

Abstract
Undifferentiated pleomorphic sarcoma of bone (UPSb), is a rare primary bone sarcoma that lacks a specific line of differentiation. There is very little information about the genetic alterations leading to tumourigenesis or malignant transformation. Distinguishing between UPSb and other malignant bone sarcomas, including dedifferentiated chondrosarcoma and osteosarcoma, can be challenging due to overlapping features. To explore the genomic and transcriptomic landscape of UPSb tumours, whole-exome sequencing (WES) and RNA Sequencing (RNA-Seq) were performed on UPSb tumours. All tumours lacked hotspot mutations in IDH1/2 132 or 172 codons, thereby excluding the diagnosis of dedifferentiated chondrosarcoma. Recurrent somatic mutations in TP53 were identified in 4/14 samples (29%). Moreover, recurrent mutations in histone chromatin remodelling genes, including H3F3A, ATRX and DOT1L, were identified in 5/14 samples (36%), highlighting the potential role of deregulated chromatin remodelling pathways in UPSb tumourigenesis. The majority of recurrent mutations in chromatin remodelling genes identified here are reported in COSMIC, including the H3F3A G35 and K36 hotspot residues. Copy number alteration analysis identified gains and losses in genes that have been previously altered in UPSb or UPS of soft tissue. Eight somatic gene fusions were identified by RNA-Seq, two of which, CLTC-VMP1 and FARP1-STK24, were reported previously in multiple cancers. Five gene fusions were genomically characterised. Hierarchical clustering analysis, using RNA-Seq data, distinctly clustered UPSb tumours from osteosarcoma and other sarcomas, thus molecularly distinguishing UPSb from other sarcomas. RNA-Seq expression profiling analysis and quantitative RT-PCR showed an elevated expression in FGF23 which can be a potential molecular biomarker in UPSb. To our knowledge, this study represents the first comprehensive WES and RNA-Seq analysis of UPSb tumours revealing novel protein-coding recurrent gene mutations, gene fusions and identifying a potential UPSb molecular biomarker, thereby broadening the understanding of the pathogenic mechanisms and highlighting the possibility of developing novel targeted therapeutics.
Keywords: whole exome sequencing, RNA sequencing, undifferentiated pleomorphic sarcoma of the bone, chromatin remodelling genes, gene fusions, sarcomas, FGF23, CNV, gene expression and hierarchical clustering analyses
Introduction
Undifferentiated high-grade pleomorphic sarcoma of bone (UPSb) is a rare aggressive bone sarcoma that lacks a specific line or pattern of differentiation [1]. These tumours represent <2% of all primary malignant bone neoplasms and rarely occur in young adults [1,2]. Tumours commonly arise in long bones of lower extremities, particularly the femur followed by tibia and humerus, with a metastatic rate of at least 50%, especially to lungs [2,3]. The morphological appearance of the tumours is heterogeneous, consisting of atypical spindle and pleomorphic cells that lack matrix production. As UPSb is a diagnosis exclusion, thorough and extensive sampling of the tumour to rule out osteosarcoma or dedifferentiated chondrosarcoma is mandatory [4]. This might pose diagnostic difficulties, particularly in a limited biopsy sample. The recommended treatment generally involves neoadjuvant therapy followed by wide surgical excision [5]. The chemosensitivity and survival rate of UPSb are similar to osteosarcoma but distinction from dedifferentiated chondrosarcoma, which has a dismal prognosis, is important [5]. To date, there are no molecular studies or stringent diagnostic criteria to distinguish between these bone sarcomas. The genetics of UPSb is poorly understood. Previous studies have reported low frequency of TP53 mutations, MDM2 amplification [6], and various genomic gains and losses, including CDKN2A, RB1 and TP53 [2]. Nevertheless, no extensive high-throughput studies have been conducted to achieve a comprehensive understanding of the aetiology of these tumours. To gain further comprehensive insights into the molecular landscape and pathogenic mechanisms of UPSb, we performed integrative analysis using whole exome sequencing (WES) and RNA sequencing (RNA-Seq).
Materials and methods
Tumour samples
A retrospective search of the pathology database at the Royal Orthopaedic Hospital for resected samples of UPSb was carried out. All samples were obtained from the Royal Orthopaedic Hospital NHS Foundation Trust Tumour Bank with informed consent from the patient and ethical approval from institutional and local research committee boards. Prior to the study, all patient samples were anonymised and used in alliance with the ethical rules and regulations presented in the Declaration of Helsinki. In total, fourteen cases with the diagnosis of UPSb were identified (additional details are provided in supplementary materials, Supplementary materials and methods).
Whole exome sequencing and copy number alteration analysis
DNA from fresh frozen and FFPE (10 × 10 μm sections) tissues was extracted and purified using the DNA Isolation (Roche Diagnostic Ltd, Burgess Hill, UK) and Arcturus PicoPure DNA Extraction kits (ThermoFisher Scientific, Gloucester, UK), respectively, following the manufacturer’s instructions. A total of 1–3 µg of DNA from 12 tumours and 9 corresponding normal tissues were sent to Oxford Gene Technology (OGT, Oxford, UK) for WES. Exons were captured using the Agilent SureSelect Human All Exon V5 kit (Agilent, Santa Clara, USA), following manufacturer’s protocol, and were massively sequenced using either the Illumina HiSeq 2000 (100-bp paired-end) or NextSeq500 (150-bp paired-end) platforms (Illumina, San Diego, CA, USA). Two tumours and one corresponding normal sample were exome-sequenced at SMCL Next Generation Sequencing (NGS) Hub (Cambridge, UK). Exons were captured by Illumina Nextera Rapid Capture Exome kit and sequenced using Illumina HiSeq 4000, generating 2 X 150 bp reads. Additional details are provided in supplementary materials, Supplementary materials and methods.
CNV analysis (from WES data) was performed on ten normal-paired UPSb tumours, using CNVkit (San Francisco, CA, USA), applying the tool’s default settings (https://cnvkit.readthedocs.io/en/stable/).
RNA Sequencing experiment
Total RNA from eight fresh frozen tissues was extracted using either standard TRIzol-chloroform method or Qiagen RNeasy Mini kit (Qiagen, Manchester, UK) following the manufacturer’s instructions. The quality and concentration of RNA was assessed using Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). A total of 50–100ng of DNase-treated RNA from each tumour was massively sequenced at the Genomics Birmingham facility (Institute of Cancer & Genomic Sciences, University of Birmingham, Birmingham, UK). Additional details are provided in supplementary materials, Supplementary materials and methods.
