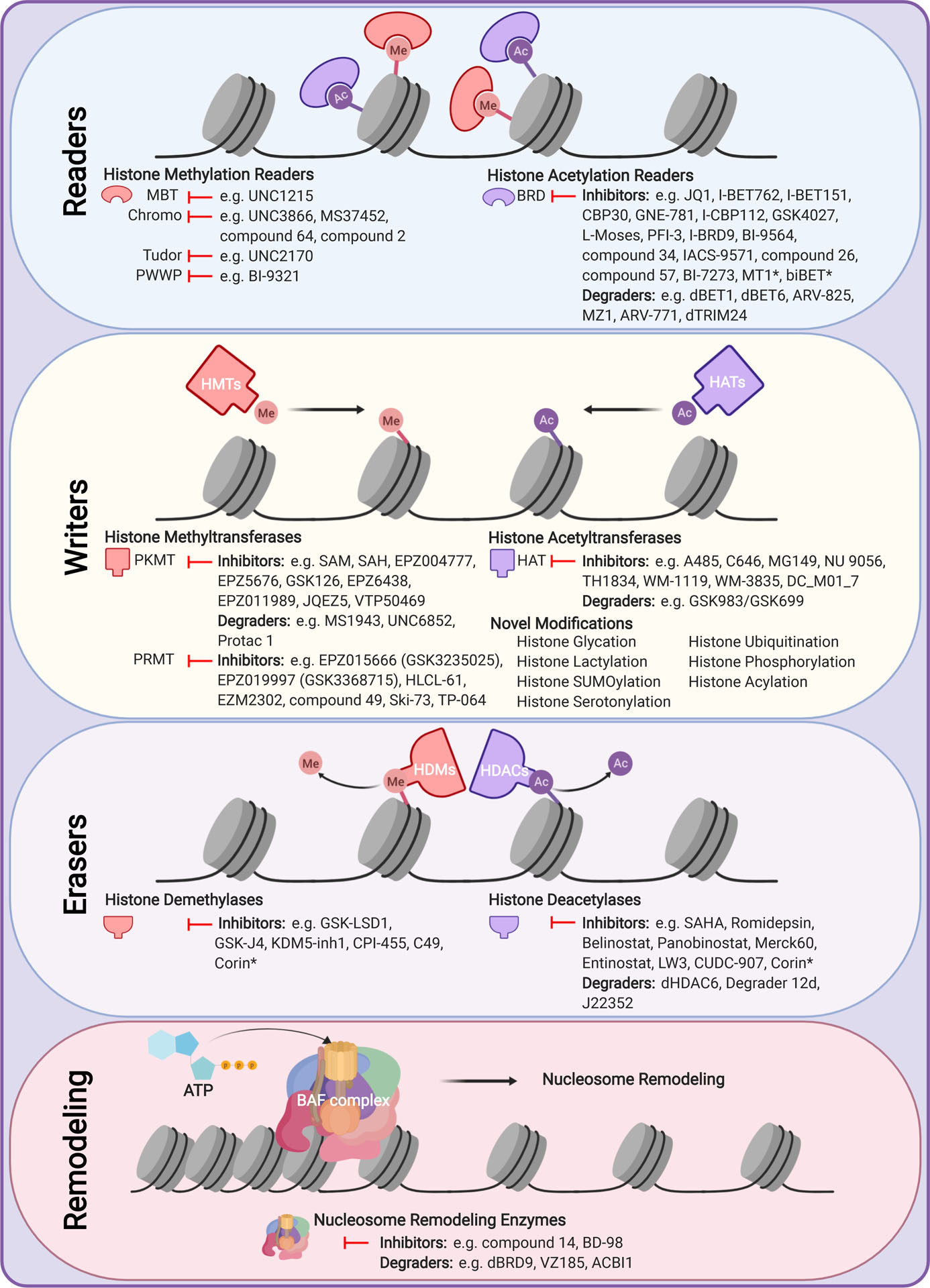
Demonstrated are common classes of histone readers, writers and erasers, as well as nucleosome remodeling complexes, and their different classes of inhibitors. MBT - malignant brain tumor; PWWP - Proline-Tryptophan-Tryptophan-Proline; PKMT - protein lysine methyltransferase; PRMT - protein arginine methyltransferase; BRD - bromodomain; HAT - histone acetyltransferase; HDM - histone demethylase; HDAC - histone eacetylase; * - bifunctional inhibitor.
Targeting Epigenetic Reader Proteins
Chemical targeting of epigenetic reader proteins was initially demonstrated in two independent studies that reported potent and selective monovalent inhibitors of BET bromodomain family reader proteins— JQ1 and I-BET762 (Filippakopoulos et al., 2010; Nicodeme et al., 2010). In these and subsequent studies, mono or bivalent inhibition of BET bromodomain proteins resulted in striking effects on mRNA transcription. Observations such as these have provided insights into the mechanism of how epigenetic protein reader inhibition manipulates transcription through regulation of key gene transcription, including c-MYC, BCL2 and CDK6 in different cancer subtypes (Dawson et al., 2011; Delmore et al., 2011). As a result, BET reader inhibition has become the model for a new class of epigenetic-targeted agents, and has stimulated interest in other epigenetic targets for the development of tool compounds and pharmacologic agents.
