

Abstract
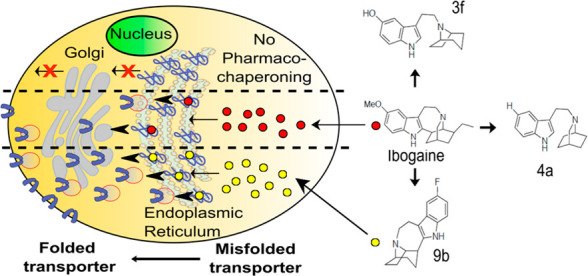
Missense mutations that give rise to protein misfolding are rare, but collectively, defective protein folding diseases are consequential. Folding deficiencies are amenable to pharmacological correction (pharmacochaperoning), but the underlying mechanisms remain enigmatic. Ibogaine and its active metabolite noribogaine correct folding defects in the dopamine transporter (DAT), but they rescue only a very limited number of folding-deficient DAT mutant proteins, which give rise to infantile Parkinsonism and dystonia. Herein, a series of analogs was generated by reconfiguring the complex ibogaine ring system and exploring the structural requirements for binding to wild-type transporters, as well as for rescuing two equivalent synthetic folding-deficient mutants, SERT-PG601,602AA and DAT-PG584,585AA. The most active tropane-based analog (9b) was also an effective pharmacochaperone in vivo in Drosophila harboring the DAT-PG584,585AA mutation and rescued 6 out of 13 disease-associated human DAT mutant proteins in vitro. Hence, a novel lead pharmacochaperone has been identified that demonstrates medication development potential for patients harboring DAT mutations.
Keywords: solute carrier-6, dopamine transporter, serotonin transporter, misfolding, ibogaine analogs, pharmacochaperoning
Ibogaine, one of many alkaloids first isolated from the shrub Tabernanthe iboga in 1901,1 has three interesting pharmacological properties: (i) It is hallucinogenic, presumably the reason why it has been used for centuries by West African tribes in rites of passage.2,3 (ii) It has been reported to mitigate substance use disorders.4−6 This action has also been recapitulated in experimental paradigms, where animals are given the opportunity to self-administer or express their preference for morphine and psychostimulants.7,8 (iii) Ibogaine and its principal active metabolite noribogaine (12-hydroxyibogamine),9 bind to the transporters for the monoamines serotonin (SER), dopamine (DA), and norepinephrine (NE).9,10 Moreover, ibogaine and noribogaine were the first compounds that were shown to rescue folding-deficient versions of SERT11−13 and dopamine transporter (DAT).14−16 This pharmacochaperoning action of ibogaine and noribogaine is of therapeutic interest because folding-deficient mutations of DAT give rise to the dopamine transporter deficiency syndrome (DTDS).17,18 DTDS is a hyperkinetic movement disorder in DAT-deficient patients that progresses into Parkinsonism and dystonia. This disease manifests generally in the first 6 months post-birth (infantile) or occasionally during childhood, adolescence, or adulthood (juvenile). The cell surface levels of DAT variants associated with DTDS are typically nondetectable (in infantile cases) or severely reduced (in juvenile cases) due to their misfolding and subsequent retention within the endoplasmic reticulum (ER). While the use of ibogaine or noribogaine as a medication for DTDS is warranted, its hallucinogenic profile will likely preclude its therapeutic usefulness in this patient population.2
The closely related monoamine transporters, SE transporter (SERT, SLC6A4), DAT (SLC6A3), and NE transporter (NET, SLC6A2), form a branch of the solute carrier-6 (SLC6) family of secondary active transporters.19 They modulate monoaminergic neurotransmission by retrieving their eponymous substrates from the synaptic cleft, which supports replenishing of vesicular stores. SERT, and to a lesser extent NET, are the most important targets for antidepressants which act as inhibitors. For example, selective serotonin reuptake inhibitors (SSRIs) are used to treat major depression, obsessive–compulsive disorders, and general anxiety disorders. The therapeutic indication for DAT inhibition is more restricted (e.g., methylphenidate for attention-deficit-hyperactivity disorder or modafinil for sleep disorders). However, DAT is a prominent target for illicit drugs (e.g., cocaine and amphetamines). This is also true to some extent for SERT, which is the target of 3,4-methylene-dioxymethamphetamine (MDMA, ecstasy) and its congeners. Thus, the chemical space for these transporter targets has been explored.20 Of note, monoamine transporter ligands can range from full substrates to typical inhibitors, as well as atypical inhibitors, depending on chemical structure and transporter conformation.21−23 Typical inhibitors (e.g., cocaine and most antidepressants) bind to and trap the transporter in the outward-facing state, thus precluding any subsequent conformational transition required for entry of the protein into a transport mode. In contrast, the substrate-bound transporter enters an occluded state. Substrate translocation is initiated by the opening of an inner gate, which releases the substrate together with cosubstrate ions (Na+ and Cl–) on the intracellular side.20 Full substrates allow the transporter to undergo its transport cycle in a manner indistinguishable from cognate neurotransmitter. Full substrates, which differ from neurotransmitters in their cooperative interaction with the cotransported sodium, act as amphetamine-like releasers by driving the transporter into an exchange mode.24 Partial substrates/releasers support the transport cycle/exchange mode, albeit less efficiently than full substrates/releasers, because they bind tightly to conformational intermediates and thus preclude rapid transitions.25 Atypical inhibitors trap the transporter in conformations other than the outward-facing state. Ibogaine is an atypical inhibitor that binds the inward-facing state of SERT,26−29 and presumably, of DAT and NET. Several arguments support the conjecture that the folding trajectory of monoamine transporters proceeds through the inward-facing state,30 thus providing a mechanistic basis for rationalizing the pharmacochaperoning action of ibogaine11−16 and of other compounds.25
Ibogaine and its metabolite noribogaine are the most efficacious pharmacochaperones identified to date for folding-deficient versions of SERT and DAT and provide templates to generate promising new leads. In this study, we explored the chemistry of ibogaine to broaden the efficacy profile for this drug in rescuing misfolded SERT and DAT mutations. Ibogaine has a complex and rigid structure, which until recently has been largely unexplored.31,32 Here, we generated a series of novel ibogaine analogs by investigating the chemical space surrounding the parent molecule using a two-pronged approach: (1) Deconstructing ibogaine by introducing flexible hydrocarbon linkers that connect the indole ring to either a isoquinuclidine ring or a tropane ring. (2) Reconfiguring and completely substituting the isoquinuclidine ring of ibogaine with the tropane ring system. This allowed for defining structural determinants required for high-affinity binding to wild-type (WT) SERT and DAT and for pharmacochaperoning two synthetic folding-deficient mutations: SERT-PG601,602AA and the orthologous DAT-PG584,585AA. On the basis of these experiments, we identified a novel fluorinated tropane-based analog that was more potent and more efficacious than the parent compound in rescuing misfolded versions of SERT and DAT, including disease-relevant DTDS mutations. Importantly, this compound was active in vivo and restored sleep to flies harboring a misfolded DAT mutant protein.
Results
Synthesis
Ibogaine can be viewed as a serotonin analog, where the basic nitrogen is fixed in space by a fused bicyclic ring structure (cf. Table 1). Two strategic approaches were undertaken that yielded a number of unique products to the ibogaine series in relatively few synthetic steps. The first approach (Scheme 1 A) involved a deconstructive strategy, whereby the indole ring was disconnected from the isoquinuclidine ring structure, thereby offering greater flexibility along with a comparable number of hydrocarbon atoms by either retaining the isoquinuclidine ring (4a–c) or replacing it with a tropane ring (3a–f). The second approach (Scheme 1B) aimed to reconfigure and completely substitute the fused isoquinuclidine ring of ibogaine with the tropane ring system conferring the novel intermediate amides (8a–c) and their reduced tertiary amine analogs (9a–d). Notably, the tropane ring is a hallmark structure in many classical monoamine transporter inhibitors (e.g., cocaine and benztropine) and thus was envisioned to potentially retain binding affinities at DAT and SERT. In both approaches, the ethyl group of ibogaine was eliminated to save on synthetic effort. In addition, we surmised that the ethyl group was immaterial for binding affinity (see below).
Table 1. Radioligand Uptake and Binding Data at DAT and SERTa.

