

Abstract
Serotonergic psychedelics are defined as compounds having serotonin 2A receptor (5-HT2AR) activation as an important pharmacological mechanism. These compounds include the phenylalkylamine class, containing substances with e.g. 2C-X structures (phenethylamines) or their N-methoxybenzyl analogues (NBOMes). Besides their abuse potential, psychedelics are increasingly recognized for having therapeutic benefits. However, many psychedelics remain incompletely characterized, even concerning their structure–activity relationships. Here, five positional isomers of 25H-NBOMe, with two methoxy groups on the different positions of the phenyl ring of the phenethylamine moiety, were subjected to split-nanoluciferase assays assessing the in vitro recruitment of cytosolic proteins to the 5-HT2AR. Furthermore, molecular docking at the 5-HT2AR allowed estimation of which residues interact with the specific isomers’ methoxy groups. Although the optimal substitution pattern of N-unsubstituted phenylalkylamines has been extensively studied, this is the first comparative evaluation of the functional effects of the positioning of the methoxy groups in the phenethylamine moiety of NBOMes.
Keywords: bioassay, structure−activity relationship, serotonin receptor, molecular docking, psychedelic, new psychoactive substances
Serotonergic psychedelics are defined as substances that have activation of the serotonin 2A receptor (5-HT2AR) as a main pharmacological mechanism.1 Within the group of serotonergic psychedelics, substances displaying a broad structural variety can be retrieved, both traditionally known substances and new psychoactive substances (NPS), belonging to three different subgroups: ergolines (such as the prototypical psychedelic substance LSD, lysergic acid diethylamide), tryptamines (such as psilocybin), and phenylalkylamines (such as the naturally occurring mescaline).2 Within the latter group, compounds can be categorized into subgroups, among which are 2C-X (phenethylamines), DOx (phenylisopropylamines), and 25X-NBOMes (N-benzyl derivatives of the 2C-X substances).3 Serotonergic psychedelics comprise a substantial portion of the 950 individual NPS that had been cumulatively reported by the beginning of 2020.4
The complex mechanism of action of psychedelics has caused this group of substances to be incompletely characterized. On the one hand, psychedelic substances are consumed for the induction of mystical experiences, empathic feelings, and alterations in consciousness, potentially resulting in severe side effects such as agitation, hyperthermia, rhabdomyolysis, and even death.1,5,6 On the other hand, psychedelics are increasingly recognized for their potential therapeutic effects. This translates into clinical trials for the treatment of addictions, mood and anxiety disorders, and for the relief of distress concerning death, mainly with LSD and psilocybin.7
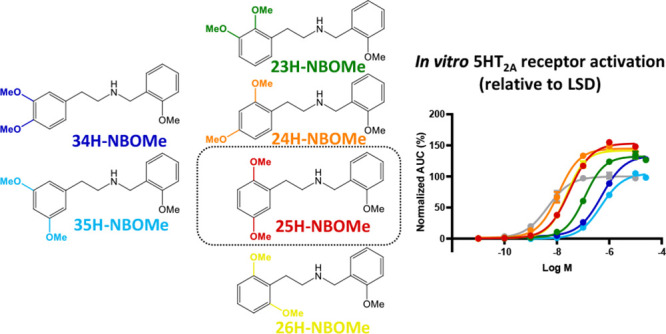
Substantial efforts have been invested in the characterization of psychedelic substances, their structure–activity relationships, and their receptor interaction(s). Very recently, structural data became available for the 5-HT2AR interacting with different ligands.8,9 Interestingly, these structures hint at differential binding modes for the prototypical psychedelic substance LSD and the N-benzyl substituted phenethylamine 25CN-NBOH, a substance highly similar to those in the recently emerging NBOMe class.8 Despite extensive data on several aspects of the structure–activity relationship of phenylalkylamine psychedelics, an in-depth examination of the influence of the position of the methoxy groups on the phenethylamine moiety in NBOMes, while leaving the N-methoxybenzyl group intact, has remained unexplored.2 In this study, we compared the potential of a set of five isomers of 25H-NBOMe (of which the structures are given in Figure 1) to induce similar effects to the “conventional” 25X-NBOMes at a molecular level. In the name of the positional isomers, the number (i.e., 25, 23, 24, 26, 34, and 35) indicates the positions at which methoxy groups were introduced, e.g. 25H-NBOMe has methoxy groups at positions 2 and 5 of the phenyl ring of the phenethylamine moiety of the molecule. Recently, the synthesis and spectral characterization of these five positional isomers of 25H-NBOMe has been described.10 Functional characterization was performed by assessing the potential of these isomers to induce recruitment of cytosolic proteins to the activated 5-HT2AR. To this end, we employed previously established bioassays, expressing either β-arrestin 2 (βarr2) or miniGαq with the 5-HT2AR in the NanoBiT system, in which a split nanoluciferase is functionally complemented following recruitment of the cytosolic protein to the activated 5-HT2AR.11,12 Additionally, a molecular model was used, based on adaptations of the above-mentioned published cryo-EM structure,8 for the molecular docking of each of the tested substances into the 5-HT2AR, suggesting interactions between the methoxy groups on different positions and specific receptor residues.
Figure 1.

Structures of the substances tested in the 5-HT2AR bioassays: the reference substances LSD and serotonin and the “conventionally substituted” 25H-NBOMe, together with the five tested positional isomers. The numbers in the name of the positional isomer correspond to the methoxy group positions in the phenethylamine fragment, e.g. 25H-NBOMe has one methoxy group at position 2 and one methoxy group at position 5.