The bromodomain (BRD) functions as a reader domain for acetylated histones (Owen et al., 2000). In recent years, inhibitors targeting other bromodomain-containing proteins have been developed, such as TRIM24, BRD9, SMARCA4, CBP, and ATAD2 (Table S1). These inhibitors bind to the bromodomains of these individual proteins, and interrupt protein binding to acetylated lysine residues (Kac). Importantly, the effects of these inhibitors on transcriptional regulation is limited, suggesting that interrupted binding of these proteins to acetylated histones alone is insufficient to inhibit the regulatory function of the whole protein. Additionally, using PROTAC technology, bromodomain inhibitors have been used as anchors for degrader development. By this strategy, loss of the full length protein, rather than focal inhibition of the bromodomain is achieved, which targets all functions of these proteins and yields increased effects on transcription. Paralleling inhibitor development, multiple bromodomain based degraders have been recently reported, including those targeting TRIM24, BRD9, and SMARCA4 (Table S1). These compounds, in comparison with parental bromodomain inhibitors, display striking differences in activity, highlighting the differences between bromodomain protein reader function inhibition and protein loss. These newly developed degraders are therefore high priority tools for studying multi-domain protein function beyond the focused inhibition of one single domain.
Histone lysine and arginine methylation are other important and widespread PTMs associated with alteration in chromatin architecture, TF recruitment and transcriptional initiation and elongation. These PTMs may function to either activate or repress transcription (Jambhekar et al., 2019). Lysines can be mono-(me1), di- (me2) or tri- (me3) methylated on their ε-amine group, whereas arginines can be mono-methylated (me1), di-methylated symmetrically (me2s) or asymmetrically (me2a) on their guanidinyl group (Jambhekar et al., 2019). Individual histone methylation reader proteins have preference for distinct mono, di or trimethylated consensus sequences, imparting specificity to their binding.
Methyl-lysine (Kme) readers comprise more than 200 reader domains clustered in several families including malignant brain tumor (MBT), chromo, Tudor, PWWP and PHD domains (Arrowsmith and Schapira, 2019). The recognition of methyl-arginine (Rme) is predominantly performed by Tudor domains (Sprangers et al., 2003). Drugging Kme reader domains represents a greater challenge compared to KAc reader domains, due to the variability of Kme reader domain volume, enclosure, and binding pocket hydrophobicity (Santiago et al., 2011). Tudor domains frequently occur in tandem arrangements and recognize Kme through canonical aromatic cage pockets, while the isolated Tudor domain reads Rme2s and Rme2a in such a way that the guanidinium group of the arginine is sandwiched in a π-stacking arrangement between two aromatic side chains (Arrowsmith and Schapira, 2019). The K/Rme binding pockets found in Tudor domains are typically small, which pose a challenge for identifying potent ligands targeting the pockets. UNC2170, which binds to dimeric Tudor domains by engaging a larger protein surface at the interface of two p53-binding protein 1 (53BP1) Tudor domains, can compete with H4K20me2 peptides for binding (Perfetti et al., 2015). An alternative and perhaps more promising strategy for targeting tandem Tudor domains may be to disrupt the interface between them (Arrowsmith and Schapira, 2019). Compounds targeting the MBT domain that recognizes Kme1 and Kme2 (Nady et al., 2012) and the chromodomain that recognizes Kme3 have been reported. In the case of chromodomain containing proteins, success has largely been derived using peptidomimetic inhibitors such as UNC3866 (Table S1). Less active, nonpeptidomimetic inhibitors such as MS37452 (Ren et al., 2015) have also been developed and show cellular activity by derepressing the polycomb regulated INK4a/ARF locus at high concentrations. This variability in activity between classical small molecule compounds and peptidomimetics likely reflects the contribution of steric constraints and bonding between compound and the chromodomain. Strategies for targeting other methylation reader domains are still lacking potent and selective molecules that can efficiently dissect the mechanism of these readers.
The Kme binding reader PWWP domain has specificity for H3K36me2/3 or H4K20me residues (Arrowsmith and Schapira, 2019). PWWP domains possess an aromatic cage for Kme binding, flanked by a basic surface that binds DNA (Arrowsmith and Schapira, 2019). BI-9321 is the first potent and selective inhibitor of the PWWP domain, and inhibits the PWWP-domain containing H3K36 methyltransferase, NSD3, resulting in downregulation of c-MYC expression in acute myeloid leukemia (AML) cell lines (Böttcher et al., 2019). Chemical interrogation of histone methylation readers is a burgeoning field that is likely to yield key insights, however given the limited progress thus far in generating potent and specific inhibitors, future studies using alternative methodologies such as peptidomimetics, PROTACs and interface binding molecules will be of great interest.
Targeting Epigenetic Writer Proteins
Dynamic and plastic histone modifications created by writer enzymes are responsible for establishing the epigenetic landscape. These marks are regulated by a delicately balanced crosstalk between readers, writers and erasers that when altered can promote pathogenesis. Here we focus on targeting more transient marks that are deposited by histone writer proteins and their role in transcription, rather than more traditionally heritable marks that may be stable across cell division. Other less well characterized modifications, such as reversible histone serotonylation, crotonylation, glycation, phosphorylation, acylation, and others listed in Fig 2 will not be discussed here due to their limited inhibitor development.
Post-translational acetylation of histones was initially associated with transcription by the experimental finding that the positive charge on acetylated histone tails caused DNA to unravel from histones (Allfrey et al., 1964). This is thought to permit access for TFs and RNA polymerase to bind and promote transcription. While several histone acetylation sites have been thoroughly characterized, the acetylation of histone H3, lysine-27 (H3K27ac) has been extensively studied as a mark of active promoter and enhancer elements that regulate specific gene expression programs (Bradner et al., 2017). As a result, several histone acetyltransferase (HAT) inhibitors have been developed against the HATs that catalyze this mark, aiming to kinetically tease apart the role of this particular acetylation mark on transcription and disease pathogenesis (Dancy and Cole, 2015; Lasko et al., 2017).