RNA-Seq differential gene expression profiling and hierarchical clustering analyses
The 149 RNAseq samples from the CINSARC dataset [7] were retrieved from the Sequence Read Archive Bioproject PRJNA282597 (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP057793) and 16 osteosarcoma samples were randomly chosen from the SRA bioproject PRJNA345424 (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP090849) [8]. Gene expression values were extracted using Kallisto v0.42.5 [9] with the GRCh38 release 79 genome annotation and transformed into log2(tpm+2) prior to sample aggregation and normalisation using the quantile method of the limma R package, within the R version 3.1.2 [10]. Clustering was computed using the Cluster v2.0.3 package. Pairwise comparisons of expression profiles were performed using F-Test variance comparison prior to Student’s t-test statistics with a Bonferroni correction. The pairwise comparisons of UPSb versus all other tumours were performed iteratively with 20 random sampling of 10 samples from the larger group. Gene ontology analyses were performed with DAVID v6.7 [11].
Results
Exome sequencing identifies recurrent mutations in TP53 and histone remodelling genes
To investigate the mutational landscape of UPSb, WES was performed on 14 tumours and ten matched-normal tissue samples, aiming to achieve a coverage depth of at least 50X in each sample. [WES and RNA-seq data have been deposited in Sequence Read Archive (SRA) on Monday 24th of September 2018 (submission number:SUB4532940). Accession number to be added at the Proof Stage please] Using stringent criteria to call high-confidence somatic mutations (see Materials and methods), 794 single nucleotide variants (SNVs), representing the majority of mutations (85%), and 138 indels (insertions and deletions) were detected, corresponding to a median of 2.2 mutations (range from 0 to 10.4) per coding megabase (supplementary material, Table S1 and Figure S1). This overall somatic mutation burden is comparable with a low mutation burden average of 1.06 per megabase identified in 206 soft tissue sarcomas, including 44 UPS of soft tissue (UPSst) [12]. Somatic SNVs comprised of: 748 nonsynonymous substitutions, 33 nonsense and 13 splice sites mutations. A total of 123 SNVs and indels were validated by Sanger sequencing, achieving a 96.6% validation rate. To identify potential cancer driver genes, we focused on somatic recurrent genes that were found mutated in more than one tumour. A total of 31 recurrent genes harbouring heterozygous somatic mutations were identified in 2/14 (14%) tumours, except for TP53 which was mutated in 4/14 (29%) samples (Figure 1A and Figure 2A). The majority of recurrent genes were confirmed by Sanger sequencing (examples in supplementary material, Figure S2). All four TP53 somatic missense substitutions (R158H, V216M, Y236C C242G) are described ‘somatic’ in COSMIC database and predicted deleterious by two independent in silico tools (supplementary material, Table S2). The V216M missense substitution has been reported previously in a UPSb tumour with a progressive disease behaviour [6]. In addition to mutations in TP53 gene, which participates in the integrity of histone remodelling complexes [13], recurrent somatic mutations in histone remodelling genes including H3F3A, ATRX and DOT1L were detected in 5/14 (36%) samples, suggesting a potential role of defective chromatin remodelling genes in UPSb tumourigenesis (Figure 2 B–D, supplementary material, Table S2). Somatic G34V missense substitution and V35_K36insL in-frame insertion in the previously known H3F3A hotspot residues were identified in two UPSb tumours. Mutations in ATRX and DOT1L were missense substitutions. One ATRX mutation, E351V, is reported somatic in COSMIC database. We identified two mutations in DOT1L, G307V and Q595H occurring in Histone-lysine N-methyltransferase catalytic DOT1 domain and STAT1 binding motifs, respectively. To further investigate the cancer-related genes, all recurrent candidate genes were assessed against COSMIC Cancer Gene Census (CCGC) and InTOgen cancer driver genes databases. Seven genes (TP53, ATRX, H3F3A, ZFHX3, CSMD3, PRPRT, TRIO) were classified as cancer drivers (Figure 1A). Eight recurrent genes (TP53, ATRX, DOT1L, GCGR, COL4A2, KCNQ3, PKLR, SLC12A1) were identified as potential druggable genes by the drug–gene interaction database (DGIdb) (Figure 1A).
Figure 1. The mutational and gene fusion landscape of UPSb.

(A) Recurrent genes identified by whole exome sequencing in 14 tumours, grouped according to their biological pathway and function. (B) Gene fusions identified by RNA sequencing in eight tumours. CCCG: COSMIC Cancer Census Gene; DGIdb: The Drug Gene Interaction database. Gene described ‘cancer driving gene’ in CCGC or druggable in DGIdb are shaded in grey. †: TRIO is classified ‘cancer driver gene’ by IntOGen.
Figure 2. Recurrent somatic mutations in TP53, H3F3A, ATRX and DOT1L.