Pharmacological Characterization
To assess the functionality of the NBOMe positional isomers, the Nanoluciferase Binary Technology (NanoBiT) was employed. This technique was specifically developed for the real-time monitoring of protein–protein interactions in live cells. To this end, two nonfunctional parts of the nanoluciferase are each fused to one of the potentially interacting proteins, in this case the 5-HT2AR and a cytosolic protein. When an agonist activates the receptor, the cytosolic protein is recruited, leading to association of the split parts, which can be monitored via a luminescent readout in the presence of the enzyme’s substrate.13 We previously established a bioassay to monitor the recruitment of βarr2, later complemented with an analogous miniGαq recruitment assay, allowing us to generate concentration–response curves.11,12 These can be used to derive the potency and efficacy of the test compounds, as compared to a reference agonist, of which the efficacy is arbitrarily set at 100%. For the functional characterization of the described positional isomers, several conditions were taken into consideration to exclude the possibility of making presumptuous conclusions based on the assay chosen, the method of analysis (with kinetic implications), or the included reference agonist. Therefore, besides performing the analyses with our standard βarr2 recruitment assay with LSD as a reference agonist, we also conducted a miniGαq recruitment assay. In both assays, both serotonin and LSD were included in all experiments as reference agonists. Additionally, data analysis was conducted using the area under the curve (AUC) of either the first 30 min of the real-time activation profile or the full (standard) 2 h activation profile. The EC50 (as a measure of potency) and Emax (as a measure of efficacy) values for all assessed conditions are summarized in Tables 1 (βarr2) and 2 (miniGαq). Figure 2 provides concentration–response curves for all substances, calculated for the 2 h activation profile, with either LSD (A and B) or serotonin (C and D) as the reference agonist.
Table 1. Summary of the Potency (EC50) and Efficacy (Emax, Where the Emax of Either Serotonin or LSD is Set to 100%) Values of All the Tested Compounds in the βarr2 Assay, Calculated Using Either the Full 120 Min or the First 30 Min Activation Profilesa.
| 120 min activation profiles | ||||
|---|---|---|---|---|
| reference
agonist LSD |
reference agonist serotonin |
|||
| compound | EC50 (nM, CI) | Emax (%, CI) | EC50 (nM, CI) | Emax (%, CI) |
| LSD | 7.43 (3.54–12.9) | 100 (91–109) | 7.29 (3.50–12.7) | 87.1 (79.4–95.4) |
| serotonin | 7.62 (4.43–13.2) | 116 (107–126) | 7.24 (4.43–11.9) | 100 (93.9–108) |
| 23H-NBOMe | 33.6 (19.8–57.3) | 123 (115–131) | 33.7 (19.7–57.9) | 115 (107–123) |
| 24H-NBOMe | 3.88 (2.33–6.37) | 145 (136–154) | 3.89 (2.30–6.45) | 136 (127–145) |
| 25H-NBOMe | 11.4 (6.36–20.4) | 164 (151–180) | 11.0 (5.60–21.5) | 134 (121–148) |
| 26H-NBOMe | 8.70 (5.81–12.5) | 156 (147–165) | 8.74 (5.91–12.4) | 146 (138–154) |
| 34H-NBOMe | 248 (111–646) | 147 (129–182) | 238 (101–691) | 120 (104–152) |
| 35H-NBOMe | 343 (187–645) | 118 (107–135) | 310 (165–593) | 94.1 (83.8–108) |
| 30 min activation profiles | ||||
|---|---|---|---|---|
| reference agonist LSD |
reference agonist serotonin |
|||
| compound | EC50 (nM, CI) | Emax (%, CI) | EC50 (nM, CI) | Emax (%, CI) |
| LSD | 24.3 (ND) | 100 (91–110) | 23.8 (ND) | 79.4 (72.0–87.3) |
| serotonin | 11.5 (7.13–18.9) | 128 (119–139) | 10.9 (7.60–15.8) | 100 (94.8–106) |
| 23H-NBOMe | 74.4 (55.8–95.9) | 133 (128–139) | 74.5 (53.9–98.8) | 114 (109–119) |
| 24H-NBOMe | 8.34 (6.11–11.1) | 154 (147–161) | 8.32 (5.85–11.5) | 132 (125–139) |
| 25H-NBOMe | 24.3 (13.4–45.0) | 179 (163–199) | 22.9 (12.8–41.8) | 130 (118–143) |
| 26H-NBOMe | 16.3 (13.0–20.8) | 165 (159–172) | 16.4 (12.4–22.0) | 142 (135–149) |
| 34H-NBOMe | 488 (226–1707) | 162 (142–218) | 458 (191–2287) | 118 (101–171) |
| 35H-NBOMe | 678 (385–1295) | 137 (123–162) | 625 (353–1148) | 98.5 (88.8–115) |
Table 2. Summary of the Potency (EC50) and Efficacy (Emax, Where the Emax of Either Serotonin or LSD is Set to 100%) Values of All the Tested Compounds in the miniGαq Recruitment Assay, Calculated Using Either the Full 120 Min or the First 30 Min Activation Profilesa.
