

Abstract
Oncogenic mutations in the KRAS gene are well-established drivers of cancer. While the recently developed KRASG12C inhibitors offer a targeted KRAS therapy and have shown success in the clinic, KRASG12C represents only 11% of all KRAS mutations. Current therapeutic approaches for all other KRAS mutations are both indirect and nonmutant-selective, largely focusing on inhibition of downstream KRAS effectors such as MAP kinases. Inhibition of KRAS downstream signaling results in a system-wide down-modulation of the respective targets, raising concerns about systemic cell toxicity. Here, we describe a custom short interfering RNA oligonucleotide (EFTX-D1) designed to preferentially bind mRNA of the most commonly occurring KRAS missense mutations in codons 12 and 13. We determined that EFTX-D1 preferentially reduced the mutant KRAS sequence versus wild-type at the levels of both transcription and translation and reversed oncogenic KRAS-induced morphologic and growth transformation. Furthermore, EFTX-D1 significantly impaired the proliferation of several KRAS mutant cancer cell lines in 2-D as well as 3-D assays. Taken together, our data indicate a novel use of RNA interference to target oncogenic KRAS-driven cancers specifically.
Keywords: KRAS, RAS, siRNA, mutant-selective, lung cancer
The KRAS oncogene is among the most frequent and lethal drivers of cancers in the world and has a mutation frequency of approximately 31% in lung, 45% in colorectal, and 98% in pancreatic cancers.1,2 Mutations in one of three distinct hot spot codons (12, 13, or 61) impair KRAS GTPase activity and thereby lock it in a GTP-bound active state.3 In the active state, KRAS continuously stimulates downstream effector signaling, resulting in increased proliferation and cell survival.4
While the KRAS oncogene is considered a primary drug target in anticancer drug discovery, KRAS poses unique structural and functional challenges which have made this protein particularly difficult to target. For example, attempts to develop GTP-competitive inhibitors of RAS (inspired by the success of ATP-competitive inhibitors for other targets) have failed due to the many-fold higher affinity of GTP for KRAS, which is difficult to outcompete.1 Structural analysis of the KRAS protein has revealed that there are no deep hydrophobic pockets readily available for binding, making the development of potent small molecule inhibitors challenging.1,5 Alternatively, attempts to inhibit key downstream effectors of KRAS, predominantly in the RAF–MEK–ERK and PI3K–AKT–mTOR pathways, have been tempered by the significant crosstalk between pathways, activation of compensatory mechanisms, and functional redundancy (i.e., multiple isoforms and pathways can contribute to tumor progression).6,7 Thus, combination approaches to inhibit multiple pathways are needed to effectively shut down KRAS oncogenic activity;8 however, this raises concerns for widespread inhibition of other pathways and resultant unwanted toxicity.1
The first true advancement in direct KRAS inhibition has been made only recently with the development of novel drugs currently in clinical trials, including AMG510 and MRTX849, which covalently bind to KRASG12C to lock it in its inactive GDP-bound state.9−12 However, this is unlikely to be a silver bullet approach for all KRAS mutations, as G12C presents some vulnerabilities unique to this mutation. For example, the lysine residue (Lys-16) in the binding pocket catalyzes covalent binding, and the G12C protein has one of the fastest turnover rates of all the KRAS mutants. These qualities allow for more frequent drug occupancy in the inactive GDP-bound state.13 Therefore, although the recent advance of these G12C inhibitors has aroused excitement in the KRAS community, the need for effective inhibition strategies for the other predominant KRAS mutations remains a critical unmet need, given that G12C accounts for only 11% of all KRAS mutant cancers. In addition, it is important to note that even for G12C, the success of these inhibitors is still tempered by limited efficacy in the clinic, due in part to the emergence of resistance mechanisms. For example, some cells that enter quiescence upon KRAS G12C inhibition can stimulate a compensatory increase in production of new KRAS G12C protein that maintains its active (GTP-bound), drug-insensitive state, thereby evading drug inhibition.14 Thus, an alternative strategy that inhibits protein production (as opposed to protein function), such as RNA interference (RNAi), may be an efficacious approach that abrogates this type of resistance to targeting KRAS. Of note, there has also been interest in the use of proteolysis targeting chimeras (PROTACs), which function by recruiting endogenous E3 ubiquitin ligase for targeted degradation, for inhibition of KRAS protein expression. However, efforts to develop PROTACs against KRAS G12C in lung and pancreatic cancer have been shown to fail due to shortcomings such as an inability to successfully promote polyubiquitination of endogenous KRAS G12C.15
Therapeutic RNAi refers to the delivery into cells of synthetic oligonucleotides complementary to specific target RNAs, which then utilize the endogenous RNA-induced silencing complex (RISC) to cleave those messenger RNA (mRNA) sequences or to inhibit their translation.16 Previous work from our lab and others has investigated the possibility of therapeutically down-modulating KRAS mRNA expression via siRNA17−19 or antisense oligonucleotides.20 However, one shortcoming of these approaches is their lack of mutant versus wild-type (WT) specificity. Systemic equal silencing of both mutant and WT KRAS mRNAs may give rise to toxicity due to the loss of KRAS WT in healthy tissues. Although the consequences of toxicity following KRAS loss in healthy adult tissues remain to be evaluated, selective inhibition of tumor-specific mutant KRAS could have significant advantages for an optimal therapeutic window. Here, we report the design and development of EFTX-D1, a mutant-selective, WT-sparing siRNA that has a higher affinity for several of the most common oncogenic KRAS mutations compared to WT KRAS.
Results
EFTX-D1 is Designed to Target the Most Common KRAS Codon 12 and Codon 13 Mutations but Spare Wild-type KRAS
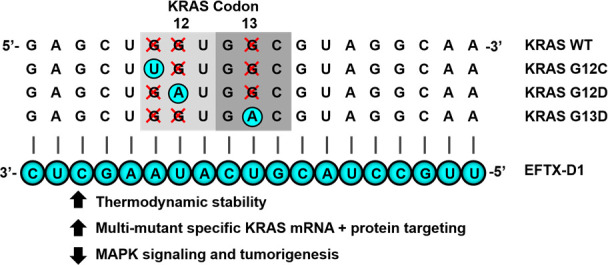
Codon 12 and codon 13 mutations are the most frequently occurring KRAS mutations in cancer, and G12D, G12V, G12C, and G13D account for about 80% of all KRAS mutations (Figure 1a). Due to the close proximity of these mutational hotspots, we sought to determine whether an antisense sequence of customized siRNAs could target several of these mutations yet spare the wild-type sequence. Using the Block-iT RNAi Designer tool (Invitrogen), we designed a library of siRNAs that target an engineered, artificial KRAS mRNA sequence that simultaneously contains the point mutations of G12C, G12D, and G13D (Figure 1b, c). We termed the siRNAs targeting this sequence EFTX-D siRNAs. Because G12D and G12V missense mutations occur in the same nucleotide of codon 12, we also engineered an artificial KRAS mRNA sense sequence carrying the G12C, G12V, and G13D mutations, which served as a target for EFTX-V siRNAs (Figure 1b, Figure S-1). Previous literature suggests there is a 3-nucleotide mismatch tolerance threshold for 19 base pair siRNA efficacy of mRNA target recognition and knockdown.21 EFTX-D and EFTX-V siRNAs were designed to have only two mismatches with any of their target KRAS mutation sequences (e.g., EFTX-D siRNAs have two mismatches with a KRAS G12C, G12D, or G13D mRNA transcript), while siRNA binding to the WT KRAS mRNA contains three mismatches (Figure 1c, Figure S-1). We then evaluated whether this imbalance of two versus three mismatches was sufficient to create preferential inhibition of the target KRAS mutation while sparing the WT sequence.
