

Abstract
Cortical neuron atrophy is a hallmark of depression and includes neurite retraction, dendritic spine loss, and decreased synaptic density. Psychoplastogens, small molecules capable of rapidly promoting cortical neuron growth, have been hypothesized to produce long-lasting positive effects on behavior by rectifying these deleterious structural and functional changes. Here we demonstrate that ketamine and LSD, psychoplastogens from two structurally distinct chemical classes, promote sustained growth of cortical neurons after only short periods of stimulation. Furthermore, we show that psychoplastogen-induced cortical neuron growth can be divided into two distinct epochs: an initial stimulation phase requiring TrkB activation and a growth period involving sustained mTOR and AMPA receptor activation. Our results provide important temporal details concerning the molecular mechanisms by which next-generation antidepressants produce persistent changes in cortical neuron structure, and they suggest that rapidly excreted psychoplastogens might still be effective neurotherapeutics with unique advantages over compounds like ketamine and LSD.
Keywords: neural plasticity, psychoplastogen, psychedelic, LSD, ketamine, BDNF
Depression is among the leading causes of disability worldwide, affecting over 300 million people.1,2 Current treatments, such as the selective serotonin reuptake inhibitor (SSRI) fluoxetine, are only moderately effective, require daily administration for 2–4 weeks before producing beneficial effects, and are associated with a number of side effects leading to discontinuation of treatment regimens.3,4 Moreover, approximately one-third of patients are unresponsive to these medicines,4 highlighting the urgent need for new treatment approaches. A better understanding of depression pathophysiology will be necessary to rationally devise more effective therapeutics.
In recent years, it has become clear that depression results from deleterious structural and functional changes in key brain circuits. These include the retraction of dendrites, the elimination of dendritic spines, and the loss of excitatory synapses in the prefrontal cortex (PFC).5−8 Psychoplastogens, small molecules that promote the rapid regrowth of atrophied cortical dendritic arbors,9 represent the leading edge of antidepressant neurotherapeutics. They include ketamine,10−13 scopolamine,14−17 and serotonergic psychedelics.18−25 These compounds rapidly promote structural and functional neural plasticity in the cortex26−28 and produce long-lasting (>24 h) changes in mood and behavior without the need for chronic dosing,29−34 presumably due to their ability to rewire pathological neural circuitry.
Induced plasticity (iPlasticity) has been proposed as a potential unifying mechanism to explain the efficacy of antidepressants from different chemical classes.35,36 Traditional antidepressants like fluoxetine are believed to promote cortical neuron growth through transactivation of the tropomyosin receptor kinase B (TrkB),37−39 the high-affinity receptor for brain-derived neurotrophic factor (BDNF). Psychoplastogens produce more rapid changes in cortical neuron structure, but their mechanisms of action are still opaque. Understanding how psychoplastogens produce enduring changes in cortical neuron structure, despite being rapidly cleared from the body, will be critical if we are to rationally engineer safer and more efficacious medicines for treating depression.
Previously, we demonstrated that psychedelics could increase cortical neuron growth when treated for extended periods of time (24–72 h). Here, we establish that very short stimulation periods (15 min to 6 h) are sufficient for ketamine and lysergic acid diethylamide (LSD), two psychoplastogens from distinct chemical classes, to initiate a neuronal growth response characterized by sustained α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and mammalian target of rapamycin (mTOR) activation. This response persists even after the removal of the stimulating ligand. Our results indicate that ketamine and serotonergic psychoplastogens may lead to enduring changes in neuronal structure through a common downstream mechanism of action. Moreover, our results have important implications for central nervous system (CNS) drug development, as they suggest that intentional engineering of neurotherapeutics to be rapidly cleared from the body might be an effective strategy for maintaining efficacy while minimizing side effects.