Acetylation of H3K27 is performed by the acetylation writer proteins, the E1A Binding Protein P300 (EP300) and CREB-Binding Protein (CREBBP, CBP). Due to their varied roles in the maintenance of cellular homeostasis and proliferation, and their role in pathogenesis of diseases including cancer, these proteins and their function are heavily studied (Dancy and Cole, 2015). In addition to the H3K27ac mark, these proteins also catalyze acetylation of many other histone and non-histone substrates, such as H3K56ac and others (Dancy and Cole, 2015; Weinert et al., 2018). Synthetic chemical probes have been developed that target their catalytic activity, including C646, a pyrazolone and A485, a spiro oxazolidinedione (Table S1). Additionally, inhibitors including CBP30, I-CBP112, and GNE-781, targeting the Kac reader bromodomain function of EP300/CBP were also developed (Table S1). With next-generation, higher potency molecules such as the HAT inhibitor A485, EP300/CBP were recognized as potential therapeutic targets in hematological malignancies and androgen receptor positive prostate cancer (Lasko et al., 2017; Michaelides et al., 2018). However, targeting distinct domains of these proteins appears to have distinct effects. While targeting the EP300/CBP catalytic HAT domain prevents differentiation, targeting the bromodomain promotes differentiation by disrupting cell-type-specific gene expression and cell identity (Ebrahimi et al., 2019). Further, while targeting the bromodomain alone results in loss of H3K27Ac at enhancers, it maintains EP300/CBP occupancy at chromatin. This strategy disrupts enhancer RNA production which is known to suppress EP300/CBP activity (Bose et al., 2017), and, as a result, suppresses the transcription of enhancer regulated genes (Raisner et al., 2018). Intriguingly, targeting both domains simultaneously dramatically delocalizes EP300/CBP from chromatin at promoters to synergistically inhibit prostate cancer cell proliferation (Zucconi et al., 2019). These intriguing studies highlight the distinct functional roles of each domain within EP300 and CBP, and the utility of targeting multidomain proteins using distinct inhibitors targeted against individual domains. Further, the use of a PROTAC agent derived against either domain would obviate the need for selective inhibitors, by degrading the full-length protein.
Although progress has been made in understanding the individual effects of these paralogous proteins on chromatin structure and transcription, little is understood about the distinct roles of each paralog in cancer, in part because developing selective small molecules is challenging with their great structural homology (Dancy and Cole, 2015). Increasing genetic evidence indicates these proteins may be responsible for the control of distinct gene networks (Ramos et al., 2010; Sen et al., 2019). Chemical and mutational strategies to dissect the functions of these large, multi-domain proteins are underway by several groups, and should provide clarification to the roles of these HAT proteins.
In addition to EP300 and CBP, chemical probes targeting P300/CBP-associated factor (PCAF) and the closely related general control nonderepressible 5 (GCN5) have proven promising for the understanding of cancer. PCAF appears to play context-dependent roles as tumor growth repressor or promoter (Jia et al., 2016; Malatesta et al., 2013). With the advent of several potent and selective bromodomain inhibitors including the pyridazinone GSK4027 and triazolopthalazine-based L-Moses (Table S1), the field has moved towards the development of PROTACs to clarify key questions about protein function. After bromodomain inhibition proved unsuccessful, the first PROTAC for PCAF, GSK983/GSK699 was developed (Table S1). Novel inhibitors of other acetyltransferases, including the KAT6 and KAT7 members TIP60, MOZ and HBO1 have been described (Table S1). While studies have helped in clarifying the roles of acetylation on transcription in cancer and other diseases (Baell et al., 2018), much is left to be discovered.
Histone acetylation is opposed, in part, by histone methylation. Histone methylation was thought to be irreversible until the initial discovery of the first demethylase, LSD1/KDM1A (Shi et al., 2004). Methylation has been associated with both promotion and suppression of transcription, enhancer and promoter function and modulation of signaling pathways, and has been reviewed in detail (Jambhekar et al., 2019). Here, we discuss recent findings with key histone methyltransferases and how chemical perturbation has uncovered insights into transcriptional regulation in cancer.
Advancements in small molecule agents against protein lysine methyltransferases (PKMTs) have been developed in the past few years (Morera et al., 2016). EPZ0014777, the first reported inhibitor against the H3K79 histone methyltransferase DOT1-like Histone Lysine Methyltransferase (DOT1L), has been used extensively in studies of MLL-rearranged (MLL-r) leukemia (Daigle et al., 2011). Its derivative with optimized pharmacokinetic properties, EPZ5676 (also known as Pinometostat), has advanced into clinical trials in MLL-r leukemias (Morera et al., 2016) and represents a hallmark of the therapeutic potential of targeting PKMTs. The mechanism of EPZ5676 treatment in leukemia cells was narrowed to implicate a functional role for H3K79me2/me3 in chromatin accessibility, acetylation and binding of TFs at H3K79me2/me3-marked enhancers (Godfrey et al., 2019). A separate approach to targeting the MLL complex by targeting the MLL interaction with Menin with small molecules has recently been reported. This results in potent loss of H3K4me3 and inhibition of AML cell growth (Krivtsov et al., 2019). The approach of targeting an interaction domain on MLL to disrupt context-dependent function required significant structural insights, and represents a powerful example for alternatives to classical enzyme inhibition as a mechanism of focused disruption (Krivtsov et al., 2019).