Figure 1.

Design and selection of a KRAS mutant, WT-sparing siRNA. (a) Most common KRAS mutations in all cancers (gray), lung cancer (green), colon cancer (blue), and pancreatic cancer (purple). (b) Sequence alignment of KRAS codons 12 and 13 in the wild-type (WT) form with the 4 most commonly mutated variants (G12C, G12D, G12V, and G13D) as well as EFTX-D siRNAs (siRs) and EFTX-V siRs (with each class of siRs targeting a hypermutated KRAS sequence carrying three oncogenic mutations). Mutated nucleotides are in red. (c) Pairing of the EFTX-D1 siR antisense strand with KRAS WT, G12C, G12D, G12V, and G13D mRNA transcripts. Non-Watson–Crick base pairings (i.e., mismatches) are indicated by a green x. (d) qPCR of NIH/3T3 cells stably expressing KRAS WT, G12C, G12D, G12V, or G13D and transiently transfected with negative control (NC), Seq2, Seq3, EFTX-D, or EFTX-V siRs at a dose of 20 nM. Cells were analyzed 24 h post transfection, and qPCR was run in technical triplicate. Results were normalized to NC siR transfection. (e) Revisualization of panel d by comparing level of WT expression (x-axis) vs expression of each KRAS mutant (y-axis) after transfection of each siR assessed in panel d. Results in the top half above the horizontal dotted line indicate poor knockdown efficacy of mutant KRAS. Results in the bottom left indicate effective knockdown of mutant KRAS but poor WT sparing (i.e., concomitant knockdown of mutant and WT KRAS expression). Lead siRs appear in the bottom right quadrant and demonstrate potent mutant inhibition and optimal WT sparing. Symbol color indicates siR treatment; symbol shape indicates KRAS mutation assessed. Results were normalized to NC siR transfection. Error bars represent standard error of the mean.
Candidate EFTX-D and EFTX-V siRNAs were transiently transfected into NIH/3T3 mouse fibroblasts stably expressing human KRAS WT, G12C, G12D, G12V, or G13D, and real-time qPCR was used to screen for their ability to potently inhibit expression of mutant KRAS while sparing WT (Figure 1d). Knockdown efficacy was compared to those of our previously published pan-KRAS siRNA sequences Seq2 and Seq3. These bind to a nonmutated, conserved region of KRAS mRNA far downstream of codon 13 (0 mismatches with G12 and G13 mutants, and with WT), rendering them nonspecific for mutant versus WT KRAS. We have previously demonstrated that Seq2 and Seq3 potently inhibit KRAS expression in vitro and exert therapeutic inhibition of KRAS-driven tumor progression in lung and colon cancer models.18 Both EFTX-D1 and EFTX-D4 demonstrated high mutant-specificity and potency of knockdown relative to negative control (NC) siRNA. However, EFTX-D1 exhibited a superior ability to consistently inhibit expression of the targeted mutants while maintaining WT sparing (Figure 1e). Unexpectedly, EFTX-D1 also demonstrated activity against the G12V mutant, which has three base pair mismatches (Figure 1d, e). Six additional EFTX-D siRNA sequences, with all possible starting positions between EFTX-D1 and EFTX-D4, were also tested for their ability to reduce mutant KRAS, but all proved to be inferior to the KRAS silencing potency of EFTX-D1 (Figure S-2).
WT KRAS Sparing Relative to Mutant KRAS Targeting Is Related to Reduced Thermodynamic Stability of the WT KRAS-EFTX-D1 Complex
We predicted that the WT sparing feature of EFTX-D1 would at least partially be due to differences in the binding energy of EFTX-D1 to its two-mismatch mutant target transcripts versus the three-mismatch WT target. To test this possibility, we characterized the melting points of EFTX-D1 complexed with differently synthesized KRAS mRNA mimics (Figure 2a, b). This allowed us to determine the binding strength between WT or mutant KRAS mRNA and EFTX-D1. We observed that the average melting temperature was lower (reflecting reduced thermodynamic stability of binding) for pairing reactions containing KRAS WT (67.7 °C) or KRAS mutations with three mismatches (G12V – 67.3 °C, G12S – 67.9 °C, G12A – 65.8 °C, and G12R – 68.7 °C) compared to the reactions containing KRAS mutations with two mismatches on EFTX-D1 (G12C – 72.3 °C, G12D – 69.0 °C, and G13D – 70.2 °C).
Figure 2.

Thermodynamic stability of EFTX-D1 and KRAS mRNA association. (a) Melting temperatures Tm of EFTX-D1 siRNA paired with each indicated target KRAS mRNA synthetic sequence. WT and gray mutants carry three mismatches with EFTX-D1; red mutants carry two mismatches with EFTX-D1. (b) Melting curves of EFTX-D1 with each indicated target KRAS mRNA synthetic sequence. Thirteen technical replicates were run for WT and 6–7 replicates for each mutant. Statistical significance was measured by a one-way ANOVA test; p-values are indicated as ***p < 0.001. Error bars represent standard error of the mean.
EFTX-D1 Selectively Reduces Oncogenic KRAS mRNA
To further characterize whether EFTX-D1 has a significant difference in downmodulation of mutant, oncogenic KRAS mRNA over WT KRAS mRNA, we performed transient knockdown experiments in our NIH/3T3 cell lines stably expressing human KRAS WT, G12C, G12D, G12V, or the codon 61 mutation Q61H (Figure 3a). EFTX-D1 has only a two base pair mismatch with G12C and G12D and a three base pair mismatch with WT and G12V. It also has a three base pair mismatch with the Q61H mutant, as the nucleotide sequence of codons 12 and 13 in this mutant is the same as in WT. We therefore anticipated that EFTX-D1 would downmodulate Q61H mRNA to the same extent as WT mRNA. Comparing the KRAS knockdown efficacy of EFTX-D1 to that of the pan KRAS (i.e., both WT and mutant-targeting) siRNAs Seq2 and Seq3 after 24 h, we observed that Seq2 and Seq3 caused the most potent downmodulation (86–95% knockdown relative to NC siRNA transfection) of all KRAS mRNAs, presumably due to the zero mismatches of these siRNAs with all the target mRNAs. As expected, Seq2 and Seq3 also demonstrated no selectivity for oncogenic mutant KRAS mRNA over WT. In contrast, EFTX-D1 demonstrated more potent inhibition of the codon 12 and 13 mutations (G12C – 79%, G12D – 85%, G12V – 85%, and G13D – 88% knockdown relative to NC siRNA), whereas it reduced WT mRNA by only 60% and Q61H mRNA by only 49%, relative to NC siRNA transfection.