Results
Transient Stimulation with Psychoplastogens Increases Dendritogenesis, Spinogenesis, and Synaptogenesis
Previously, we demonstrated that treatment of immature cortical cultures (3 days in vitro; DIV3) with psychoplastogens for 72 h significantly increased dendritic arbor complexity as measured via Sholl analysis.28 However, most psychoplastogens are rapidly cleared from the body (i.e., they have half-lives of several hours),40,41 with compounds such as N,N-dimethyltryptamine (DMT) and 5-MeO-DMT having half-lives less than 15 min.42,43 To determine the shortest period of stimulation necessary to induce neuronal growth, we treated DIV3 cortical cultures with either ketamine or LSD for varying lengths of time, removed the drug, and let the cultures mature until DIV6 (Figure 1A). Remarkably, both drugs were able to robustly increase dendritic arbor complexity after only 15 min of stimulation (Figure 1D), while exogenous BDNF (50 ng/mL) required a full hour to produce effects (Figure S1A). Treatment of cortical cultures with psychoplastogens for 1 h followed by a 71 h growth period resulted in Nmax values comparable to what we had previously observed using a 72 h treatment paradigm (Figure 1B,C). Concentration–response studies revealed that a concentration of 10 μM produced maximal effects on dendritic growth (Figure S2). When cortical cultures were fixed immediately following 1 h of treatment with psychoplastogens, no changes in dendritic arborization were observed (Figure 1E) indicating that there are distinct stimulation and growth phases.
Figure 1.
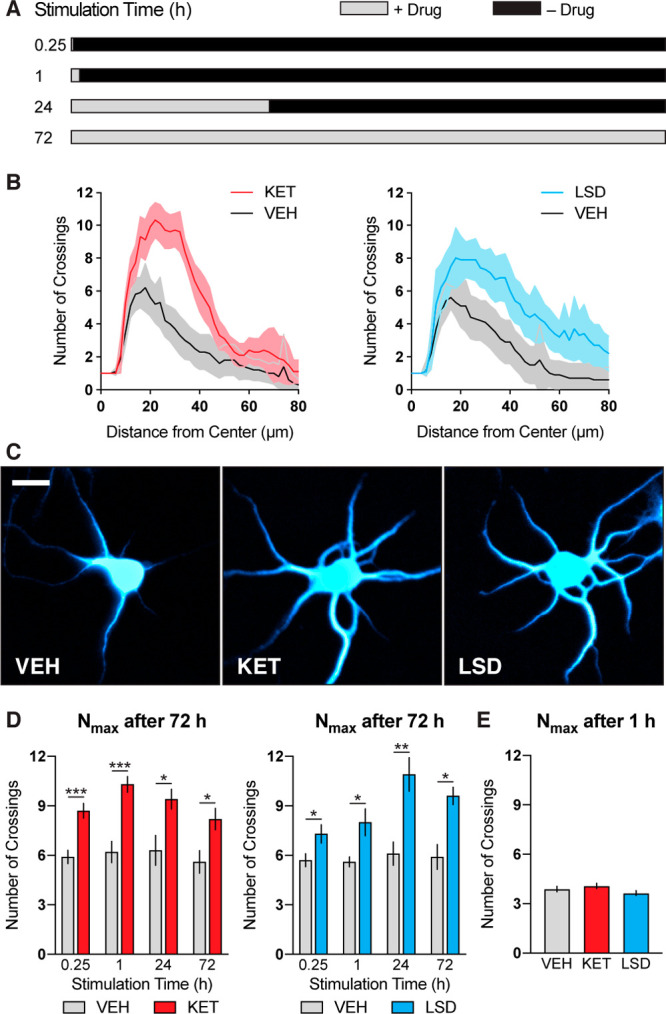
Transient stimulation with psychoplastogens is sufficient to induce dendritic growth. (A) Schematic illustrating experimental design. Cortical cultures (DIV3) were treated with psychoplastogens (10 μM) for short periods of time and then allowed to grow for a total of 72 h. (B) Sholl plots of DIV3 neurons stimulated with psychoplastogens for 1 h and then allowed to grow for 71 h. Shadings indicate 95% CI (C) Representative images of neurons used to make the Sholl plots in B. Scale bar = 10 μm. (D–E) Maximum number of crossings (Nmax) of the Sholl plots obtained from neurons that experienced both transient stimulation and growth phases (D, n = 10 neurons) or only a 1 h stimulation (E, n = 37 neurons). Data are represented as mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, as compared to VEH control. For D, multiple t tests were performed, and p values were corrected for multiple comparisons using the Holm-Sidak method. For E, a one-way analysis of variance with Dunnett’s post hoc test was utilized. VEH = vehicle; KET = ketamine; LSD = lysergic acid diethylamide. See also Figure S1.
