

1. Introduction
The sheer complexity of the nervous system, the clinical and biological heterogeneity of affected patients, and limited approaches to access relevant tissues for disease modeling have together made the investigation of schizophrenia persistently daunting. New hope has been generated by the still recent advent of human induced pluripotent stem cell (hiPSC) models of neuropsychiatric conditions. The reprogramming of hiPSCs into defined cell types is a technical advancement that has made available living neural tissues derived from patients and controls for studying neurobiological abnormalities driving disease pathology, modeling the impact of genetic and non-genetic risk factors, and testing interventions—whether genetic or pharmacologic—to prevent or reverse disease-associated phenotypes. As a result, cell reprogramming enables both patient-specific study of genetic disease and in vitro modeling of the complex genetic risk factors underlying neurological disorders such as schizophrenia.
In this chapter, we preview the syndromal presentations of schizophrenia and its treatment to provide the reader with clinically relevant information regarding both the severity of the disorder and the challenges associated with its management. As needed for integrating findings discussed later in the chapter, we highlight key genomic and biological processes disrupted in schizophrenia, with an emphasis on the essential role of neurodevelopmental perturbations in disease etiology. After previewing relevant methodological considerations, we discuss many of the studies that have employed hiPSC-based models to provide fundamental insights into mechanisms of disease in schizophrenia. We end with an overview of innovations that will facilitate the continued usefulness of hiPSCs for studying schizophrenia and for ultimately improving the lives of those affected by it.
2. Overview of Schizophrenia
2.1. Clinical Presentations of Schizophrenia
Schizophrenia is a neuropsychiatric condition that can have devastating impacts on the lives of patients and those around them. As is the case with all psychiatric disorders, diagnosis of schizophrenia is made based upon the presence of specific signs and symptoms that have persisted for a specified period of time, are not attributable to organic disease or substance use, and that cause significant distress, disability, and/or impairment in functioning. Unfortunately, there are no biologically informed tests or criteria that can be used to confirm the presence of schizophrenia, and there is notable heterogeneity among patients despite sharing an identical diagnosis.
Phenomenological descriptions of schizophrenia have varied throughout history and by clinical assessment methodologies. The development of symptom-rating scales and their widespread application across numerous patient samples and contexts served as key empirical foundations for disease description. Factor analyses [1–4] originally clustered the clinical features of schizophrenia into three subsyndromes, each relating to a specific group of defined symptoms: (1) psychotic symptoms (e.g., delusions and hallucinations), (2) negative symptoms (e.g., anhedonia, alogia, social withdrawal, blunted or flat affect), and (3) disorganized symptoms (e.g., thought disorder, bizarre behaviors). In recent years, particularly with the advent and extensive application of the Positive and Negative Syndrome Scale (PANSS) [5], robust support has been provided for an expanded Five Factor Model [6, 7] of schizophrenia that includes the addition of two more dimensions: (1) depression and anxiety and (2) agitation. These two different syndromal models of schizophrenia and their constituent signs and symptoms are listed and defined in Tables 1 and 2.
Table 1.
Definition of factor item on the positive and negative syndrome scale (PANSS)
| Factor item | Definition |
|---|---|
| Delusions | Beliefs which are unfounded, unrealistic, and idiosyncratic |
| Hallucinations | Verbal report or behavior indicating perceptions which are not generated by external stimuli. May occur in the auditory, visual, olfactory, or somatic realms |
| Grandiosity | Exaggerated self-opinion and unrealistic convictions of superiority, including delusions of extraordinary abilities, wealth, knowledge, fame, power, and moral righteousness |
| Suspiciousness/persecution | Unrealistic or exaggerated ideas of persecution, as reflected in guardedness, a distrustful attitude, suspicious hypervigilance, or frank delusions that others mean one harm |
| Unusual thought content | Thinking characterized by strange, fantastic, or bizarre ideas, ranging from those which are remote or atypical to those which are distorted, illogical, and patently absurd |
| Blunted affect | Diminished emotional responsiveness as characterized by a reduction in facial expression, modulation of feelings, and communicative gestures |
| Emotional withdrawal | Lack of interest in, involvement with, and affective commitment to life’s events |
| Poor rapport | Lack of interpersonal empathy, openness in conversation, and sense of closeness, interest, or involvement with the interviewer |
| Passive/apathetic social withdrawal | Diminished interest or initiative in social interactions due to passivity, apathy, anergy, or avolition. This leads to reduced interpersonal involvement and neglect of activities of daily living |
| Lack of spontaneity and flow of conversation | Reduction in the normal flow of communication associated with apathy, avolition, defensiveness, or cognitive deficit. This is manifested by diminished fluidity and productivity of the verbal- interactional process |
| Motor retardation | Reduction in motor activity as reflected in slowing or lessening of movements and speech, diminished responsiveness to stimuli, and reduced body tone |
| Conceptual disorganization | Disorganized process of thinking characterized by disruption of goal-directed sequencing, e.g., circumstantiality, tangentiality, loose associations, non-sequiturs, gross illogicality, or thought block |
| Difficulty in abstract thinking | Impairment in the use of the abstract-symbolic mode of thinking, as evidenced by difficulty in classification, forming generalizations, and proceeding beyond concrete or egocentric thinking in problem- solving tasks |
| Abnormal mannerisms and posturing | Unnatural movements or posture as characterized by an awkward, stilted, disorganized, or bizarre appearance |
| Poor attention | Failure in focused alertness manifested by poor concentration, distractibility from internal and external stimuli, and difficulty in harnessing, sustaining, or shifting focus to new stimuli |
| Excitement | Hyperactivity as reflected in accelerated motor behavior, heightened responsivity to stimuli, hypervigilance, or excessive mood lability |
| Hostility | Verbal and nonverbal expressions of anger and resentment, including sarcasm, passive-aggressive behavior, verbal abuse, and assaultiveness |
| Uncooperativeness | Active refusal to comply with the will of significant others, including the interviewer, hospital staff, or family, which may be associated with distrust, defensiveness, stubbornness, negativism, rejection of authority, hostility, or belligerence |
| Poor impulse control | Disordered regulation and control of action on inner urges resulting in sudden, unmodulated, arbitrary, or misdirected discharge of tension and emotions without concern about consequences |
| Anxiety | Subjective experience of nervousness, worry, apprehension, or restlessness, ranging from excessive concern about the present or future to feelings of panic |
| Feelings of guilt | Sense or remorse or self-blame for real or imagined misdeeds in the past |
Table 2.
Dimensional models of schizophrenia: the three factor and five factor models
| Three factor model | ||||
| Positive | Negative | Disorganized | ||
| Delusions | Blunted affect | Conceptual disorganization | ||
| Hallucinations | Emotional withdrawal | Difficulty in abstract thinking | ||
| Grandiosity | Poor rapport | Abnormal mannerisms and posturing | ||
| Suspiciousness/persecution | Passive/apathetic social withdrawal | Poor attention | ||
| Unusual thought content | Lack of spontaneity and flow of conversation | |||
| Motor retardation | ||||
| Five factor model | ||||
| Positive | Negative | Disorganized | Agitation and hostility | Depression and anxiety |
| Delusions | Blunted affect | Conceptual disorganization | Excitement | Anxiety |
| Hallucinations | Emotional Withdrawal | Difficulty in abstract thinking | Hostility | Feelings of guilt |
| Grandiosity | Poor rapport | Poor attention | Uncooperativeness | Depression |
| Unusual thought content | Passive/apathetic social withdrawal | Poor impulse control | ||
| Lack of spontaneity and flow of conversation | ||||
Notwithstanding the recent advances in understanding the full spectrum of symptom dimensions in schizophrenia, criteria for the disorder employ a categorical approach for diagnosis. According to the criteria outlined in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) [8], the patient must present with two or more of the following for a majority of the time for a period of at least 1 month: delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behavior, and negative symptoms. Furthermore, the DSM-5 specifies that one of those two (or more) must include one of the first three (i.e., delusions, hallucinations, disorganized speech). Overall symptoms, including the month of those outlined above along with prodromal and/or residual signs and symptoms, must persist for at least 6 months and cause significant functional impairment in one or more domains of life such as work, education, or interpersonal relationships. Finally, schizoaffective disorder, bipolar disorder, and major depressive disorder with psychotic features must be ruled out, and the symptoms cannot be attributable to substance abuse or medical illness (Table 3).
Table 3.
Diagnostic criteria for schizophrenia in the DSM-5
| 1. Two or more of the following for at least a one-month (or longer) period of time, and at least one of them must be 1, 2, or 3: |
| • Delusions |
| • Hallucinations |
| • Disorganized speech |
| • Grossly disorganized or catatonic behavior |
| • Negative symptoms, such as diminished emotional expression |
| 2. Impairment in one of the major areas of functioning for a significant period of time since the onset of the disturbance: Work, interpersonal relations, or self-care |
| 3. Some signs of the disorder must last for a continuous period of at least 6 months. This six-month period must include at least 1 month of symptoms (or less if treated) that meet criterion A (active phase symptoms) and may include periods of residual symptoms. During residual periods, only negative symptoms may be present |
| 4. Schizoaffective disorder and bipolar or depressive disorder with psychotic features have been ruled out: |
| • No major depressive or manic episodes occurred concurrently with active phase symptoms |
| • If mood episodes (depressive or manic) have occurred during active phase symptoms, they have been present for a minority of the total duration of the active and residual phases of the illness |
| 5. The disturbance is not caused by the effects of a substance or another medical condition |
| 6. If there is a history of autism spectrum disorder or a communication disorder (childhood onset), the diagnosis of schizophrenia is only made if prominent delusions or hallucinations, along with other symptoms, are present for at least 1 month |
2.2. Strategies for the Treatment of Schizophrenia
Since the mid-twentieth century the mainstay of pharmacologic treatment of schizophrenia has been dopaminergic receptor antagonism with antipsychotic medication. The incidental discovery by French surgeon Henri Laborit that chlorpromazine had strikingly calming effects on patients preparing to undergo surgical procedures was the impetus to the medication’s widespread use in psychotic patients beginning in 1952 [9]. Soon after, several other antipsychotics were synthesized, and it was discovered that the potency of these agents was mediated by blockade of dopamine 2 receptors [10] and that there was a direct, positive correlation between an antipsychotic’s efficacy and its affinity for the D2 receptor [11, 12]. In the decades that followed, another type of antipsychotic was discovered that had affinity for both D2 and serotonergic receptors [13], leading to the widespread, although over-simplified and partially inaccurate, grouping of these medications into “first-” versus “second- generation antipsychotics.” Large comparative studies have demonstrated that first- and second-generation antipsychotics have statistically indistinguishable effects on positive symptoms; instead, the two medication classes vary in their side effect profiles, with first-generation antipsychotics causing more extra-pyramidal symptoms and second-generation antipsychotics causing more notable metabolic side effects [14, 15]. One medication that stood out in its effectiveness for treatment-resistant schizophrenia was clozapine, but its substantial burden of side effects, particularly with the relatively common occurrence of agranulocytosis, has limited its use to refractory patients and to those with schizophrenia complicated by suicidality [16]. While the numerous antipsychotic medications available have clear benefit in the amelioration of the positive symptoms of schizophrenia, both first- and second- generation antipsychotics show, at most, only slight improvement in the negative and disorganization symptom dimensions [17, 18], and adverse effects from medications are a leading cause of non-adherence to them [19]. Furthermore, efforts to develop effective antipsychotics acting via other neurotransmitter systems have been disappointing (e.g., the mGluR2/3 agonist pomaglumetad methionil (LY2140023) [20]). These painful realities, along with the enormous burden of schizophrenia for patients, their families, and society, drive the urgent demand for novel approaches to understanding disease biology and therapeutic innovation.
2.3. Outcomes in Schizophrenia
Schizophrenia is associated with an expansive array of adverse outcomes. Despite treatment, at least one-third of patients in developed countries and about 60% of patients in developing countries do not achieve a satisfactory level of remission [21]. As a group, patients with schizophrenia have a life expectancy that is 15–20 years shorter than average [22] due to a multitude of factors. Rates of suicide range from 5 [23] to 13 [24] percent, and in a large study of Chinese patients, individuals with schizophrenia were 23 times more likely to die by suicide [25].
Accordingly, suicide has been shown to account for 28% of the excess mortality in patients with schizophrenia [26]. Additional likely contributing factors are adverse effects of chronic medication use [27], increased risk of several communicable diseases [28], cardiovascular disease and mortality [29], comorbid substance abuse [30], and markedly elevated rates of abuse and assault [31]. Overall, patients with schizophrenia carry a remarkable burden across domains of life, and advances in the understanding of the disease and its treatment are imperative.
3. Neurobiological Considerations of Schizophrenia
Having provided an overview of the clinical aspects of schizophrenia, we will now turn to relevant neurobiological considerations of the disorder. We begin with a discussion of the neurodevelopmental theory of schizophrenia, drawing upon numerous lines of evidence that support this line of thinking. Then, we focus on risk factors for schizophrenia, mentioning in brief those environmental or non-genetic factors, and then focusing more in-depth on the genetics of schizophrenia.
3.1. Schizophrenia as a Disorder of Neurodevelopment
Numerous lines of evidence implicate several abnormalities in neurodevelopment as contributing to schizophrenia (for review, see: [32, 33]). In addition to the impact of risk factors mentioned below that affect in utero development, birth, and early childhood, much additional data point to a “Neurodevelopmental Hypothesis of Schizophrenia.” In this section, we provide a concise review of studies that have generated convincing support of this broader hypothesis.
Findings in Patients During First Episode of Psychosis
Brain development and maturation is a dynamic process beginning in early fetal development and extending up to the third decade of life [34]. Abnormalities documented in the brains of patients with schizophrenia relate strongly to biological processes most active during neurodevelopment, such as neuronal migration [35], proliferation [36], specification [37], maturation [38], and pruning [39]. These facts, coupled with the longitudinal course of brain development, make it exceedingly unlikely that the factors that ultimately contribute to schizophrenia take place in close temporal proximity to disease onset by current diagnostic formulations [38].
In patients presenting with their “First Episode of Psychosis,” extensive reports of widespread brain abnormalities are consistent with a prolonged disease process that ultimately culminates in the manifestation of symptoms warranting acute clinical attention. First episode patients show volumetric abnormalities in the temporal lobe [40]; enlargement of CSF spaces [41]; smaller thalamic nuclei [42]; decreased grey matter in the prefrontal and temporal cortices [43, 44]; diminished grey matter in the frontal and hippocampal gyri [45]; progressively worsening reduction in overall cortical grey matter [46, 47] and in the cingulate and insular cortices [48–50]; abnormal functional connectivity [51]; and finally, disrupted maturation in several white matter tracts [52–55]. Importantly, many of these findings correlate positively with disease progression [47, 50, 52] and symptom severity [43, 45, 51], further supporting the relevance and longitudinal nature of these abnormalities. In sum, these data highly suggest that biological processes underlying schizophrenia begin many years prior to diagnosis, and as shown below, clinical studies of patients prior to diagnosis reveal many corresponding changes in psychological functioning.
Diagnosis of Schizophrenia Is Preceded by a Significant Prodromal Syndrome
Although schizophrenia is most commonly diagnosed in the late adolescent to early adult years, decades of clinical research have described the presence of substantial but non-specific psychopathology prior to disease diagnosis [56]. Regardless of prior risk determination, prodromal patients show markedly high rates of sleep alterations, anxious symptoms, suspiciousness, and non-hallucinogenic perceptual disturbances [57], as well as behavioral changes including decline in academic performance, impaired concentration, and social withdrawal [58–61], accompanied by early neurocognitive deficits (e.g., [62, 63]) that often precede illness diagnosis by many years. Of note, the duration of the prodromal syndrome correlates positively with the magnitude of grey matter volume reduction in several brain regions [64].
The heterogeneity and non-specificity of these signs and symptoms have prompted substantial research efforts into the prediction of later disease onset, particularly among children deemed “high risk” or “ultra-high risk” for schizophrenia based upon family history of the illness and/or early prodromal symptoms [65]. Presymptomatic “high-risk” individuals frequently show social, motor, and cognitive deficits from early age. In infants, delayed attainment of developmental milestones significantly increases risk of later development of schizophrenia in a dose-dependent manner and with additive effects when combined with obstetric complications [66]. Abnormalities beginning as early as age 4 in social behavior, affect, and motor development predicted later diagnosis of schizophrenia [67]. Consistent with this, IQ decline from ages 4 to 7 [68], poorer performance on intelligence and memory tasks at later developmental stages [69, 70] and decreased performance on IQ measures by age 13 predicted also predict schizophrenia [71]. A study of children aged 7–12 years who later developed schizophrenia documented substantial differences in several standardized behavioral metrics compared to children who did not develop the disease, and demonstrated that among these differences, performance in attention, memory, and motor skills predicted later onset of schizophrenia [72]. Moreover, higher rates of social maladjustment are observed in those who later received a diagnosis of schizophrenia [73]. Social and cognitive functioning deficits predict schizophrenia in adolescent males (aged 16–17) up to 10 years following initial testing [74]. Using extant data, investigators [75] have developed risk prediction tools that have high sensitivity (98%), but with less specificity (59%), for later conversion to schizophrenia, with a mean time of 4.3 years for females and 6.7 years for males between age of prodrome onset and diagnosis of schizophrenia [75]. Taken together, the data from these abundant reports show that formal diagnosis of schizophrenia is preceded by deficits across functional domains that often occur several years before onset of the classical disorder. Further strengthening the relevance of these findings are reports of alterations in neuronal functioning that concur with early psychological phenotypes, a topic to which we now turn.
Prodromal Symptoms of Schizophrenia Occur Concomitantly with Brain Abnormalities
Longitudinal neuroimaging studies of patients have documented several abnormalities that precede the diagnosis of schizophrenia. In high-risk persons, smaller grey matter volumes in several brain areas predicted onset of psychosis 1–2 years later [76]. Similar approaches have replicated and expanded on these findings, further implicating differences in subregions of the frontal, temporal, and parietal lobes [77]; thinning of the anterior cingulate cortex [78]; reduced grey matter volume in the insular cortex, [79], the superior temporal gyrus [80], and parahippocampal gyrus [81]; lower prefrontal cortex activation during a working memory task [82]; and generalized cortical thinning [83]. Strikingly, volumetric brain abnormalities have even been documented in the offspring of mothers with schizophrenia at the prenatal stage [84], providing a disease-relevant example of the high degree of heritability of several brain volumes [85]. Reduction in white matter tracts is also seen in patients showing prodromal symptoms, and the severity of the reduction predicts later diagnosis of schizophrenia [86]. Finally, high-risk patients who later transition to psychosis display abnormal white matter integrity up to 2 years prior to diagnosis [87].