| uptake
inhibition |
binding inhibition |
ratio |
pharmacochaperoning |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 ± SD (μM) |
Ki ± SD (μM) |
IC50/Ki |
EC50 ± SD (μM) | Vmax (%) | EC50 ± SD (μM) | Vmax (%) | ||||
| WT-SERT | WT-DAT | WT-SERT | WT-DAT | WT-SERT | WT-DAT | SERT-PG601,602AA | DAT-PG584,585AA | |||
| ibogaine | 8.2 ± 3.5 | 22.1 ± 6.3 | 7.4 ± 0.7 | 5.3 ± 1.7 | 1.1 | 4.2 | 2.6 ± 1.9 | 103 ± 16 | 19.6 ± 4.9 | 88 ± 16 |
| noribogaine | 1.2 ± 0.5 | 15.5 ± 7.8 | 0.6 ± 0.1 | 4.0 ± 1.8 | 2.1 | 3.9 | 4.6 ± 1.2 | 100 ± 8 | 17.1 ± 6.1 | 100 ± 13 |
| 3a | 9.1 ± 3.1 | 25.6 ± 10.2 | 1.2 ± 0.2 | 3.1 ± 0.9 | 7.8 | 8.2 | NC* | 19 ± 6* | NC* | 15 ± 10* |
| 3b | 6.5 ± 5.2 | 24.4 ± 17.9 | 1.85 ± 0.3 | 1.7 ± 0.1 | 3.5 | 14.4 | 12.9 ± 6.3 | 60 ± 17 | NC* | 15 ± 8* |
| 3c | 0.5 ± 0.1 | 9.09 ± 1.64 | 0.19 ± 0.03 | 4.4 ± 1.4 | 2.4 | 2.1 | 2.7 ± 0.78 | 57 ± 13 | 17.1 ± 5.6 | 47 ± 18 |
| 3d | 11.9 ± 1.6 | 58.2 ± 41.4 | 4.5 ± 0.4 | 8.8 ± 4.9 | 2.6 | 6.6 | NC* | 11 ± 3* | NC* | 20 ± 15* |
| 3e | 21.6 ± 17.4 | 179 ± 94 | 13.0 ± 2.7 | 10.2 ± 3.8 | 1.7 | 17.6 | NC* | 38 ± 19* | NC* | 15 ± 9* |
| 3f | 0.2 ± 0.1 | 78.0 ± 27.9 | 0.12 ± 0.02 | 21.8 ± 9.4 | 1.6 | 3.6 | NC* | 8 ± 2* | NC* | 13 ± 10* |
| 4a | 4.6 ± 2.63 | 6.5 ± 2.1 | 0.81 ± 0.03 | 1.9 ± 0.2 | 5.7 | 3.5 | NC* | 25 ± 6* | NC* | 15 ± 9* |
| 4b | 8.2 ± 1.0 | 11.9 ± 1.6 | 1.7 ± 0.3 | 1.5 ± 0.3 | 4.8 | 7.7 | NC* | 29 ± 8* | NC* | 10 ± 6* |
| 4c | 0.3 ± 0.2 | 11.8 ± 9.1 | 0.13 ± 0.34 | 2.6 ± 0.7 | 2.5 | 4.5 | 0.7 ± 0.3 | 68 ± 16 | NC* | 24 ± 8* |
| 8a | ND | ND | >100 | 39.7 ± 27.9 | ND | ND | ND | ND | ND | ND |
| 8b | ND | ND | >100 | 51.2 ± 14.6 | ND | ND | ND | ND | ND | ND |
| 8c | ND | ND | >100 | 45.78 ± 6.5 | ND | ND | ND | ND | ND | ND |
| 9a | 6.9 ± 3.9 | 16.7 ± 11.2 | 4.4 ± 0.2 | 3.6 ± 0.2 | 1.6 | 4.6 | 3.05 ± 2.3 | 94 ± 23 | 18.9 ± 2.9 | 51 ± 21 |
| 9b | 0.4 ± 0.2 | 7.2 ± 3.8 | 0.18 ± 0.01 | 0.8 ± 0.3 | 2.3 | 8.9 | 0.06 ± 0.04 | 100 ± 6 | 25.7 ± 6.3 | 673 ± 245 |
| 9c | 29.9 ± 16.4 | 10.8 ± 9.3 | 7.3 ± 0.5 | 3.1 ± 0.3 | 4.1 | 3.5 | 2.1 ± 0.8 | 81 ± 20 | 19.2 ± 8.7 | 53 ± 13 |
| 9d | 0.9 ± 0.5 | 15.2 ± 9.7 | 0.78 ± 0.04 | 2.9 ± 0.3 | 1.2 | 5.3 | 1.4 ± 0.8 | 89 ± 19 | 5.8 ± 1.9 | 49 ± 30 |
Each IC50, Ki, Km and Vmax value represents data from at least 3 independent experiments, each performed in triplicate. Vmax refers to the maximum transport restored by noribogaine, which was set at 100% to account for interassay variation. Experimental procedures are described in detail in the “Materials and Methods” section. NC*: not calculable, a reliable EC50 could not be estimated, because functional rescue was too low; in this instance Vmax* refers to the activity restored by 30 μM of the compound. ND: not determined.
Scheme 1. Synthetic Scheme of the Ibogaine Analogs.
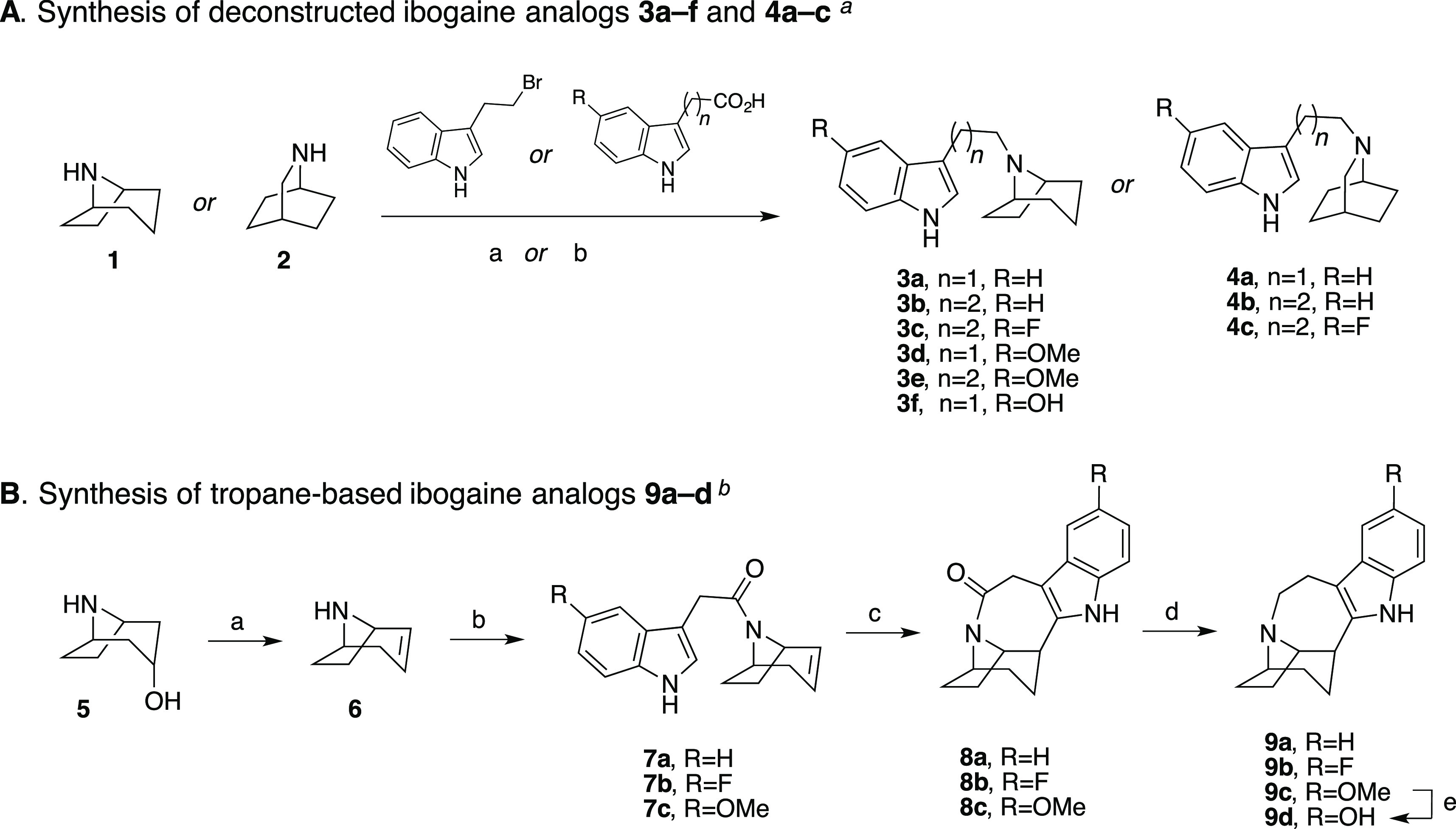
Reagents and conditions: (a) 3-(2-bromoethyl)-1H-indole, 1 or 2, K2CO3, CH3CN, reflux; (b) i. appropriate 3-indoleacetic acid or 3-indolepropionic acid, CDI, THF, RT; ii. LAH, THF, RT.
Reagents and conditions: (a) H2SO4, 160 °C; (b) appropriate 3-indoleacetic acid, CDI, THF, RT; (c) (i) Pd(CH3CN)4BF4, CH3CN, (ii) NaBH4, EtOH; (d) BMS, THF, reflux; (e) 3 N HCl, reflux.
The synthetic schemes of the two approaches are shown in Scheme 1. 8-Azabicyclo[3.2.1]octane (nortropane, (1)) or 2-azabicyclo[2.2.2]octane (2) was directly linked at the N-position with various indole substituents via either nucleophilic substitution with 3-(2-bromoethyl)-1H-indole or by tandom 1,1′-carbonyldiimidazole (CDI) coupling with an appropriate 3-indoleacetic acid or 3-indolepropanoic acid followed by lithium aluminum hydride (LAH) reduction to give the desired products (3a–f, 4a–c) as free bases (Scheme 1A). The second approach (Scheme 1B) started similarly following the dehydration of nortropine (5) in sulfuric acid to give racemic nortropidene (6), which was linked with an appropriate 3-indoleacetic acid substituent via CDI coupling. Next, similar to Trost et al.33 and Kruegel et al.31 in their syntheses of ibogamine, electrophilic palladium promoted cyclization of racemic 7a–c, followed by sodium borohydride (NaBH4) reduction, gave the racemic products 8a–c. This key step produced the corresponding amides in good yields (63–81% yields, see the “Experimental Methods” section of the Supporting Information for details), without necessitating chromatographic separation. Notably, Trost et al.33 and Kruegel et al.31 reported much lower isolated yields of ibogamine (19 and 33%, respectively) with this chemistry by using the isoquinuclidine core and corresponding amine instead. Finally, the amide reduction of racemic 8a–c was accomplished with borane dimethyl sulfide complex (BMS) to give racemic 9a–d, where 9d was formed from the O-demethylation of 9c following extended stirring at reflux in 3 N HCl.