Figure 3.

KRAS mutant-specific knockdown by EFTX-D1. (a) qPCR at 24 h post transfection of NIH/3T3 cells stably expressing KRAS WT, G12C, G12D, G12V, G13D, or Q61H and transiently transfected with 40 nM NC, Seq2, Seq3, or EFTX-D1 siRNA (siR). The “WT” line indicates average expression level of KRAS mRNA with WT codon 12 and codon 13 sequences, in response to EFTX-D1 transfection. The “Mutant” line indicates average expression level of the KRAS mutants carrying codon 12 or codon 13 mutations, in response to EFTX-D1 transfection. qPCR was run in duplicate. (b) qPCR dose response curves of NIH/3T3 cells stably expressing KRAS WT or G12C and transiently transfected with Seq2 or EFTX-D1 siR. Results are shown 24 h post transfection and are normalized to NC siR transfection. qPCR was run in duplicate. IC50 values for siR treatment of each cell line are shown. Error bars represent standard error of the mean.
The ratios of Seq2 and Seq3 siRNA inhibition of WT mRNA to inhibition of all codon 12 and codon 13 mutants assessed are near 1 (WT:Mutant inhibition, Figure S-3), consistent with their lack of sparing WT mRNA. Conversely, the ratios for EFTX-D1 treatments are markedly lower than 1 (G12C – 0.53, G12D – 0.37, G12V – 0.38, and G13D – 0.29), highlighting EFTX-D1’s preferential silencing of the targeted KRAS mutants relative to WT. Notably, the ratio of WT inhibition to Q61H inhibition by EFTX-D1 is near 1, which is expected as EFTX-D1 is not predicted to demonstrate preferential silencing for a codon 61 mutation.
Using the NIH/3T3 KRAS WT and G12C stably expressing cell lines, we performed a dose response experiment with EFTX-D1 and Seq2 and analyzed the results by qPCR (Figure 3b). Relative KRAS inhibition for Seq2 and EFTX-D1 transfections was normalized to NC siRNA treatment at each dose assessed, and absolute IC50 values for KRAS expression inhibition were calculated for each cell line (see Methods). Seq2 demonstrated near equipotency for both WT and mutant KRAS inhibition (Seq2 WT KRAS IC50 – 7.5 nM vs KRAS G12C IC50 – 7.7 nM) whereas EFTX-D1 was markedly more potent in the mutant KRAS cell line (EFTX-D1 KRAS G12C IC50 – 13.8 nM) compared to the WT cell line, in which EFTX-D1 was did not achieve 50% KRAS inhibition at any dose (EFTX-D1 KRAS WT IC50 – not determined). Notably, Seq2 demonstrated higher potency for KRAS G12C inhibition compared to EFTX-D1, as is expected due to Seq2 having 0 mismatches on the KRAS G12C transcript and EFTX-D1 having two mismatches.
EFTX-D1 Selectively Reduces Oncogenic KRAS Protein
We next assessed whether the effects of EFTX-D1 on KRAS protein were consistent with effects on KRAS mRNA by Western blotting. We compared the effects of EFTX-D1 and the pan-KRAS Seq2 on KRAS protein in NIH/3T3 cells expressing either WT or KRAS G12C, the latter of which exhibited the highest differential thermodynamic stability (Figure 2). Relative to NC siRNA transfection, we observed that both EFTX-D1 and Seq2 inhibited KRAS G12C protein by ∼70% (Figure S-4a). However, unlike Seq2, EFTX-D1 also demonstrated an ability to partially spare WT KRAS protein expression relative to KRAS G12C.
We then used an orthogonal model system to further investigate the ability of EFTX-D1 to regulate oncogenic KRAS protein expression and spare WT in a more cancer-relevant context. Using a non-KRAS-dependent carcinoma A431 cell line, following deletion of the endogenous KRAS WT allele, we generated KRAS WT, G12C, or G12D HA-tagged expressing lines. We chose KRAS G12C and G12D due to their high prevalence (G12C: 34%, G12D: 19%) in KRAS-associated lung cancers, as well as their improved thermodynamic stability when complexed with EFTX-D1 (Figure 2). Relative to NC siRNA transfection, we observed that EFTX-D1 more potently inhibited KRAS G12C and G12D, while it partially spared WT protein expression (KRAS G12C: 78%, KRAS G12D: 72%, KRAS WT: 61% inhibition; Figure S-4b). These results indicate EFTX-D1 is capable of KRAS mutant versus WT targeting on a protein level, as shown in two different model systems. However, we observed a reduced differential targeting between mutant versus WT on a protein level, compared to what was seen with an mRNA transcript read-out (Figures 2 and 3). These findings suggest EFTX-D1 may also cause an “miRNA-like” effect by inhibiting translation of KRAS WT and mutant transcripts.
EFTX-D1 Inhibits the KRAS Mutant-Induced Transformed Phenotype of NIH/3T3 Cells
The effects of EFTX-D1 on inhibiting KRAS led us to investigate the impact of EFTX-D1 treatment on KRAS-driven transformation of NIH/3T3 cells. Seq2 siRNA, which has already demonstrated robust inhibitory effects on cancer cell growth18 was used as a positive control in all phenotypic experiments. To observe if EFTX-D1 can reverse the KRAS oncogene-induced phenotypic loss of contact inhibition and formation of foci, we transiently transfected KRAS WT or G12C NIH/3T3 cells with EFTX-D1, Seq2, or NC siRNA. We observed that knockdown of KRAS with either EFTX-D1 or Seq2 but not NC siRNA reverted the transformed morphology of stably expressing KRAS G12C NIH/3T3 cells back toward a contact-inhibited phenotype, without affecting cells expressing WT KRAS (Figure S-5).
EFTX-D1 Inhibits KRAS-Driven Cancer Cell Proliferation
G1 cell cycle arrest is a common feature of cancer cells that have undergone a reduction in mutant KRAS. Therefore, we performed cell cycle analysis following KRAS knockdowns using EFTX-D1 or Seq2 in our panel of KRAS-dependent lung cancer cell lines harboring endogenous KRAS codon 12 mutations, including two cell lines with three mismatches with EFTX-D1 (H441 – G12V and A549 – G12S) and one cell line with two mismatches with EFTX-D1 (H358 – G12C) (Figure 4a). EFTX-D1 phenocopied Seq2-mediated G1 cell cycle arrest in all three KRAS mutant cell lines. Treatment with EFTX-D1 and Seq2 increased the percentage of cells in G1 relative to NC siRNA treatment in all cell lines assessed: the percentage of H358 (G12C) cells in G1 increased by 29 or 11% upon Seq2 or EFTX-D1 transfection, respectively; of H441 (G12V) cells in G1 increased by 20 or 12%, respectively; and of A549 (G12S) cells in G1 increased by 13 or 14%, respectively.