Among the most studied methyltransferases is the catalytic unit of the polycomb repressor complex 2 (PRC2), Enhancer of Zeste Homolog 2 (EZH2), which has a variety of roles as a master regulator of transcription by catalyzing H3K27me3 (Kim and Roberts, 2016). Using EZH2 inhibitors, such as GSK126, EPZ6438, EPZ011989 and JQEZ5, EZH2’s role has been probed in several tumor types (McCabe et al., 2012; Zhang et al., 2016) (Table S1). In diffuse intrinsic pontine glioma (DIPG), which harbors the signature H3K27M mutation resulting in chromatin hyperacetylation, EZH2 activity is still necessary for cell proliferation in vitro and in vivo (Mohammad et al., 2017; Piunti et al., 2017). EZH2 inhibition targets a proneural subset of glioma stem cells in glioblastoma both in cell culture and in vivo (Jin et al., 2017) and similar inhibitors have identified a role for EZH2 in maintaining MYCN expression in MYCN-amplified neuroblastoma (Chen et al., 2018). Further, EZH2 inhibitors synergize with the HDAC inhibitor panobinostat, indicating a tractable mechanism for inhibition of tumor oncogenes through regulation of multiple histone binding proteins. To this end, targeting EZH2 in combination with BRD4 and MAPK pathway inhibitors is sufficient to overcome epigenetic cross-talk and compensatory mechanisms that promote H3K27ac in EHZ2-aberrant tumors (Huang et al., 2018). These added layers of epigenetic complexity provide mechanisms for overcoming drug resistance and driving antitumor activity through molecular synergy. Several studies have investigated targeting PRC2 component members by generating PROTACs for EZH2 and EED that have effects on EZH2, EED and SUZ12 (Table S1). These valuable additions to the chemical toolbox will be important to dissect the function of the PRC2 complex in cancer and non-neoplastic cells. Recently, the EZH2 inhibitor, Tazverik (tazemetostat, EPZ6438, Epizyme) has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of epithelioid sarcoma, further indicating the translational potential of compounds targeting epigenetic proteins.
In addition to PKMTs, protein arginine methyltransferases (PRMTs) have defined roles in cancer stemness, gene expression, mRNA splicing, and the DNA damage response and therefore represent excellent targets for small molecule development (Jarrold and Davies, 2019). PRMT5 inhibitors like EPZ015666 (GSK3235025), EPZ019997 (GSK3368715), and HLCL-61 have gained momentum due to PRMT5’s defined role in a variety of cancers including lymphomas and AML (Table S1). In mantle cell lymphoma (MCL), EPZ015666 treatment prevents SMD3 methylation, resulting in cell death (Chan-Penebre et al., 2015; Tarighat et al., 2016). Similarly, HLCL-61 increased expression of miR-29b, leading to loss of Sp1 and FLT3 expression and AML cell death (Tarighat et al., 2016). In addition, several PRMT4 inhibitors have been recently reported (Table S1). Continued investigation of the role of PRMTs, in different cancer models will be facilitated by an expansion of chemical inhibitors and degraders targeting this intriguing class of epigenetic regulators.
The enzymes that write and erase other less well characterized histone modifications, such as reversible serotonylation, crotonylation, glycation, phosphorylation, and acylation, as listed in Fig. 2 are not discussed further in this review due to their limited inhibitor development, though they remain intriguing concepts to explore through further analysis (Farrelly et al., 2019; Jain and Patel, 2019; Martire et al., 2019; Sabari et al., 2017; Shiio and Eisenman, 2003; Zhang et al., 2019; Zheng et al., 2019).
Targeting Epigenetic Eraser Proteins
Epigenetic modifications are removed by epigenetic “erasers,” including histone deacetylases (HDACs) and lysine demethylases (KDMs) (Li and Seto, 2016; Thinnes et al., 2014). HDACs, in particular, have been excellent targets for drug development, yielding multiple FDA approved drugs, including romidepsin, SAHA, belinostat and panobinostat, for a variety of hematologic malignancies (Romero, 2019) (Table S1). Much like other epigenetic proteins, different cancer subtypes display sensitivity to inhibition or genetic loss of distinct HDACs (Corsello et al., 2017; Gryder et al., 2019b; Tsherniak et al., 2017). The development of a wide array of HDAC targeted inhibitors has led to a chemical toolset to explore the disparate mechanisms of these closely related but distinct proteins in various types of cancer. Recently, we employed a set of epigenetic targeting compounds including the class I HDACs (1,2,3 but not HDAC8) inhibitors Merck60, entinostat, and LW3, to identify that class I HDACs are required for core regulatory transcription in rhabdomyosarcoma cells (Gryder et al., 2019b). HDAC function was required to avoid super-enhancer hyperacetylation, resulting in disruption of three-dimensional chromatin architecture through changes in the charge of phase condensates and loss of RNA polymerase at these sites (Gryder et al., 2019a). These findings are in addition to studies demonstrating that HDAC activity promotes processive transcriptional elongation by both eviction of NELF at promoters (Greer et al., 2015) and release of chromatin bound BRD4 (Hu et al., 2014), providing a potential mechanism for the observation that HDAC inhibitors synergize with BRD4 inhibitors to drive cell death (Fiskus et al., 2014). Several HDAC6-specific degraders have been recently reported, which provides an opportunity to study HDAC6 function by inhibition and degradation in cancer, for example, in melanoma where it promotes tumor growth, and in glioblastoma where it regulates autophagy and migration (Liu et al., 2019; Yang et al., 2018). The use of HDAC inhibitors, however, is complicated by the finding that distinct HDACs may target a variety of overlapping and distinct histone and non-histone substrates in vitro. Further, some HDAC inhibitors such as CUDC-907 and romidepsin display effects on both HDACs and the PI3K pathway (Qian et al., 2012; Saijo et al., 2012). One potential solution to this issue would be an unbiased comparative proteomic time-based resolution of HDAC inhibitor function by global acetylome profiling, as has recently been demonstrated for acetyltransferase inhibitors (Weinert et al., 2018).