Widespread reports of both psychological and biologic phenotypes preceding schizophrenia onset suggest strongly that processes driving disease production occur long before its formal diagnosis. When viewed within the context of the longitudinal course of brain development—spanning about three decades in length— the data discussed here constitute the foundation of a theory in which abnormal neurodevelopmental processes both predate and contribute to the likelihood of schizophrenia manifestation, particularly in those with elevated genetic risk. Although the early emergence of phenotypes associated with future disease incidence is well documented, the extent to which innate (i.e., genetic) and environmental factors contribute causally to their occurrence awaits clarification. This is highly non-trivial distinction to make: while variation in disease states, as well as normal traits, is almost always driven by variable combinations of genetic and non-genetic factors, an understanding of the relative etiologic contributions of each is a critical informant of appropriate and meaningful strategies to prevent or alleviate disease. For those risk factors driven by a primary environmental insult, which of course may interact with genetic predisposition, there is ample opportunity—and, indeed, an imperative need—for early intervention to remove the risk factor or diminish its impact. Conversely, a better understanding of how innate, genetic pathways contribute to the development of schizophrenia may yield innovative treatment approaches that correct or mitigate the consequences of these inborn processes in a manner that improves the chances of averting disease presentation. While expanded and aggressive efforts to improve treatment strategies of bona fide schizophrenia remain imperative, the potential benefits of preventing its occurrence in the first place are unbounded.
3.2. Epidemiology and Risk Factors for Schizophrenia
Risk Factors for Schizophrenia: Environmental Factors
Highly consistent with the neurodevelopmental theory of schizophrenia are reports of numerous non-heritable factors can impact schizophrenia risk, particularly during early childhood and adolescent developmental periods. Several pregnancy- and birth-related complications increase disease risk, including maternal infection [88,89], hypoxic events [90], maternal nutritional deficiency [91, 92], pre-term birth [93], and both low [94, 95] and high [96, 97] birth weight. In the Northern hemisphere, birth in the winter months is consistently associated with a small but significant increase in schizophrenia risk [98]. Additionally, early-life stressors and childhood traumas also increase risk [99, 100]. Although suggested causal relationships remain speculative, adolescent use of cannabis, especially early and heavy abuse, has been repeatedly associated with earlier-onset and/or increased severity of schizophrenia symptoms [101, 102]. Less well understood are reports that living in more urban than rural environments can increase schizophrenia risk [103]. The extent to which these environmental effects differentially impact individuals with high versus low genetic risk for schizophrenia remains unclear, and so there is a critical need to clarify these potential “gene x environment” interactions.
Risk Factors for Schizophrenia: Heritability
Schizophrenia has an estimated lifetime prevalence of 0.7% [104]. Family studies comparing the probability of having the disorder in those with an affected relative to those without have found an odds ratio about 10 [105]. Concordance rates among monozygotic twins are estimated in the range of 41–65% [106], indicating a substantial contribution of genetics to disease risk. In line with these findings are results from meta-analyses that are able to derive a “heritability estimate,” the percent of variation in disease frequency attributable to heritable factors; as of this writing, the most recent assessment generated a heritability estimate of 79% for schizophrenia [107]. In the subsequent section, we explore the structure and putative impacts of genetic variants that drive this high level of heritability.
3.3. The Form and Function of Genetic Variants Driving
Schizophrenia Heritability
In this section of this chapter, we focus on the underlying biological nature of the genetic factors driving the substantial heritability of schizophrenia. Understanding both the types of variants contributing to disease transmission and the degree to which they influence longitudinal outcomes has essential relevance to knowledge of the neuropathology of the disorder and towards development of biologically informed treatment interventions. Broadly, the genetic contribution to schizophrenia risk consists of a combination of both rare and common variants [108]. Below, we provide an overview of current understanding of both types of variants and their relative contributions to disease risk.
Rare Variants Account for a Portion of Schizophrenia Heritability
A Role for De Novo Mutations
Reduced fecundity in patients with schizophrenia [109] poses a challenge to those studying the genetics of the disorder: How is this reality compatible with the high estimates of heritability? Because genetic variants leading to decreased reproductive fitness are subject to negative selection, the lack of evidence suggesting decreased disease prevalence renders genetic transmission alone an insufficient explanation for disease occurrence [110]. Indeed, partial explanation of this phenomenon lies in the importance of de novo mutations (DNMs). Consistent with a role of DNMs in the genetic etiology of schizophrenia are observations that risk of schizophrenia correlates positively with increasing paternal age [111], that transmission of DNMs to offspring increases with paternal age in general [112] and in schizophrenia in particular [113, 114], and that almost 80% of DNMs documented in exome-sequencing studies of schizophrenia are found on the paternal chromosome [115].
Studying trios of schizophrenia patients and their unaffected parents is one approach to identifying DNMs associated with incident cases. The first report using this design described increased DNMs in schizophrenia cases among a panel of several hundred synaptic genes [116]; targeted exome sequencing of a small number of such trios confirmed an increased rate of DNMs in schizophrenia cases [117]. Subsequent studies with expanded sample sizes found similarly increased rates of putatively damaging DNMs, including a report of substantial enrichment of the schizophrenia-associated DNMs in genes preferentially expressed in fetal but not post-natal brain [113, 114] as well as postsynaptic protein complexes and binding targets of the Fragile X Mental Retardation Protein (FMRP) [115].
Exome-sequencing studies may also be conducted using a more standard case- control sampling plan. While variants in any single gene were not associated with schizophrenia exome-wide, combined gene sets showed increased rates of rare variants in cases versus controls, with highest effects sizes observed for gene sets directly involved in PSD-95 synaptic signaling and calcium channels [118]. While rates of disruptive, “ultra-rare” variants affecting protein-coding sequence appear to be increased in schizophrenia, [119], no single variant alone with exome-wide significance has been identified, possibly due to inadequate sample sizes, the rarity of such mutations in the general population, and the lack of transmission of exonic mutations of large effect sizes. Nevertheless, gene sets specific to brain tissue and neurons in general were highly enriched in such ultra-rare variants, while those in other tissues and non-neuronal cell types were not [120]. For these reasons, there is undoubtedly an increased burden of DNMs in single genes among patients with schizophrenia.
Rates of Copy Number Variants Are Increased in Patients with Schizophrenia
In addition to rare, de novo variants affecting a single gene, there is also an increased rate of large copy number variants (CNVs) in patients with schizophrenia compared to controls [121], and genes located within associated regions implicate disruption in synaptic pathways and neurodevelopment [122]. The largest and most recent analysis found eight CNVs that reached a strict genome-wide significance level, with an additional eight CNVs at a more relaxed threshold [123]. Strikingly, psychotic disorders are present in about 40% of adults carrying the 22q11.2 deletion [124], and this CNV is the most common genetic lesion associated with schizophrenia [125]. Although further research is needed to clarify this relationship, there appears to be an interaction between disease-associated CNVs and common variants in mediating risk variability and resultant phenotypes [126, 127]. In sum, whether they involve a single gene or multiple, there is clearly an important contribution of rare variants to schizophrenia heritability, and additional work to determine the utility of targeting implicated pathways for therapeutic intervention will be essential to the overall efforts to improve outcomes among affected patients.
Common Variation Contributes Substantially to Schizophrenia Heritability
Although rare variants constitute a portion of schizophrenia heritability, most of the genetic basis of the disease lies in the transmission of numerous (and presumably interacting) common variants that each confers a relatively small risk of schizophrenia on their own. In this section, we highlight the significant findings on the role of common variants contributing to schizophrenia risk as well as an exploration of their potential etiologic mechanisms.
The Polygenic Nature of Schizophrenia Heritability Points to Common Variants of Small Effect Size
Long before the advent of genome-wide association studies (GWAS), investigators posited that schizophrenia genetic risk was fundamentally polygenic in nature [128]). Pilot GWAS in several thousands of cases and controls demonstrated that substantial genetic risk for schizophrenia was conferred by single-nucleotide polymorphisms (SNPs), including several in the Major Histocompatibility Complex (MHC) region [129], that were both common and of small effect sizes individually [130–132]. Subsequent GWAS further expanded sample sizes and identified additional loci, including one with variants in MIR-137 and its predicted targets [133]; several loci containing genes implicated in calcium signaling and numerous others containing long non-coding intergenic RNAs [134]; regions enriched in synaptic genes and genes involved in dopaminergic and glutamatergic neurotransmission [135]; and loci implicated in abnormal behavioral phenotypes, long-term potentiation, and targets of FMRP [136]. As seen in Fig. 1, the number of loci discovered is directly proportional to the sample size of the study. Importantly, these findings have been extensively replicated in cohorts of Han Chinese ancestry [138, 139] and among putatively homogenous cohorts of schizophrenia patients taking clozapine [134, 136]. To date, the contribution of common variants to schizophrenia heritability is estimated to be about one-third [136], thus confirming the value of GWAS in assessing the genetic architecture of schizophrenia. Despite the utility of this approach in identifying disease-associated variants, the demonstration of a causal relationship between any given variant and occurrence of schizophrenia is not addressed by GWAS, let alone the mechanisms by which loci negatively impact normal human biology. In the following section, we explore nascent but promising avenues to assess the potential causal impacts of genic variants to schizophrenia development.
Fig. 1.
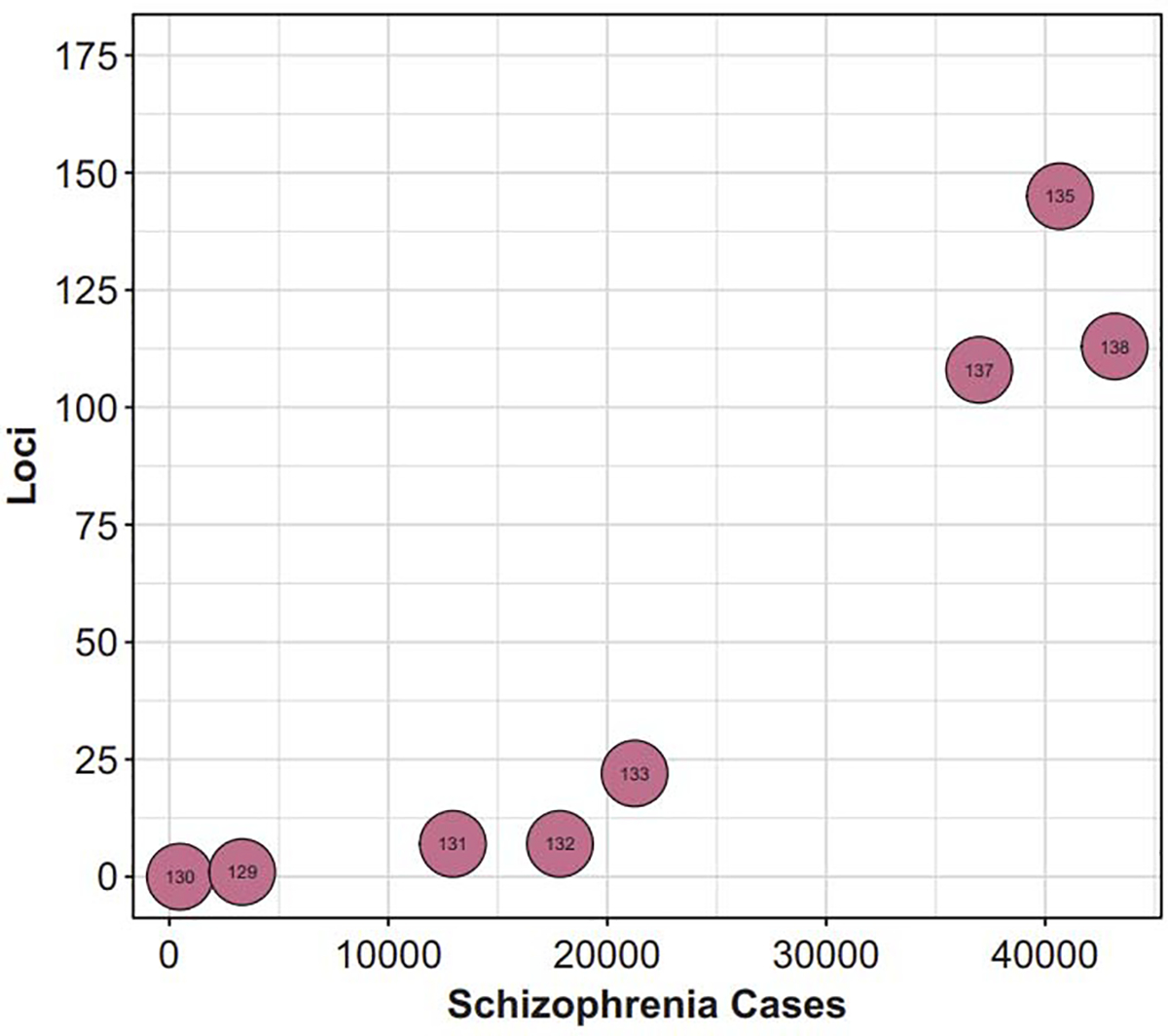
The number of loci associated with schizophrenia through GWAS has dramatically expanded in the past decade, largely as a function of increasing sample sizes. Numbers overlapping each bubble correspond to the reference number: O’Donovan et al. [131] The International Schizophrenia Consortium [130] Stefansson et al. [132] The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium [133]; Ripke et al. [134] Schizophrenia Working Group of the Psychiatrics Genomics Consortium [137]; Li et al. [138] Pardanas et al. [136]
3.4. Functional Genomics of Schizophrenia
The common variants associated with schizophrenia risk are predicted to affect patterns of gene expression [137]. More generally, disease-associated variants are enriched in regions predicted to have gene-regulatory functions [140, 141]. Such variants may alter gene expression through differential affinities of proteins facilitating transcription [142], alterations of various DNA and histone post-translational modifications [143], differential splicing events leading to changes in isoform abundances [144], and/or through changing the three-dimensional regulatory architecture of chromatin [145]. In this section, we review findings of altered gene expression profiles in schizophrenia and studies using various approaches to identify the mechanisms by which risk variants adversely affect gene expression.
Schizophrenia Risk Variants Are Enriched in Expression Quantitative Trait Loci
SNPs implicated in schizophrenia are enriched in genes expressed in post-mortem brain tissue [146]. A substantial portion of these variants are associated with changes in the expression of at least one gene and are thus termed “expression Quantitative Trait Loci” (eQTL) [147]. Furthermore, disease-associated variants are enriched in chromatin-regulatory peaks among genes that drive cortical neogenesis [148]. Strikingly, the most recent analysis concluded that about 42% of GWAS variants associated with schizophrenia contain eQTLs in regions converging on gene regulation [149]. With this in mind, it is no surprise that there are widespread differences in gene expression patterns between cases and controls, as reviewed below.
Post-Mortem Gene Expression Analyses Highlight Neurodevelopmental Pathways and Specific Brain Regions and Cell Types
Numerous investigators have sought to characterize abnormalities in nervous system gene expression in schizophrenia in order to begin yielding mechanistic insights on molecular perturbations driving disease pathologies. With the discovery that common variants associated with schizophrenia are preferentially enriched in putative gene-regulatory regions and the advent of more high-throughput technologies for measuring gene expression, these efforts have expanded substantially. Here, we highlight some of the earlier reports of altered brain gene expression with a focus on highly replicated findings with enduring relevance to the mechanisms of schizophrenia. We then turn to an assessment of large datasets generated via modern RNA-sequencing and computational platforms. As shown below, gene expression abnormalities have begun to converge on several key neuronal processes, especially those critically regulating neurodevelopment and cell type and regional specification. With further developments in computational techniques and increasing sample sizes, studies of disrupted gene expression patterns in schizophrenia will continue to generate key insights connecting disease-associated genetic variants to etiologic molecular perturbations driving symptom manifestation.
Regardless of disease status, comparative evolutionary genomics studies have revealed that brain-related genes most recently acquired by Homo sapiens are preferentially expressed in fetal and infant but not adult neocortex [150], and data from large-scale genomics approaches indicate that variants associated with schizophrenia risk preferentially impact early neurodevelopmental pathways. Initial studies of patient post-mortem brain tissue documented cellular and anatomical defects that suggest disrupted cortical neuron development and migration [151, 152]. Expression analyses of genes driving GABAergic neuron specification found that the most dramatic differences in patient brain tissue were in transcripts whose expression undergoes the most significant changes in early rather than later life [153], and that patient brains exhibit more immature and under-developed expression patterns in GABAergic genes [154]. Meta-analysis of post-mortem microarray studies implicated a variety of cell types and gene sets involved in synaptic transmission [155], as well as disruption in the expression of genes most predominantly regulated during neurodevelopment [156]. Subsequent review of findings that specifically assessed data from post-mortem prefrontal cortex found enrichment in pathways related to synaptic transmission [157], as well as immune-related processes, myelination, and oxidative phosphorylation [158]. Consistent with the reports described below [159–161], similar analysis that included other brain regions found enrichment in modules specific to certain cell types and neurotransmitter systems (i.e., glutamatergic and inhibitory) and furthermore discovered diminished expression in genes that distinguish separate brain regions [162].
RNA-sequencing platforms and increasingly sophisticated statistical techniques enabled broader assessments of disease-associated changes in gene expression. Data from the Common Mind Consortium revealed subtle alterations in mRNA levels among hundreds of genes in dorsal lateral prefrontal cortex, and network analysis highlighted a gene “node” characterized by enrichment in genes importantly involved in synaptic transmission, neuronal subtype markers, and targets of FMRP [147]. An expanded analysis found that genes differentially expressed in schizophrenia were those enriched in neurodevelopmental regulation of neuronal cell type and discovered a dramatic shift in developmentally regulated isoform expression among genes involved in dopaminergic and glutamatergic synaptic functions [149]. Hierarchical clustering of gene expression in a different case-control sample revealed similar enrichment in modules defined by neurodevelopmental regulated genes, synaptic function, and cell type-specific markers [163]. In the largest transcriptomic study of psychiatric disorders to date, investigators discovered expansive alterations in isoform expression, particularly among non-coding RNAs and gene sets involved in synaptic function, cell-type markers, and the immune system [144].
Presently, the substantial cost of RNA-sequencing and the limited number of post-mortem samples, coupled with small effect sizes of individual loci, hinder the identification of robust gene expression changes associating with a particular trait or disease. As an alternative approach, transcriptome-wide association studies (TWAS) generate predictive models relating specific genotypes to gene expression phenotypes through imputation of available datasets describing gene expression changes associated with a disease risk variant [164]. In this way, leveraging large eQTL datasets with post-mortem RNA-sequencing from case-control studies enables the generation of models that predict alterations in gene expression from given disease-associated variants. Thus far, schizophrenia TWAS efforts have identified over 150 genes whose expression differs between cases and controls [160] [165]). Of note, sets of variants discovered through TWAS are overrepresented in neurodevelopmentally regulated genes [160] and impact the expression of several hundred of them in a remarkably site-specific and temporally regulated manner across many brain regions [165]. Interestingly, pathway analysis of the implicated genes documented strong enrichment in processes related to porphyric disorders and hexosaminidase-A deficiency [165]. A different study similarly documented over one-hundred TWAS hits; among the most significant were chromatin regulators and long intergenic non-coding RNAs [144]. By integrating gene expression taxonomies derived from single-cell RNA-sequencing datasets with schizophrenia GWAS signatures, specific neuronal cell types, including interneurons, pyramidal cells, and striatal medium spiny neurons, have been emphasized in driving disease risk [161]. In a related approach, analysis of the overlap between trait- and disease-associated loci and genes exhibiting expression specificity in numerous tissues and cell types found that schizophrenia loci were overrepresented in genes expressed in neurons, particularly glutamatergic neurons, but not in non-neural tissue [159]. In sum, these studies reveal a cell-type specific disruption of gene expression, particularly among sets key to neurodevelopment.