Figure 4.

EFTX-D1 inhibits mutant KRAS-driven cancer cell growth. (a) Flow cytometry cell cycle analysis of H358 (KRAS G12C), H441 (KRAS G12V), and A549 (KRAS G12S) lung cancer cells 72 h post transient transfection with NC, Seq2, or EFTX-D1 siR at 20 nM. (b) Western blot analysis of KRAS, pERK, and total ERK expression in H358 (KRAS G12C), H441 (KRAS G12V), and A549 (KRAS G12S) lung cancer cells transiently transfected with NC, Seq2, or EFTX-D1 siR at 20 nM. Cells were analyzed 36 h post transfection. (c) 2-D proliferation of H358, H441, and A549 lung cancer cells transiently transfected with NC, Seq2, or EFTX-D1 siR at 20 nM. Cells were quantified after 7 days of growth. Samples were run in at least triplicate, and cell counts are normalized to NC. (d) 3-D growth of A549 and H441 lung cancer cells embedded in Matrigel 24 h after transient transfection with NC, Seq2, or EFTX-D1 siR at 20 nM. Cells were imaged and quantified using OrganoSeg software after 7 days of growth, and conditions were run in at least triplicate; scale bar 400 μm. Statistical significance was measured by a one-way ANOVA with a Tukey’s multiple comparisons test; p-values are indicated as **p < 0.01, ***p < 0.001. Error bars represent standard error of the mean.
As the ERK MAPK pathway is one of the major downstream effectors of KRAS signaling, we asked if EFTX-D1 treatment can also inhibit phosphorylation of ERK. Western blot analysis in H358, H441, and A549 cell lines showed a robust reduction in phosphorylated ERK upon EFTX-D1 transfection in comparison to NC siRNA transfection, similar to the effects of Seq2 (Figure 4b).
To address if KRAS inhibition-mediated G1 arrest corresponds with reduced cellular proliferation, we characterized the effects of EFTX-D1 and Seq2 treatment on 2-D and 3-D cancer cell proliferation (Figure 4c and d). Relative to NC siRNA treatment, EFTX-D1 and Seq2 reduced the growth on plastic of H358 cells by approximately 60% at day 7, of H441 cells by approximately 50%, and of A549 cells by approximately 25% (Figure 4c). Finally, a single treatment of either EFTX-D1 or Seq2 also significantly inhibited H441 and A549 spheroid growth relative to NC siRNA treatment in a Matrigel 3-D spheroid assay (Figure 4d).
We evaluated whether the effects of EFTX-D1 on oncogenic phenotypes are specific to mutant KRAS inhibition by testing 2-D and 3-D proliferation assays in the H1299 KRAS WT cancer cell line. Relative to NC siRNA transfection, EFTX-D1 modestly reduced H1299 viability at the 96 h time point (Figure S-6a), however, no significant differences in spheroid surface area were observed in a 3-D Matrigel assay (Figure S-6b).
Discussion
The KRAS oncogene has been a major target for cancer drug discovery efforts for nearly four decades. Here, we describe a novel approach to target three of the most common KRAS mutations (G12C, G12D, and G13D, together comprising 57% of KRAS mutations in cancer) with one siRNA, which also preferentially spares WT KRAS. Our approach is more specific for mutant KRAS than others that impair oncogenic KRAS signaling by inhibition of key downstream kinases. The latter approach is not exclusively tailored to a cancer cell mutation, but rather to kinases that are typically ubiquitously expressed in both cancer and normal tissues. Several of these kinases, such as the kinases of the ERK MAPK pathway and the PI3K pathway, have pivotal roles in critical cellular processes, and their inhibition at higher doses or as combination therapy are reported to have side effects such as diarrhea, nausea, vomiting, reduced appetite, rash, pyrexia, fatigue, and hyperglycemia.22,23
Thus far, the most promising direct KRAS inhibitors are targeted specifically to KRAS G12C.9−12 These inhibitors covalently bind the mutated cysteine at position 12, which is not present in wild-type KRAS. KRAS G12C inhibitors are currently leading to exciting responses in clinical trials, particularly in lung cancer,11,12 and are likely to become the first FDA-approved direct KRAS inhibitors. These successes provide proof-of-principle that direct mutant-specific inhibition of oncogenic KRAS is highly attractive and safe. However, G12C represents only 11% of all KRAS mutant cancers. An advantage of EFTX-D1 is its ability to simultaneously silence multiple KRAS G12/G13 mutations (with the most potent effects on G12C, G12D, and G13D) while partially sparing WT KRAS. This gives EFTX-D1 applicability to a much broader cohort of KRAS mutant cancers beyond only those harboring G12C, including cancers driven by KRAS G12D (33% of all KRAS mutant cancers and 19% of KRAS mutant lung cancer) and G13D (13% of all KRAS mutant cancers). In addition, targeting more than one mutation within the same tumor at the same time may help prevent the outgrowth of resistant subpopulations driven by different coexisting or de novo KRAS mutations.24−26
Although EFTX-D1 had the most potent binding affinity for KRAS G12C, G12D, and G13D as predicted, it still retained the capability to form a duplex with the other mutants with some level of thermodynamic stability. This suggests that there may still be sufficient interaction to enable knockdown of other mutant KRAS mRNA targets and subsequent oncogenic inhibition. This is further supported by the observation that the EFTX-D siRNAs demonstrated potent knockdown of G12V relative to WT (Figure 1d, e), even though both G12V and WT engage in a three-mismatch pairing with EFTX-D siRNAs (Figure 1c). These results highlight the potential broad applicability for EFTX siRNAs to inhibit the expression of other KRAS mutants beyond G12C, G12D, G12V, and G13D and suggest that a three base pair mismatch does not necessarily preclude efficacy of target knockdown. In addition to the two vs three base pair mismatch imbalance, the position and nature of the mismatch(es) may also contribute to siRNA:target duplex thermodynamic stability and to knockdown efficacy.27 For example, perhaps the ability of EFTX-D1 to distinguish between WT and G12V more stringently can be attributed to a lower tolerance for a U–G mismatch compared to a U–U mismatch at position 13 on the antisense strand. In addition, it is important to note that although differences in thermostability and target base pairing can account for some of an siRNA’s enhanced targeting of specific transcripts, there are other factors that influence targeting as well, including efficiency of RISC loading and processing of the antisense strand, target mRNA transcript abundance and stability, and intracellular distribution and longevity of the siRNA.