Histone lysine demethylases (KDMs) regulate the epigenetic landscape in multiple tumor subtypes, including neuroblastoma, AML breast cancer, and Ewing sarcoma (Thinnes et al., 2014). These occur through direct regulation of histone methylsubstrates, including H3K4me1/2 for KDM1, H3K4me3 for KDM5, H3K27me3 for KDM6 and H3K9me2/3 for KDM4 proteins, among others (Zhao and Shilatifard, 2019). While KDM1 proteins use FAD as a cofactor to catalyze demethylation of a substrate, KDM2–7 perform this function using a homologous Jumonji C-terminal domain (JmjC), which uses elemental iron and ⍺-ketoglutarate as cofactors to produce a demethylation reaction (Thinnes et al., 2014). LSD1/KDM1A inhibitors, such as GSK-LSD1, have entered clinical trials (Fu et al., 2017). LSD1 inhibitors, however, display off target toxicities on the hematopoietic system, including thrombocytopenia and neutropenia, which has limited their use (Fang et al., 2019).
Many more KDM2–7 protein inhibitors, including pan and selective inhibitors have been reported, however, unlike GSK-LSD1, their use to interrogate cancer cell growth has been limited by a lack of effective cellular activity or in vivo activity. Unique to this group are inhibitors of KDM5, such as CPI-455, that along with inhibiting KDM5, reduce the viability of drug tolerant persister cells (Vinogradova et al., 2016). Of all KDM5 inhibitors, however, compound C49 and its derivatives are the most commonly used chemical probes (Table S1). These inhibitors have been used for targeting transcription in cancer, and with an increasing understanding of KDM5’s role in regulating oncogenesis, its validity as a target for therapeutic intervention has only increased. Using KDM5 inhibitors, roles have been identified for the protein in driving lineage-specific cancer cell growth and chemoresistance (Paroni et al., 2019). Similarly, KDM6 inhibitors, such as GSK-J4 that targets KDM6A (UTX) and KDM6B (JMJD3), displays preclinical activity in multiple tumor models (Table S1) (Hong et al., 2019; Lochmann et al., 2018). Although targeting KDMs by small molecule inhibition has been shown to have important effects, there has been relatively limited development of compounds targeting KDM proteins outside of inhibitors. This suggests alternative methodologies, such as peptidomimetics and degraders, as a potential avenue of interest.
Targeting Nucleosome Remodeling Factors
A critical early step in the establishment of DNA that is accessible to TFs and epigenetic modifying enzymes depends on the activity of ATP-dependent chromatin remodeling complexes that act to pack and unpack chromatin structure. These complexes, including the SWI/SNF (BAF), ISWI, NuRD/Mi-2/CHD, INO80 complexes, modulate chromatin by removing, translocating and restructuring the nucleosome, such that DNA becomes accessible to proteins including TFs, co-activator proteins and the RNA polymerase complex (Wang et al., 2007). In addition to nucleosome remodeling, the ISWI and BAF complexes also mediate binding of CTCF and CoREST complexes to the three-dimensional chromatin architecture (Aitken et al., 2018; Inoue et al., 2019), making them key targets as these processes are dysfunctional in cancer. This has resulted in increased attention on the BAF complex in recent years, fueling a deeper understanding of structure, and therefore, increased potential for the development of inhibitors.
Mammalian BAF is a multisubunit enzyme containing complex including helicases and ATPases, along with subunits that coordinate complex formation. Distinct BAF complexes display different subunit compositions, resulting in an opportunity to target different complexes through targeted chemical inhibition of specific subunits. The BAF core complexes contain SMARCA2 and SMARCA4, homologous proteins with both ATPase activity and bromodomains. The development of the potent and selective SMARCA4 bromodomain inhibitor PFI-3 (Fedorov et al., 2015) identified that the bromodomain of SMARCA2/4 is not essential for BAF complex function in these cells (Vangamudi et al., 2015). On the other hand, a recently reported inhibitor, compound 14, blocks the ATPase catalytic function of SMARCA2/4, resulting in broad and striking effects on cancer cell growth (Papillon et al., 2018). These data emphasize the relative importance of the ATPase domain to BAF complex function. Recently, a PROTAC directed toward the bromodomain of SMARCA4, ACBI1, was developed that has potent effects on SMARCA2/4-dependent leukemia cell lines and no effects on SMARCA2/4-deficient cell lines, indicating on-target activity (Farnaby et al., 2019).
Independent of targeting the catalytic units of the BAF complex, newer compounds such as the 12-membered macrolactam compounds like BRD-K98645985 (BD-98) are useful for binding ARID1A-containing BAF complexes, resulting in physical displacement of these complexes from chromatin in ES cells and HCT116 colorectal carcinoma cells (Chory et al., 2019; Marian et al., 2018). Further examination of the effects of BD-98 on ARID1A-mediated chromatin interactions and BAF complex assembly are necessary to fully decipher its mechanism of action. These findings have provided deep insights into BAF complex function through interrogation of individual complex members, and sparked significant interest in further exploration.
In addition to ARID1A and SMARCA2/4, BRD7 and BRD9 of the BAF complex play crucial roles in cancer. Recently, selective BRD9 bromodomain inhibitors have been reported, including BI-9564, and I-BRD9 (Table S1). In AML cells, selective inhibition of BRD9 caused suppression of c-MYC transcription, cell cycle arrest and differentiation, with limited effects on other BAF dependent cell lines (Hohmann et al., 2016). The BRD9 inhibitor BI-9564 was further used as the substrate for a PROTAC, dBRD9. Though it has promising activity, the antitumor effect of dBRD9 is not as severe as SMARCA4 knockout (Brien et al., 2018a), highlighting the key role of the ATPase function of BAF complexes. Despite these findings, synovial sarcoma and malignant rhabdoid tumor cell lines that are characterized by dysfunctional BAF complexes are uniquely sensitive to loss of BRD9, through either genetic means or selective degradation with dBRD9 (Michel et al., 2018). These data suggest that alterations in BAF complex structure may yield unique dependency on non-catalytic subunits. Independently, malignant rhabdoid tumor cell lines were demonstrated to be sensitive to the combined BRD7 and BRD9 degrader VZ185, further supporting these findings (Zoppi et al., 2019). With the recent cryo-EM structure of nucleosome-bound BAF complexes being solved, deep mechanistic insights into the results of inhibition or degradation of specific subunits are likely to be identified (He et al., 2020).