Next, we determined whether transient psychoplastogen stimulation was sufficient to induce dendritic spine growth, as we have often observed parallels between compound-induced changes in spine density and dendritic arbor complexity. As with dendritic growth, transient stimulation of cortical neurons (DIV19) with ketamine or LSD followed by a growth period in the absence of drug was sufficient to induce large increases in dendritic spine density (Figure 2), with LSD producing effects similar in magnitude to those produced by BDNF (Figure S1B) and ketamine inducing more modest changes. A 6 h stimulation period appears to be optimal for increasing dendritic spine density. In general, longer stimulation periods produce smaller changes in both dendrite and spine growth, which we hypothesize might be due to homeostatic mechanisms resulting from excessive neuronal excitation.44
Figure 2.
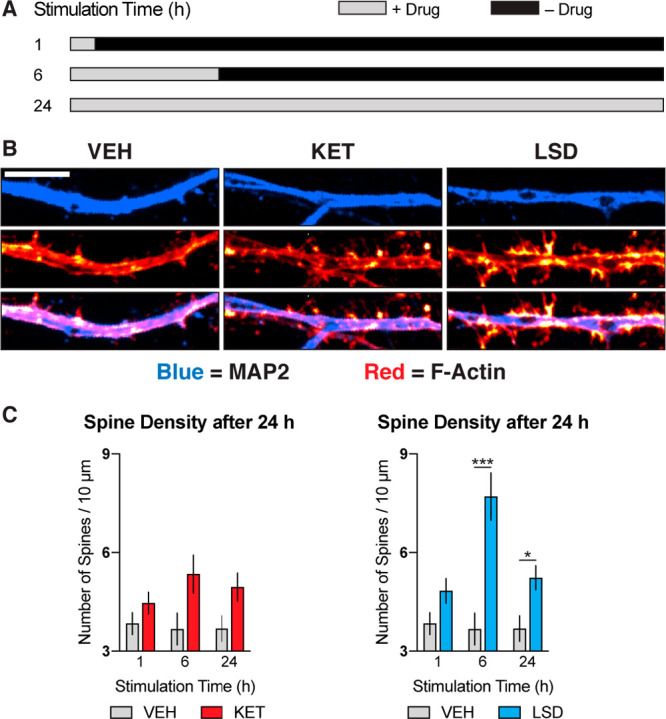
Transient stimulation with psychoplastogens is sufficient to induce spinogenesis. (A) Schematic illustrating experimental design. Cortical cultures (DIV19) were treated with psychoplastogens (10 μM) for short periods of time and then allowed to grow for a total of 24 h. (B) Representative images of secondary dendritic branches stimulated with psychoplastogens for 6 h and then allowed to grow for 18 h. Dendrites and F-actin were imaged using an anti-MAP2 antibody and fluorescent phalloidin conjugate, respectively. In the colocalization channel, dendritic spines can be visualized as red F-actin-rich protrusions emanating from blue dendrites. Scale bar = 5 μm. (C) Quantification of dendritic spine density. Data are represented as mean ± SEM (n = 11–30 neurons). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, as compared to VEH control following multiple t tests and correction of p values for multiple comparisons using the Holm–Sidak method. VEH = vehicle; KET = ketamine; LSD = lysergic acid diethylamide. See also Figure S1.