EFTX-D1 demonstrated the ability to selectively inhibit mutant KRAS and spare WT expression relative to the pan-KRAS targeting siRNA Seq2. In all experiments, Seq2 and/or Seq3 siRNA transfection served as a positive control representing maximal knockdown independent of KRAS mutational status. EFTX-D1 mediated knockdown was consistently comparable to Seq2/Seq3-mediated knockdown in KRAS mutant cells but inferior to Seq2/Seq3-mediated knockdown in WT cells.
Although the initial focus of the EFTX-D1 design was to reduce off-target silencing of WT KRAS in all nontumor tissues, it is also interesting to consider effects on the silencing ratio of WT versus mutant KRAS alleles within a given cancer cell. Recent reports have suggested that KRAS WT may harbor tumor suppressive roles in particular molecular contexts.28 Loss of heterozygosity of wild-type KRAS has frequently been observed in the presence of mutant KRAS during tumor progression, suggesting loss of wild-type KRAS may be required to enable tumor development.29,30 Similarly, restoration of wild-type KRAS expression has been demonstrated to hinder tumor progression.31 Therefore, EFTX-D1 as a therapeutic may have the added benefit of mitigating pro-tumorigenic effects of WT KRAS loss.
Over the past few years, the RNAi field has seen a rapid surge in therapeutic promise, and several siRNA therapeutics have received FDA approval in genetic disorders.32,33 Recent state-of-the-art chemical modifications of siRNAs, and conjugation of siRNAs to targeting moieties, has revolutionized the field of therapeutic RNAi.34,35 Although EFTX-D1 siRNA likely has some off-target effects and may even have “miRNA-like” properties that result in suppressed KRAS WT mRNA translation, it may serve as a template design to incorporate strategic chemical modifications that improve immune-stealth, resistance to nuclease-degradation, enhanced mutant specificity and mitigation of potentially toxic off-target effects. However, it remains to be seen whether nanoparticle-based or ligand-conjugated siRNA treatments will be effective in oncology. Looking forward, it is important to note that EFTX-D1 targets a region of KRAS mRNA that is fully conserved in both humans and mice; thus, future toxicity studies conducted in mice will yield information with direct relevance to human health. Taken together, our results demonstrate a novel opportunity to target oncogenic KRAS signaling and suppress cancer growth with EFTX-D1, a WT sparing, multimutational targeting siRNA. We believe this siRNA offers a new therapeutic tool compound in the fight against the KRAS oncogene.
Materials and Methods
Cell Lines and Culture
NIH/3T3 mouse fibroblast cells were obtained from ATCC, and KRAS-transduced NIH/3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Colorado calf serum, 1% penicillin–streptomycin (Pen–Strep), and 1 μg/mL puromycin. A431 epidermoid carcinoma cells were obtained from ATCC, and KRAS isoform transduced A431 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% Pen–Strep, and 1 μg/mL puromycin. Human embryonic kidney (HEK)293T cells and A549 lung adenocarcinoma cells were obtained from ATCC. H358 bronchioalveolar carcinoma cells and H441 lung adenocarcinoma cells were obtained from NCI. HEK293T cells were maintained in DMEM; H358, H441, A549, and H1299 cells were maintained in RPMI 1640 medium. All media were supplemented with 10% FBS and 1% Pen–Strep. All cell lines were maintained in a humidified chamber with 5% CO2/95% air at 37 °C. Cell lines were monitored for mycoplasma contamination, and all in vitro experiments were conducted with 60–80% confluent cultures.
NIH/3T3 cells stably expressing HA-tagged human KRAS WT, G12C, G12D, G12V, or G13D were generated as follows: first, retroviral particles were generated by cotransfecting 1.25 μg of pBABE-puromycin retroviral vectors expressing WT KRAS or each KRAS mutant with 1.25 μg/μL PCL10A pack vector using 6.25 μg of Lipofectamine 2000 (ThermoFisher Scientific) into HEK293T cells seeded in a 6 cm cell culture plate per the manufacturer’s instructions. At 24 h post transfection, HEK293T cells were changed into fresh media. Viral supernatant was collected 24 h later and stored on ice. Fresh media was added to the cells and again viral supernatant was collected 24 h later. The two batches of viral supernatant were combined and filtered. NIH/3T3 cells were then seeded in a 6-well plate at a density of 100 000 cells/well and 250 μL of virus was added to the cells along with 10 μg/mL Polybrene. Cells were spinoculated at 1500g for 1 h and then transduced for another 24 h at 37 °C. Virus was removed from the cells, and 24 h later, 2 μg/mL puromycin selection media was added. Cells were considered selected once all nontransduced cells in a control well were killed by the selection media.
A431 cells overexpressing HA-tagged human KRAS WT, G12C and G12D were generated by first removing the endogenous WT KRAS gene by CRISPR and then transducing A431 cells with retroviral particles packaged with the same pBABE-puro-KRAS plasmids as described above. A plasmid expressing KRAS sgRNA (atccGTAGTTGGAGCTGGTGGCGTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT, GeneCopoeia, catalogue # HCP288420-SG01-1-10) and a plasmid expressing Cas9 (Addgene Plasmid 49535) were cotransfected into A431 cells via the Neon Electroporation Transfection system (Thermo Fisher Scientific) as per the manufacturer’s instructions. CRISPR clones were generated using single cell cloning and screened via PCR and sequencing to identify clones with successful removal of the endogenous KRAS gene. One such clone (Clone 2–10) was transduced with retrovirus expressing either KRAS WT, G12C, or G12D. Retroviral particles were generated as follows: 1.25 μg pBABE-puromycin retroviral vectors overexpressing each KRAS isoform was cotransfected with 1.25 μg/μL PCL10A pack vector using 6.25 μg of Lipofectamine 2000 (ThermoFisher Scientific) into HEK293T cells seeded in a 6 cm cell culture plate per the manufacturer’s instructions. At 24 h post transfection, the media on the HEK293T cells was changed to fresh media. Viral supernatant was collected 24 h later and filtered. A431 cells were seeded in 6-well plates, and 1 mL of virus was added along with 10 μg/mL Polybrene to each well to transduce them for 48 h at 37 °C. Virus was removed and 24 h later selection media containing 1 μg/mL puromycin was added to the cells. Cells were considered selected once all nontransduced cells in a control well were killed by the selection media.