Structure–Activity Relationship of Ibogaine Analogs at SERT and DAT
Ligand affinities at DAT and SERT can be determined by measuring their ability to displace a radioligand from the transporter or to block substrate uptake. During substrate translocation, transporters undergo a conformational cycle. Radioligands are high-affinity inhibitors which bind to the outward-facing state. In addition, both the membrane potential and the asymmetric ion distribution are absent in binding assays with membrane preparations. Hence, affinity estimates for some ligands can differ substantially, if inhibition of substrate uptake and displacement of substrate are assessed. Because of conformational trapping in the absence of an ion gradient, these differences can reach several orders of magnitude.25 Accordingly, we determined the affinity of our novel analogs by measuring their ability to both, displace a radioligand in membranes prepared from rat brain stem (Figure S-1, SERT) and rat striatum (Figure S-2, DAT) and inhibit cellular uptake in transfected cells (Figures S-3 and S-4 for SERT and DAT, respectively). Noribogaine is known to bind SERT with higher affinity, in both binding and uptake inhibition assays, over the methoxylated parent compound, ibogaine, and our data confirm this in Table 1. However, as previously reported,9 ibogaine and noribogaine have similar affinities for DAT (also seen in Table 1). It is evident from the data in Table 1 that the C-12 substitutions primarily determine affinity to SERT in all analogs tested. Methoxy- (3d, 3e, and 9c) or hydroxy-substitution (3f and 9d) in this 12-position has little effect on DAT affinity as compared to the unsubstituted analogs (3a, 3b, 4a, 4b, 8a, and 9a), but the hydroxy-substituted analogs have higher affinity at SERT (3f, and 9d). Interestingly, when the hydroxyl group is substituted with fluorine, compounds 3c, 4c, and 9b also exhibit similar affinities at SERT to the hydroxy-analogs. Only 9b showed submicromolar affinity for DAT, suggesting a potentially pivotal role for fluorine substitution. In addition, compounds 9c and 9d are ibogaine and noribogaine analogs, respectively, in which the 2-ethyl group has been removed and the isoquinuclidine core has been replaced with a tropane ring. The affinities of these compound pairs were similar at DAT and SERT (9c/ibogaine v. 9d/noribogaine). The rigidity imparted by the azepine-ring (compounds 9a–d, ibogaine, and noribogaine) is a minor determinant of affinity. Indeed, the affinity of compounds 3c and 9b, for instance, were comparable.
The amide analogs, in which the basic amine was neutralized, had substantially reduced affinities (cf. compounds 8a–c and 9a–c in Table 1) demonstrating the necessity for a protonatable nitrogen. In general, the affinities obtained from binding displacement were higher than those estimated from uptake inhibition. Despite this difference, in the two affinity estimates in SERT, the rank order of potency determined by uptake inhibition was reasonably similar to that determined by binding: analogs with hydroxy and fluorine substituents were more potent in blocking substrate uptake by SERT than the methoxy-substituted and unsubstituted counterparts (Table 1).
In DAT, all fluorinated analogs (3c, 4c, and 9b) also ranked among the most potent blockers of DAT uptake (Table 1). However, affinity estimates from binding and uptake inhibition differed in part substantially with IC50/Ki ratios ranging from 2 to 14 (Table 1). We verified that ibogaine analogs bound to DAT in rat striatal membranes and human DAT heterologously expressed in HEK 293 cells with comparable affinity (Figure S-2). Thus, variations in the ratio of IC50/Ki (see Table 1) are indicative of differences in conformational trapping25 rather than of species differences (rat vs human transporters). More importantly, conformational trapping is suggestive of a pharmacochaperoning action.25 Accordingly, we determined if some of these analogs were capable of rescuing folding-deficient mutations of SERT and DAT.
Pharmacochaperoning Action of the Ibogaine Analogs on SERT-PG601,602AA and on DAT-PG584,585AA
Several folding-deficient versions of SERT have been generated and characterized to understand the actions of pharmacochaperones.11−13 The mutant protein SERT-PG601,602AA is stalled at an early stage of the folding trajectory.12 Hence, on the basis of its severe phenotype, we selected this mutation to examine and compare the ability of our ibogaine analogs (i.e., compounds 3a–3f, 4a–4c, and 9a–9d) with ibogaine and noribogaine to act as pharmacochaperones. Misfolded transporters are retained in the ER and are thus unavailable to accomplish their eponymous actions, that is, the cellular uptake of substrate. Pretreatment of cells with a pharmacochaperone restores folding of the protein and thus its subsequent delivery to the cell surface.11−16 This can be readily monitored by measuring cellular substrate uptake. As shown in Figure 1A, in cells expressing SERT-PG601,602AA, there is negligible uptake of [3H]5-HT (≪10%) when compared to those expressing WT SERT.
Figure 1.

[3H]5-HT uptake in HEK293 cells expressing hSERT-PG601,602AA after preincubation with ibogaine analogs. (A) Comparison of [3H]5-HT uptake by HEK293 cells transiently expressing WT hSERT or hSERT-PG601,602AA. Each symbol represents the result from an individual experiment (done in triplicate); means ± SD are also shown. The statistical comparison was done by a Mann–Whitney test (p = 0.0002). (B) hSERT-PG601,602AA expressing cells were incubated in the presence of the indicated compounds (30 μM in all instances but bupropion, 100 μM). After 24 h, [3H]5-HT uptake was determined as outlined in the “Materials and Methods” section. Values from individual experiments (done in triplicate) are shown as dots; box plots show the median and the interquartile range. The statistical comparison was done via one-way ANOVA with a post hoc Dunnett’s multiple comparison to confirm a statistically significant difference between untreated control cells and those exposed to individual compounds (**, p < 0.01; ***, p < 0.001). (C) Concentration–response curves for pharmacochaperoning by the indicated compounds (selected as positive hits from panel B). Rescued uptake was normalized to that achieved by 30 μM noribogaine (= 100%) to account for interexperimental variations. ***, Emax for 9b was achieved at 3 μM; uptake inhibition was observed after preincubation with higher concentrations. The solid lines were drawn by fitting the data to the equation for a rectangular hyperbola (for EC50 and Emax of mutation rescue see Table 1). Data were obtained in at least three independent experiments carried out in triplicate. The error bars indicate SD.
In cells which were preincubated for 24 h in the presence of 30 μM ibogaine, noribogaine, and other pharmacochaperones (i.e., the naphthyl-propanamine series PAL-287, PAL-1045, and PAL-104625,34 and 100 μM bupropion15), substrate uptake supported by SERT-PG601,602AA increased to levels which corresponded to 25–40% of the transport activity of WT SERT (Figure 1B). Similarly, if the cells were preincubated in the presence of 30 μM of the new ibogaine analogs, then appreciable levels of transport were seen with compounds 3b, 3c, 4c, 9a, 9c, and 9d (Figure 1B). Additional experiments (not shown) confirmed that compound 9b was more effective at concentrations < 10 μM than at 30 μM. Hence, it was also included in the list of positive hits which were further investigated to determine their potency and their efficacy as pharmacochaperones. We compared their action in individual transient transfections by normalizing their pharmacochaperoning activity to the transport activity restored by 30 μM noribogaine (Figure 1C). Known pharmacochaperones (PAL-1045, bupropion, ibogaine, and noribogaine) were also examined as reference compounds. All compounds belonging to series 9a–9d were as effective as noribogaine, but compound 9b had an EC50 of ∼60 nM (curve represented as *** in Figure 1C). The other fluoro-analog, 4c, had an EC50 of ∼600 nM but was less efficacious (Emax ∼ 70% of that of noribogaine). Other compounds, including noribogaine (EC50 = ∼3 μM), rescued SERT-PG601,602AA with EC50 values in the low to mid micromolar range (Table 1).
Our data suggests 9b to be the most potent of all compounds tested in rescuing SERT-PG601,602AA. We visualized the glycosylation state of SERT-PG601,602AA by immunoblotting to confirm the pharmacochaperoning action of 9b by an independent approach. During their synthesis in the ER, membrane proteins undergo N-linked core glycosylation; this core glycan, which can be removed by endoglycosidase H, is thus present on ER-retained misfolded mutant proteins. Rescued mutant proteins are exported to the Golgi, where they acquire additional sugar moieties. The resulting complex glycan structure is resistant to cleavage by endoglycosidase H. The core glycosylated protein is homogeneous and smaller in size than the mature glycosylated version, which is heterogeneous due to the stochastic nature of complex glycosylation. Accordingly, in lysates prepared from transiently transfected cells, WT SERT was visualized by immunoblotting as a band migrating at 75 kDa and broad smear coalescing from a collection of bands in the range of 90 to 110 kDa (left-hand lane, Figure 2A). The size of the lower band was reduced after cleavage by endoglycosidase H (cf. lanes 1 and 2 in Figure 2C) confirming that it corresponded to the core glycosylated band (“C”). In contrast, the migration of the collection of upper bands was insensitive to cleavage by endoglycosidase H, confirming that they had acquired the mature glycan (cf. bands “M” in lanes 1 and 2 in Figure 2C). In contrast, lysates from cells expressing SERT-PG601,602AA contain predominantly the core glycosylated protein (second lane in Figure 2A showing control cells). As predicted from its ability to restore substrate uptake in pretreated cells (Figure 1B,C), 9b induced the appearance of slowly migrating forms of SERT-PG601,602AA in lysates prepared after preincubation of the cells in a concentration-dependent manner (lanes 3–8 in Figure 2A). Plotting this concentration response revealed an EC50 of 134 ± 30 nM (Figure 2B), which is in excellent agreement with that calculated from restoration of substrate uptake (Figure 1C and Table 1). We confirmed that the mature glycosylated species of the rescued mutant protein is resistant to cleavage by endoglycosidase H (shown for lysates from cells compound 9b-pretreated cells in Figure 2C).