| 120 min activation profiles | ||||
|---|---|---|---|---|
| reference agonist LSD |
reference agonist serotonin |
|||
| compound | EC50 (nM, CI) | Emax (%, CI) | EC50 (nM, CI) | Emax (%, CI) |
| LSD | 7.40 (1.82–17.7) | 100 (84–115) | 7.23 (1.73–17.5) | 39.6 (33.7–46.0) |
| serotonin | 66.4 (35.5–151) | 256 (230–301) | 63.5 (33.9–143) | 103 (92.5–121) |
| 23H-NBOMe | 147 (71.8–436) | 81.2 (70–102) | 146 (73.8–408) | 31.9 (27.5–39.6) |
| 24H-NBOMe | 22.1 (12.1–42.1) | 144 (129–161) | 22.2 (12.4–41.6) | 56.9 (51.2–63.4) |
| 25H-NBOMe | 49.6 (27.2–84.8) | 141 (128–155) | 49.2 (26.6–85.4) | 58.8 (53.3–64.9) |
| 26H-NBOMe | 40.9 (23.7–68.2) | 136 (124–148) | 40.9 (23.9–67.7) | 53.3 (48.8–58.2) |
| 34H-NBOMe | 974 (ND) | 134 (ND) | 947 (ND) | 55.3 (45.6–297) |
| 35H-NBOMe | 1097 (ND) | 89.1 (74–219) | 1087 (ND) | 37.1 (30.5–112) |
| 30 min activation profiles | ||||
|---|---|---|---|---|
| reference
agonist LSD |
reference agonist serotonin |
|||
| compound | EC50 (nM, CI) | Emax (%, CI) | EC50 (nM, CI) | Emax (%, CI) |
| LSD | 17.7 (10.1–38.6) | 98.2 (85–114) | 16.3 (8.59–38.7) | 31.9 (27.4–37.2) |
| serotonin | 88.5 (46.1–213) | 305 (272–365) | 62.2 (22.5–269) | 87.9 (74.4–121) |
| 23H-NBOMe | 236 (ND) | 89.6 (75–112) | 191 (ND) | 27.1 (22.6–33.3) |
| 24H-NBOMe | 37.4 (20.6–66.4) | 171 (155–189) | 35.1 (17.5–68.7) | 54.3 (48.5–61.2) |
| 25H-NBOMe | 86.2 (57.0–124) | 181 (167–196) | 129 (69.2–263) | 74.8 (65.6–88.8) |
| 26H-NBOMe | 68.0 (38.6–115) | 162 (147–181) | 68.3 (34.0–129) | 51.9 (46.0–59.4) |
| 34H-NBOMe | 1321 (629–33 780) | 156 (130–340) | 1334 (ND) | 51.8 (ND) |
| 35H-NBOMe | 1393 (ND) | 107 (94–163) | 1296 (683–10 760) | 48.3 (41.0–83.6) |
Data are from three independent experiments. CI: 95% confidence interval. ND: confidence interval not determined.
Figure 2.

Concentration–response curves obtained by stimulation of the 5-HT2AR followed by recruitment of (A and C) βarr2 or (B and D) miniGαq constructs in the NanoBiT system. Data points are given as the mean of three independent experiments (each performed in duplicate) ± SEM (standard error of the mean). The AUC is normalized in each independent experiment for the maximum response (100%) of the reference agonist LSD (A and B) or serotonin (C and D).
As a “standard” setup, we routinely apply the βarr2 recruitment assay with LSD as a reference agonist and using the 2 h activation profiles for the calculation of the AUC. Table 1 and the corresponding Figure 2A readily show that there are substantial differences in the potency and efficacy of the compounds that are diversely substituted. The “conventionally substituted” 25H-NBOMe serves as the point of comparison for the diversely substituted isomers, yielding a low nanomolar potency (an EC50 value of 11.4 nM) in the bioassay and a high efficacy relative to LSD (an Emax of 164%). Only one of the isomers is slightly more potent than 25H-NBOMe: 24H-NBOMe yielded an EC50 value of 3.88 nM in the βarr2 recruitment assay. The potency of 26H-NBOMe (8.70 nM) is similar to that of 25H-NBOMe. The highest EC50 value (33.6 nM) of all isomers with a 2-methoxy substituent, and hence the lowest potency, was observed for 23H-NBOMe. The least potent substances in the βarr2 recruitment assay appeared to be 34H-NBOMe and 35H-NBOMe, both lacking the 2-methoxy substituent. This omission resulted in a markedly reduced in vitro potency, with EC50 values in the higher nanomolar range (248 and 343 nM, respectively). Concerning the efficacies of the compounds, a more narrow range is obtained. 25H-NBOMe appears to have the highest efficacy (164%) of all tested substances, although its 95% confidence interval is overlapping with those of 24H-, 26H-, and 34H-NBOMe, with efficacies of 145, 156, and 147%, respectively. Although the efficacies obtained for 23H-NBOMe (123%) and 35H-NBOMe (118%) were lower than that of 25H-NBOMe, it is remarkable that all substances could still be categorized as (at least) equally efficacious as LSD and serotonin.
The potency for the reference psychedelic substance LSD, yielding an EC50 value of 7.43 nM, closely corresponds with our previously reported values (5.95 nM, 5.96 nM, 6.41 nM and 5.95 nM), obtained by measuring βarr2 recruitment to the 5-HT2AR in the NanoBiT system.11,12,14
Also when using serotonin as a reference and/or when using a 30 min rather than a 2 h read-out for the βarr2 recruitment assay (Table 1), the derived potency values for the different substances yielded the same conclusions as those from our “standard” setup. Despite an upward shift in the absolute EC50 values when using a shorter read-out time, the ranking order of potencies of the assessed isomers remained unaltered, irrespective of whether serotonin or LSD was used as the reference agonist, and whether the 30 min or 2 h AUC was employed for calculation. A change in reference agonist or analysis time also did not yield different conclusions when considering the efficacies of the different isomers. With a single exception (35H-NBOMe; Emax 94.1 and 98.5 %, with serotonin as a reference), the efficacies of all different isomers superseded those of both LSD and serotonin. All methods of analysis classified 24H-, 25H-, 26H-, and 34H-NBOMe as the more efficacious substances and 23H- and 35H-NBOMe as the less efficacious substances.
As different assays could potentially lead to different outcomes, additionally, a different but highly analogous bioassay was taken, monitoring the recruitment of miniGαq (of which the engineering was described by Nehmé et al.15) to the 5-HT2AR in the NanoBiT system, as previously described.12 A first prominent observation here is that in this assay setup, the efficacy of serotonin exceeds that of LSD 2.5- to 3-fold. This finding matches the description of LSD as a less efficacious 5-HT2AR agonist than serotonin.1,16−18 When comparing the results in Table 2 with those in Table 1, and with each other, an upward shift in EC50 values can be observed, and when considering serotonin as a reference, a downward shift in the Emax of all NBOMes is observed, with the efficacy of none of the NBOMes exceeding that of serotonin in this assay format. Yet, despite these global differences between both assays, comparative analysis of the different NBOMe isomers revealed that, also here, the rank order of potencies remained unaltered, as did the classification of the isomers into more and less efficacious substances. Interestingly, the differences in efficacies were somewhat more pronounced in this assay format; the efficacy of 24H-, 25H-, 26H-, and 34H-NBOMe consistently exceeded that of LSD, while that of 23H-NBOMe was consistently lower. 35H-NBOMe also showed a trend toward efficacy values near to or lower than that of LSD, although its low potency did not allow the setup of ideal concentration–response curves.