UV Melting Experiments
RNA duplexes were prepared by mixing solutions with equimolar concentrations of two strands (1.0 μM) in 1× PBS buffer, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4, pH 7.4. Prior to running UV melting experiments, strands were annealed by heating samples in a water bath to 70 °C for 2 min, followed by slow cooling to room temperature and refrigeration overnight. All measurements were made using a Cary 100 Bio UV–vis spectrophotometer (Agilent Technologies Inc., Santa Clara, CA), equipped with a temperature controlled multicell holder (6 × 6) and a Cary temperature controller. Absorbance versus temperature profiles were acquired by monitoring the absorbance at 260 nm (A260) between 20 and 90 °C with a ramp rate of 1 °C per minute. A260 values were measured at 0.2 °C intervals, and melting temperatures Tm were extracted as the maxima of the first derivatives of smoothened melting curves (filter 5) using the Cary WinUV software (Version 3.0, Agilent Technologies Inc.).
siRNA Reverse Transfection and mRNA Isolation
Sequences for the negative control (NC) and positive pan-KRAS control (Seq2, Seq3) siRNAs were as previously described.18 All siRNAs were obtained from Sigma. At the time of transfection, KRAS-infected NIH/3T3 cells were plated either in 24-well plates a density of 60 000–120 000 cells per 500 μL media per well or in 12-well plates at 240 000 cells per 1 mL media per well. Cells were incubated in a 5:1 mixture of complete medium and serum-free medium, along with 20 nM siRNA (unless dose is otherwise indicated) in a 1:2 ratio with Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) for 16–24 h at 37 °C in 5% CO2.
Quantitative Real-Time PCR
For mRNA quantification, total RNA was extracted from cultured cells using the Quick RNA MiniPrep Zymo Research Kit (Genesee Scientific, #11–328) or the RNeasy Mini Kit (Qiagen, # 74104), and purified RNA was quantified using a spectrophotometer (Nanodrop). cDNA was synthesized using an iScript cDNA Synthesis Kit (Bio-Rad, #1708891) per the manufacturer’s instructions. Analysis of mRNA levels was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems). Specific primers for KRAS [set #1 (forward)- TGACCTGCTGTGTCGAGAAT, (reverse)- TTGTGGACGAATATGATCCAA or set #2 (forward)- TCCAACAATAGAGGATTCCTACAG, (reverse)- CCCTCATTGCACTGTACTCCT] were used for SYBR Green-based real-time PCR, and 18S rRNA was used as a housekeeping gene. PCR was done with reverse-transcribed RNA, 1 μL each of 20 μM forward and reverse primers, and 2X PowerUp SYBR Green Master Mix (Life Technologies, #100029284) in a total volume of 25 μL. For SYBR PCR, each cycle consisted of 15 s of denaturation at 95 °C and 1 min of annealing and extension at 60 °C (40 cycles). Reactions were run in duplicate or triplicate. Relative quantitation (RQ) values were calculated using the ΔΔCt method.
Dose Response Curves
Cells were transfected as described above at a range of siRNA doses from 0.74 to 60 nM. Least squares fit dose response curves were generated using the GraphPad Prism nonlinear regression “log[inhibitor] vs response – variable slope (four parameters).” Absolute IC50s were calculated by determining the concentration of siRNA at which 50% KRAS mRNA expression was attained. Fifty percent expression was defined as the halfway point between the arbitrarily defined top plateau of a qPCR RQ value of 1 (i.e., no knockdown) and bottom plateau of a qPCR RQ value of 0 (i.e., total knockdown).
Immunoblotting
Cells were washed with ice cold PBS, lysed in RIPA buffer supplemented with phosphatase (Sigma) and protease (Roche) inhibitors, scraped, and collected in prechilled tubes. Lysates were then cleared by centrifugation at 15 000g for 10 min at 4 °C, and protein concentration was determined using a Bradford assay (Bio-Rad). Standard immunoblotting procedures were followed. Membranes were blocked in Rockland blocking buffer diluted in TBS + 0.05% Tween 20 (TBST). Antibodies: Rabbit monoclonal anti-HA-Tag KRAS (Cell Signaling Technology Cat #3724), Mouse monoclonal anti-KRAS (Millipore Cat #OP24), Rabbit polyclonal antiphospho-p44/42 MAPK (pERK1/2) (Thr202/Tyr204) (Cell Signaling Technology Cat #4370), Rabbit polyclonal anti-p44/42 MAPK (ERK1/2) (Cell Signaling Technology Cat #9102), Mouse monoclonal anti-β-Actin (Sigma-Aldrich Cat #5441), and Mouse monoclonal antivinculin (Sigma-Aldrich Cat #V9131).
Focus Formation Assay
Three thousand cells were seeded into a 96-well clear bottom plate (Corning #3904), reverse transfected as described above, and imaged after 3 days with the Molecular Devices SpectraMax i3x MiniMax 300 imaging cytometer.
2-D Proliferation Assay
Three thousand cells per well were seeded into Corning 96-well clear bottom plates (Corning #3904) and reverse transfected as described above. After the indicated days or time points, cells were stained with Calcein-AM (Invitrogen #C3100MP) and imaged with the Molecular Devices SpectraMax i3x MiniMax 300 imaging cytometer. Alternatively, cells were quantified via CellTiter-Glo 2.0 Assay (Promega #G9243) according to the manufacturer’s instructions at a ratio of 50 μL of reagent to 50 μL of media in each well.
3-D Proliferation Assay
Cells were reversed transfected as described above, and after 24 h, 5000 cells were seeded into a 50 μL Matrigel (Corning) dome in a 24-well plate. After solidifying, 750 μL of medium was added. Cells were cultured at 37 °C in 5% CO2. Cells were imaged using a Leica DMi8 inverted microscope 7 days after transfection. Alternatively, cells were reversed transfected as described above, and after 24 h, 1000 cells were seeded in 150 μL of 10% growth factor reduced Matrigel (Corning) + 90% culture medium into 96-well clear flat bottom plates (Corning) coated with poly(2-hydroxyethyl methacrylate) (Sigma-Aldrich). Eight days after transfection, spheres were imaged with a Molecular Devices SpectraMax i3x MiniMax 300 imaging cytometer. Spheroid surface area was measured and quantified using OrganoSeg software.36
Cell Cycle Analysis
Two-hundred thousand cells were seeded into a 6 cm plate and reverse transfected with 20 nM of indicated siRNAs. After 3 days, cells were washed with PBS and harvested using TrypLE. After harvesting cells, were washed once with PBS and centrifuged at 400g and fixed with ice cold 70% ethanol overnight. Cell pellets were resuspended in 1× PBS before adding 9 volumes of ice cold 70% ethanol dropwise and fixed overnight at 4 °C. The next day, cells were washed with PBS and resuspended in cell cycle solution (1× PBS + 20 μg/mL propidium iodide and 100 μg/mL RNase) for 3 h at 37 °C. PI intensity was measured with the Beckman Coulter Cytoflex. The data was gated and analyzed with FCS Express Express 6 Flow Software by gating for cells (gate 1) and singlets (gate 2) before performing cell cycle analysis with FCS Express.
Statistical Analyses
All statistical tests were performed using GraphPad Prism 7 and 8 (GraphPad Software, Inc., San Diego, CA). All line and bar graphs represent mean values, and all error bars represent standard error of the mean.