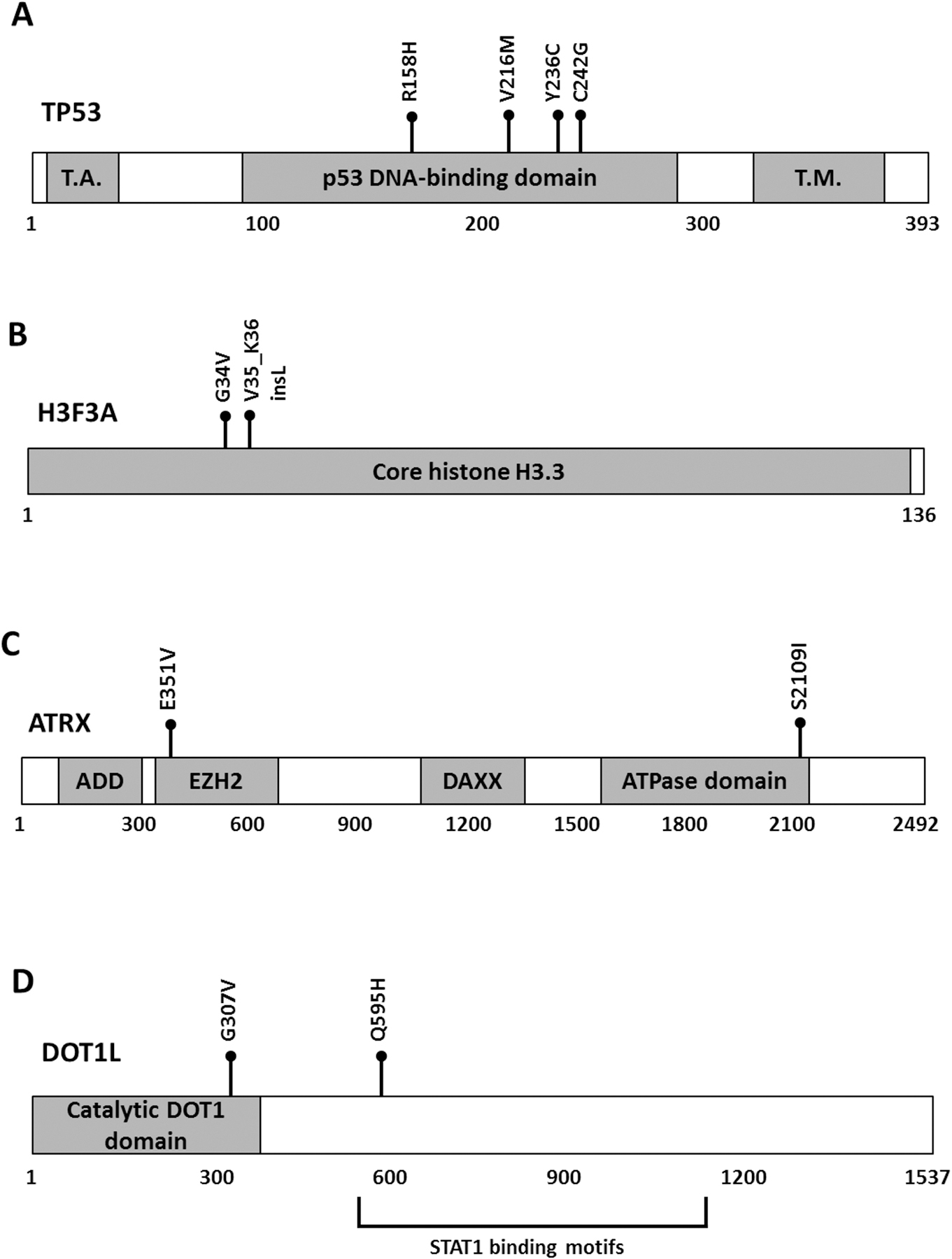
All the mutations (black balls) are missenses except for one an in-frame insertion in H3F3A. The relative positions of mutations are shown in the predicted protein sequence of (A) TP53, (B) H3F3A, (C) ATRX and (D) DOT1L. T.A.: p53 transactivation domain; T.M.: p53 tetramerization domain; ADD: ATRX-DNMT3-DNMT3L domain; EZH2: enhancer zeste homologue 2 protein (EZH2) interacting region; DAXX: death domain-associated protein interacting domain; ATPase: Helicase adenosine triphosphatase domain; Catalytic DOT1 domain: Histone-lysine N-methyltransferase DOT1 domain; STAT1: signal transducer and activator of transcription 1 binding motifs.
Somatic copy number alteration analysis using WES data
Somatic copy number variation (CNV) analysis was performed on ten normal-tumour UPSb samples. Supplementary material, Figure S3 shows the overall somatic CNV heatmap of UPSb tumours. In comparison with a study by Niini et al. [2] that assessed the CNV profile in UPSb, we comparingly identified CNV changes in the following genes: ING1, CGK4, MDM2, MYC, PDGFRA, KIT, KDR, PDGFA, PDGFB, VEGFA (supplementary material, Table S3). We also found somatic CNV losses in RB1 in 5/10 (50%) cases, gains and losses in VGLL3 and CDKN2A, losses in YAP1 as well as loss and gain in CCNE1 (supplementary material, Table S3), genes that been previously implicated in UPSst [12].
RNA-Seq identifies two previously reported and six novel gene fusions
RNA-Seq was conducted on eight tumours, achieving a mean of 59,722,616 reads with 90.07% successful alignment rate to reference genome. Using two bioinformatic fusion callers and following a series of filtering steps (see Materials and methods), eight gene fusion candidates were identified in four tumours, two of which were previously described in the literature (CLTC-VMP1 and FARP1-STK24) [14,15], whereas the remaining fusions are novel (Figure 1B and supplementary material, Table S4). In addition, the previously reported CTSC-RAB38 read-through chimera in cancer and non-cancer tissues [16] was identified in 2/8 tumours (25%). Using RT-PCR, all eight fusions were detected in the tumour cDNA but not in corresponding normal tissue samples, suggesting that the fusions are highly tumour-specific. Focusing on gene fusions with potential druggability and/or involvement in tumourigenesis, genomic analysis breakpoints using LR-PCR were performed on five out of the eight fusions to determine the precise genomic breakpoints (supplementary material, Table S4). Using LR-PCR, all five fusions were genomically characterised and validated in the tumour DNA but were not detected in corresponding normal tissues, confirming the somatic status of the fusions. The APOL1-MYH9 and PKNOX2-MMP20 occur as a result of paracentric chromosome inversion whereas ASAP2-ADAM17 forms by an interstitial chromosomal deletion of ~97 kilobase. The two gene fusions previously reported in other cancer samples and cell lines are CLTC-VMP1 and FARP1-STK24 (supplementary material, Figure S4 and S5). CLTC-VMP1 has been described previously in BT-549 and HCC1954 breast cancer cell lines [14], hypopharynx tumour [17] and large-cell lung carcinoma [18]. The CLTC-VMP1 gene fusion results from joining the first 14 exons of CLTC to the last two exons of VMP1 (supplementary material, Figure S4 A,B). Genomic breakpoint analysis revealed a ~158 kilobase interstitial deletion within CLTC and VMP1 genes, leading to a complete deletion of PTRH2 gene residing within the fusion gene partners (supplementary material, Figure S4 C,D). The CLTC-VMP1 chimeric transcript is an out-of-frame fusion, for which the predicted translation product includes the first 764 amino acids of CLTC gene that extends to the beginning of exon 11 of VMP1 (at codon 324) and reaching a premature stop codon after 75 amino acids (Figure 3A). The FARP1-STK24 chimeric fusion is formed as a result of a ~219 kilobase interstitial deletion within gene partners, linking the first three and five exons of FARP1 and STK24, respectively (supplementary material, Figure S5 A–D). The chimeric transcript is in-frame, consisting of the first 88 and 211 amino acid residues of FARP1 and STK24, respectively (Figure 3B). In FARP1-STK24, the majority of the STK24 protein kinase domain is retained yet missing the STK24 regulatory region (Figure 3B). This gene fusion has been previously described in an invasive breast cancer tumour [15]. To infer the potential biological relevance of the novel chimeric transcripts, gene fusion partner genes were checked against CCCG and InTOgen database, classifying CLTC and MYH9 (in APOL1-MYH9) as ‘cancer driver genes’ (Figure 1B). DGIdb identified three drug-gene interactions for three fusions partner genes (underlined) in the following gene fusions: FARP1-STK24, ASAP2-ADAM17, PKNOX2-MMP20 (Figure 1B).
Figure 3. The impact of the CLTC-VMP1 and FARP1-STK24 gene fusions on protein domain organization.