Overall, this extensive set of experiments led us to conclude that neither the functional assay used, nor the reference agonist, nor the time point of analysis influenced the conclusions drawn for this set of NBOMe positional isomers, i.e. that 24H-NBOMe is the most potent positional isomer, followed by 26H- and 25H-NBOMe, with a lower potency for 23H-NBOMe, and even lower potencies for 34H and 35H-NBOMe. Smaller differences were observed between the isomers in terms of efficacy, classifying 25H-, 26H-, 24H-, and 34H-NBOMe as the more efficacious substances and 23H- and 35H-NBOMe as the less efficacious substances in the set.
Little to no pharmacological information is available on the tested positional isomers, with four of these (23H-, 26H-, 34H-, and 35H-NBOMe) never having been functionally evaluated before.10 Rickli et al. reported a potency value of 490 nM for 25H-NBOMe, employing a FLIPR assay, while Eshleman et al. obtained an EC50 value of 40 nM by measuring inositol-phosphate 1 formation.16,17 Braden et al. found a slightly increased potency for 24H-NBOMe (4.0 nM) as compared to 25H-NBOMe (15.3 nM), employing an inositol phosphate accumulation assay, with similar efficacies for the two compounds.18 From this limited amount of pharmacological data, and as also confirmed by our experiments, it is clear that the use of different signaling events as a readout method can severely impact the obtained numbers, thereby hampering straightforward interpretation and comparability of results obtained in different assays. However, apart from the findings obtained with the FLIPR assay, our findings and the order of magnitude of the obtained potency values correspond with the literature.
The optimal substitution pattern of the phenyl group of the N-unsubstituted phenylalkylamine psychedelics has been studied extensively, both in vitro and in vivo. In PiHKAL, the most-explored patterns are 2,4,5-substitution of the phenyl ring and 3,4,5-substitution, with the former being concluded to be the most effective. It has been suggested that the 2,5-dimethoxy pattern is to be kept intact for optimal psychedelic activity as well as receptor activity and affinity, while the substituent at position 4 can be modified. Furthermore, it is hypothesized that the 2,4,6-substitution pattern could involve potentially active substances. On the other hand, receptor mutation studies have suggested that the 5-substituent is of lesser importance for phenethylamines than for phenylisopropylamines. This emphasizes the dramatic impact of the position of the methoxy groups on the psychedelic activity and receptor binding/activation of an individual substance.2,19,20 In a recent study, the 2- and 5-desmethoxy analogues of 2C–B and DOB were individually tested both in an in vitro Ca2+ release assay and in vivo through the head twitch response in mice. This study reported modestly reduced binding affinities and functional potencies at the 5-HT2AR and 5-HT2CR, with the removal of the 2-methoxy group having a more severe impact than that of the 5-methoxy group. Removal of either, however, appears to have a more dramatic impact on the in vivo head twitch response.21 Similarly, the employed in vitro recruitment assays also hint at the importance of the 2-methoxy group for strong activation of the 5-HT2AR in the group of NBOMes. Not only do we find markedly decreased potencies for the 34H- and 35H-NBOMe isomers, we also find that 24H- and 26H-NBOMe are (at least) equally as potent as 25H-NBOMe with, in comparison, a decreased potency of 23H-NBOMe. This underscores the importance of the 2-methoxy group and, at the same time, suggests that a substituent at position 3 on the phenyl group of the phenethylamine moiety reduces in vitro receptor activation, or that the introduction of a methoxy group at the position next to position 2 may negatively impact the positive effect of that methoxy group. Distinct interactions of the distinctly substituted NBOMes at the 5-HT2AR are indeed supported by molecular docking data, as discussed further.
While multiple reports are available on the substitution pattern of the phenyl group of N-unsubstituted phenylalkylamine psychedelics (e.g., 2C-X and DOx), this effect is largely unstudied in their N-methoxybenzyl counterparts. The introduction of a methoxybenzyl group at the N-position of the phenethylamine results in the NBOMe group of substances, with higher affinities and potencies reported for 25H-NBOMe than for the unsubstituted counterpart 2C–H.2,14,17,18 Literature indicates that the 4-substitution, albeit very important for the activity of 2C-X and DOx substances, is not as essential for the receptor activation by NBOMes.3,14 Furthermore, in an attempt to reduce the high first-pass metabolism of NBOMes, several substitutions of the methoxy group at position 5, reportedly the metabolic “soft spot” of NBOMes, have been explored. When assessing the functionality of these molecules, it was concluded that the 2,5-dimethoxy motif is not necessarily as imperative for the in vitro functionality as it was in the N-unsubstituted psychedelics, finding reduced affinity but comparable potency and efficacy in a Ca2+/Fluo-4 assay upon omission of the 5-methoxy group of 25B-NBOMe.22 Our findings are consistent with the aforementioned literature, as changing the position of the 5-methoxy group in additionally 2-methoxy substituted NBOMes does not necessarily result in severely altered potencies and efficacies, at least in vitro. In addition, in this study, we find the 2,4-dimethoxy pattern to result in a higher potency and similar efficacy as the 2,5-dimethoxy pattern in NBOMes with no further phenethylamine moiety substituents.