To determine the effect of transient psychoplastogen stimulation on synapse formation, we performed colocalization experiments of pre- (VGLUT1) and postsynaptic (PSD-95) markers. Psychoplastogen-induced increases in synapse density mirrored changes in spine density with a 6 h stimulation/18 h growth period producing the greatest effects (Figure 3). Stopping the experiment after 6 h of stimulation resulted in a much smaller change (Figure 3D), indicating that a growth phase following stimulation is necessary to achieve maximal increases in synapse density. While the effects of psychoplastogens on cortical structural plasticity (i.e., dendrite and spine growth) closely resemble those induced by BDNF, their effects on synaptogenesis are markedly distinct. BDNF induces a noticeably larger (∼6-fold increase) and more rapid (15 min stimulation) increase in synaptogenesis (Figure S1C,D) than do psychoplastogens (∼2-fold increase, 6 h stimulation). These results have important implications for the therapeutic properties of current and next-generation psychoplastogens. First, they suggest that psychoplastogens (10 μM) and BDNF (50 ng/mL) produce maximal increases in cortical structural plasticity (dendrite and spine growth) at these concentrations. Our previous work supports such a ceiling effect, as a combination of BDNF and a psychoplastogen did not result in additive or synergistic effects on neuronal growth.28 Second, these results suggest that a psychoplastogen with BDNF-like synaptogenic efficacy has yet to be discovered, highlighting an important area for future drug discovery. As synaptogenic effects can be observed prior to substantial increases in spine growth (Figure S1), it appears that these two processes are distinct. This conclusion is further supported by the recent discovery of compounds capable of increasing synapse density while concomitantly decreasing spine density.45
Figure 3.
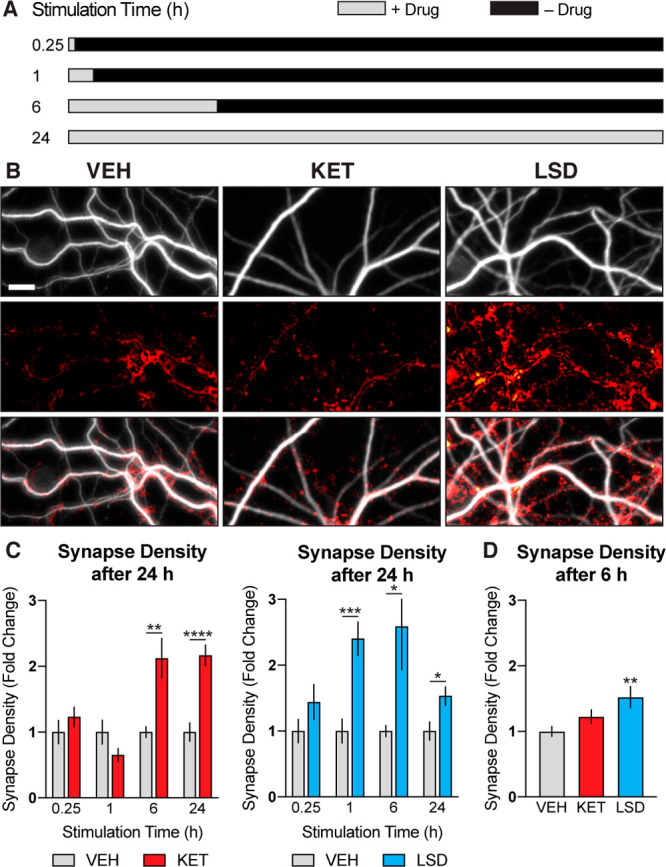
Transient stimulation with psychoplastogens is sufficient to induce synaptogenesis. (A) Schematic illustrating experimental design. Cortical cultures (DIV19) were treated with psychoplastogens (10 μM) for short periods of time and then allowed to grow for a total of 24 h. (B) Representative images of dendritic branches stimulated with psychoplastogens for 6 h and then allowed to grow for 18 h. Dendrites were imaged using an anti-MAP2 antibody (gray). Synapses are represented as colocalization of VGLUT1 and PSD-95 channels (red). Scale bar = 10 μm. (C) Quantification of synapse density (n = 25–27 neurons) from neurons that experienced both transient stimulation and growth phases. (D) Quantification of synapse density (n = 50–54 neurons) from neurons that experienced a 6 h stimulation only. Data are represented as mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, as compared to VEH control. For C, multiple t tests were performed, and p values were corrected for multiple comparisons using the Holm–Sidak method. For D, a one-way analysis of variance with Dunnett’s post hoc test was utilized. VEH = vehicle; KET = ketamine; LSD = lysergic acid diethylamide. See also Figure S1.