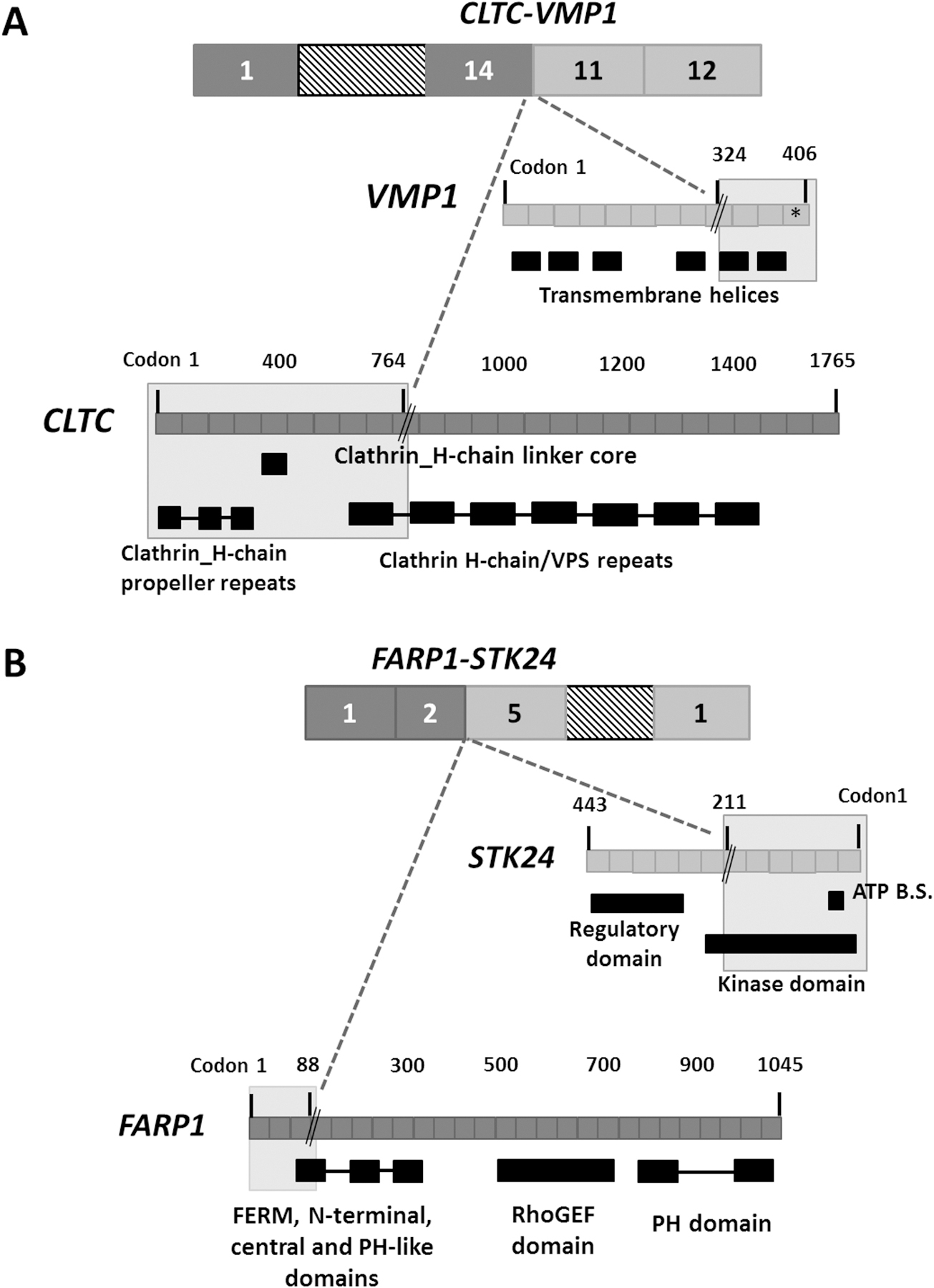
The grey shaded area represents the retained protein domains of the fused exons of (A) CLTC-VMP1 and (B) FARP1-STK24. The gene fusion breakpoints are denoted by a black double slash. The CLTC-VMP1 is out of frame, reaching a premature stop (denoted by *) at codon 399 of VMP1 gene. The FARP1-STK24 is in-frame, resulting from joining the first 88 and 211 amino acids of FARP1 and STK24, respectively. ATP B.S: ATP binding site.
Hierarchical clustering and expression profiling analyses using RNA-Seq data
Using RNA-Seq data, unsupervised hierarchical clustering analysis of UPSb tumours, other sarcomas and SRP090849 datasets distinctly clustered the UPSb tumours together in two groups, UPSb-G1 and UPSb-G2, thus molecularly distinguishing UPSb from osteosarcoma and other sarcomas (Figure 4A). Checking the clinicopathological information of the two UPSb groups, UPSb-G2 (T1, T2, T5, T6) primarily has spindle cell morphology, in comparison with UPSb-G1 (T9, T10, T13, T14), which shows a mixture of spindle, pleomorphic and epithelioid morphology (an example of each group in Supplementary material, Figure S6). Supervised expression analysis of UPSb versus other sarcoma subtypes highlighted FGF23, fibroblast growth factor 23, as a specifically expressed gene in UPSb tumours (Figure 4B). David Gene Ontology analyses of specific UPSb genes identified a strong enrichment of immune response genes, suggesting their potential involvement in UPSb tumourigenesis (Figure 4C).
Figure 4. Unsupervised and supervised analyses of UPSb, other sarcomas and SRP090849 datasets using RNA-Seq data.