Combination and Chimaeric Inhibitors
One key issue with compounds targeting epigenetic proteins, however, remains the rapidity over which they act -often requiring several cell cycles in order to elicit their maximal effects. Thus, interest has turned to combination of compounds to elicit stronger and more rapid effects. In addition to combination of agents, an alternative approach is to develop inhibitors that target multiple proteins with a single molecule. To this end, one bifunctional molecule has been reported termed Corin, that targets both LSD1 and HDAC1 simultaneously (Anastas et al., 2019; Kalin et al., 2018) (Table S1). This molecule blocks the function of two members of the CoREST complex, and has striking effects in models of glioma and melanoma. While preliminary, this strategy represents a novel and potentially useful method for inhibiting multiple subunits within a single complex, or in addition, between complexes, each of which may be selectively required for tumor cell growth.
Conclusions and Future Directions
Exciting advances remain on the horizon for the generation of compounds targeting epigenetic readers, writers and erasers. Along with the central role of epigenetic proteins in regulating cell state, differentiation and growth has been the recognition that they are often dysregulated in disease states, and therefore act as key mediators of tumor cell growth, survival and apoptosis. In parallel, by harnessing structural information to elicit focused efforts by many groups to derive targeted chemical inhibitors with selectivity for one member of a protein class over all others, there is now a preliminary “toolbox” of compounds for use in a variety of tumor states. The power of genome-scale functional genomics, coupled with new chemical methodologies, including structural-based peptidomimetics and PROTAC technology, has permitted rapid advances in pharmacological modeling of knockout methods, in addition to inhibition, and this has yielded deep insights into the differential importance of full length protein products, compared to inhibition of specific domains.
Despite these advances, several key tools remain missing and are priorities for development. For many described classes of epigenetic enzymes, even tool compounds for selective inhibition are currently lacking. Large-scale, non-profit groups focused on targeting these epigenetic enzymes with open-source chemical probes, such as the Structural Genomics Consortium and Chemical Probe Portal (Muller et al., 2018), have set the tone for appropriate resource sharing. With the rising use of functional genomics to define the genes required for tumor-specific cell growth, we are poised to define important targets for inhibition or degradation. Thus, key future directions include focused derivation of a toolbox of compounds to the targets most highly relevant to individual tumor states, which may include MYC family oncogenes, multiple subunits of key regulatory complexes such as the BAF complex in synovial sarcoma or the MLL complex in leukemia, in addition to the aberrant transcriptional complexes formed by chimaeric fusion oncoproteins, such as PAX3-FOXO1, EWS-FLI1, TMPRSS2-ERG, ETV6-NTRK3 in rhabdomyosarcoma, Ewing sarcoma, prostatic carcinoma and infantile fibrosarcoma, among many others. Finally, as described, the role of novel regulators yet to be defined that perform only recently described histone modifications including serotonylation, crotonylation, lactylation, phosphorylation and ubiquitination/SUMOylation remain to be explored, and chemical targeting of these modifications will be a useful adjunct to genetic studies to identify the role of these and other future epigenetic post-translational modifications.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank Dr. Yan Zi Au and Dr. Milka Kostic for their valuable comments. We apologize to authors of studies which, for reasons of text space, could not be referenced within this work. This work was supported in part by grants from the National Cancer Institute (NCI) P01-CA066996-19, U54HD093540-03, and P50-CA100707-15 (to J.Q.), Lymphoma Leukemia Society TRP award (to J. Q.), the Alex’s Lemonade Stand Foundation (J.Q., A.D.D), the Damon-Runyon Cancer Research Foundation (DRSG-24-18), Rally Foundation for Childhood Cancer Research, CureSearch For Children’s Cancer Foundation, American Society for Clinical Oncology and the Joey O’Neil Fund (A.D.D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
All chemical structures in this review and their associated references are listed in Supplemental Table 1. Supplementary discussion is provided in a supplementary note.