Psychoplastogen-Induced Growth Requires Sustained AMPA Receptor and mTOR Activation
Having firmly established that transient stimulation with ketamine and LSD is sufficient to induce cortical neuron growth, we next used several pharmacological inhibitors to probe which biochemical signaling pathways are essential during the stimulation and growth periods (Figure 4A). The leading hypothesis concerning the mechanism of antidepressant psychoplastogens involves a compound-induced glutamate burst in the cortex resulting in BDNF secretion, TrkB stimulation, and ultimately mTOR activation.28,46 Duman and co-workers have proposed that ketamine and scopolamine initiate this glutamate burst by blocking NMDA and muscarinic receptors on inhibitory neurons, respectively,47 while Aghajanian and co-workers have shown that psychedelics increase cortical glutamate levels by activating 5-HT2A receptors on excitatory neurons.48 Treating cortical neurons with DNQX, an AMPA receptor blocker, during the initial stimulation phase blocked dendritic growth, as expected (Figure 4B). However, we were surprised to find that treatment with DNQX during the growth period had a similar effect, indicating that sustained AMPA receptor activation is necessary for both the initiation and maintenance of psychoplastogen-induced neuronal growth.
Figure 4.
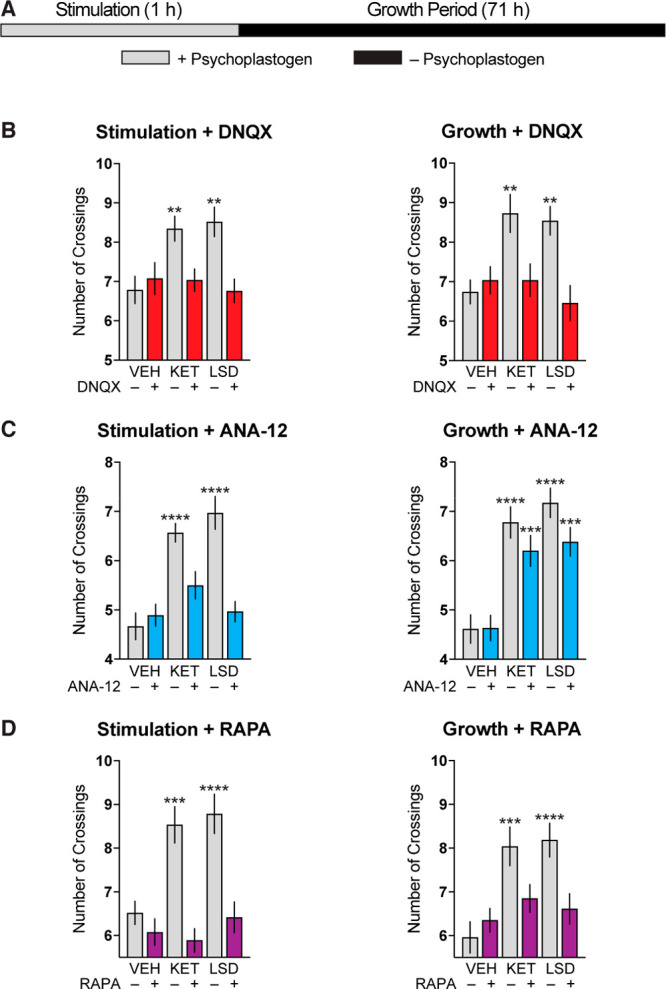
Sustained AMPA receptor and mTOR stimulation is necessary for psychoplastogen-induced dendritic growth. (A) Schematic illustrating experimental design. Cortical cultures (DIV3) were treated with psychoplastogens (10 μM) for 1 h (stimulation phase) and then allowed to grow in the absence of psychoplastogens for 71 h (growth phase). (B–D) Maximum number of crossings (Nmax) of the Sholl plots obtained after the stimulation and growth phases. Pharmacological inhibitors of AMPA receptors (B), TrkB receptors (C), and mTOR (D) were applied during either the stimulation or growth phases. Data are represented as mean ± SEM (n = 22–37 neurons). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, as compared to VEH control without inhibitor following a one-way analysis of variance with Dunnett’s post hoc test. VEH = vehicle; KET = ketamine; LSD = lysergic acid diethylamide; DNQX = 6,7-dinitroquinoxaline-2,3-dione; RAPA = rapamycin. See also Figure S2.