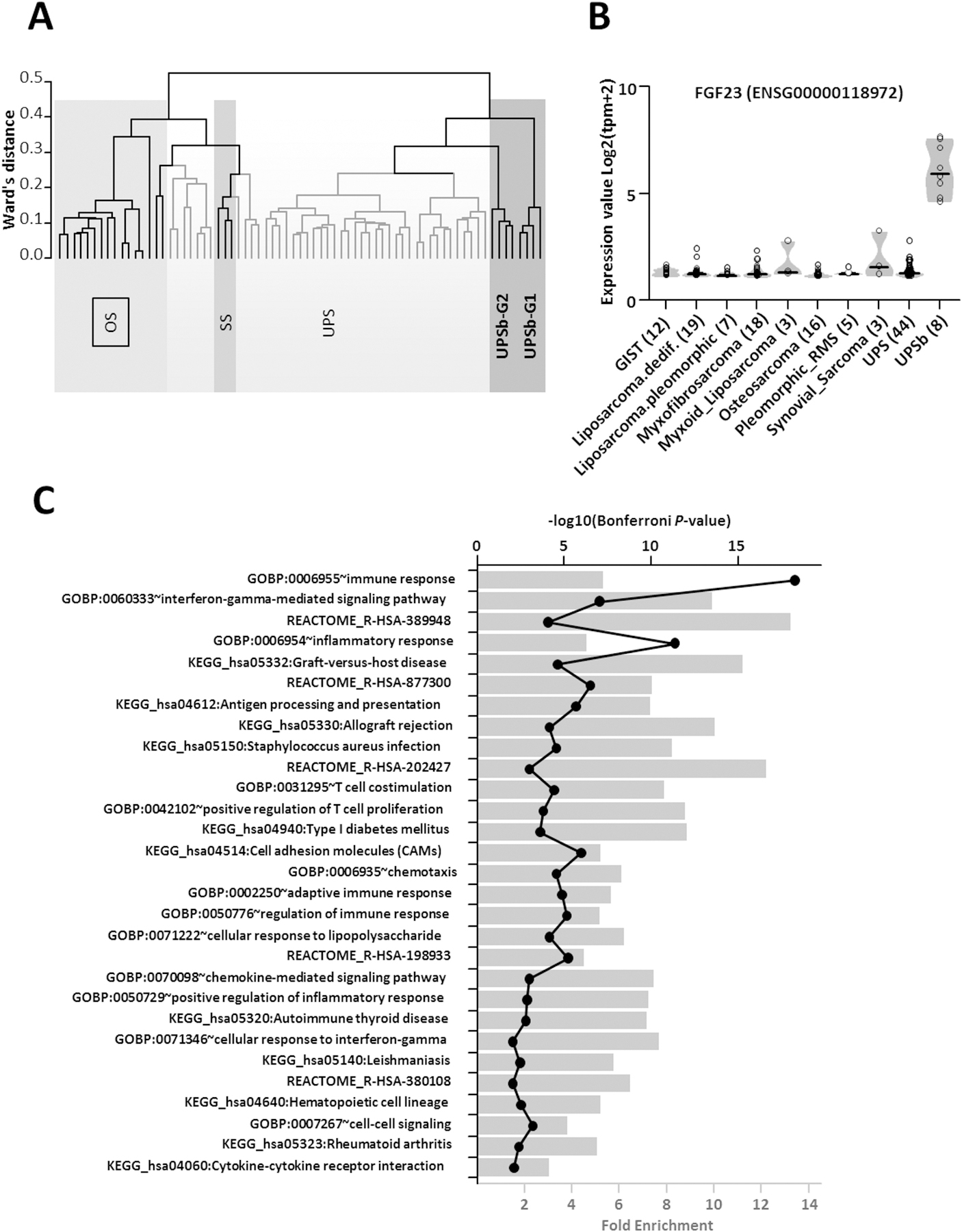
(A) Unsupervised clustering analysis using Ward’s distance and 1-pearson correlation highlighted two groups of UPSb tumors (dark grey). UPSb_G1 samples are clearly separated from classical UPS (light grey) and synovial sarcomas from the SRP057793 dataset and from osteosarcomas from the SRP090849 dataset. UPSb_G2 samples are closer to classical UPS but remain clearly distinct. (B) Supervised analysis showing a violin plot of the FGF23 gene expression across UPSb and the different tumor types present in the SRP057793 dataset and in the osteosarcoma SRP090849 dataset. Individual samples are shown in circle and group median is represented as a black bar. (C) David Gene Ontology analyses of specific UPSb genes indicating a strong enrichment of immune response genes. Fold enrichment (bar chart) and the –log10 of the hypergeometric test P-value corrected by Bonferroni (black line with closed circles) are shown.
Confirmation of elevated FGF23 expression in UPSb using RT-qPCR
To confirm the elevated expression of FGF23 by RNA-Seq, RT-qPCR was carried out on four UPSb tumours and four normal tissue controls. An elevated expression of FGF23 was significantly observed in all tumours, comparing to low FGF23 expression levels in normal samples (p=0.0286, supplementary material, Figure S7).
Discussion
In this study, we examined the somatic genetic alterations present in UPSb tumours using WES and RNA-Seq technologies as well as comparing transcriptomic profiles of UPSb with other sarcomas. We performed WES on 14 tumours and focused on genes that are altered in more than one tumour, identifying a total of 31 recurrent genes. TP53 was the most frequently mutated gene (29% of tumours). In 36% of the tumours, we identified mutations in chromatin remodelling genes (H3F3A, ATRX, DOT1L) which have not been previously described in the UPSb subtype. A significant co-occurrence of G34 H3F3A mutation with ATRX/DAXX and TP53 mutations has been observed in nearly 100% of glioblastoma tumours [19]. Notably, no correlation of H3F3A mutations was observed in UPSb tumours harbouring TP53, ATRX or DOT1L mutations.
Somatic CNV analysis on ten tumours revealed alterations in genes that have been previously implicated in UPSb [2] and UPSst [12], including MDM2, ING1, RB1, CDKN2A, VGLL3, YAP1 and CCNE1. Deep deletions in RB1 and CDKN2A has been identified recently in 16% and 20% of UPSst, respectively; whereas high-level amplifications were present in VGLL3, YAP1 and CCNE1 in 11%, 3% and 10% of UPSst [12].
Heterozygous R132 and R172 hotspot point mutations in IDH1 and IDH2, respectively, are commonly present in 61–87% of chondrosarcoma cases, including dedifferentiated chondrosarcoma [4,20]; by contrast, these changes are absent in 222 osteosarcoma samples [21]. In an earlier study by Chen et al. [4], we investigated the IDH1/2 mutation status in the 14 UPSb tumours used in this study and found no R132 and R172 mutations in any tumour, ruling out a diagnosis of dedifferentiated chondrosarcoma. Unlike the established association of IDH1/2 in chondrosarcoma, the somatic genomic profile of osteosarcoma is complex and involves multiple genes, mainly TP53 and RB1 [22]. These genes are well-known cancer driver genes implicated in multiple cancers and therefore cannot be sensitively used to exclude the diagnosis of osteosarcoma.
The TP53 gene is the most frequently mutated gene in various human cancers and 90% of TP53 mutations are missense changes with potential gain-of-function characteristics [23,24]. Previously, TP53 mutations were identified in 22% of UPSb tumours by conventional PCR and Sanger sequencing [6]. In this study, using massive-parallel sequencing for the first time on this tumour subtype, four TP53 missense mutations (R158H, COSMIC ID: COSM1640853; V216M, COSM10667; Y236C, COSM10731; C242G, COSM3717645), all occurring in the DNA-binding domain of p53 protein (Figure 2A), were identified. Mutations in the p53 DNA-binding domain can reduce the protein’s binding specificity to DNA sequence motifs in p53-regulated genes. The R158H, V216M and Y236C mutations are reported among the 50 most common somatic missense mutations in TP53, highlighting their potential pathological role in tumourigenesis [23]. We could not find significant differences in the total number of mutations in samples harbouring TP53 mutations (n=4) comparing to the remaining samples (n=10). Correlations between TP53 mutations and clinical implications are difficult to establish due to the clinical heterogeneity of the patients and small sample size; hence, additional investigations to elucidate their prognostic information are required.