Acknowledgments
Support was provided by grants from the National Cancer Institute (NCI) to A.D.C. and/or C.J.D. from the NCI (R21CA179193, R01CA42978, R01CA175747, R01CA223775, P50CA196510, U01CA199235, P01CA203657, and R35CA232113), and from the Pancreatic Cancer Action Network/AACR (15-90-25-DER), Department of Defense (W81XWH-15-1-0611), and the Lustgarten Foundation (388222) to C.J.D. B.P. was supported by the Deutsche Forschungsgemeinschaft (DFG PA 3051/1-1). C.V.P. was supported in part by the National Institutes of Health (NIH) R01CA215075 and 1R41CA246848, a Mentored Research Scholar Grants in Applied and Clinical Research (MRSG-14-222-01-RMC) from the American Cancer Society, the Jimmy V Foundation Scholar Award, the Stuart Scott V Foundation, the Lung Cancer Research Foundation, the Free to Breathe Metastasis Research Award, and a North Carolina Biotechnology Translation Research Grant (NCBC TRG). S.H.A. was supported in part by an NIH 1R41CA246848 and an NCBC TRG award. P.S.P. was supported by the Volkswagen Stiftung (Project Az 92 768).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00165.
Figures showing EFTX-V siRNA targeting of KRAS transcripts, EFTX-D1 sparing of WT, investigation of other EFTX-D candidate siRNAs, EFTX-D1 mediated KRAS mutant protein inhibition and WT protein sparing, EFTX-D1 inhibits KRAS mutant-induced 3T3 transformation, and effects of EFTX-D1 on KRAS WT H1299 cancer phenotype (PDF)
Author Contributions
□ B.P. and S.H.A. are cofirst authors.
The authors declare the following competing financial interest(s): S.H.A., A.D.C., C.J.D., and C.V.P. hold intellectual property interests and/or an issued U.S. patent on this work. C.V.P. is the founder of EnFuego Therapeutics, Inc. and holds equity in the company. S.H.A. and H.H. are employees of EnFuego Therapeutics. C.J.D. is an advisory board member for Anchiano Therapeutics, Deciphera Pharmaceuticals, and Mirati Therapeutics. C.J.D. has received research funding support from Deciphera Pharmaceuticals, Mirati Therapeutics, and SpringWorks Therapeutics. C.J.D. has consulted for Axon Advisors LLC, Eli Lilly, Jazz Therapeutics, Ribometrix, Sanofi, SVB Leerink, SmartAnalyst, Third Bridge, and Turning Point Therapeutics. A.D.C. has consulted for Eli Lilly, Mirati Therapeutics, and SpringWorks Therapeutics and has received research funding support from Mirati Therapeutics and SpringWorks Therapeutics.
Supplementary Material
References
- Cox A. D.; Fesik S. W.; Kimmelman A. C.; Luo J.; Der C. J. (2014) Drugging the undruggable RAS: Mission possible?. Nat. Rev. Drug Discovery 13 (11), 828–51. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Jemal A. (2020) Cancer statistics, 2020. Ca-Cancer J. Clin. 70 (1), 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Hobbs G. A.; Der C. J.; Rossman K. L. (2016) RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129 (7), 1287–92. 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen A. G.; Esposito D.; Bagni R. K.; McCormick F. (2014) Dragging ras back in the ring. Cancer Cell 25 (3), 272–81. 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Lukman S.; Hocker H. J.; Sayyah J.; Brown J. H.; McCammon J. A.; Gorfe A. A. (2011) Novel allosteric sites on Ras for lead generation. PLoS One 6 (10), e25711 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke B.; Der C. J. (2017) Drugging RAS: Know the enemy. Science 355 (6330), 1158–1163. 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- Waters A. M.; Der C. J. (2018) KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 8 (9), a031435. 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Chen L.; Tan X.; Crosby K.; Guimaraes A. R.; Upadhyay R.; Maira M.; McNamara K.; Perera S. A.; Song Y.; Chirieac L. R.; Kaur R.; Lightbown A.; Simendinger J.; Li T.; Padera R. F.; Garcia-Echeverria C.; Weissleder R.; Mahmood U.; Cantley L. C.; Wong K. K. (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14 (12), 1351–6. 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L. S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; Feng J.; Chen J. H.; Li S.; Li S.; Long Y. O.; Thach C.; Liu Y.; Zarieh A.; Ely T.; Kucharski J. M.; Kessler L. V.; Wu T.; Yu K.; Wang Y.; Yao Y.; Deng X.; Zarrinkar P. P.; Brehmer D.; Dhanak D.; Lorenzi M. V.; Hu-Lowe D.; Patricelli M. P.; Ren P.; Liu Y. (2018) Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172 (3), 578–589. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503 (7477), 548–51. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J.; Rex K.; Saiki A. Y.; Mohr C.; Cooke K.; Bagal D.; Gaida K.; Holt T.; Knutson C. G.; Koppada N.; Lanman B. A.; Werner J.; Rapaport A. S.; San Miguel T.; Ortiz R.; Osgood T.; Sun J. R.; Zhu X.; McCarter J. D.; Volak L. P.; Houk B. E.; Fakih M. G.; O’Neil B. H.; Price T. J.; Falchook G. S.; Desai J.; Kuo J.; Govindan R.; Hong D. S.; Ouyang W.; Henary H.; Arvedson T.; Cee V. J.; Lipford J. R. (2019) The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575 (7781), 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Hallin J.; Engstrom L. D.; Hargis L.; Calinisan A.; Aranda R.; Briere D. M.; Sudhakar N.; Bowcut V.; Baer B. R.; Ballard J. A.; Burkard M. R.; Fell J. B.; Fischer J. P.; Vigers G. P.; Xue Y.; Gatto S.; Fernandez-Banet J.; Pavlicek A.; Velastagui K.; Chao R. C.; Barton J.; Pierobon M.; Baldelli E.; Patricoin E. F. 3rd; Cassidy D. P.; Marx M. A.; Rybkin I. I.; Johnson M. L.; Ou S. I.; Lito P.; Papadopoulos K. P.; Janne P. A.; Olson P.; Christensen J. G. (2020) The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 10 (1), 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F. (2020) Sticking it to KRAS: Covalent Inhibitors Enter the Clinic. Cancer Cell 37 (1), 3–4. 10.1016/j.ccell.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J. Y.; Zhao Y.; Aronowitz J.; Mai T. T.; Vides A.; Qeriqi B.; Kim D.; Li C.; de Stanchina E.; Mazutis L.; Risso D.; Lito P. (2020) Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577 (7790), 421–425. 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; Xiong Y.; Safaee N.; Nowak R. P.; Donovan K. A.; Yuan C. J.; Nabet B.; Gero T. W.; Feru F.; Li L.; Gondi S.; Ombelets L. J.; Quan C.; Janne P. A.; Kostic M.; Scott D. A.; Westover K. D.; Fischer E. S.; Gray N. S. (2020) Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 27 (1), 19–31. 