Molecular Docking
Attempting to provide an explanation for the differences in potencies/efficacies observed for the isomers, molecular docking was performed, with a model based on adaptations of the recently published cryo-EM structure of the 5-HT2AR in complex with 25CN-NBOH (PDB: 6WHA).8Figure 3A shows the position of 24H-NBOMe in the orthosteric binding pocket of the receptor, in which all of the isomers bind. Figure 3B and 3C focus on the proposed interactions of the methoxy groups on the phenethylamine moiety of 24H-NBOMe and 35H-NBOMe with the amino acid residues of the receptor, providing insight into the modeled interaction of specific residues with the methoxy groups. The analogous figures for the other isomers and 25CN-NBOH are provided in Supplementary Data. Tables 3 and 4 show the calculated interaction energies between the molecules and specific 5-HT2AR residues, with the latter specifically focusing on the interactions of the methoxy groups on the phenethylamine moiety of the isomers. The values in these Tables reflect the model-derived strength of the interactions between a certain amino acid residue of the binding pocket and the respective ligand. The more negative the given energy, the stronger the proposed interaction would be. These values are obtained through Prime MM-GBSA (Molecular Mechanics-Generalized-Born Surface Area), a tool for which the calculated values have previously been shown to provide a good estimate for the relative binding affinities of a set of ligands.23,24 The model showed that all NBOMe isomers can be docked in the same binding pocket as 25CN-NBOH, despite the change of the hydroxyl group on the N-benzyl moiety into a methoxy group. This latter group specifically stabilizes the molecule through an H-bond with S1593.36 (as also reflected by the interaction energies, lying between −8.5 and −11.5 kcal/mol), and through hydrophobic interaction with S1593.36, W3366.48 and S3737.46. The nitrogen atom is observed to form a salt bridge with D1553.32, which has been previously defined as critical for receptor interaction.8 The strongly negative values in Table 3 (between −18 and −20 kcal/mol) are indeed consistent with a strong salt bridge-type interaction with D1553.32. The N-benzyl moiety is stabilized in the binding pocket by hydrophobic interactions with W3366.48 and F3396.51, and both the N-benzyl moiety and the phenyl group of the phenethylamine function are stabilized by interaction with F3406.52. Additionally, the strongly negative values obtained for F3396.51 and F3406.52 confirm the strength of this interaction, consistent with pi stacking interactions. The reference compound LSD interacts with S2425.46 stronger than NBOMe compounds due to an H-bond between the indole on LSD and S2425.46, in which NBOMes cannot participate (Table 4).
Figure 3.

Visual representations of the NBOMe isomers docked into the binding pocket of the 5-HT2AR (based on PDB: 6WHA).8 (A) 24H-NBOMe, as seen from the perspective of the N-benzyl moiety; (B) 24H-NBOMe and (C) 35H-NBOMe bound to the 5-HT2AR looking from the perspective of the phenyl group, with specific mentioning of the residues proposed to interact with the methoxy groups on the phenethylamine moiety.
Table 3. Summary of the Interaction Energies (kcal/mol) between Compounds and the 5-HT2AR.
| compound | S1593.36 | D1553.32 | V1563.33 | W3366.48 | F3396.51 | F3406.52 |
|---|---|---|---|---|---|---|
| LSD | –3.50 | –18.5 | –13.5 | –2.30 | –10.49 | –6.39 |
| 25CN-NBOH | –9.34 | –18.3 | –10.7 | –7.30 | –8.76 | –7.73 |
| 23H-NBOMe | –11.5 | –19.8 | –11.3 | –9.06 | –9.41 | –8.72 |
| 24H-NBOMe | –9.86 | –19.5 | –11.2 | –8.66 | –9.35 | –8.36 |
| 25H-NBOMe | –9.90 | –19.3 | –11.0 | –8.58 | –9.01 | –7.54 |
| 26H-NBOMe | –9.77 | –20.7 | –12.9 | –8.70 | –9.16 | –7.89 |
| 34H-NBOMe | –8.59 | –20.7 | –10.6 | –8.62 | –9.30 | –8.39 |
| 35H-NBOMe | –8.72 | –20.49 | –10.1 | –8.54 | –10.27 | –7.25 |
Table 4. Summary of the Interaction Energies (kcal/mol) Involved in the Proposed Interaction between the Methoxy Groups on the Phenyl Ring and Nearby Amino Acid Residues on the 5-HT2AR.
| compound | T1603.37 | G2385.42 | S2425.46 | S2395.43 | V2355.39 | N3436.55 | L229ECL2 |
|---|---|---|---|---|---|---|---|
| LSD | –1.25 | –1.54 | –6.46 | –4.01 | –2.28 | –5.02 | –9.91 |
| 25CN-NBOH | –2.38 | –3.07 | –1.90 | –2.13 | –0.68 | –1.69 | –2.98 |
| 23H-NBOMe | –2.77 | –2.04 | –4.86 | –1.29 | –0.57 | –0.99 | –0.56 |
| 24H-NBOMe | –2.14 | –1.07 | –1.74 | –2.18 | –3.38 | –2.25 | –1.13 |
| 25H-NBOMe | –2.43 | –0.82 | –1.81 | –0.59 | –0.93 | –1.61 | –2.25 |
| 26H-NBOMe | –2.51 | –0.93 | –1.70 | –0.60 | –0.58 | –0.73 | –1.76 |
| 34H-NBOMe | –0.47 | –2.69 | –3.23 | –2.29 | –3.35 | –0.95 | –1.09 |
| 35H-NBOMe | –0.21 | –2.26 | –2.57 | –1.46 | –1.60 | –3.18 | –2.46 |
Because of the ability of the utilized model to predict to a reasonable extent the interactions of LSD and 25CN-NBOH with the 5-HT2AR, as described by Kim et al.,8 it was additionally used to assess the interactions with the methoxy groups of the phenethylamine moiety of the panel of NBOMe positional isomers. In contrast to the energies presented in Table 3, the interaction energies in Table 4, representing the interactions of the respective methoxy groups with residues T1603.37, G2385.42, S2395.43, S2425.46, V2355.39, N3436.55, and L229ECL2, indicate relatively weak interactions with the isomers. This Table, however, shows strong interaction energies between LSD and receptor residues S2425.46 and L229ECL2, consistent with observations by Kim et al.8 While it is not straightforward to speculate on the significance of the interaction energies between the receptor residues and the different isomers, as depicted in Table 4, some patterns do seem to emerge. When all substances carrying a methoxy group at a certain position have a stronger interaction energy (more negative value), this suggests an interaction between the methoxy group at that position and the concerned receptor residue. Important here is to additionally consider the steric effects that are relevant when two methoxy groups are placed on adjacent positions (e.g., 23H-NBOMe and 34H-NBOMe). Overall, this approach allows to propose an explanation for certain trends in interaction energies. A hydrophobic interaction between a methoxy group on position 2 of the phenyl ring of the phenethylamine moiety (as is the case in 23H-, 24H-, 25H-, and 26H-NBOMe) with T1603.37, S1593.36, and V1563.33 is proposed. The model also indicates hydrophobic interactions between G2385.42 and S2425.46 and a methoxy group at position 3, as reflected by a weaker interaction of these residues with substances lacking a 3-methoxy group. For 25CN-NBOH, G2385.42 would interact with the cyano group rather than with a methoxy group through a weak electrostatic interaction. The occurrence of a methoxy group at position 4 appears to be linked to a stronger hydrophobic interaction (lower interaction energies) with residues S2395.43 and V2355.39. Overall, substances containing a 5-methoxy group show a relatively stronger hydrophobic interaction with residues N3436.55 and L229ECL2 than substances lacking that group, with the exception of 24H-NBOMe, where the 4-methoxy group may also interact with N3436.55. However, the obtained values for these latter two residues indicate substantially weaker interactions with these residues than those observed for LSD. Lastly, a methoxy group at position 6 appears to contribute to the already strong interaction with V1563.33 and D1553.32 and weakly interacts with L229ECL2. Based on the interactions proposed here, it is clear that there is no trivial or “single-residue” explanation for the observed lower potencies and efficacies of 23H-, 34H-, and 35H-NBOMe, suggesting a concerted impact of the interaction with several residues. It must also be taken into consideration that this model was adapted from a miniGαq bound 5-HT2AR structure and that the presence of βarr2 could differentially impact ligand interaction. Within the context of this study, it was not possible to generate binding data; however, such data may help to explain some of the observed differences in receptor activation by different positional isomers.