DECLARATIONS OF INTERESTS
J. Q. is scientific co-founder and consultant for Epiphanes, and share holder of Zentalis.
REFERENCES
- Aitken SJ, Ibarra-Soria X, Kentepozidou E, Flicek P, Feig C, Marioni JC, and Odom DT (2018). CTCF maintains regulatory homeostasis of cancer pathways. Genome Biol 19, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, and Mirsky AE (1964). Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, Blanco MA, Giera S, Gillespie SM, Das J, et al. (2019). Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 36, 528–544 e510. [DOI] [PubMed] [Google Scholar]
- Arrowsmith CH, and Schapira M (2019). Targeting non-bromodomain chromatin readers. Nature structural & molecular biology, 1–7. [DOI] [PubMed] [Google Scholar]
- Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, Nguyen N, Wichmann J, McRae HM, Yang Y, et al. (2018). Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. [DOI] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149 e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher J, Dilworth D, Reiser U, Neumüller RA, Schleicher M, Petronczki M, Zeeb M, Mischerikow N, Allali-Hassani A, Szewczyk MM, et al. (2019). Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nature chemical biology 15, 822–829. [DOI] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, Dillon ET, Cagney G, Van Mierlo G, Baltissen MP, et al. (2018a). Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Stegmaier K, Armstrong SA. Targeting Chromatin Complexes in Fusion Protein-Driven Malignancies. Nat Rev Cancer. 2019. May; 19 (5): 255–269. [DOI] [PubMed] [Google Scholar]
- Burslem GM, and Crews CM (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437. [DOI] [PubMed] [Google Scholar]
- Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, Wang EJ, Veschi V, Lam N, Qi J, et al. (2018). CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest 128, 446–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chory EJ, Kirkland JG, Chang C-Y, D’Andrea VD, Gourinsankar S, Dykhuizen EC, and Crabtree GR (2019). Inhibition of a Selective SWI/SNF Function Synergizes with ATR Inhibitors in Cancer Cell Killing. bioRxiv 660456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, et al. (2017). The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 23, 405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy BM, and Cole PA (2015). Protein lysine acetylation by p300/CBP. Chem Rev 115, 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, Robson SC, Chung C.-w., Hopf C, Savitski MM, et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi A, Sevinc K, Gurhan Sevinc G, Cribbs AP, Philpott M, Uyulur F, Morova T, Dunford JE, Goklemez S, Ari S, et al. (2019). Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat Chem Biol 15, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Liao G, and Yu B (2019). LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol 12, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, et al. (2019). BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nature Chemical Biology 15, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, Zhang B, Loh YE, Ramakrishnan A, Vadodaria KC, et al. (2019). Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, et al. (2015). Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci Adv 1, e1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, Peth K, Portier BP, Rodriguez M, Devaraj SG, et al. (2014). Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther 13, 1142–1154. [DOI] [PubMed] [Google Scholar]
- Fu X, Zhang P, and Yu B (2017). Advances toward LSD1 inhibitors for cancer therapy. Future Med Chem 9, 1227–1242. [DOI] [PubMed] [Google Scholar]
- Godfrey L, Crump NT, Thorne R, Lau IJ, Repapi E, Dimou D, Smith AL, Harman JR, Telenius JM, Oudelaar AM, et al. (2019). DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat Commun 10, 2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CB, Tanaka Y, Kim YJ, Xie P, Zhang MQ, Park IH, and Kim TH (2015). Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep 13, 1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian GP, Segura-Wang M, Brabetz S, et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555, 321–327. [DOI] [PubMed] [Google Scholar]
- Gryder BE, Pomella S, Sayers C, Wu XS, Song Y, Chiarella AM, Bagchi S, Chou H-C, Sinniah RS, Walton A, et al. (2019a). Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nature Genetics 51, 1714–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder BE, Wu L, Woldemichael GM, Pomella S, Quinn TR, Park PMC, Cleveland A, Stanton BZ, Song Y, Rota R, et al. (2019b). Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat Commun 10, 3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li J, Liu B, and Xu Y (2020). Structure of nucleosome-bound human BAF complex. Science 367, 875–881. [DOI] [PubMed] [Google Scholar]
- Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, McConnell D, Pearson M, Gerstberger T, et al. (2016). Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol 12, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong BJ, Park WY, Kim HR, Moon JW, Lee HY, Park JH, Kim SK, Oh Y, Roe JS, and Kim MY (2019). Oncogenic KRAS Sensitizes Lung Adenocarcinoma to GSK-J4-Induced Metabolic and Oxidative Stress. Cancer Res 79, 5849–5859. [DOI] [PubMed] [Google Scholar]
- Hu X, Lu X, Liu R, Ai N, Cao Z, Li Y, Liu J, Yu B, Liu K, Wang H, et al. (2014). Histone cross-talk connects protein phosphatase 1alpha (PP1alpha) and histone deacetylase (HDAC) pathways to regulate the functional transition of bromodomain-containing 4 (BRD4) for inducible gene expression. J Biol Chem 289, 23154–23167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, Wei R, Zheng XL, Liu Z, Zhang X, et al. (2018). Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell 175, 186–199 e119. [DOI] [PubMed] [Google Scholar]
- Inoue D, Chew GL, Liu B, Michel BC, Pangallo J, D’Avino AR, Hitchman T, North K, Lee SC, Bitner L, et al. (2019). Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 574, 432–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain PG, and Patel BD (2019). Medicinal chemistry approaches of poly ADP-Ribose polymerase 1 (PARP1) inhibitors as anticancer agents - A recent update. Eur J Med Chem 165, 198–215. [DOI] [PubMed] [Google Scholar]
- Jambhekar A, Dhall A, and Shi Y (2019). Roles and regulation of histone methylation in animal development. Nat Rev Mol Cell Biol 20, 625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrold J, and Davies CC (2019). PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med 25, 993–1009. [DOI] [PubMed] [Google Scholar]
- Jia YL, Xu M, Dou CW, Liu ZK, Xue YM, Yao BW, Ding LL, Tu KS, Zheng X, and Liu QG (2016). P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis 7, e2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, Gimple RC, Wang X, Mack SC, Miller TE, et al. (2017). Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med 23, 1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin JH, Wu M, Gomez AV, Song Y, Das J, Hayward D, Adejola N, Wu M, Panova I, Chung HJ, et al. (2018). Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, and Roberts CW (2016). Targeting EZH2 in cancer. Nat Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, Ross KN, Perner F, Olsen SN, Pritchard T, et al. (2019). A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 36, 660–673 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, and Seto E (2016). HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell TJ, Weinberg F, Morris PW, Roeder RG, and Rutter WJ (1970). Specific inhibition of nuclear RNA polymerase II by alpha-amanitin. Science 170, 447–449. [DOI] [PubMed] [Google Scholar]