Figure 2.

Enhanced mature glycosylation of hSERT-PG601,602AA or hDAT-PG584,585AA in cells preincubated with 9b. (A) HEK293 cells transiently expressing hSERT-PG601,602AA were incubated in the absence (negative control = lane 2) or presence of the indicated concentrations of 9b for 24 h. Cells expressing WT hSERT were the positive control (first labeled as hSERTWT). Membrane proteins extracted from these cells were denatured, electrophoretically resolved, and transferred onto nitrocellulose membranes. The blots were incubated overnight at 4 °C with anti-GFP (top) or anti-Gβ (bottom, loading control) antibodies. The immunoreactive bands were detected with using a horseradish peroxidase-conjugated secondary antibody. The image was captured by a CCD-camera (Fluor Chem HD2 system, Alpha Innotech). (B) Concentration–response curve generated from three independent experiments carried out as in panel A. The ratio of mature (M) to core glycosylated band (C) was quantified densitometrically, normalized to the density of Gβ (loading control) and compared with the ratio observed in each blot for untreated control cells. The density ratios for untreated control cells and those exposed to compound 9b (30 μM) were 0.38 ± 0.09 and 1.65 ± 0.14, respectively. The solid line was drawn by fitting the data to the equation for a rectangular hyperbola. The EC50 of rescue was 134 ± 30 nM. (C and D) In separate experiments, lysates were prepared from cells expressing the SERT-PG601,602AA and DAT-PG584,585AA mutations treated in the absence or presence of 30 μM 9b and subjected to enzymatic digestion by endoglycosidase H (Endo H). Endo H specifically cleaves core glycans (C) to generate lower molecular weight deglycosylated fragments (D). Mature glycosylated bands (M) are resistant to the actions of Endo H. The peroxidase reaction was visualized by film exposure.
We posited that the potency and efficacy of the ibogaine analogs in pharmacochaperoning SERT and DAT would best be compared in the equivalent folding-deficient mutations. The residues P601 and G602 in SERT are conserved in DAT at positions P584 and G585, respectively. Thus, we created DAT-PG584,585AA and verified that it was retained within the cell (not shown), accumulated predominantly as core glycosylated species (cf. lanes 3 and 4 and lanes 1 and 2 in Figure 2D), and only mediated a very low level of substrate uptake (Figure 3A). The folding deficiency of this mutation is expected not only on the basis of the homology of SERT and DAT but also as predicted from earlier work: Mutation of glycine585 to an alanine in DAT suffices to completely abrogate surface expression of the transporter.35 As shown in Figure 3B, if cells transiently expressing DAT-PG584,585AA were preincubated in the presence of 30 μM noribogaine, then there was a robust increase (∼6 fold) in substrate uptake. Of the other known pharmacochaperones, ibogaine, PAL-287, and PAL-1046 (but not PAL-1045) were also active. Of the ibogaine analogs, compounds 3c, 4c (the fluorinated open-ring analogs), 9a, 9c, and 9d rescued the function of DAT-PG584,585AA to a modest extent. In contrast, compound 9b was a very efficacious pharmacochaperone for DAT-PG584,585AA mutant protein, with transport activity exceeding by ∼4-fold the activity restored by noribogaine. Thus, after preincubation in the presence of compound 9b, cells expressing DAT-PG584,585AA recovered up to ∼40% of the uptake velocity seen in cells expressing WT DAT (cf. Figure 3A,B), and mature glycosylated protein accumulated to appreciable levels (lanes 5 and 6 in Figure 2D). We selected compounds 3c, 4c, 9a–9d, PAL-287, and PAL-1046 as positive hits. Cells transiently expressing DAT-PG584,585AA were preincubated with increasing concentrations of these compounds, and substrate uptake was subsequently determined to obtain concentration–response curves for their pharmacochaperoning activity. When compared to the reference compounds ibogaine and noribogaine, compounds 3c, 4c, 9a, 9c, 9d, and PAL-287 were all less efficacious, and the EC50 values of compounds 3c, 4c, 9a, 9d, and PAL-287 were lower than those of ibogaine and noribogaine (Figure 3C, upper panel). As predicted from the screening assays summarized in Figure 3B, compound 9b was substantially more efficacious than noribogaine or ibogaine, but the EC50 values of these three compounds were comparable (Figure 3C, lower panel; Table 1).
Figure 3.

[3H]Dopamine ([3H]DA) uptake in HEK293 cells expressing the hDAT-PG584,585AA after preincubation with ibogaine analogs. (A) Comparison of [3H]DA uptake in HEK293 cells transiently expressing WT hDAT or hDAT-PG584,585AA. Each symbols represents the result from an individual experiment (done in triplicate); means ± SD are also shown. The statistical comparison was done via a Mann–Whitney test (p < 0.0001). (B) Cells expressing hDAT-PG584,585AA were incubated in the presence of the indicated compounds (30 μM in all instances but bupropion, 100 μM). After 24 h, specific [3H]DA uptake was determined as outlined in the “Materials and Methods” section. Values from individual experiments (done in triplicate) are shown as dots; box plots show the median and the interquartile range. The statistical comparison was done by one-way ANOVA with a post hoc Dunnett’s multiple comparison to confirm a statistically significant difference between untreated control cells and those exposed to individual compounds (*, p < 0.05; ***, p < 0.001). (C, top) Concentration–response curves for pharmacochaperoning by the indicated compounds (selected as positive hits from panel B). Rescued uptake was normalized to that achieved by 30 μM noribogaine (= 100%) to account for inter-experimental variations. Rescue by 9b was plotted separately because of its very high efficacy in comparison to those of other analogs (C, bottom). The solid lines were drawn by fitting the data to the equation for a rectangular hyperbola (for EC50 and Emax of mutant protein rescue, see Table 1). Data were obtained in at least three independent experiments carried out in triplicate. The error bars indicate SD.
Rescue of DAT-PG584,585AA in Flies by In Vivo Pharmacochaperoning with Ibogaine Analog 9b
In Drosophila, dopaminergic projections into the fan-shaped body are required to maintain a wake/sleep cycle.36,37 In the absence of functional DAT, flies are hyperactive and have abnormal sleep regulation. Accordingly, Drosophila in which the endogenous DAT gene is disrupted are referred to as fumin (i.e., Japanese for sleepless) flies.38 We previously verified that noribogaine was an effective pharmacochaperone in vivo for some folding-deficient versions of DAT in flies: When expressed in dopaminergic neurons of fumin (fmn) flies, folding-deficient mutations of human DAT were retained in the ER of the soma of PPL1 neurons but were delivered to the axonal terminals of the fan-shaped body. If flies were administered noribogaine, then concomitantly, sleep was restored.14,16 Accordingly, we tested compound 9b to determine if it was also effective as a pharmacochaperone in vivo. We generated fumin flies which harbored the human cDNA encoding DAT-PG584,585AA under the control of GAL4 driven from a tyrosine hydroxylase promoter.39 Adult flies were individually placed in transparent tubes which contained a food pellet supplemented with designated concentrations of compound 9b or of noribogaine and allowed to recover for 1 day; they then spent an additional day of a 12 h:12 h light/dark cycle (marked as yellow and black rectangles in Figure 4A) to entrain their circadian rhythm. It is evident that when subsequently released into a dark/dark cycle (marked as gray and black rectangles in Figure 4A), these flies were as hyperactive as fmn flies (cf. green and red symbols in Figure 4A). Their hyperactivity was greatly reduced by treatment with either compound 9b or noribogaine at concentrations of 30 and 100 μM, respectively, in the food pellet (cf. blue and orange symbols in Figure 4A). In fact, both compound 9b and noribogaine reduced the locomotor activity to that seen in the isogenic line w1118, which was used as control because isogenized TH-Gal4 flies and w1118 flies were previously shown to have similar sleep times.40 We quantified the effect of increasing doses of compound 9b (administered by raising its concentration in the food pellet from 10 to 100 μM) on sleep time (Figure 4B): On average this increased in a dose-dependent manner by about 2.5-fold (i.e., from 300 min in the absence of any pharmacochaperone in the food pellet to 750 min with 100 μM of compound 9b in the food pellet). Sleep duration was comparable to that seen after administration of feed containing 100 μM noribogaine and approached that seen in w1118 flies. This indicates that pharmacochaperoning by compound 9b rescued DAT-PG584,585AA in amounts sufficient to restore clearance of dopamine from the synapse and to thus reinstate normal dopaminergic transmission. Finally, the effect of compound 9b (and of noribogaine) was specific: Sleep duration was affected by compound 9b (or noribogaine) in neither w1118 flies harboring an intact DAT nor DAT-deficient fumin flies (Figure 4C).
Figure 4.