Even though in vitro data on receptor activation provide valuable information on newly synthesized substances, the extrapolation to the in vivo effects in humans remains difficult. On the one hand, the actual mechanism inducing psychedelic effects (on a molecular level) remains elusive. Contributing factors could involve biased agonism, receptor dimerization, and activation of receptors other than the 5-HT2AR.25,26 Several additional factors besides receptor activation need to be taken into account. An example of such a factor is the potential (first pass) metabolism of these substances, as NBOMes are prone to a high intrinsic clearance. The 5-methoxy group of NBOMes has been defined to be the metabolic soft spot of the molecule, and the omission of this group was found to decrease the intrinsic clearance. The effect of the reintroduction of this methoxy group at a different position has not been assessed.22,27 Additionally, the potencies and efficacies reported here only reflect the ability of the compounds to recruit certain cytosolic proteins to the receptor. From these data, it is not possible to derive receptor binding affinities of the substance, because efficacy and potency do not necessarily correlate with the affinity. It is therefore theoretically possible that certain isomers would have higher receptor affinities than others, but would yield a “less effective” receptor configuration, and thus a lower potency and/or efficacy for a given pathway. In addition, it remains elusive how the methoxy groups on different positions of the phenyl group of the phenethylamine moiety of NBOMes will impact the pharmacokinetic and pharmacodynamic properties of the molecules in vivo. Besides that, the use of artificial systems such as cell lines expressing assay components, as used here, may cause another layer of difficulty in the interpretation of obtained in vitro results.28
In conclusion, we report on the functional characterization of a set of positional isomers of 25H-NBOMe, in which the methoxy groups are placed in different positions of the phenethylamine moiety. More specifically, the impact of the structural changes on the ability of the activated 5-HT2AR to recruit cytosolic proteins was assessed. To this end, bioassays monitoring the recruitment of either βarr2 or miniGαq to the 5-HT2AR were employed, yielding a luminescent readout upon functional complementation of the assay components. The results show clear differences in the EC50 (as a measure of potency) and Emax (as a measure of efficacy) values of the differentially substituted isomers. Overall, the isomers with a methoxy group on position 2 were more potent than those that did not have this substituent, with 24H-NBOMe being the most potent substance tested, and 23H-NBOMe being slightly less potent than 25- and 26H-NBOMe. The two isomers lacking the 2-methoxy group, 34- and 35H-NBOMe, were substantially less potent than the others. In terms of efficacy, the differences are less evident, with 23H- and 35H-NBOMe being markedly less efficacious than all other isomers, although they remained approximately as efficacious as LSD. The results showed that nor the reference agonist, nor the assay employed, nor the method of analysis influenced these findings. Data obtained from molecular docking of these substances into a 5-HT2AR model suggest specific residues that interact with the specific methoxy groups on the phenyl moiety of the phenethylamine part of the molecule which, in a concerted manner, result in the observed differential receptor activation potential of the differently substituted isomers. While the methoxy pattern on the phenylalkylamine moiety has been described extensively for N-unsubstituted psychedelics, this is the first report comparatively assessing the functional effects of this isomerization in their NBOMe counterparts.