Recurrent mutations in H3F3A, ATRX and DOT1L were detected in 5/14 tumours. Highly specific cancer-driving hotspot mutations in H3F3A (G34) and H3F3B (K36) were identified in 92% of giant cell tumours of the bone and 95% of chondroblastoma cases, respectively [25]. In this study, we identified recurrent somatic mutations (G34V, COSM502595 and V35_K36insL, COSM5574356) in H3F3A affecting the previously reported amino acid residues (Figure 2B). These hotspot sites are well-conserved amino acid residues of the amino-terminus tail that undergoes post-translational modifications [19]. Lysine 36 is a principal methylation site that typically promotes gene transcription when methylated or acetylated [19]. Histone 3.3 lysine to methionine substitution (K36M) reduces the methylation of lysine residue through inhibition of SET domain-containing enzymes [26]. This reduction of methylation at K36 was also observed in cell lines carrying the G34V substitution [26]. Because V35_K36insL is an-frame insertion and not a methionine substitution, further investigations will be necessary to elucidate any pathogenic mechanism of this in-frame insertion.
H3F3A/H3F3B driver mutations were described in giant cell tumour of the bone and chondroblastoma which are considered benign tumours or benign but locally aggressive tumours, respectively [25,27] as well as in malignant giant cell tumour of the bone [28,29]. A recent study by Amary et al. [28] identified H3F3A G34 substitutions in 13/385 (3.37%) of primary malignant bone tumours, classified as either osteosarcoma or malignant giant cell tumour of bone. In this study, we report recurrent H3F3A alterations in 2/14 (14.3%) UPSb tumours, a higher percentage than in previously reported malignant tumours, with the caveat of a smaller cohort size. Hence, the possibility of a malignant phenotype or evolution should be considered in tumours harbouring H3F3A alterations.
ATRX is a member of the SWI/SNF2 (SWItch/Sucrose Non-Fermentable) ATP-dependent chromatin remodelling protein complex [30] which regulates the expression of thousands of genes through remodelling of chromatin structure [31,32]. ATRX has an established role in the carcinogenesis of multiple cancers including gliomas [30], small cell lung cancers [33] and six adult soft tissue sarcomas (including UPS, leiomyosarcoma, dedifferentiated liposarcoma) [12]. Studies have demonstrated a regulatory role for ATRX, along with DAXX (death domain-associated protein) and other histone chaperone complex proteins, in the enrichment of histone H3.3 in telomeres and heterochromatin regions [34,35]. Dysfunction ATRX/DAXX is associated with the alternative lengthening of telomeres (ALT), a phenomenon observed in 10–15% of cancers of mesenchymal origin (e.g. UPSb) resulting in widespread genomic destabilisation [36,37]. In this study, we identified two missense mutations in ATRX: S2109I and E351V (COSMIC ID: COSM6608613) occurring in the Helicase/ adenosine triphosphatase (ATPase) conserved C-terminus and Enhancer zeste homologue 2 (EZH2) interacting region of the ATRX protein, respectively (Figure 2C). The helicase/ATPase subunit is the catalytic core of ATRX protein [38] which, along with other SNF2 proteins, is involved in ATP hydrolysis responsible for chromatin structural conformations [39]. The ATRX EZH2 interaction region is involved in the interaction of ATRX with (polycomb repressive complex 2) PRC2 protein complex, including EZH2 catalytic subunit [40,41]. Although further studies are required, mutations in these two functionally important domains can disrupt ATRX protein function, affecting the integrity of the chromatin structure. PRC2/EZH2 inhibitors are currently in clinical trials [40,42]; however, the complex and dual oncogenic and tumour-suppressing role of PRC2 in cancers requires detailed mechanistic insights before establishing the efficacy of these inhibitors [40].
In the second part of this study, we aimed to identify genome-wide gene fusions arising from chromosomal aberrations in eight UPSb tumour using RNA-Seq. A total of eight highly tumour-specific gene fusions were validated by RT-PCR, five of which were genomically characterised by LR-PCR. We identified two gene fusions previously reported in other cancers, CLTC-VMP1 and FARP1-STK24.
The CLTC-VMP1 gene fusion is identified in T13, lacking any WES alterations in TP53 or, H3F3A, ATRX, DOT1L chromatin remodelling genes. The cDNA breakpoint of CLTC identified here is different to those previously reported; however, the breakpoint position in VMP1 in UPSb is the same as reported for other tumours. The genomic interstitial deletion at 17q23.1 locus, revealed by genomic breakpoint analysis, leads to a complete deletion of PTRH2 gene and a loss of six repeats of clathrin heavy chain/VPS and four transmembrane helices of CLTC and VMP1, respectively (Figure 3A). The clathrin protein is involved in chromosome segregation and Golgi reassembly during mitosis and protein-protein interactions [43]. Rearrangements involving CLTC-PTRH2-VMP1 locus have been observed in multiple tumour types, including glioblastoma, lung cancer, breast cancer and leukaemias [14]. VMP1 encodes an autophagy-related protein that promotes apoptosis in pancreatic cancer cells [44]. PTRH2 is a mitochondrial protein that induces apoptosis by regulation of the function of Groucho family transcriptional regulators [45]. Knockdown of PTRH2/BIT1 in adherent cells decreased cell survival and promoted staurosporine and serum-deprivation apoptosis of cells, consistent with tumour suppressive role [46]. Altogether and as Giacomini et al. [14] suggested, the rearrangement involving CLTC-PTRH2-VMP1 and the CLTC-VMP1 gene fusion being out-of-frame are indicative of a disruption of the tumour suppressor activity.
The FARP1-STK24 gene fusion is formed by joining the two gene partners in opposing (sense-to-antisense) orientations, known as 5’-to-5’ gene fusions (Figure 3B). 5’-5’ gene fusions have previously been reported in breast cancers [47]. Since the 5’ transcription regulatory apparatus of both FARP1 and STK24 is retained, theoretically, both sense (FARP1) and antisense (STK24) genes can start transcription that extends into the other gene partner, or vice versa. Gene fusions involving FARP1 have been reported in multiple cancers, including lung adenocarcinoma, breast adenocarcinoma and lower grade glioma (http://www.tumorfusions.org). STK24 is serine/threonine protein kinase belonging to the mammalian Sterile20-like (MST) kinase family, key signalling molecules that regulate cell division cycle, cell morphogenesis, apoptosis and oncogenic transformation [48,49]. A caspase-dependent apoptotic role has been identified for STK24 protein [50]. The STK24 protein is cleaved and activated by caspase-3 protein, through the STK24 regulatory domain, and translocated into the nucleus to promote apoptotic responses [48]. The loss of STK24 regulatory domain can subsequently interfere with STK24 activation and nuclear localisation. The FARP1-STK24 forms an in-frame fusion protein with a potential constitutive activation of a kinase, a phenomenon observed in gene fusions exerting oncogenic functionality [51]. Altogether, FARP1-STK24 may be associated with oncogenic properties but further investigations are required.