DNA and Histone Post-Translational Modifications Are Altered in Schizophrenia
Efforts to understand the mechanisms by which schizophrenia-associated risk variants alter gene expression have discovered key roles for several transcriptional regulatory processes. Assessment of the excess association of GWAS SNPs for several psychiatric disorders in particular biological pathways found that, for the combined datasets of schizophrenia, bipolar disorder, and depression data sets, histone H3K3me methylation showed the strongest enrichment overall, while the top pathways among schizophrenia SNPs were postsynaptic density and membrane, dendritic spine, H3K4 methylation, and axon part [166]. Across diseases and traits, associated SNPs are globally enriched in regions marked by histone post- translational modifications (PTMs), such as H3K4me3 and H3K9ac, indicating active enhancer, in a cell-type specific manner [167]. Using publicly available post- mortem ChIP-sequencing datasets from, Roussos et al. [168] found that this is also the case for SNPs associated with schizophrenia in both fetal and adult brain tissue. Based upon the growing consensus that epigenomic processes drive molecular phenotypes in numerous diseases, the PsychENCODE Project was developed to study the role of DNA regulatory elements in various psychiatric disorders [169]. A recent landmark study [170] assessed the enrichment of SNPs for several brain- and non-brain-related diseases and traits among specific regulatory chromatin marks in both brain tissue homogenate and neuron-enriched and neuron-depleted populations of cells. SNPs associated with schizophrenia demonstrated the strongest enrichment in open-chromatin peaks over all traits, including other brain-related traits and diseases; importantly, this enrichment was strengthened substantially in neuron- enriched samples over brain homogenate and non-neuronal samples, and the variable driving the most variation in histone peaks was cell-type identity (i.e., neuron-enriched versus non-depleted versus homogenate) [170]. Furthermore, many of the strongest histone QTLs (hQTLs) associated with schizophrenia were found in neuronal but not non-neuronal tissues [170]. This study highlighted, among other findings, both the strong enrichment of schizophrenia SNPs in regulatory chromatin regions and the importance of assessing potential functional roles of disease- and trait-associated variants in refined biological samples in order to more adequately capture meaningful signals. In reports of differential DNA post-translational modifications, schizophrenia SNPs are also enriched in sites of DNA methylation (meQTLs), which differ between cases and controls at genes that are strongly enriched in neuronal differentiation and neurodevelopment in frontal cortex [171], hippocampus [172], and prefrontal cortex [173].
Taken together, these data provide a mechanistic link between genetic variation and altered gene expression. The extent to which differential DNA and histone PTMs are driven by primary effects of genetic sequence variation or numerous other secondary processes that ultimately converge on alteration of these marks remains an area of significant uncertainty, but future studies will integrate the contributions of innate genetic variation and non-genetic factors in pathologically disrupting both the regulatory states and expression patterns of causal gene sets. We now turn to a budding field that extends the functional impact of schizophrenia risk loci on gene expression to include the pivotal role of three-dimensional chromatin-regulatory structures.
The Disruption of Three-Dimensional Chromatin Dynamics by Schizophrenia Risk Variants
A critical way in which DNA and histone PTMs change gene expression is through the alteration of three-dimensional (3D) chromatin structures. If variants associated with a disorder are enriched in putative participants in such structures, it follows that schizophrenia risk variants may cause pathogenic alterations in 3D chromatin architecture, thereby leading to disrupted patterns of gene expression [174]. While still a nascent field, the role of 3D genomics in schizophrenia disease biology has increasingly become come into focus for investigators. In 2013, Barhdwaj et al. identified a GABAergic neuron-specific, activity-regulated 3D chromatin interaction involving GAD1 and an upstream transcriptional regulator, likely an enhancer, that was decreased in post-mortem samples of schizophrenia patients who also had decreased GAD1 transcript levels [175]. In the following year, an activity-regulated distal regulatory element of the NMDA receptor subunit GRIN2B was identified using chromosome conformation capture 3C); this distal regulatory region was found to contain a SNP associated with schizophrenia risk, and post-mortem prefrontal cortex from patients with that SNP had lower levels of GRIN2B mRNA transcript compared to controls as well as patients without the risk allele [176]. Furthermore, studies in post-mortem brain tissue identified a remarkable overlap between regions of open chromatin and variants associated with schizophrenia risk [177–179]. Adapting 3C assays genome-wide, HiC analysis in human fetal brain tissue found enrichment of schizophrenia SNPs in 3D contacts of gene sets involved in neurogenesis, postsynaptic density, and chromatin remodeling proteins, among others [180], consistent with TWAS findings of a high degree of overlap between brain chromatin loops and signal for TWAS genes [160]. Recently, investigators demonstrated directly that risk variants for schizophrenia are enriched in 3D chromatin loops that are specific to neurons and neural progenitor cells (NPCs) [174].
Overall, genomic and functional genomic studies of schizophrenia heritability point to a model in which both rare and common genetic variants contribute to disease risk, and that common variants exert their effects in large part through altering gene-regulatory processes and thus disturbing normal neurodevelopment in specific cell types highly implicated in schizophrenia. In the next section, we begin our consideration of the role of hiPSC models in exploring schizophrenia with relevant technical considerations, and then dive into the numerous reports that have used hiPSCs to further expound disease biology.
4. Technical Considerations in Human Induced Pluripotent Stem Cells
A landmark screen of transcription factors determined that OCT4, KLF4, SOX2, and c-MYC, the now termed “Yamanaka Factors,” were sufficient to reprogram somatic cells into induced pluripotent stem cells (iPSCs), thus bypassing the need for embryonic stem cells [181]. Critical for model validity, iPSCs and ESCs have been shown to be comparable across several features [182]. Advancements in hiPSC models and related technologies have fueled a new approach to studying disease-relevant biological pathways in living neural tissue from individuals with and without a particular trait or disease (for review see [183]).
4.1. Technical Challenges and Considerations in hiPSCs Models
hiPSC-based models offer as their primary advantage the ability to analyze living neuronal tissue in a way that preserves donor gene background. Nonetheless, both genetic [184] and epigenetic [185] errors are known to occur during the reprogramming process. Moreover, certain chromosomal abnormalities identified in patient somatic cells reprogrammed into hiPSCs may not retain the abnormality through traditional differentiation methods [186, 187]. While there is indeed variation in gene expression between hiPSCs derived from different donors (and to a lesser extent between hiPSCs from the same donor), genetic heritability also impacts gene expression patterns and differentiation potential [188]. Further variability is introduced during the neuronal differentiation process: RNA-seq analysis of hiPSC-derived neurons demonstrated that inter- and intra-donor variability could be decreased by correcting for variation in cell-type composition [189]. While hiPSC-based studies remain dramatically underpowered for the study of idiopathic disease, there is a minimal yet highly significant concordance between gene expression signal in hiPSC-derived neurons and RNA-seq datasets generated from the two largest post-mortem studies [189]. If applied appropriately, hiPSC nevertheless serves as a key modeling platform.
Brief Overview of Techniques for Generating Disease-Relevant Tissues from hiPSCs
In this section, we provide a targeted overview of techniques for generating the types of neural cell types relevant for the discussions throughout the chapter. Broadly speaking, techniques for producing neural tissue from hiPSCs may be classified as “directed differentiation” or “induction-based” approaches. Directed differentiations are those techniques that use a combination of small molecules, proteins, and other chemical factors to modulate intracellular signaling in order to recapitulate in vivo developmental pathways that give rise to the target cell type; these approaches often use varying combinations of agents at different steps of the protocol to mirror sequential phases of neurodevelopment. Approaches based upon inductions, on the other hand, employ transgenes, often packaged into viral vectors, to ectopically express transcription factors that are known to be necessary and/or sufficient to driving the hiPSC towards a specified neurodevelopmental pathway to produce the desired cell type. Each general approach has its own advantages and disadvantages, and an increasing number of techniques are employing combined strategies to improve cell type yields and enhance specific phenotypes of interest (e.g., [190]). We turn now to an overview of the protocols used to produce relevant cell types, with particular emphasis on those employed in the studies discussed herein. In both sections, we mention first the techniques for producing excitatory and inhibitory neurons, and then turn towards protocols that yield cells of other neurotransmitter system identities. For excellent and more thorough review, see Mertens et al. [191].
Directed Differentiation Approaches
A frequently used directed differentiation technique for deriving forebrain neural progenitor cells (NPCs) from hiPSCs involves the dual inhibition of SMAD signaling. The application of SMAD inhibitors Noggin and SB431542 is known to reliably produce cultures that can be specialized to several neuron phenotypes [192]. To generate a mixed population of forebrain neurons consisting primarily of excitatory neurons, stepwise patterning of embryoid bodies into neural rosettes and NPCs using developmental signals and then differentiating NPCs with defined neuronal growth factors are used [193]. Cortical interneurons can be efficiently generated from hPSCs with a combination of dual-SMAD inhibition and small-molecule application [194]. In the most frequently used directed differentiation approach for producing dopaminergic neurons, hiPSCs are first patterned into ventral midbrain floor plate progenitors and then matured into dopaminergic neurons with a standard mixture of neurotrophic factors and small molecules [195]. Serotonergic neurons may be differentiated from hiPSCs through sequential dual-SMAD inhibition and modulation of WNT, Sonic Hedgehog (SHH), and FGF4signaling [196]. Using sequential application of small-molecule inhibitors and morphogens, hippocampal progenitor cells and dentate granule neurons can be produced [197], and a related technique enriches for CA3 hippocampal neurons [198]. Combinatorial small-molecule approaches are also capable of producing sensory neurons [199, 200]. Often, co-culturing developing neurons with astrocytes during directed differentiation yields more mature, functional neurons. A study conducted by Kuijlaars and colleagues demonstrated enhanced synchronized synaptic activity in a population of GABAergic and glutamatergic neurons differentiated in a co-culture of astrocytes via a small-molecule technique [201]. In a simplified approach, astrocytes and neurons are produced together from the same starting hiPSCs with modified differentiation strategies [202].
Induction Approaches
Fibroblasts can be reprogrammed into functional induced neuronal (iN) cells via overexpression of the transcription factors ASCL1, BRN2, and MYTL1 [203]; the same three factors yield human neurons when combined with the transcription factor NEUROD1[204]. Greatly simplifying the ability of investigators to produce cortical excitatory neurons was the approached developed by Zhang et al. [205] involving the overexpression of NGN2 alone in antibiotic-selected hiPSCs. This technique works similarly when starting from NPCs [206] and was recently modified to include the addition of small molecules in order to improve functional maturation [190]. Several cell-type specific induction techniques have now been developed. GABAergic neurons of varying subtype identities and maturities can be produced through the overexpression of different combinations of transcription factors [207–209]. In order to produce midbrain dopaminergic neurons, hiPSCs can be transduced with a combination of ASCL1, LMX1a/b, and NURR1 [210, 211], among others. Production of serotonergic neurons with induction approaches has been achieved from fibroblasts using a combination of either LMX1b, FOXA2, ASCL1, and FEV [212] or LMX1b, FEV, NKX2.2, GATA2, ASCL1, and NGN2 [213]. See Fig. 2 for an overview of these techniques.
Fig. 2.
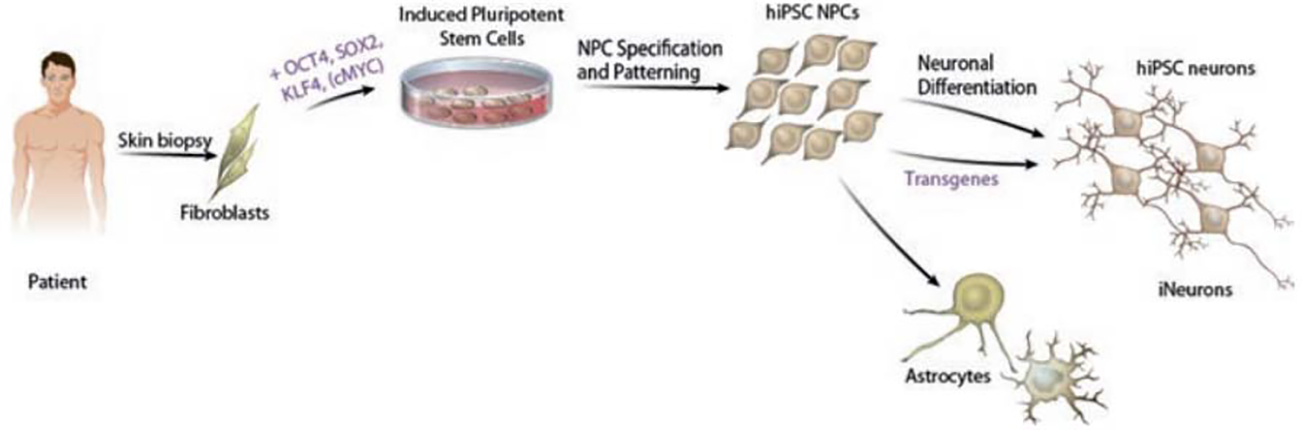
Generalized schematic of hiPSC-based models
Overall, these approaches enable the production of numerous cell types relevant to schizophrenia. Continued work to improve neuronal maturity and subtype specificity will only bolster the field, and efforts to render approaches more scalable and reproducible across donors and laboratories will increase the attractiveness of hiPSC models of neuropsychiatric disease to many investigators (Fig. 3).
Figure 3.
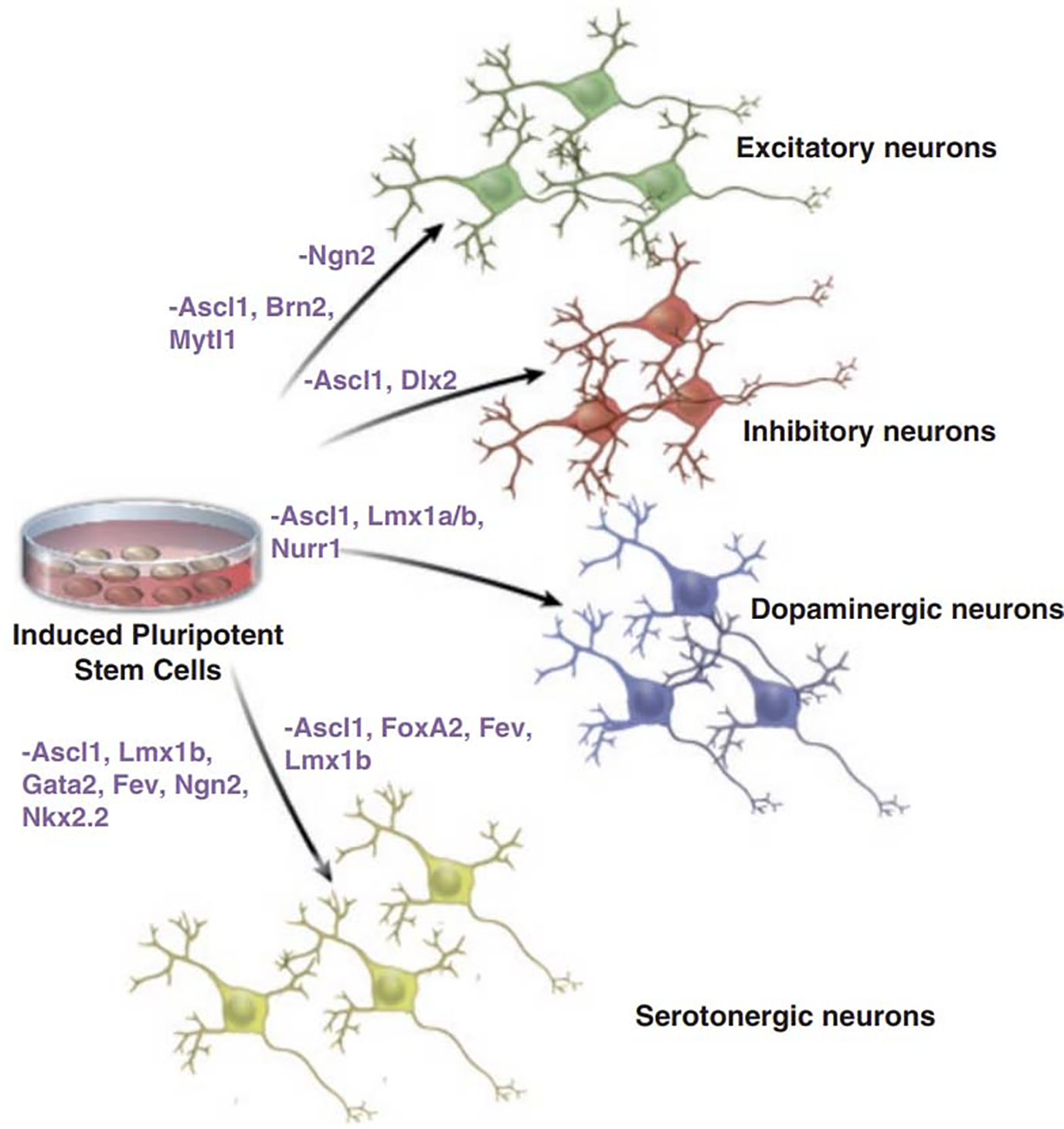
Transduction approaches to generate samples of defined neurotransmitter system identities
5. Investigation of Schizophrenia with Human Induced Pluripotent Stem Cells
We turn now to the primary focus of this chapter. Here, we discuss studies that have explored various aspects of schizophrenia biology using human induced pluripotent stem cells (hiPSCs). We begin with pioneering reports that ignited the field and highlight cellular and molecular phenotypes that were documented in patient cells. Then, we evaluate studies that have assessed the impact of both common and rare genetic variants associated with schizophrenia neuronal phenotypes. Afterwards, we share results of approaches that have used hiPSC models to explore the impact of non-genetic factors implicated in schizophrenia. We end with a discussion of recent advances in using hiPSC platforms for drug screening purposes and applications towards improving clinical outcomes.
5.1. Assessment of Phenotypic Differences in Schizophrenia Across Neurodevelopment with hiPSCs
The first report assessing phenotypic differences in hiPSC-neurons from patients and controls found that patient-derived forebrain neurons exhibited decreased connectivity, fewer neurites, decreased expression of the synaptic protein PSD95, and altered gene expression profiles in pathways important to WNT signaling, glutamatergic neurotransmission, and cAMP-related processes. Strikingly, the antipsychotic loxapine improved connectivity and partially reversed abnormal gene expression in patient neurons [214]. Two follow-up studies from these same hiPSCs revealed reduced excitatory synaptic activity [197] and altered dopamine release [215]. In an independent study, hiPSC-derived dopaminergic and glutamatergic neurons showed diminished ability to develop into morphologically mature neurons and displayed several mitochondrial abnormalities [216]. Experiments in schizophrenia hiPSC-derived forebrain and NGN2 neurons were coupled with two mouse genetic models of schizophrenia to provide robust, cross-model validation of perturbed expression and activity of the striatal-enriched tyrosine phosphatase isoform 61 (STEP61) [217], a postsynaptic density-enriched enzyme implicated in animal and pharmacologic models of schizophrenia [218].