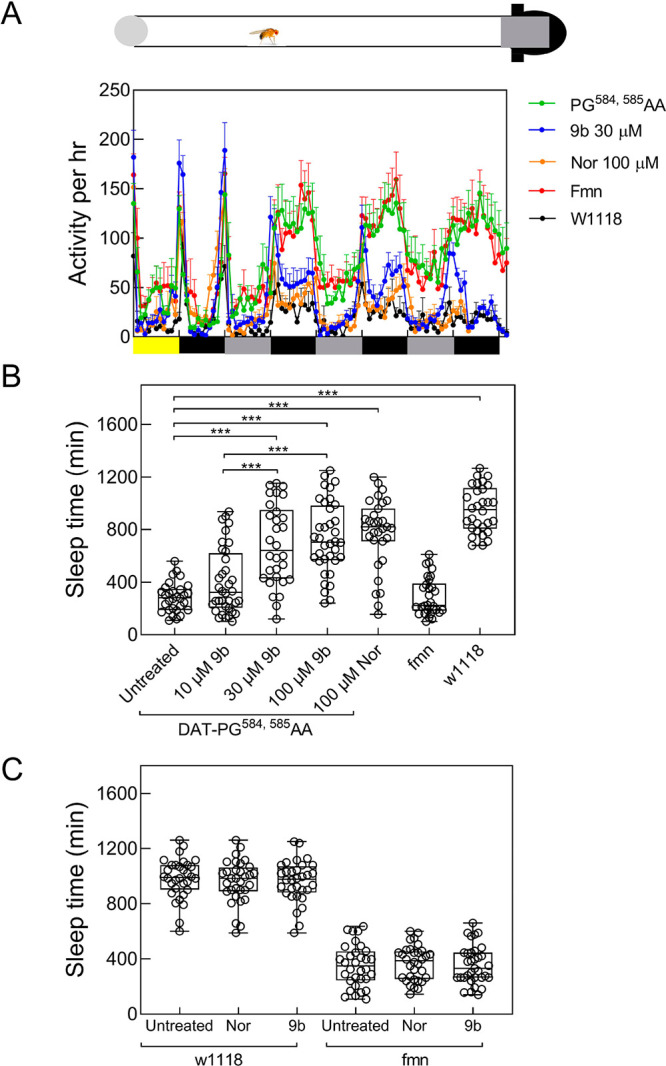
Pharmacochaperones restore sleep in flies harboring DAT-PG584,585AA. (A) Locomotor activity (measured over 1 min interval and binned into 60 min intervals) was recorded during days 2–5 using a Drosophila activity monitor system (schematically represented above the graph, for details see the “Materials and Methods” section). The data are means ± SEM from three independent experiments, which were each carried out in parallel with at least 10 flies/condition. The diurnal light cycles (light-dark on day 2, and dark–dark on days 3−5) are outlined schematically below the graph. (B and C) Sleep time of treated DAT-PG584,585AA mutant flies. Flies expressing hDAT-PG584,585AA in the fmn background were fed with food pellets containing the indicated concentrations of noribogaine and 9b. (C) w1118 and fumin (fmn) flies were used as control and were given food containing 100 μM noribogaine or 100 μM 9b. Locomotor activity on day 4 was used to quantify sleep time using pySolo software. Empty circle represents individual flies. In panel B, statistical significance of the observed differences was determined by a Kruskal–Wallis test followed by Dunn’s post hoc multiple comparison (***, p < 0.001); sleep time in fumin flies differed in a statistically significant manner from that of w1118 files and of flies harboring hDAT-PG584,585AA, which had been given food containing 30 and 100 μM 9b or 100 μM noribogaine (p < 0.001; not shown in B for the sake of clarity).
Pharmacochaperoning of Folding-Deficient DAT Mutant Proteins Associated with Human Disease by Ibogaine Analog 9b
The efficacy of compound 9b, which was seen in vivo, is promising. We therefore examined the ability of compound 9b to rescue all disease-relevant mutations of DAT. When heterologously expressed in HEK293 cells, measurable substrate influx was restored for six mutations (Figure 5); in four of these (i.e., DAT-G386R, -R521W, -P395L, and -P554L), compound 9b was more efficacious than the reference compounds ibogaine and noribogaine. In contrast, preincubation in the presence of corresponding deconstructed analogs 3c and 4c invariably failed to restore any transport activity. Compound 9b did not rescue the function of DAT-R85L, -V158F, -L224P, -G327R, -L368Q, -Y470S and -P529L in any appreciable manner (data not shown).
Figure 5.

Pharmacochaperoning of DTDS-associated DAT mutations by 9b. HEK293 cells were transiently transfected with plasmids encoding YFP-tagged WT hDAT or each of the 13 hDAT mutations discovered in patients suffering from dopamine transporter deficiency syndrome (DTDS). After 24 h, the cells were seeded onto 96-well plates for 24 h and were incubated in either the absence (untreated) or presence of ibogaine, noribogaine, or fluorinated ibogaine analogs 3c, 4c, and 9b (all at 30 μM) for another 24 h. The cells were washed 4 times with Krebs-MES buffer (pH 5.5) and once with Krebs-HEPES buffer (pH 7.4) to completely remove extracellular reservoir of the compounds. Uptake of [3H]dopamine was subsequently measured as outlined in the Material and Methods section. The mutant proteins which were pharmacochaperoned by 9b are shown in this figure. The statistical comparison was done by one-way ANOVA with a post hoc Dunnett’s multiple comparison to confirm a statistically significant difference between untreated control cells and those exposed to individual compounds (*, p < 0.05; ***, p < 0.001). The data are represented as the individual values from at least three independent experiments carried out in triplicate for WT hDAT and the DAT mutations and as box plots with the median and the interquartile range; whiskers indicate the 95% confidence interval. WT-DAT uptake values were used as a control for transfection efficiency. Compound 9b failed to restore activity in the other DAT mutations (i.e., DAT-R85L, -V158F, -L224P, -G327R, -L368Q, -Y470S, and -P529L).
Discussion
More than 60 point mutations have been identified which give rise to human disease because they lead to misfolding of a member of the SLC6 transporter family.30 Folding defects can be overcome by pharmacochaperones which reduce energy barriers in the folding trajectory: Stalled intermediates proceed to reach the native fold. Pharmacochaperoning by ibogaine was serendipitously discovered when studying the ER export of SERT.11 Ibogaine and its metabolite noribogaine effectively rescue some disease-relevant DAT mutations but with limited efficacy.15,16 Progress is contingent on understanding the attributes which account for the pharmacochaperoning action of ibogaine. Here, we relied on an orthogonal approach by interrogating the chemical space populated by variations of the ibogaine structure and by probing their effect on the folding space of SERT and DAT, using two analogous mutations and disease-associated mutations of DAT. Our observations lead to the insight that affinity for the WT transporter (i.e., the native folded state) and pharmacochaperoning activity are not tightly linked, as the structure–activity relationships differ. This conclusion is based on the following lines of evidence: The affinity for SERT is governed predominantly by substitution on the indole ring, with structural rigidity playing a minor role (Table 1). Accordingly, hydroxy- (3f and 9d) and fluoro- (3c, 4c, and 9b) analogs had higher affinities for SERT than those of methoxy- and unsubstituted analogs, but 9d had a lower affinity for SERT than that of its more flexible analogue 3f.
The 2-ethyl group of the isoquinuclidine core is immaterial for supporting binding: 9c and 9d are the tropane-based analogs of ibogaine and noribogaine, respectively; they bind with equivalent affinities to SERT and DAT. These data supported our prediction that the ethyl-group on the isoquinuclidine ring of the ibogamine structure is dispensable and that the isoquinuclidine core can be replaced by tropane. The substitution on the indole ring is a less important determinant for DAT affinity. The most important determinant for pharmacochaperoning efficacy is the structural rigidity imparted by the azepine ring, which is retained by ibogaine, noribogaine, and compounds of series 9a–9d. In contrast, this rigidity is not required for supporting moderate to high-affinity binding to SERT and DAT. Fluorinated compounds 3c and 4c, for instance, were as potent as 9b in inhibiting SERT, but they were substantially less efficacious and less potent in rescuing SERT-PG601,602AA. Similarly, these three analogs only differed modestly in their affinity for DAT, but only compound 9b was an efficacious pharmacochaperone for DAT-PG584,585AA. In fact, compound 9b meets three important criteria to be considered a significant advance in the pharmacochaperoning of DAT mutant proteins: (i) The pharmacochaperoning efficacy of compound 9b exceeded that of the parent compound noribogaine. (ii) Compound 9b had a therapeutic window in vivo (i.e., there was an effective dose range where it restored sleep in drosophilae harboring DAT-PG584,585AA). This effect was specifically linked to the pharmacochaperoning action, because compound 9b did not affect locomotion and sleep in control flies (harboring endogenous DAT) or fmn flies lacking DAT. (iii) Compound 9b restored folding to disease-associated DAT mutant proteins, which were unresponsive to noribogaine, thus doubling the number of DAT mutant proteins potentially amenable to folding correction.
The mechanistic basis underlying pharmacochaperoning is poorly understood. At least four mechanisms are conceivable: (i) stabilization of the native state or (ii) of a folding intermediate, (iii) prevention of aggregate formation, or (iv) dissolution of aggregates.41 Stabilization of the native state is posited to be the most common mechanism of action for pharmacochaperones. In this instance, chaperoning efficacy is related to binding affinity.41 Our observations are difficult to reconcile with binding to the native state, because (i) the structure–activity relationships for binding to the WT transporters (i.e., the native state) differed substantially from those for pharmacochaperoning. (ii) The EC50 values for rescuing DAT-PG584,585AA and SERT-PG601,602AA differed by 400-fold. This difference was substantially larger than variation in affinity for the native states of SERT and DAT. This indicates that compound 9b has a high and low affinity for the relevant folding intermediate(s) of SERT-PG601,602AA and of DAT-PG584,585AA, respectively. (iii) The relative pharmacochaperoning efficacies of ibogaine, noribogaine, and compound 9b were highly dependent on the nature of the DAT mutant protein: In DAT-PG584,585AA and in several disease-relevant DAT mutations (i.e., DAT-G386R, -R521W, -P395L, P554L), compound 9b was substantially more efficacious than were ibogaine and noribogaine, but this was not the case in DAT-A314V and DAT-R445C. This variation in relative efficacy is again difficult to reconcile with stabilization of the native state. (iv) Circumstantial evidence also argues that the other two proposed mechanisms of pharmacochaperoning (inhibition of aggregation and disassembly of aggregates) do not apply: Diseases arising from mutations in a SLC6 transporter can be transmitted in both a recessive and a dominant fashion. Because SLC6 transporters are exported from the ER in an oligomeric form, folding-deficient mutaions can act in a dominant-negative manner and retain the WT transporter.42,43 However, all disease-relevant human DAT mutations are transmitted as recessive alleles.17,18 Thus, their folding trajectory is stalled at a stage where they are complexed with and shielded by ER chaperones such as calnexin, which precludes oligomerization.44 Taken together, these observations indicate that binding to folding intermediate(s) is the most likely mechanism underlying the pharmacochaperoning action of compound 9b.