Gene fusions involving protein kinases are considered potential therapeutic targets. The use of kinase inhibitors in tumours harbouring kinase-related gene fusions can improve tumour prognosis and patient outcome [52]. For example, the efficacy of using ALK inhibitors in advanced non–small-cell lung cancer with ALK rearrangement is evident in clinical trials [53]. A study by Olsen et al. [54] identified 14 inhibitors, eight of which are in clinical trials or are FDA approved, that inhibited the enzymatic activity of STK24. Although further detailed investigations are required, STK24-selective inhibitors are potential cancer therapeutics in tumours harbouring STK24 rearrangements.
Unsupervised clustering analysis of the RNA-Seq data clearly distinguished the UPSb samples from classical UPS as well as synovial sarcomas and osteosarcomas. Supervised expression profiling of UPSb versus other tumour subtypes revealed elevated expression of FGF23, which was confirmed in four UPSb tumours using RT-qPCR. A study by Shiba et al. [55] showed a highly specific immunohistochemical expression of FGF23 in phosphaturic mesenchymal tumours, whereas FGF23 expression was scored negative in 46 tumours, including osteosarcoma, chondrosarcoma and synovial sarcoma. Elevated expression of FGF23 can serve as a molecular biomarker specific to UPSb which can be diagnostically utilised in clinics. However, confirmation of elevated FGF23 expression in a larger UPSb cohort is recommended. FGF family, comprised of signalling proteins, has a role in tissue repair and tumourigenesis, by regulating cell proliferation, migration and angiogenesis [56]. Promising curative results of a FGF23 monoclonal antibody drug, KRN23, was observed in patients with tumour-induced osteomalacia, a rare paraneoplastic syndrome clinically described by bone pain fractures and muscle weakness [57]. Further investigations are necessary to confirm an FGF23-targeted therapeutic opportunity in UPSb.
An enrichment of immune response gene expression was identified in UPSb, which has also been documented in other sarcomas, including UPSst [12]. The therapeutic benefit of pembrolizumab, an immune checkpoint inhibitor, has been documented in 40% of UPSst cases [58]. Although further investigations are needed, immune checkpoint inhibitors can be considered as a potential therapeutic option in UPSb patients.
In summary, this study provides a first detailed genetic and transcriptomic alterations landscape of UPSb tumours, thus providing useful insights into tumourigenesis and broadening the understanding of this tumour subtype. We identified novel recurrent gene mutations in multiple cancer-related genes, including chromatin remodelling genes, that are reported in UPSb tumours for the first time. We also identified novel and previously reported gene fusions. Several of the recurrently mutated genes and gene fusions represent potential druggable targets that can translate into clinics and improve patient prognosis. Elevated expression of FGF23 was identified, which may be a potential molecular biomarker for UPSb.
Supplementary Material
Figure S1. The number of somatic alterations identified in each UPSb tumour
Figure S2. Examples of Sanger sequencing confirmation of eight recurrent mutations identified by WES
Figure S3. Somatic copy number alteration heatmap of ten normal-tumour paired UPSb samples
Figure S4. Diagrammatic representation and validation of CLTC-VMP1 gene fusion
Figure S7. Expression of FGF23 in UPSb tumours versus normal tissue samples using RT-qPCR
Table S1. Coding mutational rate of UPSb
Table S2. Summary of the individual recurrent mutations identified in TP53 and chromatin remodelling genes
Table S3. Somatic copy number alterations status of genes for which copy number alterations have been previously reported in UPS of bone and UPS soft tissue
Table S4. Summary of the eight somatic gene fusions identified by RNA-Seq
Table S5. Primer sequences
Figure S5. Schematic representation and validation of FARP1-STK24 fusion
Figure S6. Microscopy images of T1 and T10
Acknowledgments
This research was funded in part by the Kuwait Medical Genetics Centre (KMGC), Ministry of Health, Kuwait and the Royal Orthopaedic Hospital NHS Foundation trust, Birmingham, England and by the Italian Ministry of Health (Ricerca Corrente L2029 and L4097, IRCCS Istituto Ortopedico Galeazzi).
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
WES and RNA-seq data have been deposited in Sequence Read Archive (SRA) on Monday 24th of September 2018 (submission number:SUB4532940). Accession number to be added at proof stage
*Cited only in supplementary material.