Whereas forebrain neural progenitor cells (NPCs) from patients with schizophrenia exhibited reduced neural migration [219], altered WNT signaling [220] and increased abundance of translation machinery [221], hippocampal NPCs from this same hiPSC cohort revealed decreased expression of several key markers of hippocampal neurogenesis [197]. Moreover, hippocampal CA3 neurons derived from these patients had several electrophysiological abnormalities and altered network connectivity when co-cultured with human dentate granule neurons [198]. An independent group confirmed defective migration patterns in patient NPCs, and further reported depressed expression of several angiogenic proteins and reduced angiogenic capacity [222]. These findings are intriguing in light of reports of altered angiogenesis protein expression post-mortem [223] and in living patients [224], as well as diminished vasculature in patients with schizophrenia [225], highlighting the importance of studying non-neuronal cell types in schizophrenia [226].
Evidence is gradually accumulating that abnormalities in mitochondria and reactive oxygen species (ROS) may contribute to schizophrenia etiology [227–229]. A study of ROS production in neural cells derived from a single patient with schizophrenia showed increased ROS, a phenotype that could be alleviated by exposure to the mood-stabilizer and anti-epileptic valproic acid [230]. Subsequently, proteomic analysis revealed alterations in oxidative stress pathways [219] and mitochondrial abnormalities [216] in schizophrenia hiPSC NPCs and neurons, respectively. Intriguingly, transfer of healthy control mitochondria to differentiating hiPSCs derived from schizophrenia patients ameliorated abnormalities in mitochondrial bioenergetics and neuronal differentiation [231].
A pivotal role for abnormal expression of microRNAs and their targets in schizophrenia pathology has garnered increasing levels of support [232]. Studying gene expression across dopaminergic differentiation of hiPSCs, Shi et al. [233] confirmed findings from post-mortem data on an inverse correlation between expression of the dopamine receptor 2 (DRD2) and a regulatory microRNA, miRNA-326. After establishing a key role for miR-19 in the regulation of NPC proliferation and migration, Han et al. [234] documented increased miR-19 expression and a corresponding decrease in the RNA and protein levels of one of its key regulatory targets. A broad examination of microRNA expression levels in NPCs derived from schizophrenia patients and controls identified miR-9 as the most substantially decreased transcript [235]. Subsequent analysis correlated decreased miR-9 expression levels with defective migration of patient NPCs in a neurosphere migration assay; abnormalities in NPC migration and gene expression could be partially rescued by overexpression of miR-9, and knockdown in control NPCs resulted in the production of a “SCZ NPC” gene expression and migration phenotype [235]. Separately, GWAS studies identified SNPs associated with miR-9 targets to be enriched in schizophrenia [236], independently validating this hiPSC-based discovery.
Taken together, these reports indicate the pivotal role of hiPSC-based models in parsing altered molecular pathways in living neurons from patients. As shown, the generation of isogenic neural tissue from patients and appropriate controls has been a fruitful approach to further assessing cell and molecular phenotypes observed in schizophrenia. Next, we focus on studies that have used hiPSC-derived tissue to test specific hypotheses regarding putative roles of genetic variants in disease biology.
5.2. Application of hiPSC Models to Study the Effects of Common Variants
Analyses of the contribution of common genetic variation to schizophrenia indicate that hundreds of single-nucleotide polymorphisms (SNPs) may be involved in disease risk [136] and that common variation may account for one-third to a half of the genetic risk for schizophrenia [130, 134]. Individually, each implicated SNP has an incredibly small effect size, with odds ratios (OR) typically ranging from about 1.05 to 1.20 [137]. The remarkably polygenic nature of common variants in schizophrenia, coupled with their low effect sizes, indicates that studying any single, individual SNP in a case-control fashion is highly unlikely to yield meaningful results. While the sample sizes required to detect phenotypic differences attributable to common variants in hiPSC-based models remain undetermined, it is noteworthy that a supplementary analysis conducted by members of the Common Mind Consortium [147] found that the median sample size needed to detect a genome- wide significant difference in the expression of a gene (among 10,000 genes and assuming a mean allele frequency similar to that found in existing data) [147] to be 28,500. Despite this sobering reality, hiPSCs models may still be used to test specific hypotheses on the impacts of a given genetic variant on gene expression, chromatin biology, and cell phenotypes, and they offer the advantage of exploring these effects in cells of a defined genetic background. Below, we discuss promising findings that have used this approach to make important contributions to understanding of the neurobiology of schizophrenia.
Rather than causing damaging mutations in protein-coding genes, common variants for schizophrenia are thought to contribute to disease risk through alteration of gene expression [137]. Increasingly, investigations into the biological effects of disease-associated quantitative trait loci on gene expression, alternative splicing, and chromatin features are employing hiPSC-based models [200, 237]. For example, hiPSC platforms have been used to assess activity-dependent differences in gene expression in patient and control lines [238]. At the present time, hiPSCs models are beginning to serve as a viable platform to study the actual mechanisms of gene expression alteration by schizophrenia risk loci. Ascertainment of hiPSC lines from individuals homozygous for a schizophrenia risk allele or the protective allele, as well as heterozygotes, for a voltage-gated calcium channel (CACNA1C) found altered CACNA1C gene expression and electrophysiological properties upon conversion to induced neurons [239]. Additionally, hiPSC-derived NPCs were employed to show that knockdown of FURIN, a gene for which CMC analysis found a highly significant eQTL, depresses normal migration in neurospheres [147]. Further supporting the relevance of microRNAs to disease biology, longitudinal ATAC-Seq analysis of neurons differentiating from hiPSCs found enrichment of schizophrenia risk loci in open chromatin regions in neurons, and genetic alteration of that a risk allele within an open chromatin region at miRNA137 led to altered dendritic morphology and synaptic maturity [240]. Additionally, CRISPR/Cas9 approaches enable the manipulation of gene sequence or expression with exquisite precision, and applications of CRISPR-based tools are beginning to make fundamental contributions to the study of schizophrenia genetic risk variants in hiPSC models [241]. In a landmark report, Ho et al. [242] provided the first demonstration of altering expression of specific schizophrenia risk genes with CRISPR-dCas9 in hiPSC-derived NPCs, astrocytes, and neurons. Continued development of this approach and related techniques to improve reproducibility, scalability, and bi- directionality will likely yield a key platform for exploring the functional impacts of disease-associated gene expression abnormalities.
In investigations of the potential role of specific chromatin structures in mediating schizophrenia susceptibility, hiPSC-derived NPCs and neurons have been a key model system to further validate findings in post-mortem and animal model studies and to test the effects of manipulating such structures in living human brain tissue [243]. Intriguingly, chromosome conformation capture (3C) assessment of patient- derived neurons revealed increased contact frequency between a schizophrenia SNP predicted to affect gene expression of CACNA1C, confirming a finding that had also seen in post-mortem samples [168]. In a recent report, Rajarajan et al. [244] profiled global patterns of three-dimensional chromatin architecture in hiPSC-derived excitatory neurons, NPCs, and glial cells. Among their findings included the discovery of about double the localization of schizophrenia risk loci in chromatin loops specific to neurons and NPCs over those specific to glial cells [244], thus indicating the pivotal role of cell-type identity in assessing the participation of risk loci gene-regulatory regions. Of note, CRISPR/dCas9-mediated targeting of transcriptional effectors to putative gene-regulatory structures, often separated by several hundred kilobases of DNA, confirmed functional capabilities of selected loops [244]. Future studies will further expand these findings to additional cell types relevant to schizophrenia pathophysiology to provide a comprehensive assessment of the potential disruption of 3D gene-regulatory structures by disease-associated loci.
5.3. Abnormalities in hiPSC-Derived Neurons Harboring Rare Schizophrenia Risk Variants
The higher penetrance typically seen with rare variants associated with schizophrenia makes them feasible contexts to analyze in a case-control fashion. In this section, we discuss reports highlighting such differences in lines containing rare variants implicated in schizophrenia.
(I). Copy Number Variants
Copy number variants (CNVs) are often duplications or deletions ranging from 50 bp to several hundred kilobases in length [245]. The Psychiatric Genomics Consortium has performed the largest genome-wide analysis of CNVs in schizophrenia to date and identified eight loci associated with disease risk [123]. Furthermore, there is a particularly high incidence of CNVs associated with cases of COS [246]. In the text that follows, we discuss investigations of the impact of schizophrenia-associated CNVs on neuronal phenotype using hiPSCs (reviewed in [247].
22q11 Deletion
The 22q11.2 microdeletion is the strongest genetic risk factor for schizophrenia [123], and several prospective studies have shown that at least one-third of such patients develop some sort of psychotic disorder [248–250]. Derivation of hiPSCs from patients with the 22q11.2 deletion was one of the first published reports on in vitro models of schizophrenia [251]. Transcriptomic profiling of neurons derived from 22q11.2 deletion hiPSCs provided a list of hundreds of genes that were differentially expressed between patients and controls; GO analysis of top hits strongly implicated molecular pathways regulating apoptosis, the cell cycle, and neural proliferation [252]. Importantly, assessment of these processes in NPCs generated from the same lines revealed diminished proliferation [252]. 22q11.2 hiPSCs generated from SCZ patients demonstrate reductions in neurosphere size upon differentiation without a decrease in the total number of neurospheres and several morphological abnormalities [252]. Given the location of DCG8, a microRNA-regulating protein, in the 22q11.2 deletion band, Zhao et al. [253] sought to assess the impact of this CNV on microRNA expression in lines derived from patients carrying the deletion; they found that several microRNAs and their targets were perturbed by the deletion [253]. Furthermore, gene expression analysis from 22q11.2 neural tissue confirmed substantial alterations in microRNA levels across numerous genes [254]. These data confirm an important role for disruption in microRNA pathways, particularly those involved in neurodevelopment [255], in 22q11.2 deletion carriers with schizophrenia. Future studies will further dissect the molecular pathways by which 22q11.2 deletion contributes to cell-type specific abnormalities to further inform knowledge of disease mechanisms and reveal plausible targets of therapeutic intervention.
15q11.2
An additional CNV implicated in schizophrenia is 15q11.2 [123]. Neural rosettes generated from hiPSC lines harboring a 15q11.2 heterozygous microdeletion displayed abnormal polarity and adherens junction distributions, and complementation experiments in rosettes and NPCs demonstrated that haploinsufficiency of CYFIP1, located within the 15q11.2 region, drove these alterations in a process dependent on WAVE [256], a member of a complex regulating actin cytoskeletal organization. Importantly, the in vitro and rodent in vivo findings in this study led the authors to perform a targeted eQTL analysis to examine interactions between components of the WAVE pathway, even though individual variants by themselves did not show GWAS-level association with schizophrenia risk [256], and thus demonstrated the power of observations in pre-clinical models to inform larger-scale human genomics studies to discover findings not observed from a relatively unbiased—“omics” approach.
16p11.2
Copy number variations in the 16p11.2 band are also associated with schizophrenia [257]. In a remarkable demonstration of the utility of hiPSC-derived neuron models in recapitulating clinical phenotypes and revealing corresponding disease mechanisms, Deshpande et al. reported opposing cell morphological abnormalities in 16p11.2 duplication and deletion forebrain neurons that mirrored clinical phenotypes of micro- and macrocephaly, respectively. Subsequent functional studies revealed a mechanism connecting altered cell morphologies to specific electrophysiological and synaptic abnormalities in these neurons [258].
(II). Other Variants
Neurexins and Their Loci
Mutations in Neurexin1 have been implicated in several neurodevelopmental disorders, including schizophrenia [259], and RNA-seq analysis on neural stem cells was employed to investigate the effects of NRXN1 knockdown on the expression of several genes potentially important in disease processes [260]. Generation of human ESC lines with conditional knockout of NRXN1 and their subsequent conversion to NGN2 neurons found impaired functional synaptic activity but not structure [261]. By using lines from patients with childhood-onset schizophrenia with specific deletions in NRNX1, Flaherty et al. (2019) [262] discovered alterations in neurexin isoform expression and neuronal phenotypes associated with defined deletions and demonstrated the remarkable utility of hiPSC platforms to elucidate disease biology.
Assessment of the Role of DISC1 in Schizophrenia with hiPSCs
The origin of investigations into the so-called disrupted in schizophrenia I (DISC1) gene lies in the identification of a balanced t(1;11) (q42;q14) translocation in a Scottish adolescent boy discovered as part of a cytogenetic survey of detainees in an juvenile delinquent center [263]. The proband had a diagnosis of conduct disorder, and subsequent assessment of family members found exceptionally high rates of several mental illnesses among carriers of the translocation, a minority of which met criteria for schizophrenia [264]. Although only five of the 34 members who carried the translocation (77 in pedigree as a whole) had a diagnosis of schizophrenia or schizoaffective disorder, the protein-coding gene altered by the translocation was named disrupted in schizophrenia 1 (DISC1) [265].
After initial publication of the original DISC1 translocation found in the Scottish pedigree, a subsequent report documented a four base-pair deletion in DISC1 in a proband with schizophrenia that co-segregated with other family members who had the same condition or schizoaffective disorder [266]. Although this mutation was not found to associated with risk for schizophrenia in a larger follow-up study [267], it served as a valuable platform for modeling disease biology in patients with defined genetic lesions. In fact, the first report of the generation of hiPSCs from patients with schizophrenia was the publication of integration-free hiPSC from two patients with this particular DISC1 mutation [268]. In a landmark study, investigators [269] derived hiPSCs from four members of the same family. Forebrain neurons generated from mutant patient lines expressed substantially less wild type DISC1 protein and had defective glutamatergic synapses [269]. Strikingly, TALEN-mediated introduction of the mutant form of DISC1 in isogenic lines from unaffected family members as well as correction of the mutation in an affected cell line confirmed a causal role for the DISC1 mutation in production of abnormal synapses. Finally, global transcription profiling confirmed robust alteration in the expression of genes essential in synaptic processes and neural development, as well as numerous genes previously implicated in schizophrenia and other psychiatric disorders [269]. In a follow-up report, neural stem cells derived from the same DISC1 mutation and isogenic control lines were used to provide evidence for a relationship between altered DISC1 expression and a microRNA pathway that was shown to regulate neural stem cell proliferation; both patient and mutant-edited isogenic displayed alterations in this pathway and differentiation abnormalities [270]. Most recently, Yalla et al. [271] explored the role of the ubiquitin-proteasome system in regulating levels of DISC1 protein and identified a crucial regulator of DISC1 turnover whose disruption could increase the abnormally low levels of DISC1 in mutant NPC lines. Generation of medium spiny neuron-like cells [272] from hiPSC lines was used as a platform to functionally validate the role of a schizophrenia-associated protein and DISC1 interaction partner, TRAX1, in neuroprotection and DNA damage repair [273]. On the other hand, generation of hiPSCs with TALEN-mediated deletion of DISC1 exons relevant to the Scottish translocation and comparison with isogenic controls revealed altered expression of cell fate markers, neurodevelopment, Wnt signaling, and schizophrenia-related genes by mutant DISC1 [274].
Evidence suggests that DISC1 exerts many of its neuronal effects through its protein-binding partners [275–278]. In a post-mortem gene expression analysis of cases and controls, a small group of SNPs in DISC1 were associated with altered expression of DISC1 isoforms in brain tissue of schizophrenia patients [279]. A recent report [280] documented generation of an hiPSC line with CRISPR/Cas9-mediated insertion of a 3X FLAG tag downstream of endogenous DISC1 and characterized DISC1-interaction proteomes across multiple cell types; NPCs and astrocytes showed cell-type specificity in DISC1 binding patterns, and the “DISC1 interactome” of NPCs was enriched in genes previously identified as candidate de novo variants associated with SCZ [281]. Elegant biochemical experiments confirmed an interaction between a specific residue of DISC1 protein and NDEL1, a component of the dynein complex that is highly important in the brain [282], and disruption of this interaction altered cell cycle progression in radial glial cells in developing murine cortex [283]. Furthermore, the importance of the DISC1-NDEL1 interaction was confirmed when its disruption reduced neural stem cell proliferation in a human cerebral organoid model; strikingly, cerebral organoids derived from a schizophrenia patient carrying the 4 bp DISC1 deletion in the NDEL1-interacting residue exhibited delayed cell-cycle progression in radial glial cells and confirmed the key role of DISC1-NDEL1 interaction in regulation of neural proliferation [283].
In total, hiPSC-based models of DISC1 mutation have generated important insights about the functions of DISC1 protein in normal neuronal biology and have certainly yielded data on mechanistic pathways contributing to disease in those patients harboring lesions in this gene. The number of patients carrying the defined lesions highlighted, however, is thus far limited to the few studies in which they were originally identified. The importance of DISC1 variants to the vast majority of “idiopathic” cases of schizophrenia overall is at best unclear and at worst increasingly doubtful. Genetic studies of both common [136, 137, 284] and rare [119, 123,285] variants have all failed to detect genome-wide significant associations between DISC1 and risk of schizophrenia. As aptly pointed out already [286, 287], the fact that repeated studies evidence neuronal functions for DISC1 is both unsurprising given its effects in the (very) small number of people carrying DISC1 mutations, and insufficient justification alone to assert its relevance to schizophrenia as a whole, as a substantial proportion of all genes could plausibly lead to neuronal phenotypes upon their disruption.
We turn now to mention remaining reports on hiPSC models of selected genetic mutations. Studies of NPCs, oligodendrocyte precursor cells (OPCs), and neurons from a trio containing a patient with an exonic deletion in contactin-associated protein-like 2 (CNTNAP2) revealed altered migration patterns [288] and expression patterns in synaptic genes [289]. Derivation of hiPSCs from a family with a missense mutation in CSPG4, a gene highly enriched in OPCs [290], revealed that OPCs from carriers of the mutation demonstrated altered patterns of CSPG4 protein subcellular localization and ratios of modified versus unmodified protein as well as reduced viability and oligodendrogenesis [290]. Remarkably, diffusion tensor imaging of affected patients compared to sibling controls demonstrated white matter abnormalities, connecting cellular phenotypes observed in vitro to abnormalities in vivo [290]. Taken together, investigations seeking to uncover the impact of genetic variants associated with schizophrenia have benefited tremendously from hiPSC models, and we expect substantial expansion of this field going forward.
5.4. Exploring the Impact of Non-Genetic Risk Factors with hiPSCs
As enumerated above, several non-genetic factors have been implicated in schizophrenia risk. In several cases, risk factors that are thought to be proximal causes of disease initiation and/or exacerbation can be isolated and studied upon the exposure of hiPSCs to them throughout experimental time points. In this section, we highlight studies that have assessed the impact of environmental risk factors for schizophrenia using hiPSCs-based modes.