For most compounds, the Ki for binding inhibition was lower than the IC50 for uptake inhibition. This is not surprising, because uptake inhibition is measured in intact cells, where the transport cycle is driven by ionic gradients and multiple conformations are visited. In contrast, in membrane binding assays, the inward- and the outward-facing state are confronted with the same ionic concentrations. This allows for conformational trapping to be more pronounced.24,25 The ratio of IC50 (for uptake inhibition) over Ki varied by up to 8-fold. However, it was not a useful guide to identify pharmacochaperones: In hDAT, for instance, the IC50/Ki ratios of compounds 3a and 3b were comparable to that of compound 9b, but their pharmacochaperoning efficacies was >40-fold lower (cf. Table 1). The fact that large IC50/Ki ratios fail to identify effective pharmacochaperones is not surprising: While it is indicative of conformational trapping, the trapped conformation need not be relevant to progress in the folding trajectory.
Individual folding-deficient mutant proteins of SERT and DAT are stalled at different points of their folding trajectory and differ in their susceptibility to pharmacochaperoning.12,13,15,16,30 Our approach relied on using the two analogous folding-deficient mutations SERT-PG601,602AA and DAT-PG584,585AA by assuming that these two proteins are stalled at related positions of their folding trajectory. However, compound 9b was more than 100-fold more potent in rescuing SERT-PG601,602AA than DAT-PG584,585AA. A similar discrepancy was seen in the naphthylamine series of pharmacochaperones:25 PAL-287 was more effective than PAL-1045 in rescuing DAT-PG584,585AA; the reverse was true for SERT-PG601,602AA (cf. Figure 3 and ref (25)). Taken together, these observations are again consistent with the conjecture that the compounds act as pharmacochaperones by binding to folding intermediates rather than by stabilizing the native state. The hypothetical model posits that in the folding trajectory of WT SLC6 transporters there is a large isoenergetic conformational search space. Mutations convert this smooth surface into a rugged landscape. Accordingly, they create multiple traps which reside at different locations in the peripheral ring of the champagne-glass-like energy landscape.45 Some of these traps can be overcome by pharmacochaperoning, but they differ, even for closely related folding-deficient mutations, in their position in the conformational search space.14 Hence, the folding intermediates in the vicinity of the trap(s) differ in their ability to bind to and respond to a pharmacochaperoning ligand. General rules which govern folding of helical membrane proteins have been inferred, but the details are obscure.41,42 Progress is hampered because the nature of the folding intermediate(s) is poorly understood. The affinity of compound 9b for the folding intermediate(s) of SERT-PG601,602AA was estimated to be in the submicromolar range. Hence, compound 9b may be useful as a starting point to develop probes to address the folding trajectory of SERT-PG601,602AA and other misfolded SERT variants. We anticipate that the resulting insights will also advance the search for additional pharmacochaperones. These are needed because the majority of disease-associated DAT mutations are still not remedied by the available pharmacochaperones. Moreover, compound 9b is a significant breakthrough, because it not only expands the number of rescued DAT mutant proteins but also restores the functional activity of DAT-G386R essentially to WT levels. Thus, compound 9b provides proof-of-principle that it is possible to fully correct the folding defect of a mutant protein by pharmacochaperoning.
Material and Methods
Cell Culture and Materials
Cells were propagated Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 units·100 mL–1 penicillin and 100 units·100 mL–1 streptomycin. The medium used in the maintenance of stable lines (see below) was also supplemented with 50 μg mL–1 Geneticin (G418) for selection. HEK293 cells were transfected by combining plasmid DNA with PEI (linear 25 kDa polyethylenimine; Santa Cruz, SC-360988A) at a ratio of 1:3 (w/w) in serum-free DMEM. The plasmids encoding YFP-tagged human wild-type and mutant versions of SERT12 and of DAT14,16 were previously described; YFP-tagged DAT-PG584,585AA was created by introducing the substitutions created with the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA), using wild-type YFP-tagged DAT as the template. The mutations were confirmed by automatic DNA sequencing (LGC Labor GmbH Augsburg, Germany). For the pharmacochaperoning experiments, noribogaine was purchased from Cfm Oskar Tropitzsch GmbH (Marktredwitz, Germany). [3H]5-HT (serotonin, 41.3 Ci/mmol) and [3H]dopamine ([3H]DA, 39.6 Ci/mmol) were purchased from PerkinElmer Life Sciences. Scintillation mixture (Rotiszint eco plus) was purchased from Carl Roth GmbH (Karlsruhe, Germany). Cell culture media and antibiotics were obtained from Sigma and Invitrogen, respectively. Anti-GFP antibody (rabbit, ab290) was from Abcam (Cambridge, UK). An antibody raised against an N-terminal peptide of the G protein β subunit was used to verify comparable loading of lanes.46 Horseradish-peroxidase-linked anti-rabbit IgG1 antibody was purchased from Amersham Biosciences. All other chemicals were analytical-grade.
Radioligand Binding Studies
For DAT binding assays, frozen striata previously dissected from freshly harvested male Sprague–Dawley rat brains (supplied on ice by Bioreclamation, Hicksville, NY) were homogenized in 20 volumes (w/v) of ice-cold modified sucrose phosphate buffer (0.32 M sucrose, 7.74 mM Na2HPO4, and 2.26 mM NaH2PO4, pH adjusted to 7.4) using a Brinkman Polytron (Setting 6 for 20 s) and centrifuged at 48 400g for 10 min at 4 °C. The resulting pellet was washed by resuspension in buffer. The suspension was centrifuged again, and the final pellet was resuspended in ice-cold buffer to a concentration of 20 mg/mL (original wet weight/volume, OWW/V). Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentrations of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of sucrose phosphate buffer, 50 μL of [3H]WIN 35 428 (final concentration 1.5 nM; KD = 28.2 nM; PerkinElmer Life Sciences, Waltham, MA), and 100 μL of tissue (2.0 mg/well OWW). All compound dilutions were tested in triplicate, and the competition reactions were started with the addition of tissue. The plates were incubated for 120 min at 0–4 °C. Nonspecific binding was determined using 10 μM indatraline. Alternatively, ibogaine was allowed to displace binding of [3H]WIN 35 428 (final concentration 10 nM) to membranes (8–10 μg/assay), which were prepared from HEK293 cells stably expressing hDAT under conditions outlined previously.47
For SERT binding assays, frozen brain stem tissue previously dissected from freshly harvested male Sprague–Dawley rat brains (supplied on ice by Bioreclamation, Hicksville, NY) was homogenized in 20 volumes (w/v) of 50 mM Tris buffer (120 mM NaCl and 5 mM KCl, adjusted to pH 7.4) at 25 °C using a Brinkman Polytron (at setting 6 for 20 s) and centrifuged at 48 400g for 10 min at 4 °C. The resulting pellet was resuspended in buffer. The suspension was centrifuged, and the final pellet was suspended in buffer again to a concentration of 20 mg/mL (OWW/V). Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentration solutions of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of Tris buffer, 50 μL of [3H]citalopram (final concentration 1.5 nM; KD = 6.91 nM; PerkinElmer Life Sciences, Waltham, MA), and 100 μL of tissue (2.0 mg/well OWW). All compound dilutions were tested in triplicate, and the competition reactions started with the addition of tissue, and the plates were incubated for 60 min at room temperature. Nonspecific binding was determined using 10 μM fluoxetine.
For all binding assays, incubations were terminated by rapid filtration through PerkinElmer Uni-Filter-96 GF/B presoaked in either 0.3% (SERT) or 0.05% (DAT) polyethylenimine, using a Brandel 96-well harvester manifold or Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed a total of 3 times with 3 mL (3 × 1 mL/well or 3 × 1 mL/tube) of ice-cold binding buffer. PerkinElmer MicroScint 20 Scintillation Cocktail (65 μL) was added to each filter well. Radioactivity was counted in a PerkinElmer MicroBeta Microplate Counter. IC50 values for each compound were determined from inhibition curves and Ki values were calculated using the Cheng-Prusoff equation. Kd values for the radioligands were determined via separate homologous competitive binding or radioligand binding saturation experiments. Ki values were determined from at least 3 independent experiments performed in triplicate and are reported as means ± SD.
[3H]5-HT and [3H]Dopamine Uptake Assays
For uptake inhibition assays, HEK293 cells stably expressing either wild-type human YFP-tagged hSERT or YFP-tagged hDAT were seeded on poly-d-lysine-coated 96-well plates at a density of ∼20 000 cells/well. After 24 h, the medium in each well was aspirated, and the cells were washed once with Krebs-HEPES buffer (10 mM HEPES·NaOH, pH 7.4, 120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 2 mM glucose). Cells were preincubated in buffer containing logarithmically spaced concentrations (0.003–300 μM) of ibogaine analogs for 10 min. Subsequently, the reaction was started by addition of substrate (0.4 μM of either [3H]5-HT or [3H]dopamine) at constant concentrations of ibogaine analogs for 1 min. The reaction was terminated by aspirating the reaction medium followed by a wash with ice-cold buffer. The cells were lysed with 1% SDS, and the released radioactivity was quantified by liquid scintillation counting.