- Liu JR, Yu CW, Hung PY, Hsin LW, and Chern JW (2019). High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem Pharmacol 163, 458–471. [DOI] [PubMed] [Google Scholar]
- Lochmann TL, Powell KM, Ham J, Floros KV, Heisey DAR, Kurupi RIJ, Calbert ML, Ghotra MS, Greninger P, Dozmorov M, et al. (2018). Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, Zhou X, Li Y, Rusch MC, Easton J, et al. (2018). Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, and Helin K (2013). Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res 73, 6323–6333. [DOI] [PubMed] [Google Scholar]
- Marian CA, Stoszko M, Wang L, Leighty MW, de Crignis E, Maschinot CA, Gatchalian J, Carter BC, Chowdhury B, Hargreaves DC, et al. (2018). Small Molecule Targeting of Specific BAF (mSWI/SNF) Complexes for HIV Latency Reversal. Cell Chem Biol 25, 1443–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC, Tastemel M, and Banaszynski LA (2019). Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat Genet 51, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Michaelides MR, Kluge A, Patane M, Van Drie JH, Wang C, Hansen TM, Risi RM, Mantei R, Hertel C, Karukurichi K, et al. (2018). Discovery of Spiro Oxazolidinediones as Selective, Orally Bioavailable Inhibitors of p300/CBP Histone Acetyltransferases. ACS Med Chem Lett 9, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, Valencia AM, Zhou Q, Bocker M, Soares LMM, et al. (2018). A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol 20, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Morera L, Lubbert M, and Jung M (2016). Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Ackloo S, Arrowsmith CH, Bauser M, Baryza JL, Blagg J, Bottcher J, Bountra C, Brown PJ, Bunnage ME, et al. (2018). Donated chemical probes for open science. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nady N, Krichevsky L, Zhong N, Duan S, Tempel W, Amaya MF, Ravichandran M, and Arrowsmith CH (2012). Histone Recognition by Human Malignant Brain Tumor Domains. Journal of Molecular Biology 423, 702–718. [DOI] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C. w., Chandwani R, Marazzi I, Wilson P, Coste H, et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, and Travers AA (2000). The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J 19, 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon JPN, Nakajima K, Adair CD, Hempel J, Jouk AO, Karki RG, Mathieu S, Mobitz H, Ntaganda R, Smith T, et al. (2018). Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J Med Chem 61, 10155–10172. [DOI] [PubMed] [Google Scholar]
- Paroni G, Bolis M, Zanetti A, Ubezio P, Helin K, Staller P, Gerlach LO, Fratelli M, Neve RM, Terao M, et al. (2019). HER2-positive breast-cancer cell lines are sensitive to KDM5 inhibition: definition of a gene-expression model for the selection of sensitive cases. Oncogene 38, 2675–2689. [DOI] [PubMed] [Google Scholar]
- Perfetti MT, Baughman BM, Dickson BM, Mu Y, Cui G, Mader P, Dong A, Norris JL, Rothbart SB, Strahl BD, et al. (2015). Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53BP1. ACS chemical biology 10, 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, Atoyan R, Qu H, Yin L, Samson M, et al. (2012). Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res 18, 4104–4113. [DOI] [PubMed] [Google Scholar]
- Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, Haverty PM, Bainer R, Cheung T, Arnott D, et al. (2018). Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep 24, 1722–1729. [DOI] [PubMed] [Google Scholar]
- Ramos YF, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, van Dam H, van Ommen GJ, den Dunnen JT, Zantema A, et al. (2010). Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res 38, 5396–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, et al. (2015). Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chemistry & biology 22, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D (2019). HDAC inhibitors tested in phase III trial. Nat Rev Clin Oncol 16, 465. [DOI] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, and Zhao Y (2017). Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Katoh T, Shimodaira H, Oda A, Takahashi O, and Ishioka C (2012). Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors. Cancer Sci 103, 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago C, Nguyen K, and Schapira M (2011). Druggability of methyl-lysine binding sites. J Comput Aided Mol Des 25, 1171–1178. [DOI] [PubMed] [Google Scholar]
- Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, Frederick B, Chen Q, Luense LJ, Garcia BA, et al. (2019). Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol Cell 73, 684–698 e688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shiio Y, and Eisenman RN (2003). Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A 100, 13225–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, and Crews CM (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Groves MR, Sinning I, and Sattler M (2003). High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J Mol Biol 327, 507–520. [DOI] [PubMed] [Google Scholar]
- Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, Wang H, Huang X, Alinari L, Walker A, et al. (2016). The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, and Hopkinson RJ (2014). Targeting histone lysine demethylases - progress, challenges, and the future. Biochim Biophys Acta 1839, 1416–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, et al. (2015). The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Research 75, 3865–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, Williamson KE, Guler GD, Gangurde P, Manieri W, et al. (2016). An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nature Chemical Biology 12, 531–538. [DOI] [PubMed] [Google Scholar]
- Wang GG, Allis CD, and Chi P (2007). Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends in Molecular Medicine 13, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Song Y, Xie H, Wu H, Wu YT, Leisten ED, and Tang W (2018). Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett 28, 2493–2497. [DOI] [PubMed] [Google Scholar]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F, Lin CY, Perry JA, Lawlor MA, Federation A, et al. (2016). Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer discovery 6, 1006–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, and Shilatifard A (2019). Epigenetic modifications of histones in cancer. Genome Biol 20, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Omans ND, Leicher R, Osunsade A, Agustinus AS, Finkin-Groner E, D’Ambrosio H, Liu B, Chandarlapaty S, Liu S, et al. (2019). Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat Commun 10, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, Spallarossa A, et al. (2019). Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J Med Chem 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucconi BE, Makofske JL, Meyers DJ, Hwang Y, Wu M, Kuroda MI, and Cole PA (2019). Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry 58, 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.