Cannabis Exposure
Meta-analyses have found an increased risk of psychotic disorders in anyone who has ever used cannabis (OR = 1.4) and in frequent users (OR = 2.1) [102]. hiPSC- derived forebrain neurons exposed to THC showed transcriptional alterations of numerous genes implicated in schizophrenia [291], including WNT signaling pathways and mitochondrial processes, and expressed a blunted response to KCl- mediated depolarization similar to that seen in patient-derived forebrain neurons in previous studies [238]. In a related report, Obiorah et al. [292] showed that THC exposure reduced the expression of several glutamatergic receptor subunits in hiPSC-derived neurons. Future studies will integrate the ability of hiPSCs to model both typical neurodevelopment and disease phenotypes to continue shedding light on the mechanisms by which cannabis and other drugs of abuse contribute to schizophrenia risk.
Immunologic Processes and Schizophrenia
Another non-genetic risk factor implicated in schizophrenia is maternal infection [293]. However, data in humans is largely based upon cohort and case-control studies [294], and such study designs do not allow the establishment of causation. While animal models may serve as important tools in exploring the effects of maternal infections on brain development and disease, many infectious agents provoke immune responses that differ among host organisms [295]. For these reasons, hiPSC models of neurodevelopment are an attractive approach for mechanistic studies on the effects of infectious agents on brain development and disease-associated phenotypes.
The increased risk of schizophrenia associated with maternal infection may be due at least in part to the elevation of maternal cytokines and other stress-related cellular processes [296]. Exposure of in vitro generated neural aggregates to heat shock altered the expression of several genes implicated in schizophrenia and autism [297]. After demonstrating a role for heat shock protein (HSP)-mediated processes in protection of developing cortex in response to subthreshold exposure to environmental toxins, investigators [298] also documented increased variance in HSP expression levels in response to the same toxins in NPCs derived from patient hiPSC lines. Follow-up analysis confirmed in both animal models and NPCs that exposure to environmental toxins induces HSP signaling in a probabilistic manner, generating significant mosaicism among affected cells [299]. These studies serve as notable examples of combining animal in vivo approaches with human in vitro neural models to provide robust exploration of non-genetic risk factors for schizophrenia [135].
6. Future Directions
6.1. Further Elucidation of Risk Factor Biology in Schizophrenia
While studies of cannabis use and risk of schizophrenia are numerous, similar approaches to assessing the impact of other drugs of abuse on schizophrenia risk are comparatively sparse. Several drugs of abuse can produce a psychotic state during acute intoxication and may also increase risk for later onset of schizophrenia. Psychosis was documented in 6.3% (n = 329) of 5529 hospital drug intoxication cases, particularly among those intoxicated with cannabis (25.9%), amphetamine (25.0%), and cocaine (16.1%) [300]. Patients admitted for substance use disorder to methamphetamine had the highest risk of schizophrenia (hazard ratio = 9.37), followed by cannabis (Hazard ratio = 8.16), cocaine (HR = 5.84), alcohol (HR = 5.56), and opioids (HR = 3.60) [301]. Overall, diagnosis of any substance abuse disorder significantly elevated risk of schizophrenia (HR = 6.04) [302], but most strongly alcohol or cannabis use disorders (HR = 3.38 and 5.20, respectively) [302]. Similar findings have been reported for tobacco smoking (HR of 5.9 [303]. Because cannabis alone is not the only substance associated with schizophrenia risk, and in light of recent genomic findings suggesting a reverse direction of affect [304], hiPSC models should be used to explore the impact of other drugs of abuse on the nervous system in general and on potentially increasing risk of schizophrenia in particular. The differentiation of NPCs to forebrain neurons was used to model the effects of nicotine exposure on neurodevelopment [305], and we anticipate that future studies will assess the effects of other drugs of abuse as well.
6.2. Screening for New Schizophrenia Therapeutics
Progress in understanding disease biology must be coupled with therapeutic innovations derived from emerging discoveries. Considerable interest has been generated in using hiPSC-derived tissues to assess responses of disease-relevant cells to interventions seeking to alleviate disease processes the so-called precision medicine. hiPSC- neurons from cohorts of patients with a given psychiatric diagnosis who exhibit differential responses to treatment will serve as an invaluable strategy to correlate patient genotypes with phenotypic differences in in vitro neural tissue; data derived from such cohorts may be employed to predict treatment responses of psychiatric conditions to specific medications, a longstanding but nevertheless unobtained goal of psychiatric medicine [306]. Comparisons of gene sets impacted by different pharmacologic agents confirmed overlap between genes enriched in schizophrenia SNPs and those involved in antipsychotic response [307], and early-stage studies are now investigating the potential utility of schizophrenia genetic risk variants to predict response to specific antipsychotic medications (e.g., [308]). Further supporting the relationship between schizophrenia risk variants and treatment response, RNA-sequencing analysis of brain tissue of mice chronically treated with the antipsychotic Haloperidol found that differentially expressed genes (DEGs) were enriched in schizophrenia risk loci and biological pathways implicated in the disease [309]. Recently, a screen of over 100 compounds predicted to have relevance to schizophrenia treatment and biology in NPCs and cancer cell lines (CCLs) demonstrated the advantages of using disease-relevant cell types in drug screens and identified compounds that reversed abnormal gene expression signature documented in post-mortem brain samples [310]. Further demonstrating the future utility of hiPSC-based drug screening, high-throughput screening has identified hit compounds that inhibit Zika virus replication [311], infection [312] and reverse Zika-induced neurodevelopmental phenotypes [313]. Taken together, these studies garner much optimism to the prospect of using more disease-relevant and patient-specific approaches toward driving therapeutic innovation.
7. Conclusion
hiPSC enables a broader “ex vivo” study of normal human development and disease mechanisms. Of particular importance to neuropsychiatry, hiPSC-based models produce living, functional, human brain cells accessible for characterization, manipulation, and disease modeling. Insofar as hiPSCs remain isogenic to their donors, they provide a platform for studying the impact of genetics and associated non-genetic factors on neurodevelopment, schizophrenia-associated phenotypes, and response to various interventions both genetic and pharmacological. For these reasons, the further application of hiPSC-based models and the advancement of current technologies should provide a level of hope for investigators striving to understand neuropsychiatric conditions like schizophrenia as well for those affected by them.
Contributor Information
Samuel K. Powell, Medical Scientist Training Program, Icahn School of Medicine at Mount Sinai, New York, NY, USA Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Callan P. O’Shea, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Sara Rose Shannon, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Schahram Akbarian, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Kristen J. Brennand, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
References
- 1.Carpenter WT Jr., Strauss JS, & Bartko JJ (1974). An approach to the diagnosis and understanding of schizophrenia. Introduction. Schizophrenia Bulletin (11), 35–36. 10.1093/schbul/1.11.35 [DOI] [PubMed] [Google Scholar]
- 2.Crow TJ (1985). The two-syndrome concept: origins and current status. Schizophrenia Bulletin, 11(3), 471–486. [DOI] [PubMed] [Google Scholar]
- 3.Sartorius N, Shapiro R, Kimura M, & Barrett K (1972). WHO international pilot study of schizophrenia. Psychological Medicine, 2(4), 422–425. [DOI] [PubMed] [Google Scholar]
- 4.Strauss JS, Carpenter WT Jr., & Bartko JJ (1974). The diagnosis and understanding of schizophrenia. Summary and conclusions. Schizophrenia Bulletin (11), 70–80. [PubMed] [Google Scholar]
- 5.Kay SR, Opler LA, & Lindenmayer JP (1988). Reliability and validity of the positive and negative syndrome scale for schizophrenics. Psychiatry Research, 23(1), 99–110. [DOI] [PubMed] [Google Scholar]
- 6.Lindenmayer JP, Bernstein-Hyman R, & Grochowski S (1994). A new five factor model of schizophrenia. Psychiatric Quarterly, 65(4), 299–322. [DOI] [PubMed] [Google Scholar]
- 7.Wallwork RS, Fortgang R, Hashimoto R, Weinberger DR, & Dickinson D (2012). Searching for a consensus five-factor model of the Positive and Negative Syndrome Scale for schizophrenia. Schizophrenia Research, 137(1–3), 246–250. 10.1016/j.schres.2012.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA: Author. [Google Scholar]
- 9.Staehelin JE, & Kielholz P (1953). Largactil, a new vegetative damping agent in mental disorders. Schweizerische Medizinische Wochenschrift, 83(25), 581–586. [PubMed] [Google Scholar]
- 10.Carlsson A, & Lindqvist M (1963). Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacologica et Toxicologica, 20, 140–144. [DOI] [PubMed] [Google Scholar]
- 11.Creese I, Burt DR, & Snyder SH (1976). Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science, 192(4238), 481–483. [DOI] [PubMed] [Google Scholar]
- 12.Seeman P, & Lee T (1975). Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science, 188(4194), 1217–1219. [DOI] [PubMed] [Google Scholar]
- 13.Borison RL, Pathiraja AP, Diamond BI, & Meibach RC (1992). Risperidone: clinical safety and efficacy in schizophrenia. Psychopharmacology Bulletin, 28(2), 213–218. [PubMed] [Google Scholar]
- 14.Jones PB, Barnes TR, Davies L, Dunn G, Lloyd H, Hayhurst KP, et al. (2006). Randomized controlled trial of the effect on quality of life of second- vs first-generation antipsychotic drugs in schizophrenia: cost utility of the latest antipsychotic drugs in schizophrenia study (CUtLASS 1). Archives of General Psychiatry, 63(10), 1079–1087. 10.1001/archpsyc.63.10.1079 [DOI] [PubMed] [Google Scholar]
- 15.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. The New England Journal of Medicine, 353(12), 1209–1223. 10.1056/NEJMoa051688 [DOI] [PubMed] [Google Scholar]
- 16.Kane J, Honigfeld G, Singer J, & Meltzer H (1988). Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Archives of General Psychiatry, 45(9), 789–796. [DOI] [PubMed] [Google Scholar]
- 17.Fusar-Poli P, Papanastasiou E, Stahl D, Rocchetti M, Carpenter W, Shergill S, et al. (2015). Treatments of negative symptoms in schizophrenia: meta-analysis of 168 randomized placebo-controlled trials. Schizophrenia Bulletin, 41(4), 892–899. 10.1093/schbul/sbu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. (2013). Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet, 382(9896), 951–962. 10.1016/S0140-6736(13)60733-3 [DOI] [PubMed] [Google Scholar]
- 19.Naber D, & Lambert M (2009). The CATIE and CUtLASS studies in schizophrenia: results and implications for clinicians. CNS Drugs, 23(8), 649–659. 10.2165/00023210-200923080-00002 [DOI] [PubMed] [Google Scholar]
- 20.Downing AM, Kinon BJ, Millen BA, Zhang L, Liu L, Morozova MA, et al. (2014). A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry, 14, 351. 10.1186/s12888-014-0351-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, et al. (1992). Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten-country study. Psychological Medicine. Monograph Supplement, 20, 1–97. [DOI] [PubMed] [Google Scholar]
- 22.Hjorthoj C, Sturup AE, McGrath JJ, & Nordentoft M (2017). Years of potential life lost and life expectancy in schizophrenia: a systematic review and meta-analysis. Lancet Psychiatry, 4(4), 295–301. 10.1016/S2215-0366(17)30078-0 [DOI] [PubMed] [Google Scholar]
- 23.Palmer BA, Pankratz VS, & Bostwick JM (2005). The lifetime risk of suicide in schizophrenia: a reexamination. Archives of General Psychiatry, 62(3), 247–253. 10.1001/archpsyc.62.3.247 [DOI] [PubMed] [Google Scholar]
- 24.Caldwell CB, & Gottesman II (1990). Schizophrenics kill themselves too: a review of risk factors for suicide. Schizophrenia Bulletin, 16(4), 571–589. [DOI] [PubMed] [Google Scholar]
- 25.Phillips MR, Yang G, Li S, & Li Y (2004). Suicide and the unique prevalence pattern of schizophrenia in mainland China: a retrospective observational study. Lancet, 364(9439), 1062–1068. 10.1016/S0140-6736(04)17061-X [DOI] [PubMed] [Google Scholar]
- 26.Brown S (1997). Excess mortality of schizophrenia. A meta-analysis. The British Journal of Psychiatry, 171, 502–508. [DOI] [PubMed] [Google Scholar]
- 27.Weinmann S, Read J, & Aderhold V (2009). Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophrenia Research, 113(1), 1–11. 10.1016/j.schres.2009.05.018 [DOI] [PubMed] [Google Scholar]
- 28.Nielsen PR, Laursen TM, & Agerbo E (2016). Comorbidity of schizophrenia and infection: a population-based cohort study. Social Psychiatry and Psychiatric Epidemiology, 51(12), 1581–1589. 10.1007/s00127-016-1297-1 [DOI] [PubMed] [Google Scholar]
- 29.Goff DC, Cather C, Evins AE, Henderson DC, Freudenreich O, Copeland PM, et al. (2005). Medical morbidity and mortality in schizophrenia: guidelines for psychiatrists. Journal of Clinical Psychiatry, 66(2), 183–194; quiz 147, 273–184. [DOI] [PubMed] [Google Scholar]
- 30.Winklbaur B, Ebner N, Sachs G, Thau K, & Fischer G (2006). Substance abuse in patients with schizophrenia. Dialogues in Clinical Neuroscience, 8(1), 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brekke JS, Prindle C, Bae SW, & Long JD (2001). Risks for individuals with schizophrenia who are living in the community. Psychiatric Services, 52(10), 1358–1366. 10.1176/appi.ps.52.10.1358 [DOI] [PubMed] [Google Scholar]
- 32.Rapoport JL, Addington AM, Frangou S, & Psych MR (2005). The neurodevelopmental model of schizophrenia: update 2005. Molecular Psychiatry, 10(5), 434–449. 10.1038/sj.mp.4001642 [DOI] [PubMed] [Google Scholar]
- 33.Rapoport JL, Giedd JN, & Gogtay N (2012). Neurodevelopmental model of schizophrenia: update 2012. Molecular Psychiatry, 17(12), 1228–1238. 10.1038/mp.2012.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stiles J, & Jernigan TL (2010). The basics of brain development. Neuropsychology Review, 20(4), 327–348. 10.1007/s11065-010-9148-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muraki K, & Tanigaki K (2015). Neuronal migration abnormalities and its possible implications for schizophrenia. Frontiers in Neuroscience, 9, 74. 10.3389/fnins.2015.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoenfeld TJ, & Cameron HA (2015). Adult neurogenesis and mental illness. Neuropsychopharmacology, 40(1), 113–128. 10.1038/npp.2014.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt MJ, & Mirnics K (2015). Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology, 40(1), 190–206 10.1038/npp.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartzokis G (2002). Schizophrenia: breakdown in the well-regulated lifelong process of brain development and maturation. Neuropsychopharmacology, 27(4), 672–683. 10.1016/S0893-133X(02)00364-0 [DOI] [PubMed] [Google Scholar]
- 39.Forsyth JK, & Lewis DA (2017). Mapping the consequences of impaired synaptic plasticity in schizophrenia through development: an integrative model for diverse clinical features. Trends in Cognitive Sciences, 21(10), 760–778. 10.1016/j.tics.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirayasu Y, Shenton ME, Salisbury DF, Dickey CC, Fischer IA, Mazzoni P, et al. (1998). Lower left temporal lobe MRI volumes in patients with first-episode schizophrenia compared with psychotic patients with first-episode affective disorder and normal subjects. The American Journal of Psychiatry, 155(10), 1384–1391. 10.1176/ajp.155.10.1384 [DOI] [PubMed] [Google Scholar]
- 41.Wilke M, Kaufmann C, Grabner A, Putz B, Wetter TC, & Auer DP (2001). Gray matter-changes and correlates of disease severity in schizophrenia: a statistical parametric mapping study. NeuroImage, 13(5), 814–824. 10.1006/nimg.2001.0751 [DOI] [PubMed] [Google Scholar]
- 42.Salgado-Pineda P, Baeza I, Perez-Gomez M, Vendrell P, Junque C, Bargallo N, et al. (2003). Sustained attention impairment correlates to gray matter decreases in first episode neuroleptic-naive schizophrenic patients. NeuroImage, 19(2 Pt 1), 365–375. [DOI] [PubMed] [Google Scholar]
- 43.Berge D, Carmona S, Rovira M, Bulbena A, Salgado P, & Vilarroya O (2011). Gray matter volume deficits and correlation with insight and negative symptoms in first- psychotic-episode subjects. Acta Psychiatrica Scandinavica, 123(6), 431–439. 10.1111/j.1600-0447.2010.01635.x [DOI] [PubMed] [Google Scholar]
- 44.Hirayasu Y, Tanaka S, Shenton ME, Salisbury DF, DeSantis MA, Levitt JJ, et al. (2001). Prefrontal gray matter volume reduction in first episode schizophrenia. Cerebral Cortex, 11(4), 374–381. [DOI] [PubMed] [Google Scholar]
- 45.Paillere-Martinot M, Caclin A, Artiges E, Poline JB, Joliot M, Mallet L, et al. (2001). Cerebral gray and white matter reductions and clinical correlates in patients with early onset schizophrenia. Schizophrenia Research, 50(1–2), 19–26. [DOI] [PubMed] [Google Scholar]
- 46.Crespo-Facorro B, Roiz-Santianez R, Perez-Iglesias R, Rodriguez-Sanchez JM, Mata I, Tordesillas-Gutierrez D, et al. (2011). Global and regional cortical thinning in first- episode psychosis patients: relationships with clinical and cognitive features. Psychological Medicine, 41(7), 1449–1460. 10.1017/S003329171000200X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitford TJ, Grieve SM, Farrow TF, Gomes L, Brennan J, Harris AW, et al. (2006). Progressive grey matter atrophy over the first 2–3 years of illness in first-episode schizophrenia: a tensor-based morphometry study. NeuroImage, 32(2), 511–519. 10.1016/j.neuroimage.2006.03.041 [DOI] [PubMed] [Google Scholar]
- 48.Hirayasu Y, Shenton ME, Salisbury DF, Kwon JS, Wible CG, Fischer IA, et al. (1999). Subgenual cingulate cortex volume in first-episode psychosis. The American Journal of Psychiatry, 156(7), 1091–1093. 10.1176/ajp.156.7.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasai K, Shenton ME, Salisbury DF, Onitsuka T, Toner SK, Yurgelun-Todd D, et al. (2003). Differences and similarities in insular and temporal pole MRI gray matter volume abnormalities in first-episode schizophrenia and affective psychosis. Archives of General Psychiatry, 60(11), 1069–1077. 10.1001/archpsyc.60.11.1069 [DOI] [PubMed] [Google Scholar]
- 50.Rothlisberger M, Riecher-Rossler A, Aston J, Fusar-Poli P, Radu EW, & Borgwardt S (2012). Cingulate volume abnormalities in emerging psychosis. Current Pharmaceutical Design, 18(4), 495–504. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Pearlson G, Windemuth A, Ruano G, Perrone-Bizzozero NI, & Calhoun V (2009). Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Human Brain Mapping, 30(1), 241–255. 10.1002/hbm.20508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carpenter DM, Tang CY, Friedman JI, Hof PR, Stewart DG, Buchsbaum MS, et al. (2008). Temporal characteristics of tract-specific anisotropy abnormalities in schizophrenia. Neuroreport, 19(14), 1369–1372. 10.1097/WNR.0b013e32830abc35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karlsgodt KH, van Erp TG, Poldrack RA, Bearden CE, Nuechterlein KH, & Cannon TD (2008). Diffusion tensor imaging of the superior longitudinal fasciculus and working memory in recent-onset schizophrenia. Biological Psychiatry, 63(5), 512–518. 10.1016/j.biopsych.2007.06.017 [DOI] [PubMed] [Google Scholar]