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Fire A.; Xu S.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391 (6669), 806–11. 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Yuan T. L.; Fellmann C.; Lee C. S.; Ritchie C. D.; Thapar V.; Lee L. C.; Hsu D. J.; Grace D.; Carver J. O.; Zuber J.; Luo J.; McCormick F.; Lowe S. W. (2014) Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discovery 4 (10), 1182–1197. 10.1158/2159-8290.CD-13-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot C. V.; Wu S. Y.; Bellister S.; Filant J.; Rupaimoole R.; Hisamatsu T.; Bhattacharya R.; Maharaj A.; Azam S.; Rodriguez-Aguayo C.; Nagaraja A. S.; Morelli M. P.; Gharpure K. M.; Waugh T. A.; Gonzalez-Villasana V.; Zand B.; Dalton H. J.; Kopetz S.; Lopez-Berestein G.; Ellis L. M.; Sood A. K. (2014) Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Ther. 13 (12), 2876–85. 10.1158/1535-7163.MCT-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W.; Dahlman J. E.; Tammela T.; Khan O. F.; Sood S.; Dave A.; Cai W.; Chirino L. M.; Yang G. R.; Bronson R.; Crowley D. G.; Sahay G.; Schroeder A.; Langer R.; Anderson D. G.; Jacks T. (2014) Small RNA combination therapy for lung cancer. Proc. Natl. Acad. Sci. U. S. A. 111 (34), E3553–61. 10.1073/pnas.1412686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S. J.; Revenko A. S.; Hanson L. L.; Ellston R.; Staniszewska A.; Whalley N.; Pandey S. K.; Revill M.; Rooney C.; Buckett L. K.; Klein S. K.; Hudson K.; Monia B. P.; Zinda M.; Blakey D. C.; Lyne P. D.; Macleod A. R. (2017) Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 9 (394), eaal5253. 10.1126/scitranslmed.aal5253. [DOI] [PubMed] [Google Scholar]
- Naito Y.; Yamada T.; Ui-Tei K.; Morishita S.; Saigo K. (2004) siDirect: highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Res. 32, W124–W129. 10.1093/nar/gkh442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancuceanu R.; Dinu M.; Furtunescu F.; Boda D. (2019) An inventory of medicinal products causing skin rash: Clinical and regulatory lessons. Exp. Ther. Med. 18 (6), 5061–5071. 10.3892/etm.2019.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen E.; Koivunen J. P. (2015) MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther. Adv. Med. Oncol. 7 (3), 170–80. 10.1177/1758834015571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improta G.; Zupa A.; Possidente L.; Tartarone A.; Pedicini P.; Nappi A.; Molinari S.; Fraggetta F.; Vita G. (2013) Coexistence of two different mutations in codon 12 of the Kras gene in colorectal cancer: Report of a case supporting the concept of tumoral heterogeneity. Oncol. Lett. 5 (5), 1741–1743. 10.3892/ol.2013.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo M. P.; Andrade L. D. B.; Coudry R.; Crespo R.; Gomes M.; Lisboa B. C. G.; Aguiar S.; Soares F. A.; Carraro D. M.; Cunha I. W. (2011) Multiple mutations in the Kras gene in colorectal cancer: review of the literature with two case reports. Int. J. Colorectal Dis 26 (10), 1241–1248. 10.1007/s00384-011-1238-0. [DOI] [PubMed] [Google Scholar]
- (2017) Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32 (2), 185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q.; Thonberg H.; Wang J.; Wahlestedt C.; Liang Z. (2005) A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res. 33 (5), 1671–7. 10.1093/nar/gki312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Der C. J.; Cox A. D. (2016) The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol. 58, 60–9. 10.1016/j.semcdb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zhang Z.; Dai Z.; Plass C.; Morrison C.; Wang Y.; Wiest J. S.; Anderson M. W.; You M. (2003) LOH of chromosome 12p correlates with Kras2 mutation in non-small cell lung cancer. Oncogene 22 (8), 1243–6. 10.1038/sj.onc.1206192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W.; Sahin F.; Iacobuzio-Donahue C. A.; Garcia-Carracedo D.; Wang W. M.; Kuo C. Y.; Chen D.; Arking D. E.; Lowy A. M.; Hruban R. H.; Remotti H. E.; Su G. H. (2011) Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2 (11), 862–73. 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staffas A.; Karlsson C.; Persson M.; Palmqvist L.; Bergo M. O. (2015) Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia 29 (5), 1032–40. 10.1038/leu.2014.315. [DOI] [PubMed] [Google Scholar]
- Balwani M.; Sardh E.; Ventura P.; Peiro P. A.; Rees D. C.; Stolzel U.; Bissell D. M.; Bonkovsky H. L.; Windyga J.; Anderson K. E.; Parker C.; Silver S. M.; Keel S. B.; Wang J. D.; Stein P. E.; Harper P.; Vassiliou D.; Wang B.; Phillips J.; Ivanova A.; Langendonk J. G.; Kauppinen R.; Minder E.; Horie Y.; Penz C.; Chen J.; Liu S.; Ko J. J.; Sweetser M. T.; Garg P.; Vaishnaw A.; Kim J. B.; Simon A. R.; Gouya L.; Investigators E. (2020) Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 382 (24), 2289–2301. 10.1056/NEJMoa1913147. [DOI] [PubMed] [Google Scholar]
- Adams D.; Gonzalez-Duarte A.; O’Riordan W. D.; Yang C. C.; Ueda M.; Kristen A. V.; Tournev I.; Schmidt H. H.; Coelho T.; Berk J. L.; Lin K. P.; Vita G.; Attarian S.; Plante-Bordeneuve V.; Mezei M. M.; Campistol J. M.; Buades J.; Brannagan T. H. 3rd; Kim B. J.; Oh J.; Parman Y.; Sekijima Y.; Hawkins P. N.; Solomon S. D.; Polydefkis M.; Dyck P. J.; Gandhi P. J.; Goyal S.; Chen J.; Strahs A. L.; Nochur S. V.; Sweetser M. T.; Garg P. P.; Vaishnaw A. K.; Gollob J. A.; Suhr O. B. (2018) Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 379 (1), 11–21. 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- Khvorova A.; Watts J. K. (2017) The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35 (3), 238–248. 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M.; Manoharan M. (2019) Re-Engineering RNA Molecules into Therapeutic Agents. Acc. Chem. Res. 52 (4), 1036–1047. 10.1021/acs.accounts.8b00650. [DOI] [PubMed] [Google Scholar]
- Borten M. A.; Bajikar S. S.; Sasaki N.; Clevers H.; Janes K. A. (2018) Automated brightfield morphometry of 3D organoid populations by OrganoSeg. Sci. Rep. 8 (1), 5319. 10.1038/s41598-017-18815-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.