Materials and Methods
Chemicals and Reagents
Dulbecco’s modified Eagle’s medium (DMEM, supplemented with GlutaMAX), Hank’s Balanced Salt Solution (HBSS), and penicillin/streptomycin (10 000 IU/mL and 10 000 μg/mL) were purchased from Thermo Fisher Scientific (Pittsburgh, PA, United States). The FuGENE HD Transfection Reagent, Nano-Glo Live Cell reagent, and the Nano-Glo LCS Dilution buffer were procured from Promega (Madison, WI, United States). Fetal bovine serum (FBS), poly-d-lysine hydrobromide, methanol, and the analytical standards of LSD (lysergic acid diethylamide) and serotonin were bought from Sigma-Aldrich (Overijse, Belgium). The analytical standard of 25H-NBOMe hydrochloride 2-(2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethanamine was from Chiron AS (Trondheim, Norway). The positional isomers of 25H-NBOMe, depicted in Figure 1, were synthesized as described previously and dissolved in methanol.10
Cell Culture and the 5-HT2AR Activation Assays
Human Embryonic Kidney (HEK) 293T cells were routinely cultured in DMEM (GlutaMAX, supplemented with 10% heat-inactivated FBS, 100 IU/mL of penicillin, 0.25 μg/mL amphotericin B and 100 μg/mL streptomycin) in a humidified atmosphere of 37 °C and 5% CO2. For the 5-HT2AR activation assays, the cells were seeded in 6-well plates at a density of 500 000 cells per well, following the protocol as described before.11,12 Following overnight incubation, the cells were transfected with 1.65 μg of both the 5-HT2AR and the cytosolic protein (either βarr2 or the miniGαq protein) in the NanoBiT system (NanoLuc Binary Technology), employing FuGENE in a 3:1 FuGENE:DNA ratio, according to the manufacturer’s protocol. After 24 h, the cells were seeded in poly-d-lysine coated 96-well plates at a density of 50 000 cells per well and incubated overnight. The cells were then rinsed twice with HBSS, and 100 μL of HBSS was added to each of the wells. To this, 25 μL of Nano-Glo Live Cell Substrate was added (diluted 1/20 in the Nano-Glo LCS Dilution buffer, according to the manufacturer’s protocol), and the plate was transferred to the Tristar2 LB 942 multimode microplate reader (Berthold Technologies GmbH & Co, Germany). After the equilibration phase, 10 μL of the 13.5× concentrated agonist solution was added, and the luminescent signal was monitored for 2 h. Each experiment was performed in duplicate with at least three independent experiments per compound. For the purpose of normalization of the data, on each 96-well plate, a concentration curve was run of the reference agonists LSD and serotonin. Appropriate solvent controls were included per condition.
Data Processing and Analysis
The obtained time-luminescence profiles were corrected for interwell variability and used for the calculation of the AUC, using either the full activation profiles of 2 h or only the first 30 min, as described previously in more detail.29 After subtraction of the AUC of the solvent control, the data were used for the fitting of a sigmoidal concentration–response curve through the four-parametric nonlinear regression model in GraphPad Prism software (San Diego, CA, United States). For each separate experiment, the data were normalized to the maximal response of the reference agonist, either LSD or serotonin, alternately set at 100% for a comparison between the obtained results. The data of all individual experiments were then pooled to determine the total EC50 and Emax values per substance.
Molecular Docking
The recently published cryo-EM structure (PDW: 6WHA) of the 5-HT2AR in complex with miniGαq and the psychedelic substance 25CN-NBOH was used as a starting template.8 Each of the structures of the -NBOMe isomers and LSD and 25CN-NBOH (as controls) was built and optimized in Spartan ’18 Parallel Suite (Wave function, Irvine, CA, United States) and molecularly docked, employing induced fit docking (Schrödinger, NY, United States). To reduce atom clashing, ligand–receptor complexes were minimized using the OPLS3 force field in Prime (Schrödinger). Prime MM-GBSA (Schrödinger) was used for the calculation of the amino acid interaction energy with the individual residues.
Acknowledgments
C.P.S. acknowledges funding by the Research Foundation-Flanders (FWO) [G069419N and G0B8817N] and the Ghent University Special Research Fund (BOF) [01J15517]. O.V.K. acknowledges support by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (Project 0671-2020-0058). A.L.B. and R.B.L. acknowledge support by a partnership grant from GlaxoSmithKline and the Canadian Institutes of Health Research (CIHR) [RN323670-386247]. V.A.S. acknowledges the Russian Science Foundation for financially supporting this work, Project 20-13-00089. The authors specifically acknowledge the reviewers for their helpful assessment of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00189.
Figure S1: visual representation of the docking of 25CN-NBOH and 23H-, 25H-, 26H-, and 34H-NBOMe in the binding pocket of the 5-HT2AR (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nichols D. E. (2004) Hallucinogens. Pharmacol. Ther. 101, 131–181. 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Nichols D. E. (2017) Chemistry and Structure-Activity Relationships of Psychedelics. Curr. Top. Behav. Neurosci. 36, 1–43. 10.1007/7854_2017_475. [DOI] [PubMed] [Google Scholar]
- Poulie C. B. M.; Jensen A. A.; Halberstadt A. L.; Kristensen J. L. (2020) DARK Classics in Chemical Neuroscience: NBOMes. ACS Chem. Neurosci. 11, 3860. 10.1021/acschemneuro.9b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNODC . Current NPS Threats, 2020. https://www.unodc.org/unodc/en/scientists/current-nps-threats.html (accessed September 2020).