- 54.Perez-Iglesias R, Tordesillas-Gutierrez D, Barker GJ, McGuire PK, Roiz-Santianez R, Mata I, et al. (2010). White matter defects in first episode psychosis patients: a voxelwise analysis of diffusion tensor imaging. NeuroImage, 49(1), 199–204. 10.1016/j.neuroimage.2009.07.016 [DOI] [PubMed] [Google Scholar]
- 55.Ruef A, Curtis L, Moy G, Bessero S, Badan Ba M, Lazeyras F, et al. (2012). Magnetic resonance imaging correlates of first-episode psychosis in young adult male patients: combined analysis of grey and white matter. Journal of Psychiatry & Neuroscience, 37(5), 305–312. 10.1503/jpn.110057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White T, Anjum A, & Schulz SC (2006). The schizophrenia prodrome. The American Journal of Psychiatry, 163(3), 376–380. 10.1176/appi.ajp.163.3.376 [DOI] [PubMed] [Google Scholar]
- 57.Yung AR, & McGorry PD (1996a). The initial prodrome in psychosis: descriptive and qualitative aspects. The Australian and New Zealand Journal of Psychiatry, 30(5), 587–599. 10.3109/00048679609062654 [DOI] [PubMed] [Google Scholar]
- 58.Beiser M, Erickson D, Fleming JA, & Iacono WG (1993). Establishing the onset of psychotic illness. The American Journal of Psychiatry, 150(9), 1349–1354. 10.1176/ajp.150.9.1349 [DOI] [PubMed] [Google Scholar]
- 59.Lencz T, Cornblatt B, & Bilder RM (2001). Neurodevelopmental models of schizophrenia: pathophysiologic synthesis and directions for intervention research. Psychopharmacology Bulletin, 35(1), 95–125. [PubMed] [Google Scholar]
- 60.Tsuang MT, Faraone SV, Bingham S, Young K, Prabhudesai S, Haverstock SL, et al. (2000). Department of Veterans Affairs Cooperative Studies Program genetic linkage study of schizophrenia: ascertainment methods and sample description. American Journal of Medical Genetics, 96(3), 342–347. [DOI] [PubMed] [Google Scholar]
- 61.Yung AR, & McGorry PD (1996b). The prodromal phase of first-episode psychosis: past and current conceptualizations. Schizophrenia Bulletin, 22(2), 353–370. [DOI] [PubMed] [Google Scholar]
- 62.Cornblatt B, Lencz T, & Obuchowski M (2002). The schizophrenia prodrome: treatment and high-risk perspectives. Schizophrenia Research, 54(1–2), 177–186. [DOI] [PubMed] [Google Scholar]
- 63.Cornblatt B, Obuchowski M, Roberts S, Pollack S, & Erlenmeyer-Kimling L (1999). Cognitive and behavioral precursors of schizophrenia. Development and Psychopathology, 11(3), 487–508. [DOI] [PubMed] [Google Scholar]
- 64.Lappin JM, Dazzan P, Morgan K, Morgan C, Chitnis X, Suckling J, et al. (2007). Duration of prodromal phase and severity of volumetric abnormalities in first-episode psychosis. The British Journal of Psychiatry. Supplement, 51, s123–s127. 10.1192/bjp.191.51.s123 [DOI] [PubMed] [Google Scholar]
- 65.Fusar-Poli P, Tantardini M, De Simone S, Ramella-Cravaro V, Oliver D, Kingdon J, et al. (2017). Deconstructing vulnerability for psychosis: meta-analysis of environmental risk factors for psychosis in subjects at ultra high-risk. European Psychiatry, 40, 65–75. 10.1016/j.eurpsy.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 66.Clarke MC, Tanskanen A, Huttunen M, Leon DA, Murray RM, Jones PB, et al. (2011). Increased risk of schizophrenia from additive interaction between infant motor developmental delay and obstetric complications: evidence from a population-based longitudinal study. The American Journal of Psychiatry, 168(12), 1295–1302. 10.1176/appi.ajp.2011.11010011 [DOI] [PubMed] [Google Scholar]
- 67.Jones P, Rodgers B, Murray R, & Marmot M (1994). Child development risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet, 344(8934), 1398–1402. [DOI] [PubMed] [Google Scholar]
- 68.Kremen WS, Buka SL, Seidman LJ, Goldstein JM, Koren D, & Tsuang MT (1998). IQ decline during childhood and adult psychotic symptoms in a community sample: a 19-year longitudinal study. The American Journal of Psychiatry, 155(5), 672–677. 10.1176/ajp.155.5.672 [DOI] [PubMed] [Google Scholar]
- 69.Wood SJ, Pantelis C, Proffitt T, Phillips LJ, Stuart GW, Buchanan JA, et al. (2003). Spatial working memory ability is a marker of risk-for-psychosis. Psychological Medicine, 33(7), 1239–1247. [DOI] [PubMed] [Google Scholar]
- 70.Brewer WJ, Francey SM, Wood SJ, Jackson HJ, Pantelis C, Phillips LJ, et al. (2005). Memory impairments identified in people at ultra-high risk for psychosis who later develop first-episode psychosis. The American Journal of Psychiatry, 162(1), 71–78. 10.1176/appi.ajp.162.1.71 [DOI] [PubMed] [Google Scholar]
- 71.Dickson H, Laurens KR, Cullen AE, & Hodgins S (2012). Meta-analyses of cognitive and motor function in youth aged 16 years and younger who subsequently develop schizophrenia. Psychological Medicine, 42(4), 743–755. 10.1017/S0033291711001693 [DOI] [PubMed] [Google Scholar]
- 72.Erlenmeyer-Kimling L, Rock D, Roberts SA, Janal M, Kestenbaum C, Cornblatt B, et al. (2000). Attention, memory, and motor skills as childhood predictors of schizophrenia- related psychoses: the New York High-Risk Project. The American Journal of Psychiatry, 157(9), 1416–1422. 10.1176/appi.ajp.157.9.1416 [DOI] [PubMed] [Google Scholar]
- 73.Done DJ, Crow TJ, Johnstone EC, & Sacker A (1994). Childhood antecedents of schizophrenia and affective illness: social adjustment at ages 7 and 11. BMJ, 309(6956), 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davidson M, Reichenberg A, Rabinowitz J, Weiser M, Kaplan Z, & Mark M (1999). Behavioral and intellectual markers for schizophrenia in apparently healthy male adolescents. The American Journal of Psychiatry, 156(9), 1328–1335. 10.1176/ajp.156.9.1328 [DOI] [PubMed] [Google Scholar]
- 75.Klosterkotter J, Hellmich M, Steinmeyer EM, & Schultze-Lutter F (2001). Diagnosing schizophrenia in the initial prodromal phase. Archives of General Psychiatry, 58(2), 158–164. [DOI] [PubMed] [Google Scholar]
- 76.Pantelis C, Velakoulis D, McGorry PD, Wood SJ, Suckling J, Phillips LJ, et al. (2003). Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. Lancet, 361(9354), 281–288. 10.1016/S0140-6736(03)12323-9 [DOI] [PubMed] [Google Scholar]
- 77.Borgwardt SJ, McGuire PK, Aston J, Berger G, Dazzan P, Gschwandtner U, et al. (2007). Structural brain abnormalities in individuals with an at-risk mental state who later develop psychosis. The British Journal of Psychiatry. Supplement, 51, s69–s75. 10.1192/bjp.191.51.s69 [DOI] [PubMed] [Google Scholar]
- 78.Fornito A, Yung AR, Wood SJ, Phillips LJ, Nelson B, Cotton S, et al. (2008). Anatomic abnormalities of the anterior cingulate cortex before psychosis onset: an MRI study of ultra-high-risk individuals. Biological Psychiatry, 64(9), 758–765. 10.1016/j.biopsych.2008.05.032 [DOI] [PubMed] [Google Scholar]
- 79.Takahashi T, Wood SJ, Soulsby B, Kawasaki Y, McGorry PD, Suzuki M, et al. (2009a). An MRI study of the superior temporal subregions in first-episode patients with various psychotic disorders. Schizophrenia Research, 113(2–3), 158–166. 10.1016/j.schres.2009.06.016 [DOI] [PubMed] [Google Scholar]
- 80.Takahashi T, Wood SJ, Yung AR, Phillips LJ, Soulsby B, McGorry PD, et al. (2009b). Insular cortex gray matter changes in individuals at ultra-high-risk of developing psychosis. Schizophrenia Research, 111(1–3), 94–102. 10.1016/j.schres.2009.03.024 [DOI] [PubMed] [Google Scholar]
- 81.Mechelli A, Riecher-Rossler A, Meisenzahl EM, Tognin S, Wood SJ, Borgwardt SJ, et al. (2011). Neuroanatomical abnormalities that predate the onset of psychosis: a multicenter study. Archives of General Psychiatry, 68(5), 489–495. 10.1001/archgenpsychiatry.2011.42 [DOI] [PubMed] [Google Scholar]
- 82.Fusar-Poli P, Broome MR, Woolley JB, Johns LC, Tabraham P, Bramon E, et al. (2011). Altered brain function directly related to structural abnormalities in people at ultra high risk of psychosis: longitudinal VBM-fMRI study. Journal of Psychiatric Research, 45(2), 190–198. 10.1016/j.jpsychires.2010.05.012 [DOI] [PubMed] [Google Scholar]
- 83.Jung WH, Kim JS, Jang JH, Choi JS, Jung MH, Park JY, et al. (2011). Cortical thickness reduction in individuals at ultra-high-risk for psychosis. Schizophrenia Bulletin, 37(4), 839–849. 10.1093/schbul/sbp151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gilmore JH, Kang C, Evans DD, Wolfe HM, Smith JK, Lieberman JA, et al. (2010a). Prenatal and neonatal brain structure and white matter maturation in children at high risk for schizophrenia. The American Journal of Psychiatry, 167(9), 1083–1091. 10.1176/appi.ajp.2010.09101492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gilmore JH, Schmitt JE, Knickmeyer RC, Smith JK, Lin W, Styner M, et al. (2010b). Genetic and environmental contributions to neonatal brain structure: A twin study. Human Brain Mapping, 31(8), 1174–1182. 10.1002/hbm.20926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walterfang M, McGuire PK, Yung AR, Phillips LJ, Velakoulis D, Wood SJ, et al. (2008). White matter volume changes in people who develop psychosis. The British Journal of Psychiatry, 193(3), 210–215. 10.1192/bjp.bp.107.043463 [DOI] [PubMed] [Google Scholar]
- 87.Bloemen OJ, de Koning MB, Schmitz N, Nieman DH, Becker HE, de Haan L, et al. (2010). White-matter markers for psychosis in a prospective ultra-high-risk cohort. Psychological Medicine, 40(8), 1297–1304. 10.1017/S0033291709991711 [DOI] [PubMed] [Google Scholar]
- 88.Brown AS (2006). Prenatal infection as a risk factor for schizophrenia. Schizophrenia Bulletin, 32(2), 200–202. 10.1093/schbul/sbj052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown AS (2012). Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Developmental Neurobiology, 72(10), 1272–1276. 10.1002/dneu.22024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cannon M, Jones PB, & Murray RM (2002). Obstetric complications and schizophrenia: historical and meta-analytic review. The American Journal of Psychiatry, 159(7), 1080–1092. 10.1176/appi.ajp.159.7.1080 [DOI] [PubMed] [Google Scholar]
- 91.Picker JD, & Coyle JT (2005). Do maternal folate and homocysteine levels play a role in neurodevelopmental processes that increase risk for schizophrenia? Harvard Review of Psychiatry, 13(4), 197–205. 10.1080/10673220500243372 [DOI] [PubMed] [Google Scholar]
- 92.Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, & de Rooij SR (2011). Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas, 70(2), 141–145. 10.1016/j.maturitas.2011.06.017 [DOI] [PubMed] [Google Scholar]
- 93.Knud Larsen J, Bendsen BB, Foldager L, & Munk-Jorgensen P (2010). Prematurity and low birth weight as risk factors for the development of affective disorder, especially depression and schizophrenia: a register study. Acta Neuropsychiatrica, 22(6), 284–291. 10.1111/j.1601-5215.2010.00498.x [DOI] [PubMed] [Google Scholar]
- 94.Rifkin L, Lewis S, Jones P, Toone B, & Murray R (1994). Low birth weight and schizophrenia. The British Journal of Psychiatry, 165(3), 357–362. [DOI] [PubMed] [Google Scholar]
- 95.Wahlbeck K, Forsen T, Osmond C, Barker DJ, & Eriksson JG (2001). Association of schizophrenia with low maternal body mass index, small size at birth, and thinness during childhood. Archives of General Psychiatry, 58(1), 48–52. [DOI] [PubMed] [Google Scholar]
- 96.Torniainen M, Wegelius A, Tuulio-Henriksson A, Lonnqvist J, & Suvisaari J (2013). Both low birthweight and high birthweight are associated with cognitive impairment in persons with schizophrenia and their first-degree relatives. Psychological Medicine, 43(11), 2361–2367. 10.1017/S0033291713000032 [DOI] [PubMed] [Google Scholar]
- 97.Moilanen K, Jokelainen J, Jones PB, Hartikainen AL, Jarvelin MR, & Isohanni M (2010). Deviant intrauterine growth and risk of schizophrenia: a 34-year follow-up of the Northern Finland 1966 Birth Cohort. Schizophrenia Research, 124(1–3), 223–230. 10.1016/j.schres.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 98.Davies G, Welham J, Chant D, Torrey EF, & McGrath J (2003). A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophrenia Bulletin, 29(3), 587–593. [DOI] [PubMed] [Google Scholar]
- 99.Frissen A, Lieverse R, Drukker M, van Winkel R, Delespaul P, & Investigators G (2015). Childhood trauma and childhood urbanicity in relation to psychotic disorder. Social Psychiatry and Psychiatric Epidemiology, 50(10), 1481–1488. 10.1007/s00127-015-1049-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lataster J, Myin-Germeys I, Lieb R, Wittchen HU, & van Os J (2012). Adversity and psychosis: a 10-year prospective study investigating synergism between early and recent adversity in psychosis. Acta Psychiatrica Scandinavica, 125(5), 388–399. 10.1111/j.1600-0447.2011.01805.x [DOI] [PubMed] [Google Scholar]
- 101.Marconi A, Di Forti M, Lewis CM, Murray RM, & Vassos E (2016). Meta-analysis of the association between the level of cannabis use and risk of psychosis. Schizophrenia Bulletin, 42(5), 1262–1269. 10.1093/schbul/sbw003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M, et al. (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet, 370(9584), 319–328. 10.1016/S0140-6736(07)61162-3 [DOI] [PubMed] [Google Scholar]
- 103.Heinz A, Deserno L, & Reininghaus U (2013). Urbanicity, social adversity and psychosis. World Psychiatry, 12(3), 187–197. 10.1002/wps.20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. (2009). Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet, 373(9659), 234–239. 10.1016/S0140-6736(09)60072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lichtenstein P, Bjork C, Hultman CM, Scolnick E, Sklar P, & Sullivan PF (2006). Recurrence risks for schizophrenia in a Swedish national cohort. Psychological Medicine, 36(10), 1417–1425. 10.1017/S0033291706008385 [DOI] [PubMed] [Google Scholar]
- 106.Cardno AG, & Gottesman II (2000). Twin studies of schizophrenia: from bow-and- arrow concordances to star wars Mx and functional genomics. American Journal of Medical Genetics, 97(1), 12–17. [PubMed] [Google Scholar]
- 107.Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. (2018). Heritability of schizophrenia and schizophrenia spectrum based on the Nationwide Danish Twin Register. Biological Psychiatry, 83(6), 492–498. 10.1016/j.biopsych.2017.08.017 [DOI] [PubMed] [Google Scholar]
- 108.Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum AD, Breen G, et al. (2018). Psychiatric genomics: an update and an agenda. The American Journal of Psychiatry, 175(1), 15–27. 10.1176/appi.ajp.2017.17030283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Power RA, Kyaga S, Uher R, MacCabe JH, Langstrom N, Landen M, et al. (2013). Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry, 70(1), 22–30. 10.1001/jamapsychiatry.2013.268 [DOI] [PubMed] [Google Scholar]
- 110.Gershon ES, Alliey-Rodriguez N, & Liu C (2011). After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. The American Journal of Psychiatry, 168(3), 253–256. 10.1176/appi.ajp.2010.10091340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Malaspina D, Brown A, Goetz D, Alia-Klein N, Harkavy-Friedman J, Harlap S, et al. (2002). Schizophrenia risk and paternal age: a potential role for de novo mutations in schizophrenia vulnerability genes. CNS Spectrums, 7(1), 26–29. [DOI] [PubMed] [Google Scholar]
- 112.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature, 488(7412), 471–475. 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell, 154(3), 518–529. 10.1016/j.cell.2013.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, et al. (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nature Genetics, 44(12), 1365–1369. 10.1038/ng.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature, 506(7487), 179–184. 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, et al. (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. American Journal of Human Genetics, 87(3), 316–324. 10.1016/j.ajhg.2010.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, Jouan L, et al. (2011). Increased exonic de novo mutation rate in individuals with schizophrenia. Nature Genetics, 43(9), 860–863. 10.1038/ng.886 [DOI] [PubMed] [Google Scholar]
- 118.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature, 506(7487), 185–190. 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M, et al. (2016). Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nature Neuroscience, 19(11), 1433–1441. 10.1038/nn.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, et al. (2016) Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nature Neuroscience 19(11):1433–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, et al. (2014). Copy number variation in schizophrenia in Sweden. Molecular Psychiatry, 19(7), 762–773. 10.1038/mp.2014.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rees E, Kirov G, O’Donovan MC, & Owen MJ (2012). De novo mutation in schizophrenia. Schizophrenia Bulletin, 38(3), 377–381. 10.1093/schbul/sbs047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature Genetics, 49(1), 27–35. 10.1038/ng.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, et al. (2014). Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 deletion syndrome. The American Journal of Psychiatry, 171(6), 627–639. 10.1176/appi.ajp.2013.13070864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Van L, Boot E, & Bassett AS (2017). Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia. Current Opinion in Psychiatry, 30(3), 191–196. 10.1097/YCO.0000000000000324 [DOI] [PubMed] [Google Scholar]
- 126.Bergen SE, Ploner A, Howrigan D, CNV Analysis Group and the Schizophrenia Working Group of the Psychiatric Genomics Consortium, O’Donovan MC, Smoller JW, et al. (2018). Joint contributions of rare copy number variants and common SNPs to risk for schizophrenia. Am J Psychiatry, 176, 29. 10.1176/appi.ajp.2018.17040467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tansey KE, Rees E, Linden DE, Ripke S, Chambert KD, Moran JL, et al. (2016). Common alleles contribute to schizophrenia in CNV carriers. Molecular Psychiatry, 21(8), 1153. 10.1038/mp.2015.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gottesman II, & Shields J (1967). A polygenic theory of schizophrenia. Proceedings of the National Academy of Sciences of the United States of America, 58(1), 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature, 460(7256), 753–757. 10.1038/nature08192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460(7256), 748–752. 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nature Genetics, 40(9), 1053–1055. 10.1038/ng.201 [DOI] [PubMed] [Google Scholar]
- 132.Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. (2009). Common variants conferring risk of schizophrenia. Nature, 460(7256), 744–747. 10.1038/nature08186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, et al. (2011). Genome-wide association study identifies five new schizophrenia loci. Nature Genetics, 43(10), 969–976. 10.1038/ng.940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature Genetics, 45(10), 1150–1159. 10.1038/ng.2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schmitt A, Malchow B, Hasan A, & Falkai P (2014). The impact of environmental factors in severe psychiatric disorders. Frontiers in Neuroscience, 8, 19. 10.3389/fnins.2014.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pardinas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature Genetics, 50(3), 381–389. 10.1038/s41588-018-0059-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, et al. (2017). Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nature Genetics, 49(11), 1576–1583. 10.1038/ng.3973 [DOI] [PubMed] [Google Scholar]