For uptake assays determining the functional rescue of mutant transporters, HEK293 cells were transfected with either YFP-tagged SERT-PG601,602AA or YFP-tagged DAT-PG584,585AA plasmids. Transfected cells were seeded on poly-d-lysine-coated 96-well plates at a density of ∼60–80 000 cells/well in either the absence or presence of increasing concentrations (0.1–100 μM) of the ibogaine analogs. After 24 h, the cells were washed four times with Krebs-MES buffer (10 mM 2-(N-morpholino)ethanesulfonic acid, pH 5.5, 120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 2 mM glucose) in a 10 min interval and once with Krebs-HEPES (pH 7.4) buffer. The cells were subsequently incubated with 0.2 μM of [3H]5-HT for 1 min or [3H]dopamine for 5 min and processed as outlined above.
Immunoblotting after Pharmacochaperoning of SERT-PG601,602AA or DAT-PG584,585AA
HEK293 cells were transiently transfected with plasmids encoding either wild-type SERT, SERT-PG601,602AA, wild-type DAT, or DAT-PG584,585AA. Approximately 1.5–2 × 106 of these transfected cells were seeded either in 6-well plates or 6 cm dishes in the presence of individual candidate hits (30 μM) identified by uptake assays. After 24 h, cells were washed thrice with ice-cold phosphate-buffered saline, detached by mechanical scraping, and harvested by centrifugation at 1000g for 5 min. The cell pellet was lysed in a buffer containing Tris·HCl, pH 8.0, 150 mm NaCl, 1% dodecyl maltoside, 1 mm EDTA, and protease inhibitors (Complete, Roche Applied Science). This soluble protein lysate was separated from the detergent-insoluble material by centrifugation (16 000g for 15 min at 4 °C). An aliquot of this lysate (20 μg) was mixed with 1% SDS and 20 mM DTT containing sample buffer, denatured at 45 °C for 30 min, and resolved in denaturing polyacrylamide gels. After protein transfer onto nitrocellulose membranes, the blots were probed with an antibody against GFP (rabbit, ab290) at a 1:3000 dilution overnight. This immunoreactivity was detected using a horseradish peroxidase conjugated secondary antibody (1:5000, Amersham ECL Prime Western Blotting Detection Reagent). In separate experiments, lysates were prepared from cells treated in the absence or presence of 30 μM DG4–69; these were incubated in the presence and absence of endoglycosidase H (New England Biolabs),16 and aliquots (20 μg) were then resolved electrophoretically as described above. Densitometric analyses of individual blots were done using ImageJ.
Fly Genetics, Treatment and Locomotion Assay
The transgenic UAS reporter line for YFP-tagged hDAT-PG584, 585AA was generated using pUASg-attB vector (gift from Dr. Bischof and Dr. Basler, University of Zürich). The sequenced construct was injected into embryos from ZH-86Fb flies (Bloomington stock no 24749). Positive transformants were selected and crossed with balancer flies (Bloomington stock no. 3704). Fumin flies (fmn or DAT-KO mutant flies) were a generous gift from Dr. Kume, Nagoya City University, Japan. Tyrosine hydroxylase Gal4 (TH-Gal4, Bloomington stock no. 8848) was used to drive the expression of hDAT-PG584, 585AA in dopaminergic neurons. Isogenized fmn and w1118 flies were used as control. TH-Gal4 was isogenized in the w1118 background, and the UAS-hDAT-PG584,585AA insertion was stabilized using balancer flies (Bloomington stock no. 3704, w1118/Dp(1;Y)y[+]; CyO/Bl[1]; TM2/TM6B, Tb[1]). The genotypes of flies used in Figure 4a,b were w1118;fmn(w; roo{}DATfmn);TH-Gal4/UAS-hDAT-PG584,585AA (PG584,585AA), w1118/y;fmn(w; roo{}DATfmn)+/+ (fmn), and w1118. All flies were kept at 25 °C in a 12 h:12 h light/dark cycle, and all crosses were performed at 25 °C. As described previously,14 the locomotion assay was performed on 3–5 days old male flies using Drosophila activity monitor system (DAM2, Trikinetics, Waltham, MA). Briefly, individual flies were housed in 5 mm diameter glass tubes carrying food pellets supplemented with specified concentrations of noribogaine and 9b. Flies were first entrained for a 12 h:12 h day/night rhythm for 2 days, and locomotion activity was studied on the second day in a subsequent 12 h:12 h dark/dark phase. Locomotion activity was measured in 1 min bins, and pySolo software48 was used to quantify sleep time.
Statistics
Individual data points in dot plots represent means from triplicate determination in independent experiments. Affinity estimates (KD, EC50, IC50) were obtained by subjecting the data points to nonlinear least-squares curve fitting the appropriate equation (for a rectangular hyperbola or for a monophasic inhibition). Differences between two groups were examined for statistical significance by a t-test or by a Mann–Whitney U-test, depending on the distribution of individual values. Similarly, differences between more than two groups were evaluated by a one-way analysis of variance (ANOVA) or a Kruskal–Wallis test followed by post hoc tests (Dunn’s or Dunnett’s multiple comparisons for testing against the control or all possible combinations, respectively).
Acknowledgments
This work was supported by a grant (LSC17-026 to M.F.) from the Vienna Science and Technology Fund/WWTF.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00102.
Chemical synthesis of compounds 3a–3f, 4a–4d, 6, 7a, 7b, 8a–8c, and 9a–9d; inhibition by ibogaine analogs of [3H]citalopram binding to rat SERT; inhibition by ibogaine analogs of [3H]WIN 35,428 binding to rat and human DAT; inhibition by ibogaine analogs of [3H]5-HT uptake by hSERT; inhibition by ibogaine analogs of [3H]5-DA uptake by hDAT (PDF)
Author Contributions
# S.B. and D.A.G. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Dybowski J.; Landrin E. (1901) Plant Chemsitry. Concerning Iboga, its excitement-producing properties, its composition, and the new alkaloid it contains, ibogaine. C. R. Acad. Sci. 133, 748. [Google Scholar]
- Wasko M. J.; Witt-Enderby P. A.; Surratt C. K. (2018) DARK classics in chemical neuroscience: ibogaine. ACS Chem. Neurosci. 9, 2475–2483. 10.1021/acschemneuro.8b00294. [DOI] [PubMed] [Google Scholar]
- Corkery J. M. (2018) Ibogaine as a treatment for substance misuse: Potential benefits and practical dangers. Prog. Brain Res. 242, 217–257. 10.1016/bs.pbr.2018.08.005. [DOI] [PubMed] [Google Scholar]
- Brown T. K.; Alper K. (2018) Treatment of opioid use disorder with ibogaine: detoxification and drug use outcomes. Am. J. Drug Alcohol Abuse 44, 24–36. 10.1080/00952990.2017.1320802. [DOI] [PubMed] [Google Scholar]
- Noller G. E.; Frampton C. M.; Yazar-Klosinski B. (2018) Ibogaine treatment outcomes for opioid dependence from a twelve-month follow-up observational study. Am. J. Drug Alcohol Abuse 44, 37–46. 10.1080/00952990.2017.1310218. [DOI] [PubMed] [Google Scholar]
- Glick S. D.; Rossman K.; Steindorf S.; Maisonneuve I. M.; Carlson J. N. (1991) Effects and after effects of ibogaine on morphine self-administration in rats. Eur. J. Pharmacol. 195, 341–345. 10.1016/0014-2999(91)90474-5. [DOI] [PubMed] [Google Scholar]
- Glick S. D.; Kuehne M. E.; Raucci J.; Wilson T. E.; Larson D.; Keller R. W. Jr.; Carlson J. N. (1994) Effects of iboga alkaloids on morphine and cocaine self-administration in rats: relationship to tremorigenic effects and to effects on dopamine release in nucleus accumbens and striatum. Brain Res. 657, 14–22. 10.1016/0006-8993(94)90948-2. [DOI] [PubMed] [Google Scholar]
- Blackburn J.; Szumlinski K. K. (1997) Ibogaine effects on sweet preference and amphetamine induced locomotion: implications for drug addiction. Behav. Brain Res. 89, 99–106. 10.1016/S0166-4328(97)00050-8. [DOI] [PubMed] [Google Scholar]
- Mash D. C.; Staley J. K.; Baumann M. H.; Rothman R. B.; Hearn W. L. (1995) Identification of a primary metabolite of ibogaine that targets serotonin transporters and elevates serotonin. Life Sci. 57, PL45–PL50. 10.1016/0024-3205(95)00273-9. [DOI] [PubMed] [Google Scholar]
- Sweetnam P. M.; Lancaster J.; Snowman A.; Collins J. L.; Perschke S.; Bauer C.; Ferkany J. (1995) Receptor binding profile suggests multiple mechanisms of action are responsible for ibogaine’s putative anti-addictive activity. Psychopharmacol (Berl) 118, 369–376. 10.1007/BF02245936. [DOI] [PubMed] [Google Scholar]