- Kyriakou C.; Marinelli E.; Frati P.; Santurro A.; Afxentiou M.; Zaami S.; Busardo F. P. (2015) NBOMe: new potent hallucinogens--pharmacology, analytical methods, toxicities, fatalities: a review. Eur. Rev. Med. Pharmacol. Sci. 19, 3270–3281. [PubMed] [Google Scholar]
- Luethi D.; Liechti M. E. (2020) Designer drugs: mechanism of action and adverse effects. Arch. Toxicol. 94, 1085–1133. 10.1007/s00204-020-02693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenschutz M. P.; Ross S. (2016) Therapeutic Applications of Classic Hallucinogens. Curr. Top. Behav. Neurosci. 36, 361–391. 10.1007/7854_2016_464. [DOI] [PubMed] [Google Scholar]
- Kim K.; Che T.; Panova O.; DiBerto J. F.; Lyu J.; Krumm B. E.; Wacker D.; Robertson M. J.; Seven A. B.; Nichols D. E.; Shoichet B. K.; Skiniotis G.; Roth B. L. (2020) Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell 182, 1574–1588.e1519. 10.1016/j.cell.2020.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K. T.; Asada H.; Inoue A.; Kadji F. M. N.; Im D.; Mori C.; Arakawa T.; Hirata K.; Nomura Y.; Nomura N.; Aoki J.; Iwata S.; Shimamura T. (2019) Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 26, 121–128. 10.1038/s41594-018-0180-z. [DOI] [PubMed] [Google Scholar]
- Kupriyanova O. V.; Shevyrin V. A.; Shafran Y. M.; Lebedev A. T.; Milyukov V. A.; Rusinov V. L. (2020) Synthesis and determination of analytical characteristics and differentiation of positional isomers in the series of N-(2-methoxybenzyl)-2-(dimethoxyphenyl)ethanamine using chromatography-mass spectrometry. Drug Test. Anal. 12, 1154–1170. 10.1002/dta.2859. [DOI] [PubMed] [Google Scholar]
- Pottie E.; Cannaert A.; Van Uytfanghe K.; Stove C. P. (2019) Setup of a Serotonin 2A Receptor (5-HT2AR) Bioassay: Demonstration of Its Applicability To Functionally Characterize Hallucinogenic New Psychoactive Substances and an Explanation Why 5-HT2AR Bioassays Are Not Suited for Universal Activity-Based Screening of Biofluids for New Psychoactive Substances. Anal. Chem. 91, 15444–15452. 10.1021/acs.analchem.9b03104. [DOI] [PubMed] [Google Scholar]
- Pottie E.; Dedecker P.; Stove C. P. (2020) Identification of psychedelic new psychoactive substances (NPS) showing biased agonism at the 5-HT2AR through simultaneous use of beta-arrestin 2 and miniGalphaq bioassays. Biochem. Pharmacol. 182, 114251. 10.1016/j.bcp.2020.114251. [DOI] [PubMed] [Google Scholar]
- Dixon A. S.; Schwinn M. K.; Hall M. P.; Zimmerman K.; Otto P.; Lubben T. H.; Butler B. L.; Binkowski B. F.; Machleidt T.; Kirkland T. A.; Wood M. G.; Eggers C. T.; Encell L. P.; Wood K. V. (2016) NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 11, 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- Pottie E.; Cannaert A.; Stove C. P. (2020) In vitro structure-activity relationship determination of 30 psychedelic new psychoactive substances by means of beta-arrestin 2 recruitment to the serotonin 2A receptor. Arch. Toxicol. 94, 3449. 10.1007/s00204-020-02836-w. [DOI] [PubMed] [Google Scholar]
- Nehme R.; Carpenter B.; Singhal A.; Strege A.; Edwards P. C.; White C. F.; Du H.; Grisshammer R.; Tate C. G. (2017) Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One 12, e0175642. 10.1371/journal.pone.0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman A. J.; Wolfrum K. M.; Reed J. F.; Kim S. O.; Johnson R. A.; Janowsky A. (2018) Neurochemical pharmacology of psychoactive substituted N-benzylphenethylamines: High potency agonists at 5-HT2A receptors. Biochem. Pharmacol. 158, 27–34. 10.1016/j.bcp.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickli A.; Luethi D.; Reinisch J.; Buchy D.; Hoener M. C.; Liechti M. E. (2015) Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology 99, 546–553. 10.1016/j.neuropharm.2015.08.034. [DOI] [PubMed] [Google Scholar]
- Braden M. R.; Parrish J. C.; Naylor J. C.; Nichols D. E. (2006) Molecular interaction of serotonin 5-HT2A receptor residues Phe339(6.51) and Phe340(6.52) with superpotent N-benzyl phenethylamine agonists. Mol. Pharmacol. 70, 1956–1964. 10.1124/mol.106.028720. [DOI] [PubMed] [Google Scholar]
- Shulgin A., and Shulgin A. (1991) PiHKAL: A chemical Love Story; Transform Press, Berkeley, CA. [Google Scholar]
- Braden M. R.; Nichols D. E. (2007) Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol. Pharmacol. 72, 1200–1209. 10.1124/mol.107.039255. [DOI] [PubMed] [Google Scholar]
- Marcher-Rorsted E.; Halberstadt A. L.; Klein A. K.; Chatha M.; Jademyr S.; Jensen A. A.; Kristensen J. L. (2020) Investigation of the 2,5-Dimethoxy Motif in Phenethylamine Serotonin 2A Receptor Agonists. ACS Chem. Neurosci. 11, 1238–1244. 10.1021/acschemneuro.0c00129. [DOI] [PubMed] [Google Scholar]
- Leth-Petersen S.; Petersen I. N.; Jensen A. A.; Bundgaard C.; Baek M.; Kehler J.; Kristensen J. L. (2016) 5-HT2A/5-HT2C Receptor Pharmacology and Intrinsic Clearance of N-Benzylphenethylamines Modified at the Primary Site of Metabolism. ACS Chem. Neurosci. 7, 1614–1619. 10.1021/acschemneuro.6b00265. [DOI] [PubMed] [Google Scholar]
- Mulakala C.; Viswanadhan V. N. (2013) Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies?. J. Mol. Graphics Modell. 46, 41–51. 10.1016/j.jmgm.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D. E. (2016) Psychedelics. Pharmacol. Rev. 68, 264–355. 10.1124/pr.115.011478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal C. E. (2018) Serotonergic Psychedelics: Experimental Approaches for Assessing Mechanisms of Action. Handb. Exp. Pharmacol. 252, 227–260. 10.1007/164_2018_107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leth-Petersen S.; Gabel-Jensen C.; Gillings N.; Lehel S.; Hansen H. D.; Knudsen G. M.; Kristensen J. L. (2016) Metabolic Fate of Hallucinogenic NBOMes. Chem. Res. Toxicol. 29, 96–100. 10.1021/acs.chemrestox.5b00450. [DOI] [PubMed] [Google Scholar]
- Wouters E.; Walraed J.; Banister S. D.; Stove C. P. (2019) Insights into biased signaling at cannabinoid receptors: synthetic cannabinoid receptor agonists. Biochem. Pharmacol. 169, 113623. 10.1016/j.bcp.2019.08.025. [DOI] [PubMed] [Google Scholar]
- Pottie E.; Tosh D. K.; Gao Z. G.; Jacobson K. A.; Stove C. P. (2020) Assessment of biased agonism at the A3 adenosine receptor using beta-arrestin and miniGalphai recruitment assays. Biochem. Pharmacol. 177, 113934. 10.1016/j.bcp.2020.113934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.