- 139.Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J, et al. (2011). Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nature Genetics, 43(12), 1224–1227. 10.1038/ng.980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, et al. (2017). Genetic effects on gene expression across human tissues. Nature, 550(7675), 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science, 337(6099), 1190–1195. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Albert FW, & Kruglyak L (2015). The role of regulatory variation in complex traits and disease. Nature Reviews Genetics, 16(4), 197–212. 10.1038/nrg3891 [DOI] [PubMed] [Google Scholar]
- 143.Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. (2017). An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nature Neuroscience, 20(10), 1418–1426. 10.1038/nn.4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science, 362(6420). 10.1126/science.aat8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rajarajan P, Gil SE, Brennand KJ, & Akbarian S (2016). Spatial genome organization and cognition. Nature Reviews Neuroscience, 17(11), 681–691. 10.1038/nrn.2016.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Richards AL, Jones L, Moskvina V, Kirov G, Gejman PV, Levinson DF, et al. (2012). Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Molecular Psychiatry, 17(2), 193–201. 10.1038/mp.2011.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. (2016). Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature Neuroscience, 19(11), 1442–1453. 10.1038/nn.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, et al. (2018).The dynamic landscape of open chromatin during human cortical neurogenesis. Cell2, 172(1–2), 289–304, e218. 10.1016/j.cell.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. (2018). Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nature Neuroscience, 21(8), 1117–1125. 10.1038/s41593-018-0197-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang YE, Landback P, Vibranovski MD, & Long M (2011). Accelerated recruitment of new brain development genes into the human genome. PLoS Biology, 9(10), e1001179. 10.1371/journal.pbio.1001179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Akbarian S, Bunney WE Jr., Potkin SG, Wigal SB, Hagman JO, Sandman CA, et al. (1993). Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Archives of General Psychiatry, 50(3), 169–177. [DOI] [PubMed] [Google Scholar]
- 152.Jakob H, & Beckmann H (1986). Prenatal developmental disturbances in the limbic allocortex in schizophrenics. Journal of Neural Transmission, 65(3–4), 303–326. [DOI] [PubMed] [Google Scholar]
- 153.Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, & Weickert CS (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. The American Journal of Psychiatry, 167(12), 1479–1488. 10.1176/appi.ajp.2010.09060784 [DOI] [PubMed] [Google Scholar]
- 154.Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, et al. (2011).Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. The Journal of Neuroscience, 31(30), 11088–11095. 10.1523/JNEUROSCI.1234-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Horváth S, Janka Z, Mirnics K, (2011) Analyzing Schizophrenia by DNA Microarrays.Biological Psychiatry 69(2):157–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Torkamani A, Dean B, Schork NJ, & Thomas EA (2010). Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Research, 20(4), 403–412. 10.1101/gr.101956.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mistry M, Gillis J, & Pavlidis P (2013a). Genome-wide expression profiling of schizophrenia using a large combined cohort. Molecular Psychiatry, 18(2), 215–225. 10.1038/mp.2011.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mistry M, Gillis J, & Pavlidis P (2013b). Meta-analysis of gene coexpression networks in the post-mortem prefrontal cortex of patients with schizophrenia and unaffected controls. BMC Neuroscience, 14, 105. 10.1186/1471-2202-14-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Finucane HK, Reshef YA,Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. (2018).Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nature Genetics, 50(4), 621–629. 10.1038/s41588-018-0081-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, et al. (2018).Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nature Genetics, 50(4), 538–548. 10.1038/s41588-018-0092-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. (2018).Genetic identification of brain cell types underlying schizophrenia. Nature Genetics, 50(6), 825–833. 10.1038/s41588-018-0129-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roussos P, Katsel P, Davis KL, Siever LJ, & Haroutunian V (2012). A system-level transcriptomic analysis of schizophrenia using postmortem brain tissue samples. Archives of General Psychiatry, 69(12), 1205–1213. 10.1001/archgenpsychiatry.2012.704 [DOI] [PubMed] [Google Scholar]
- 163.Radulescu E, Jaffe AE, Straub RE, Chen Q, Shin JH, Hyde TM, et al. (2018).Identification and prioritization of gene sets associated with schizophrenia risk by co- expression network analysis in human brain. Molecular Psychiatry. 10.1038/s41380-018-0304-1 [DOI] [PubMed] [Google Scholar]
- 164.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nature Genetics, 48(3),245–252. 10.1038/ng.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardiñas AF, et al. (2019) Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nature Genetics 51(4):659–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.The Network, O’Dushlaine C, Rossin L, Lee PH, Duncan L, Parikshak NN, et al. (2015). Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience, 18, 199. 10.1038/nn.3922. https://www.nature.com/articles/nn.3922#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, et al. (2013). Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics,45(2), 124–130. 10.1038/ng.2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, et al. (2014). A role for noncoding variation in schizophrenia. Cell Reports, 9(4), 1417–1429. 10.1016/j.celrep.2014.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, et al. (2015). The PsychENCODE project. Nature Neuroscience, 18(12), 1707–1712. 10.1038/nn.4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, et al. (2018). Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nature Neuroscience, 21(8), 1126–1136. 10.1038/s41593-018-0187-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. (2016). Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nature Neuroscience, 19(1), 40–47. 10.1038/nn.4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schulz H, Ruppert AK, Herms S, Wolf C, Mirza-Schreiber N, Stegle O, et al. (2017). Genome-wide mapping of genetic determinants influencing DNA methylation and gene expression in human hippocampus. Nature Communications, 8(1), 1511. 10.1038/s41467-017-01818-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dobbyn A, Huckins LM, Boocock J, Sloofman LG, Glicksberg BS, Giambartolomei C, et al. (2018). Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. American Journal of Human Genetics, 102(6), 1169–1184. 10.1016/j.ajhg.2018.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casino C, et al. (2018a).Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science, 362(6420), eaat4311. 10.1126/science.aat4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bharadwaj R, Jiang Y, Mao W, Jakovcevski M, Dincer A, Krueger W, et al. (2013).Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. The Journal of Neuroscience, 33(29), 11839–11851. 10.1523/JNEUROSCI.1252-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY, et al. (2014).Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition.Neuron, 84(5), 997–1008. 10.1016/j.neuron.2014.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bryois J, Garrett ME, Song L, Safi A, Giusti-Rodriguez P, Johnson GD, et al. (2018). Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nature Communications, 9(1), 3121. 10.1038/s41467-018-05379-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fullard JF, Giambartolomei C, Hauberg ME, Xu K, Voloudakis G, Shao Z, et al. (2017). Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Human Molecular Genetics, 26(10), 1942–1951. 10.1093/hmg/ddx103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fullard JF, Hauberg ME, Bendl J, Egervari G, Cirnaru MD, Reach SM, et al. (2018). An atlas of chromatin accessibility in the adult human brain. Genome Research,28(8), 1243–1252. 10.1101/gr.232488.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature, 538(7626), 523–527. 10.1038/nature19847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Takahashi K, & Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), 663–676. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 182.Narsinh KH, Plews J, & Wu JC (2011). Comparison of human induced pluripotent and embryonic stem cells: fraternal or identical twins? Molecular Therapy, 19(4), 635–638. 10.1038/mt.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hoffman GE, Schrode N, Flaherty E, & Brennand KJ (2018). New considerations for hiPSC-based models of neuropsychiatric disorders. Molecular Psychiatry, 24, 49. 10.1038/s41380-018-0029-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, et al. (2011). Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell, 8(1), 106–118. 10.1016/j.stem.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. (2011).Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells.Nature, 471(7336), 68–73. 10.1038/nature09798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Julia TCW, Carvalho CMB, Yuan B, Gu S, Altheimer AN, McCarthy S, et al. (2017). Divergent levels of marker chromosomes in an hiPSC-based model of psychosis. Stem Cell Reports, 8(3), 519–528. 10.1016/j.stemcr.2017.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grochowski CM, Gu S, Yuan B, Tcw J, Brennand KJ, Sebat J, et al. (2018). Marker chromosome genomic structure and temporal origin implicate a chromoanasynthesis event in a family with pleiotropic psychiatric phenotypes. Human Mutation, 39(7), 939–946. 10.1002/humu.23537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kyttala A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK, et al. (2016). Genetic variability overrides the impact of parental cell type and determines iPSC Differentiation Potential. Stem Cell Reports, 6(2), 200–212. 10.1016/j.stemcr.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer DM, et al. (2017). Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nature Communications, 8(1), 2225. 10.1038/s41467-017-02330-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. (2018).Combining NGN2 Programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Reports, 23(8), 2509–2523. 10.1016/j.celrep.2018.04.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mertens J, Marchetto MC, Bardy C, & Gage FH (2016). Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nature Reviews Neuroscience, 17(7), 424–437. 10.1038/nrn.2016.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, & Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology, 27(3), 275–280. 10.1038/nbt.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell, 143(4), 527–539. 10.1016/j.cell.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell, 12(5), 559–572. 10.1016/j.stem.2013.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature, 480(7378), 547–551. 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lu J, Zhong X, Liu H, Hao L, Tzu-Ling Huang C, Sherafat MA, et al. (2016) Generation of serotonin neurons from human pluripotent stem cells. Nature Biotechnology 34(1):89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yu DX, Di Giorgio FP, Yao J, Marchetto MC, Brennand K, Wright R, et al. (2014).Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports,2(3), 295–310. 10.1016/j.stemcr.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sarkar A, Mei A, Paquola ACM, Stern S, Bardy C, Klug JR, et al. (2018). Efficient generation of CA3 neurons from human pluripotent stem cells enables modeling of hippocampal connectivity in vitro. Cell Stem Cell, 22(5), 684–697. e689. 10.1016/j.stem.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Qi Y, Zhang XJ, Renier N, Wu Z, Atkin T, Sun Z, et al. (2017). Combined small- molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nature Biotechnology, 35(2), 154–163. 10.1038/nbt.3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, et al. (2018). Molecular and functional variation in iPSC-derived sensory neurons. Nature Genetics, 50(1), 54–61. 10.1038/s41588-017-0005-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kuijlaars J, Oyelami T, Diels A, Rohrbacher J, Versweyveld S, Meneghello G, et al. (2016). Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Scientific Reports, 6, 36529. 10.1038/srep36529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gunhanlar N, Shpak G, van der Kroeg M, Gouty-Colomer LA, Munshi ST, Lendemeijer B, et al. (2018). A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Molecular Psychiatry, 23(5), 1336–1344. 10.1038/mp.2017.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, & Wernig M (2010).Direct conversion of fibroblasts to functional neurons by defined factors. Nature, 463(7284),1035–1041. 10.1038/nature08797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pang ZP,Yang N, Vierbuchen T, Ostermeier A, Fuentes DR,Yang TQ, et al. (2011).Induction of human neuronal cells by defined transcription factors. Nature, 476(7359),220–223. 10.1038/nature10202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. (2013). Rapid single- step induction of functional neurons from human pluripotent stem cells. Neuron, 78(5),785–798. 10.1016/j.neuron.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ho SM, Hartley BJ, Tcw J, Beaumont M, Stafford K, Slesinger PA, et al. (2016).Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells.Methods, 101, 113–124. 10.1016/j.ymeth.2015.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Colasante G, Lignani G, Rubio A, Medrihan L,Yekhlef L, Sessa A, et al. (2015). Rapid conversion of fibroblasts into functional forebrain GABAergic interneurons by direct genetic reprogramming. Cell Stem Cell, 17(6), 719–734. 10.1016/j.stem.2015.09.002 [DOI] [PubMed] [Google Scholar]
- 208.Sun AX, Yuan Q, Tan S, Xiao Y, Wang D, Khoo AT, et al. (2016). Direct induction and functional maturation of forebrain GABAergic neurons from human pluripotent stem cells. Cell Reports, 16(7), 1942–1953. 10.1016/j.celrep.2016.07.035 [DOI] [PubMed] [Google Scholar]
- 209.Yang N, Chanda S, Marro S, Ng YH, Janas JA, Haag D, et al. (2017). Generation of pure GABAergic neurons by transcription factor programming. Nature Methods, 14(6), 621–628. 10.1038/nmeth.4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature, 476(7359), 224–227. 10.1038/nature10284 [DOI] [PubMed] [Google Scholar]
- 211.Theka I, Caiazzo M, Dvoretskova E, Leo D, Ungaro F, Curreli S, et al. (2013).Rapid generation of functional dopaminergic neurons from human induced pluripotent stem cells through a single-step procedure using cell lineage transcription factors. Stem Cells Translational Medicine, 2(6), 473–479. 10.5966/sctm.2012-0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lu J, Zhong X, Liu H, Hao L, Huang CT, Sherafat MA, et al. (2016). Generation of serotonin neurons from human pluripotent stem cells. Nature Biotechnology, 34(1), 89–94. 10.1038/nbt.3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Vadodaria KC, Mertens J, Paquola A, Bardy C, Li X, Jappelli R, et al. (2016).Generation of functional human serotonergic neurons from fibroblasts. Molecular Psychiatry, 21(1), 49–61. 10.1038/mp.2015.161 [DOI] [PubMed] [Google Scholar]
- 214.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. (2011).Modelling schizophrenia using human induced pluripotent stem cells. Nature, 473(7346), 221–225. 10.1038/nature09915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hook V, Brennand KJ, Kim Y, Toneff T, Funkelstein L, Lee KC, et al. (2014).Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Reports, 3(4), 531–538. 10.1016/j.stemcr.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Robicsek O, Karry R, Petit I, Salman-Kesner N, Muller FJ, Klein E, et al. (2013).Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Molecular Psychiatry, 18(10),1067–1076. 10.1038/mp.2013.67 [DOI] [PubMed] [Google Scholar]
- 217.Xu J, Hartley BJ, Kurup P, Phillips A, Topol A, Xu M, et al. (2018). Inhibition of STEP61 ameliorates deficits in mouse and hiPSC-based schizophrenia models. Molecular Psychiatry, 23(2), 271–281. 10.1038/mp.2016.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Carty NC, Xu J, Kurup P, Brouillette J, Goebel-Goody SM, Austin DR, et al. (2012). The tyrosine phosphatase STEP: implications in schizophrenia and the molecular mechanism underlying antipsychotic medications. Translational Psychiatry, 2, e137. 10.1038/tp.2012.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. (2015).Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Molecular Psychiatry, 20(3), 361–368. 10.1038/mp.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Topol A, English JA, Flaherty E, Rajarajan P, Hartley BJ, Gupta S, et al. (2015a).Increased abundance of translation machinery in stem cell-derived neural progenitor cells from four schizophrenia patients. Translational Psychiatry, 5, e662. 10.1038/tp.2015.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Topol A, Zhu S, Tran N, Simone A, Fang G, & Brennand KJ (2015b). Altered WNT signaling in human induced pluripotent stem cell neural progenitor cells derived from four schizophrenia patients. Biological Psychiatry, 78(6), e29–e34. 10.1016/j.biopsych.2014.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Casas BS, Vitoria G, do Costa MN, Madeiro da Costa R, Trindade P, Maciel R, et al. (2018). hiPSC-derived neural stem cells from patients with schizophrenia induce an impaired angiogenesis. Translational Psychiatry, 8(1), 48. 10.1038/s41398-018-0095-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hino M, Kunii Y, Matsumoto J, Wada A, Nagaoka A, Niwa S, et al. (2016). Decreased VEGFR2 expression and increased phosphorylatedAkt1 in the prefrontal cortex of individuals with schizophrenia. Journal of Psychiatric Research, 82, 100–108. 10.1016/j.jpsychires.2016.07.018 [DOI] [PubMed] [Google Scholar]
- 224.Lee BH, Hong JP, Hwang JA, Ham BJ, Na KS, Kim WJ, et al. (2015).Alterations in plasma vascular endothelial growth factor levels in patients with schizophrenia before and after treatment. Psychiatry Research, 228(1), 95–99. 10.1016/j.psychres.2015.04.020 [DOI] [PubMed] [Google Scholar]
- 225.Lopes R, Soares R, Coelho R, & Figueiredo-Braga M (2015). Angiogenesis in the pathophysiology of schizophrenia - a comprehensive review and a conceptual hypothesis. Life Sciences, 128, 79–93. 10.1016/j.lfs.2015.02.010 [DOI] [PubMed] [Google Scholar]
- 226.Gonzalez DM, Gregory J, & Brennand KJ (2017). The importance of non-neuronal cell types in hiPSC-based disease modeling and drug screening. Frontiers in Cell and Development Biology, 5, 117. 10.3389/fcell.2017.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ben-Shachar D (2002). Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. Journal of Neurochemistry, 83(6), 1241–1251. [DOI] [PubMed] [Google Scholar]
- 228.Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, et al. (2004). Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Molecular Psychiatry, 9(7), 684–697, 643. 10.1038/sj.mp.4001511 [DOI] [PubMed] [Google Scholar]
- 229.Uguz AC, Demirci K, & Espino J (2016). The importance of melatonin and mitochondria interaction in mood disorders and schizophrenia: a current assessment. Current Medicinal Chemistry, 23(20), 2146–2158. [DOI] [PubMed] [Google Scholar]
- 230.Paulsen Bda S, de Moraes Maciel R, Galina A, Souza da Silveira M, dos Santos Souza C, Drummond H, et al. (2012). Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplantation,21(7), 1547–1559. 10.3727/096368911X600957 [DOI] [PubMed] [Google Scholar]