Originally, we hypothesized that AMPA receptor activation would lead to BDNF secretion, ultimately stimulating TrkB receptors. As BDNF is well known to enhance its own expression through positive autoregulation,49−52 we hypothesized that sustained TrkB signaling would be necessary for the long-lasting effects of transient psychoplastogen stimulation on dendritogenesis. However, this did not appear to be the case, as treatment with ANA-12, a selective TrkB antagonist,53 only blocked pyschoplastogen-induced growth when applied during the initial stimulation period and was ineffective when applied in the absence of psychoplastogen during the growth phase (Figure 4C). Conversely, blocking mTOR during either the stimulation or growth periods was sufficient to abrogate neuronal growth (Figure 4D), likely due to the critical role that mTOR plays in the synthesis of structural proteins and ion channels necessary for neural plasticity.54
Given the key role of AMPA receptor activation on both the initiation and maintenance of psychoplastogen-induced cortical neuron growth, we next sought to determine if neuronal activity in the absence of a psychoplastogen was capable of producing comparable changes in dendritogenesis. Stimulation of cortical cultures for 1 h with 40 mM KCl, a concentration known to induce glutamate release,55 did not lead to a growth phenotype comparable to that produced by psychoplastogens (Figure S3). Moreover, the combination of KCl with either ketamine or LSD did not enhance psychoplastogen-induced dendritogenesis (Figure S3). These results suggest that psychoplastogens induce neuronal growth by engaging biochemical signaling mechanisms that are distinct from those activated simply through neuronal depolarization/overexcitation.
Discussion
Accumulating evidence has suggested that activation of AMPA receptors, TrkB, and mTOR are all critical for the effects of ketamine and other psychoplastogens on both cortical structural plasticity and behavior.26,28,56−64 Recently, an elegant study by Liston and co-workers established a causal link between ketamine-induced spinogenesis in the PFC and antidepressant-like behavior in the tail suspension test.65 However, the details of psychoplastogen-initiated biochemical signaling leading to sustained changes in neuronal structure/function have remained poorly defined. Here, we demonstrate that transient stimulation with either ketamine or LSD, psychoplastogens from chemically distinct classes, trigger a growth phase that continues in the absence of drug.
Single administrations of psychoplastogens are well-known to produce effects in humans and rodents that persist long after the drug has been cleared from the body.29−34 Pharmacokinetic studies in rodents have revealed that behaviorally relevant doses of ketamine and DMT yield brain concentrations >10 μM,66,67 concentrations that are more than sufficient to trigger neuronal growth in culture. Psychoplastogen-induced growth of cortical neurons appears to require initial TrkB activation and is characterized by sustained AMPA receptor and mTOR activation. Though the exact details of this biochemical signaling network still remain elusive, it is clear that mechanisms exist for sustaining growth through an autoregulatory positive feedback loop. For example, initial activation of AMPA receptors is known to induce BDNF secretion, which in turn activates mTOR.68,69 Activation of mTOR not only leads to the translation of proteins necessary for cell growth but also produces additional BDNF. Finally, BDNF itself is known to induce glutamate release in cortical neurons via a nonexocytotic pathway,70 which could lead to sustained AMPA receptor activation. Superficially, it would appear that psychoplastogens merely catalyze this growth process by initiating a glutamate burst. However, psychoplatogen-induced neuronal growth cannot simply be the result of transient neuronal excitation, as a high concentration of potassium ions is not sufficient to produce similar changes in cortical neuron structure.