- El-Kasaby A.; Just H.; Malle E.; Stolt-Bergner P. C.; Sitte H. H.; Freissmuth M.; Kudlacek O. (2010) Mutations in the carboxyl-terminal SEC24 binding motif of the serotonin transporter impair folding of the transporter. J. Biol. Chem. 285, 39201–39210. 10.1074/jbc.M110.118000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kasaby A.; Koban F.; Sitte H. H.; Freissmuth M.; Sucic S. (2014) A cytosolic relay of heat shock proteins HSP70–1A and HSP90β monitors the folding trajectory of the serotonin transporter. J. Biol. Chem. 289, 28987–29000. 10.1074/jbc.M114.595090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koban F.; El-Kasaby A.; Häusler C.; Stockner T.; Simbrunner B. M.; Sitte H. H.; Freissmuth M.; Sucic S. (2015) A salt bridge linking the first intracellular loop with the C terminus facilitates the folding of the serotonin transporter. J. Biol. Chem. 290, 13263–13278. 10.1074/jbc.M115.641357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasture A.; El-Kasaby A.; Szöllősi D.; Asjad H. M. M.; Grimm A.; Stockner T.; Hummel T.; Freissmuth M.; Sucic S. (2016) Functional rescue of a misfolded Drosophila melanogaster dopamine transporter mutant associated with a sleepless phenotype by pharmacological chaperones. J. Biol. Chem. 291, 20876–20890. 10.1074/jbc.M116.737551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerepoot P.; Lam V. M.; Salahpour A. (2016) Pharmacological chaperones of the dopamine transporter rescue dopamine transporter deficiency syndrome mutations in heterologous cells. J. Biol. Chem. 291, 22053–22062. 10.1074/jbc.M116.749119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asjad H. M. M.; Kasture A.; El-Kasaby A.; Sackel M.; Hummel T.; Freissmuth M.; Sucic S. (2017) Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism. J. Biol. Chem. 292, 19250–19265. 10.1074/jbc.M117.797092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian M. A.; Li Y.; Zhen J.; Meyer E.; Hai N.; Christen H. J.; Hoffmann G. F.; Jardine P.; von Moers A.; Mordekar S. R.; O’Callaghan F.; Wassmer E.; Wraige E.; Dietrich C.; Lewis T.; Hyland K.; Heales S. Jr.; Sanger T.; Gissen P.; Assmann B. E.; Reith M. E.; Maher E. R. (2011) Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 10, 54–62. 10.1016/S1474-4422(10)70269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J.; Zhen J.; Meyer E.; Erreger K.; Li Y.; Kakar N.; Ahmad J.; Thiele H.; Kubisch C.; Rider N. L.; et al. (2014) Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 137, 1107–1119. 10.1093/brain/awu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen A. S.; Andersen J.; Jørgensen T. N.; Sørensen L.; Eriksen J.; Loland C. J.; Strømgaard K.; Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640. 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Sitte H. H.; Freissmuth M. (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 36, 41–50. 10.1016/j.tips.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K. C.; Rothman R. B.; Reith M. E. (2013) Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators and partial substrates. J. Pharmacol. Exp. Ther. 346, 2–10. 10.1124/jpet.111.191056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith M. E.; Blough B. E.; Hong W. C.; Jones K. T.; Schmitt K. C.; Baumann M. H.; Partilla J. S.; Rothman R. B.; Katz J. L. (2015) Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend. 147, 1–19. 10.1016/j.drugalcdep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S.; Newman A. H.; Freissmuth M. (2019) How to rescue misfolded SERT, DAT and NET: targeting conformational intermediates with atypical inhibitors and partial releasers. Biochem. Soc. Trans. 47, 861–874. 10.1042/BST20180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenhuetl P. S.; Bhat S.; Freissmuth M.; Sandtner W. (2019) Functional selectivity and partial efficacy at the monoamine transporters: a unified model of allosteric modulation and amphetamine-induced substrate release. Mol. Pharmacol. 95, 303–312. 10.1124/mol.118.114793. [DOI] [PubMed] [Google Scholar]
- Bhat S.; Hasenhuetl P. S.; Kasture A.; El-Kasaby A.; Baumann M. H.; Blough B. E.; Sucic S.; Sandtner W.; Freissmuth M. (2017) Conformational state interactions provide clues to the pharmacochaperone potential of serotonin transporter partial substrates. J. Biol. Chem. 292, 16773–16786. 10.1074/jbc.M117.794081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M. T.; Zhang Y. W.; Campbell S. D.; Rudnick G. (2007) Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J. Biol. Chem. 282, 29441–29447. 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- Bulling S.; Schicker K.; Zhang Y. W.; Steinkellner T.; Stockner T.; Gruber C. W.; Boehm S.; Freissmuth M.; Rudnick G.; Sitte H. H.; Sandtner W. (2012) The mechanistic basis for noncompetitive ibogaine inhibition of serotonin and dopamine Transporters. J. Biol. Chem. 287, 18524–18534. 10.1074/jbc.M112.343681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtscher V.; Hotka M.; Li Y.; Freissmuth M.; Sandtner W. (2018) A label-free approach to detect ligand binding to cell surface proteins in real time. eLife 7, e34944 10.7554/eLife.34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. A.; Yang D.; Zhao Z.; Wen P. C.; Yoshioka C.; Tajkhorshid E.; Gouaux E. (2019) Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 569, 141–145. 10.1038/s41586-019-1135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freissmuth M.; Stockner T.; Sucic S. (2017) SLC6 Transporter folding diseases and pharmacochaperoning. Handb. Exp. Pharmacol. 245, 249–270. 10.1007/164_2017_71. [DOI] [PubMed] [Google Scholar]
- Kruegel A. C.; Rakshit S.; Li X.; Sames D. (2015) Constructing Iboga alkaloids via C-H bond functionalization: examination of the direct and catalytic union of heteroarenes and isoquinuclidine alkenes. J. Org. Chem. 80, 2062–2071. 10.1021/jo5018102. [DOI] [PubMed] [Google Scholar]
- Gassaway M. M.; Jacques T. L.; Kruegel A. C.; Karpowicz R. J. Jr.; Li X.; Li S.; Myer Y.; Sames D. (2016) Deconstructing the iboga alkaloid skeleton: potentiation of FGF2-induced glial cell line-derived neurotrophic factor release by a novel compound. ACS Chem. Biol. 11, 77–87. 10.1021/acschembio.5b00678. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Godleski S. A.; Genet J. P. (1978) A total synthesis of racemic and optically active ibogamine. Utilization and mechanism of a new silver ion assisted palladium catalyzed cyclization. J. Am. Chem. Soc. 100, 3930–3931. 10.1021/ja00480a047. [DOI] [Google Scholar]
- Rothman R. B.; Partilla J. S.; Baumann M. H.; Lightfoot-Siordia C.; Blough B. E. (2012) Studies of the biogenic amine transporters. 14. Identification of low-efficacy “partial” substrates for the biogenic amine transporters. J. Pharmacol. Exp. Ther. 341, 251–262. 10.1124/jpet.111.188946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M.; Sorkina T.; Grammatopoulos T. N.; Zawada W. M.; Sorkin A. (2004) Multiple molecular determinants in the carboxyl terminus regulate dopamine transporter export from endoplasmic reticulum. J. Biol. Chem. 279, 30760–30770. 10.1074/jbc.M312774200. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Liu S.; Kodama L.; Driscoll M. R.; Wu M. N. (2012) Two dopaminergic neurons signal to the dorsal fan-shaped body to promote wakefulness in Drosophila. Curr. Biol. 22, 2114–2123. 10.1016/j.cub.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel D.; Donlea J. M.; Talbot C. B.; Song S. M.; Thurston A. J. F.; Miesenböck G. (2016) Operation of a homeostatic sleep switch. Nature 536, 333–337. 10.1038/nature19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K.; Kume S.; Park S. K.; Hirsh J.; Jackson F. R. (2005) Dopamine is a regulator of arousal in the fruit fly. J. Neurosci. 25, 7377–7384. 10.1523/JNEUROSCI.2048-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friggi-Grelin F.; Coulom H.; Meller M.; Gomez D.; Hirsh J.; Birman S. (2003) Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J. Neurobiol. 54, 618–627. 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Ueno T.; Tomita J.; Kume S.; Kume K. (2012) Dopamine modulates metabolic rate and temperature sensitivity in Drosophila melanogaster. PLoS One 7, e31513 10.1371/journal.pone.0031513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinko J. T.; Huang H.; Penn W. D.; Capra J. A.; Schlebach J. P.; Sanders C. R. (2019) Folding and Misfolding of Human Membrane Proteins in Health and Disease: from single molecules to Cellular proteostasis. Chem. Rev. 119, 5537–5606. 10.1021/acs.chemrev.8b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba P.; Freissmuth M.; Stockner T. (2014) Defining the blanks--pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol. Res. 83, 63–73. 10.1016/j.phrs.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Corcuera B.; Arribas-González E.; Aragón C. (2019) Hyperekplexia-associated mutations in the neuronal glycine transporter 2. Neurochem. Int. 123, 95–100. 10.1016/j.neuint.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Korkhov V. M.; Milan-Lobo L.; Zuber B.; Farhan H.; Schmid J. A.; Freissmuth M.; Sitte H. H. (2008) Peptide-based interactions with calnexin target misassembled membrane proteins into endoplasmic reticulum-derived multilamellar bodies. J. Mol. Biol. 378, 337–352. 10.1016/j.jmb.2008.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K. A.; Chan H. S. (1997) From Levinthal to pathways to funnels. Nat. Struct. Mol. Biol. 4, 10–19. 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Hohenegger M.; Mitterauer T.; Voss T.; Nanoff C.; Freissmuth M. (1996) Thiophosphorylation of the G protein β-subunit in human platelet membranes: evidence against a direct phosphate transfer reaction to Gα-subunits. Mol. Pharmacol. 49, 73–80. [PubMed] [Google Scholar]
- Sucic S.; Dallinger S.; Zdrazil B.; Weissensteiner R.; Jørgensen T. N.; Holy M.; Kudlacek O.; Seidel S.; Cha J. H.; Gether U.; Newman A. H.; Ecker G. F.; Freissmuth M.; Sitte H. H. (2010) The N terminus of monoamine transporters is a lever required for the action of amphetamines. J. Biol. Chem. 285, 10924–10938. 10.1074/jbc.M109.083154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilestro G. F.; Cirelli C. (2009) pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics 25, 1466–1467. 10.1093/bioinformatics/btp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.