- 231.Robicsek O, Ene HM, Karry R, Ytzhaki O, Asor E, McPhie D, et al. (2018). Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophrenia Bulletin, 44(2), 432–442. 10.1093/schbul/sbx077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Caputo V, Ciolfi A, Macri S, & Pizzuti A (2015). The emerging role of MicroRNA in schizophrenia. CNS & Neurological Disorders Drug Targets, 14(2), 208–221. [DOI] [PubMed] [Google Scholar]
- 233.Shi S, Leites C, He D, Schwartz D, Moy W, Shi J, et al. (2014). MicroRNA-9 and microRNA-326 regulate human dopamine D2 receptor expression, and the microRNA- mediated expression regulation is altered by a genetic variant. The Journal of Biological Chemistry, 289(19), 13434–13444. 10.1074/jbc.M113.535203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Han J, Kim HJ, Schafer ST, Paquola A, Clemenson GD, Toda T, et al. (2016).Functional implications of miR-19 in the migration of newborn neurons in the adult brain.Neuron, 91(1), 79–89. 10.1016/j.neuron.2016.05.034 [DOI] [PubMed] [Google Scholar]
- 235.Topol A, Zhu S, Hartley BJ, English J, Hauberg ME, Tran N, et al. (2016).Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Reports, 15(5), 1024–1036. 10.1016/j.celrep.2016.03.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hauberg ME, Roussos P, Grove J, Borglum AD, Mattheisen M, & Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2016). Analyzing the role of MicroRNAs in schizophrenia in the context of common genetic risk variants. JAMA Psychiatry, 73(4), 369–377. 10.1001/jamapsychiatry.2015.3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hoffman GE, & Brennand KJ (2018). Mapping regulatory variants in hiPSC models.Nature Genetics, 50(1), 1–2. 10.1038/s41588-017-0017-4 [DOI] [PubMed] [Google Scholar]
- 238.Roussos P, Guennewig B, Kaczorowski DC, Barry G, & Brennand KJ (2016).Activity-dependent changes in gene expression in schizophrenia human-induced pluripotent stem cell neurons. JAMA Psychiatry, 73(11), 1180–1188. 10.1001/jamapsychiatry.2016.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, et al. (2015). Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Molecular Psychiatry, 20(2), 162–169. 10.1038/mp.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Forrest MP, Zhang H, Moy W, McGowan H, Leites C, Dionisio LE, et al. (2017).Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell, 21(3), 305–318. e308. 10.1016/j.stem.2017.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Powell SK, Gregory J, Akbarian S, & Brennand KJ (2017). Application of CRISPR/ Cas9 to the study of brain development and neuropsychiatric disease. Molecular and Cellular Neurosciences, 82, 157–166. 10.1016/j.mcn.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ho SM, Hartley BJ, Flaherty E, Rajarajan P, Abdelaal R, Obiorah I, et al. (2017).Evaluating synthetic activation and repression of neuropsychiatric-related genes in hiPSC- derived NPCs, neurons, and astrocytes. Stem Cell Reports, 9(2), 615–628. 10.1016/j.stemcr.2017.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jiang Y, Loh YE, Rajarajan P, Hirayama T, Liao W, Kassim BS, et al. (2017). The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nature Genetics, 49(8), 1239–1250. 10.1038/ng.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rajarajan P, Jiang Y, Kassim BS, & Akbarian S (2018b). Chromosomal conformations and epigenomic regulation in schizophrenia. Progress in Molecular Biology and Translational Science, 157, 21–40. 10.1016/bs.pmbts.2017.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zarrei M, MacDonald JR, Merico D, & Scherer SW (2015). A copy number variation map of the human genome. Nature Reviews Genetics, 16(3), 172–183. 10.1038/nrg3871 [DOI] [PubMed] [Google Scholar]
- 246.Ahn K, Gotay N, Andersen TM, Anvari AA, Gochman P, Lee Y, et al. (2014). High rate of disease-related copy number variations in childhood onset schizophrenia. Molecular Psychiatry, 19(5), 568–572. 10.1038/mp.2013.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Flaherty EK, Brennand KJ, (2017) Using hiPSCs to model neuropsychiatric copy number variations (CNVs) has potential to reveal underlying disease mechanisms. Brain Research 1655:283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, et al. (2005).COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nature Neuroscience, 8(11), 1500–1502. 10.1038/nn1572 [DOI] [PubMed] [Google Scholar]
- 249.Gothelf D, Feinstein C, Thompson T, Gu E, Penniman L, Van Stone E, et al. (2007).Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. The American Journal of Psychiatry, 164(4), 663–669. 10.1176/ajp.2007.164.4.663 [DOI] [PubMed] [Google Scholar]
- 250.Murphy KC, Jones LA, & Owen MJ (1999). High rates of schizophrenia in adults with velo-cardio-facial syndrome. Archives of General Psychiatry, 56(10), 940–945. [DOI] [PubMed] [Google Scholar]
- 251.Pedrosa E, Sandler V, Shah A, Carroll R, Chang C, Rockowitz S, et al. (2011).Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. Journal of Neurogenetics, 25(3), 88–103. 10.3109/01677063.2011.597908 [DOI] [PubMed] [Google Scholar]
- 252.Lin M, Pedrosa E, Hrabovsky A, Chen J, Puliafito BR, Gilbert SR, et al. (2016).Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Systems Biology, 10(1), 105. 10.1186/s12918-016-0366-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, et al. (2015).MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 Del. PLoS One, 10(7), e0132387. 10.1371/journal.pone.0132387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Toyoshima M, Akamatsu W, Okada Y, Ohnishi T, Balan S, Hisano Y, et al. (2016).Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Translational Psychiatry, 6(11), e934. 10.1038/tp.2016.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Warnica W, Merico D, Costain G, Alfred SE, Wei J, Marshall CR, et al. (2015).Copy number variable microRNAs in schizophrenia and their neurodevelopmental gene targets. Biological Psychiatry, 77(2), 158–166. 10.1016/j.biopsych.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, et al. (2014). Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell, 15(1), 79–91. 10.1016/j.stem.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, et al. (2009) Microduplications of 16p11.2 are associated with schizophrenia. Nature Genetics 41 (11):1223–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Deshpande A,Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, et al. (2017).Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Reports, 21(10), 2678–2687. 10.1016/j.celrep.2017.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rujescu D, Ingason A, Cichon S, Pietilainen OP, Barnes MR, Toulopoulou T, et al. (2009). Disruption of the neurexin 1 gene is associated with schizophrenia. Human Molecular Genetics, 18(5), 988–996. 10.1093/hmg/ddn351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zeng L, Zhang P, Shi L, Yamamoto V, Lu W, & Wang K (2013). Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS One, 8(3), e59685. 10.1371/journal.pone.0059685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pak C, Danko T, Zhang Y, Aoto J, Anderson G, Maxeiner S, et al. (2015). Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell, 17(3), 316–328. 10.1016/j.stem.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Flaherty E, Zhu S, Barretto N, Cheng E, Michael Deans PJ, Fernando MB, et al. (2019) Neuronal impact of patient-specific aberrant NRXN1α splicing. Nature Genetics 51 (12):1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Jacobs P, Brunton M, Frackiewicz A, Newton M, Cook P, & Robson E (1970). Studies on a family with three cytogenetic markers. Annals of Human Genetics, 33, 325–336. [Google Scholar]
- 264.St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, et al. (1990).Association within a family of a balanced autosomal translocation with major mental illness. Lancet, 336(8706), 13–16. [DOI] [PubMed] [Google Scholar]
- 265.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Human Molecular Genetics, 9(9), 1415–1423. [DOI] [PubMed] [Google Scholar]
- 266.Sachs NA, Sawa A, Holmes SE, Ross CA, DeLisi LE, & Margolis RL (2005).A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Molecular Psychiatry, 10(8), 758–764. 10.1038/sj.mp.4001667 [DOI] [PubMed] [Google Scholar]
- 267.Green EK, Norton N, Peirce T, Grozeva D, Kirov G, Owen MJ, et al. (2006).Evidence that a DISC1 frame-shift deletion associated with psychosis in a single family may not be a pathogenic mutation. Molecular Psychiatry, 11(9), 798–799. 10.1038/sj.mp.4001853 [DOI] [PubMed] [Google Scholar]
- 268.Chiang CH, Su Y, Wen Z, Yoritomo N, Ross CA, Margolis RL, et al. (2011).Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Molecular Psychiatry, 16(4), 358–360. 10.1038/mp.2011.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature, 515(7527), 414–418. 10.1038/nature13716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Murai K, Sun G, Ye P, Tian E, Yang S, Cui Q, et al. (2016). The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nature Communications, 7, 10965. 10.1038/ncomms10965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yalla K, Elliott C, Day JP, Findlay J, Barratt S, Hughes ZA, et al. (2018). FBXW7 regulates DISC1 stability via the ubiquitin-proteosome system. Molecular Psychiatry, 23(5),1278–1286. 10.1038/mp.2017.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Chiu FL, Lin JT, Chuang CY, Chien T, Chen CM, Chen KH, et al. (2015).Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Human Molecular Genetics, 24(21), 6066–6079. 10.1093/hmg/ddv318 [DOI] [PubMed] [Google Scholar]
- 273.Chien T, Weng YT, Chang SY, Lai HL, Chiu FL, Kuo HC, et al. (2018).GSK3beta negatively regulates TRAX, a scaffold protein implicated in mental disorders, for NHEJ-mediated DNA repair in neurons. Molecular Psychiatry. 10.1038/s41380-017-0007-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Srikanth P, Han K, Callahan DG, Makovkina E, Muratore CR, Lalli MA, et al. (2015). Genomic DISC1 disruption in hiPSCs alters Wnt signaling and neural cell fate. Cell Reports, 12(9), 1414–1429. 10.1016/j.celrep.2015.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Bradshaw NJ, & Porteous DJ (2012). DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology, 62(3), 1230–1241. 10.1016/j.neuropharm.2010.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, et al. (2007). Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Molecular Psychiatry, 12(1), 74–86. 10.1038/sj.mp.4001880 [DOI] [PubMed] [Google Scholar]
- 277.Camargo LM, Wang Q, & Brandon NJ (2008). What can we learn from the disrupted in schizophrenia 1 interactome: lessons for target identification and disease biology? Novartis Foundation Symposium, 289, 208–216; discussion 216–221, 238–240. [DOI] [PubMed] [Google Scholar]
- 278.Teng S, Thomson PA, McCarthy S, Kramer M, Muller S, & Lihm J (2018). Rare disruptive variants in the DISC1 Interactome and Regulome: association with cognitive ability and schizophrenia. Molecular Psychiatry, 23(5), 1270–1277. 10.1038/mp.2017.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Nakata K, Lipska BK, Hyde TM, Ye T, Newburn EN, Morita Y, et al. (2009).DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proceedings of the National Academy of Sciences of the United States of America,106(37), 15873–15878. 10.1073/pnas.0903413106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wilkinson B, Evgrafov OV, Zheng D, Hartel N, Knowles JA, Graham NA, et al. (2018). Endogenous cell type-specific disrupted in schizophrenia 1 interactomes reveal protein networks associated with neurodevelopmental disorders. Biological Psychiatry, 85, 305. 10.1016/j.biopsych.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, Stessman HA, et al. (2017). denovo-db: a compendium of human de novo variants. Nucleic Acids Research,45(D1), D804–D811. 10.1093/nar/gkw865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, et al. (2011).The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. American Journal of Human Genetics, 88(5), 523–535. 10.1016/j.ajhg.2011.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ye F, Kang E, Yu C, Qian X, Jacob F, Yu C, et al. (2017). DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron, 96(5), 1041–1054. e1045. 10.1016/j.neuron.2017.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Mathieson I, Munafo MR, & Flint J (2012). Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Molecular Psychiatry, 17(6), 634–641. 10.1038/mp.2011.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Richards AL, Leonenko G, Walters JT, Kavanagh DH, Rees EG, Evans A, et al. (2016). Exome arrays capture polygenic rare variant contributions to schizophrenia. Human Molecular Genetics, 25(5), 1001–1007. 10.1093/hmg/ddv620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O’Donovan MC, et al. (2015). Evaluating historical candidate genes for schizophrenia. Molecular Psychiatry, 20(5), 555–562. 10.1038/mp.2015.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sullivan PF (2013). Questions about DISC1 as a genetic risk factor for schizophrenia. Molecular Psychiatry, 18(10), 1050–1052. 10.1038/mp.2012.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Lee IS, Carvalho CMB, Douvaras P, Ho S-M, Hartley BJ, Zuccherato LW, et al. (2015) Characterization of molecular and cellular phenotypes associated with a heterozygous CNTNAP2 deletion using patient-derived hiPSC neural cells. npj Schizophrenia 1 (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Flaherty E, Deranieh RM, Artimovich E, Lee IS, Siegel AJ, Levy DL, et al. (2017). Patient-derived hiPSC neurons with heterozygous CNTNAP2 deletions display altered neuronal gene expression and network activity. NPJ Schizophrenia, 3, 35. 10.1038/s41537-017-0033-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.de Vrij FM, Bouwkamp CG, Gunhanlar N, Shpak G, Lendemeijer B, Baghdadi M, et al. (2018). Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Molecular Psychiatry, 24, 757. 10.1038/s41380-017-0004-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Guennewig B, Bitar M, Obiorah I, Hanks J, O’Brien EA, Kaczorowski DC, et al. (2018). THC exposure of human iPSC neurons impacts genes associated with neuropsychiatric disorders. Translational Psychiatry, 8(1), 89. 10.1038/s41398-018-0137-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Obiorah IV, Muhammad H, Stafford K, Flaherty EK, & Brennand KJ (2017). THC treatment alters glutamate receptor gene expression in human stem cell-derived neurons. Molecular Neuropsychiatry, 3(2), 73–84. 10.1159/000477762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Khandaker GM, Zimbron J, Lewis G, & Jones PB (2013). Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychological Medicine, 43(2), 239–257. 10.1017/S0033291712000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, et al. (2015). Schizophrenia. Nature Reviews Disease Primers, 1, 15067. 10.1038/nrdp.2015.67 [DOI] [PubMed] [Google Scholar]
- 295.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. (2017). Humanized mouse models of clinical disease. Annual Review of Pathology, 12, 187–215. 10.1146/annurev-pathol-052016-100332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Allswede DM, Buka SL, Yolken RH, Torrey EF, & Cannon TD (2016). Elevated maternal cytokine levels at birth and risk for psychosis in adult offspring. Schizophrenia Research, 172(1–3), 41–45. 10.1016/j.schres.2016.02.022 [DOI] [PubMed] [Google Scholar]
- 297.Lin M, Zhao D, Hrabovsky A, Pedrosa E, Zheng D, & Lachman HM (2014). Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. PLoS One, 9(4), e94968. 10.1371/journal.pone.0094968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Hashimoto-Torii K, Torii M, Fujimoto M, Nakai A, El Fatimy R, Mezger V, et al. (2014). Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron, 82(3), 560–572. 10.1016/j.neuron.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ishii S, Torii M, Son AI, Rajendraprasad M, Morozov YM, Kawasawa YI, et al. (2017). Variations in brain defects result from cellular mosaicism in the activation of heat shock signalling. Nature Communications, 8, 15157. 10.1038/ncomms15157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Vallersnes OM, Dines AM, Wood DM, Yates C, Heyerdahl F, Hovda KE, et al. (2016). Psychosis associated with acute recreational drug toxicity: a European case series. BMC Psychiatry, 16, 293. 10.1186/s12888-016-1002-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Callaghan RC, Cunningham JK, Allebeck P, Arenovich T, Sajeev G, Remington G, et al. (2012). Methamphetamine use and schizophrenia: a population-based cohort study in California. The American Journal of Psychiatry, 169(4), 389–396. 10.1176/appi.ajp.2011.10070937 [DOI] [PubMed] [Google Scholar]
- 302.Nielsen SM, Toftdahl NG, Nordentoft M, & Hjorthoj C (2017). Association between alcohol, cannabis, and other illicit substance abuse and risk of developing schizophrenia: a nationwide population based register study. Psychological Medicine, 47(9), 1668–1677. 10.1017/S0033291717000162 [DOI] [PubMed] [Google Scholar]
- 303.de Leon J, & Diaz FJ (2005). A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophrenia Research, 76(2–3), 135–157. 10.1016/j.schres.2005.02.010 [DOI] [PubMed] [Google Scholar]
- 304.Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. (2018). GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nature Neuroscience, 21(9), 1161–1170. 10.1038/s41593-018-0206-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Chatterton Z, Hartley BJ, Seok MH, Mendelev N, Chen S, Milekic M, et al. (2017). In utero exposure to maternal smoking is associated with DNA methylation alterations and reduced neuronal content in the developing fetal brain. Epigenetics & Chromatin, 10, 4. 10.1186/s13072-017-0111-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Oedegaard KJ, Alda M, Anand A, Andreassen OA, Balaraman Y, Berrettini WH, et al. (2016). The pharmacogenomics of bipolar disorder study (PGBD): identification of genes for lithium response in a prospective sample. BMC Psychiatry, 16, 129. 10.1186/s12888-016-0732-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ruderfer DM, Charney AW, Readhead B, Kidd BA, Kahler AK, Kenny PJ, et al. (2016). Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry, 3(4), 350–357. 10.1016/S2215-0366(15)00553-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Li J, Yoshikawa A, Brennan MD, Ramsey TL, & Meltzer HY (2018). Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophrenia Research, 192, 194–204. 10.1016/j.schres.2017.04.009 [DOI] [PubMed] [Google Scholar]
- 309.Kim Y, Giusti-Rodriguez P, Crowley JJ, Bryois J, Nonneman RJ, Ryan AK, et al. (2018). Comparative genomic evidence for the involvement of schizophrenia risk genes in antipsychotic effects. Molecular Psychiatry, 23(3), 708–712. 10.1038/mp.2017.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Readhead B, Hartley BJ, Eastwood BJ, Collier DA, Evans D, & Farias R (2018). Expression-based drug screening of neural progenitor cells from individuals with schizophrenia. Nature Communications, 9(1), 4412. 10.1038/s41467-018-06515-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, et al. (2016). Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nature Medicine, 22(10), 1101–1107. 10.1038/nm.4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Zhou T, Tan L, Cederquist GY, Fan Y, Hartley BJ, Mukherjee S, et al. (2017). High-content screening in hPSC-neural progenitors identifies drug candidates that inhibit Zika virus infection in fetal-like organoids and adult brain. Cell Stem Cell, 21(2), 274–283. e275. 10.1016/j.stem.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Watanabe M, Buth JE, Vishlaghi N, de la Torre-Ubieta L, Taxidis J, Khakh BS, et al. (2017). Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Reports, 21(2), 517–532. 10.1016/j.celrep.2017.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]