Though we know that psychoplastogens activate mTOR, it is still unclear how compounds with distinct primary targets (NMDA receptors for ketamine and 5-HT2A receptors for LSD) are capable of activating the same biochemical pathways. One hypothesis is that ketamine and LSD both induce glutamate bursts through their respective primary targets. Ketamine is believed to increase glutamate secretion by inhibiting NMDA receptors on inhibitory interneurons,46 while serotonergic psychedelics like LSD have been shown to promote glutamate release by activating 5-HT2A receptors.47 However, given the fact that increased neuronal activity is insufficient to produce psychoplastogen-like effects on neuronal growth, it is possible that ketamine and LSD could share an unidentified common target responsible for their psychoplastogenic effects. Given recent evidence suggesting that 5-HT1A receptors may play a role in the antidepressant effects of ketamine,71 coupled with the fact that LSD has high affinity for 5-HT1A receptors, we decided to test the effects of 8-OH-DPAT (a selective 5-HT1A agonist) on dendritic growth. Treating cortical neurons with 8-OH-DPAT did not increase dendritic growth at any concentration (Figure S2), suggesting that 5-HT1A receptors do not play a role in the psychoplastogenic effects of ketamine or LSD. At this point, it seems the most likely mechanism explaining the shared plasticity-promoting properties of psychoplastogenic NMDA receptor and 5-HT2A receptor ligands is related to their shared abilities to promote glutamate secretion through different receptors.
Currently, it is unclear exactly how long psychoplastogen-induced plasticity lasts. Two-photon in vivo imaging experiments have shown that ketamine can increase the rate of dendritic spine formation for up to 2 weeks in rodents,72 with behavioral effects often observed 1 week following administration.64 However, not all psychoplastogens are equivalent, with some producing more enduring effects than others. Recently, the antidepressant-like behavioral effects of psilocybin in rodents were shown to last much longer than those produced by ketamine,73 results that mirror the human clinical data for these two compounds.
Here, we have shown that like BDNF, psychoplastogens activate positive autoregulatory feedback loops leading to sustained neuronal growth. Small molecules offer significant advantages over protein- and peptide-based activators of neuronal growth signaling, as they can readily cross the blood–brain barrier. Moreover, our work suggests the potential for a paradigm shift in CNS drug development toward molecules that are intentionally designed to reach high brain concentrations and be rapidly cleared from the body in order to minimize side effects. One of the major concerns with using drugs like psilocybin and ketamine in the clinic is related to their relatively long-lasting hallucinogenic/dissociative properties, necessitating the hospitalization of patients for extended periods of time. In this regard, shorter acting plasticity-promoting psychedelics, such as DMT,74 could offer significant advantages by reducing healthcare costs.75 With the advent of nonhallucinogenic psychoplastogens,76 it might be possible to produce a plasticity-promoting drug suitable for at home administration. The work described here suggests that infrequent administration of such compounds might be sufficient to produce sustained changes in neural circuitry, thus avoiding potential side effects from chronic daily use, such as those that have plagued patients using traditional antidepressants.
Methods
Data Analysis and Statistics
Treatments were randomized, and data were analyzed by experimenters blinded to treatment conditions. Statistical analyses were performed using GraphPad Prism (version 8.1.2). Data are represented as mean ± SEM, unless otherwise noted, with asterisks: *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. Brightness and contrast were adjusted equally for all representative images.
Data Availability
The data sets analyzed as part of this study are available via Mendeley Data: https://data.mendeley.com/datasets/6f5wf65fbf/draft?a=5311108e-7332-4df8-9d4c-86dcdd023cd0.