List of supplementary material online Information
Supplementary materials and methods YES
Supplementary table and figure legends YES
References
- 1.Fletcher CDM. WHO classification of tumours of soft tissue and bone. 4th edition. International Agency for Research on Cancer, Lyon: 2013. [Google Scholar]
- 2.Niini T, Lahti L, Michelacci F, et al. Array comparative genomic hybridization reveals frequent alterations of G1/S checkpoint genes in undifferentiated pleomorphic sarcoma of bone. Genes Chromosomes Cancer 2011; 50: 291–306. [DOI] [PubMed] [Google Scholar]
- 3.Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer 2014; 120: 1763–1774. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Fritchie K, Wei S, et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum Pathol 2017; 65: 239–246. [DOI] [PubMed] [Google Scholar]
- 5.Gerrand C, Athanasou N, Brennan B, et al. UK guidelines for the management of bone sarcomas. Clin Sarcoma Res 2016; 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawaguchi K, Oda Y, Sakamoto A, et al. Molecular analysis of p53, MDM2, and H-ras genes in osteosarcoma and malignant fibrous histiocytoma of bone in patients older than 40 years. Mod Pathol 2002; 15: 878–888. [DOI] [PubMed] [Google Scholar]
- 7.Lesluyes T, Perot G, Largeau MR, et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur J Cancer 2016; 57: 104–111. [DOI] [PubMed] [Google Scholar]
- 8.Scott MC, Temiz NA, Sarver AE, et al. Comparative Transcriptome Analysis Quantifies Immune Cell Transcript Levels, Metastatic Progression, and Survival in Osteosarcoma. Cancer Res 2018; 78: 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bray NL, Pimentel H, Melsted P, et al. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016; 34: 525–527. [DOI] [PubMed] [Google Scholar]
- 10.Team. RDC. R: A language and environment for statistical computing.[Internet]. R Foundation for Statistical Computing. Available from: http://www.R-project.org. In. (ed)^(eds): Vienna, Austria, 2017. [Google Scholar]
- 11.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network. Electronic address edsc, Cancer Genome Atlas Research N. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017; 171: 950–965 e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair SS, Kumar R. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Molecular oncology 2012; 6: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giacomini CP, Sun S, Varma S, et al. Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet 2013; 9: e1003464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veeraraghavan J, Tan Y, Cao XX, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun 2014; 5: 4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babiceanu M, Qin F, Xie Z, et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res 2016; 44: 2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair J, Jain P, Chandola U, et al. Gene and miRNA expression changes in squamous cell carcinoma of larynx and hypopharynx. Genes & cancer 2015; 6: 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Lee W, Jiang Z, et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 2012; 22: 2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482: 226–231. [DOI] [PubMed] [Google Scholar]
- 20.Kerr DA, Lopez HU, Deshpande V, et al. Molecular distinction of chondrosarcoma from chondroblastic osteosarcoma through IDH1/2 mutations. The American journal of surgical pathology 2013; 37: 787–795. [DOI] [PubMed] [Google Scholar]
- 21.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011; 224: 334–343. [DOI] [PubMed] [Google Scholar]
- 22.Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy. Nat Rev Endocrinol 2017; 13: 480–491. [DOI] [PubMed] [Google Scholar]
- 23.Baugh EH, Ke H, Levine AJ, et al. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 2018; 25: 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45: 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cleven AH, Hocker S, Briaire-de Bruijn I, et al. Mutation Analysis of H3F3A and H3F3B as a Diagnostic Tool for Giant Cell Tumor of Bone and Chondroblastoma. The American journal of surgical pathology 2015; 39: 1576–1583. [DOI] [PubMed] [Google Scholar]
- 28.Amary F, Berisha F, Ye H, et al. H3F3A (Histone 3.3) G34W Immunohistochemistry: A Reliable Marker Defining Benign and Malignant Giant Cell Tumor of Bone. The American journal of surgical pathology 2017; 41: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Righi A, Mancini I, Gambarotti M, et al. Histone 3.3 mutations in giant cell tumor and giant cell-rich sarcomas of bone. Hum Pathol 2017; 68: 128–135. [DOI] [PubMed] [Google Scholar]
- 30.Nandakumar P, Mansouri A, Das S. The Role of ATRX in Glioma Biology. Front Oncol 2017; 7: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasad P, Lennartsson A, Ekwall K. The roles of SNF2/SWI2 nucleosome remodeling enzymes in blood cell differentiation and leukemia. Biomed Res Int 2015; 2015: 347571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Progress in biophysics and molecular biology 2010; 102: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Science advances 2015; 1: e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drane P, Ouararhni K, Depaux A, et al. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 2010; 24: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong LH, McGhie JD, Sim M, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 2010; 20: 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 2010; 11: 319–330. [DOI] [PubMed] [Google Scholar]
- 37.Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011; 333: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons RJ, Wada T, Fisher CA, et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat 2008; 29: 796–802. [DOI] [PubMed] [Google Scholar]
- 39.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013; 154: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comet I, Riising EM, Leblanc B, et al. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer 2016; 16: 803–810. [DOI] [PubMed] [Google Scholar]
- 41.Ratnakumar K, Bernstein E. ATRX: the case of a peculiar chromatin remodeler. Epigenetics 2013; 8: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492: 108–112. [DOI] [PubMed] [Google Scholar]
- 43.Young A Structural insights into the clathrin coat. Semin Cell Dev Biol 2007; 18: 448–458. [DOI] [PubMed] [Google Scholar]
- 44.Pardo R, Lo Re A, Archange C, et al. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 2010; 10: 19–26. [DOI] [PubMed] [Google Scholar]
- 45.Jan Y, Matter M, Pai JT, et al. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell 2004; 116: 751–762. [DOI] [PubMed] [Google Scholar]
- 46.Griffiths GS, Grundl M, Leychenko A, et al. Bit-1 mediates integrin-dependent cell survival through activation of the NFkappaB pathway. The Journal of biological chemistry 2011; 286: 14713–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shlien A, Raine K, Fuligni F, et al. Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer. Cell Rep 2016; 16: 2032–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho CY, Lee KT, Chen WC, et al. MST3 promotes proliferation and tumorigenicity through the VAV2/Rac1 signal axis in breast cancer. Oncotarget 2016; 7: 14586–14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol 2015; 210: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuller SJ, McGuffin LJ, Marshall AK, et al. A novel non-canonical mechanism of regulation of MST3 (mammalian Sterile20-related kinase 3). Biochem J 2012; 442: 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun 2014; 5: 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamura R, Yoshihara K, Yamawaki K, et al. Novel kinase fusion transcripts found in endometrial cancer. Sci Rep 2015; 5: 18657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine 2013; 368: 2385–2394. [DOI] [PubMed] [Google Scholar]
- 54.Olesen SH, Zhu JY, Martin MP, et al. Discovery of Diverse Small-Molecule Inhibitors of Mammalian Sterile20-like Kinase 3 (MST3). ChemMedChem 2016; 11: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiba E, Matsuyama A, Shibuya R, et al. Immunohistochemical and molecular detection of the expression of FGF23 in phosphaturic mesenchymal tumors including the non-phosphaturic variant. Diagn Pathol 2016; 11: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng S, Wang J, Zhang Y, et al. FGF23 promotes prostate cancer progression. Oncotarget 2015; 6: 17291–17301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Florenzano P, Gafni RI, Collins MT. Tumor-induced osteomalacia. Bone Rep 2017; 7: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. The Lancet Oncology 2017; 18: 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *59.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *60.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *61.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *62.McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010; 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *63.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *64.Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *65.Haas B, Dobin A, Stransky N, et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The number of somatic alterations identified in each UPSb tumour
Figure S2. Examples of Sanger sequencing confirmation of eight recurrent mutations identified by WES
Figure S3. Somatic copy number alteration heatmap of ten normal-tumour paired UPSb samples
Figure S4. Diagrammatic representation and validation of CLTC-VMP1 gene fusion
Figure S7. Expression of FGF23 in UPSb tumours versus normal tissue samples using RT-qPCR
Table S1. Coding mutational rate of UPSb
Table S2. Summary of the individual recurrent mutations identified in TP53 and chromatin remodelling genes
Table S3. Somatic copy number alterations status of genes for which copy number alterations have been previously reported in UPS of bone and UPS soft tissue
Table S4. Summary of the eight somatic gene fusions identified by RNA-Seq
Table S5. Primer sequences
Figure S5. Schematic representation and validation of FARP1-STK24 fusion
Figure S6. Microscopy images of T1 and T10