Drugs
The NIH Drug Supply Program provided (+)-lysergic acid diethylamide (+)-tartrate (2:1) (LSD). Other chemicals/proteins were purchased from commercial sources such as ketamine hydrochloride (KET, Fagron), (±)-8-hydroxy-2-(dipropylamino)tetralin hydrobromide (8-OH-DPAT, Sigma), brain-derived neurotrophic factor (BDNF, Sigma-Aldrich), ANA-12 (MedChem Express), DNQX (Sigma), and rapamycin (Alfa Aesar). For cell culture experiments, VEH = 0.1% (studies assessing the effects of psychoplastogen alone) or 0.2% (studies assessing the effects of psychoplastogen + inhibitor) molecular-biology-grade dimethyl sulfoxide (Acros).
Animals
All experimental procedures involving animals were approved by the University of California, Davis, Institutional Animal Care and Use Committee (IACUC) and adhered to principles described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The University of California, Davis, is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Cell Culture
Primary cortical cell cultures were prepared as described previously.28 Briefly, pregnant Sprague Dawley dams were euthanized at embryonic day 18 (E18), and the cortices of the pups were harvested. Cells were plated on poly-d-lysine-coated plates at specific densities depending on the experiment (vide infra). Cultures were maintained at 37 °C under an atmosphere containing 5% CO2. Plating media consisted of 10% heat-inactivated fetal bovine serum (FBS; Life Technologies), 1% penicillin–streptomycin (Life Technologies), and 0.5 mM glutamine (Life Technologies) in Neurobasal (Life Technologies). After 16–24 h, media was exchanged for replacement media consisting of 1× B27 supplement (Life Technologies), 1% penicillin–streptomycin, 0.5 mM glutamine, and 12.5 μM glutamate in Neurobasal. For experiments requiring cells older than 7 days in vitro (DIV7), at 96 h postplating, 50% of media was removed, and feeding media containing 1× B27 supplement, 1% penicillin–streptomycin, 0.5 mM glutamine in Neurobasal was added with an additional 20% volume added to account for evaporation.
Dendritogenesis Experiments
The dendritogenesis experiments reported in Figures 1D and S1A were performed as previously described28 with the exception that cells were treated with psychoplastogens or BDNF for short periods of time (15 min or 1, 24, or 72 h) prior to exchanging the media for replacement media devoid of psychoplastogen/BDNF. The dendritogenesis experiments reported in Figures 1E, 4, and S2 were performed as previously described74 with the exception of the treatment conditions.
Spinogenesis Experiments
Spinogenesis experiments were performed as previously described28 with the exception that cells were treated on DIV19 with psychoplastogens (10 μM) for short periods of time (1, 6, and 24 h) prior to exchanging the media for replacement media devoid of psychoplastogens. Cells were fixed on DIV20 (24 h following treatment). Images were taken on a Nikon HCA Confocal microscope a with a 100×/NA 1.45 oil objective.
Synaptogenesis Experiments
Synaptogenesis experiments were performed using 96-well plates coated with poly-d-lysine at a density of 15 000 cells per well. The outer wells were not used to avoid edge effects. On DIV7 and DIV14, 50% of the medium was removed (100 μL) and fresh feeding media containing Neurobasal, 1× B27 supplement (Life Technologies), 1% penicillin–streptomycin, and 0.5 mM glutamine was added (70%, 140 μL). Cells were treated with drugs on DIV19. First, 10 mM DMSO stock solutions were diluted 100-fold in Neurobasal, then diluted 10-fold into each well (1:1000 dilution, final concentration of DMSO = 0.1%). Cells were incubated with drug for 0.25, 1, and 6 h, then media was removed and replaced with fresh feeding media devoid of psychoplastogens. Cells were allowed to grow until a total of 24 h had elapsed. At this time, 80% of the media (160 μL) was removed, and a 50% volume of a 4% aqueous paraformaldehyde (PFA) solution (100 μL) at room temperature was added. The fixative was applied to the cultures for 20 min at room temperature. Cells were then washed two times with Dulbecco’s phosphate-buffered saline (dPBS, Life Technologies) and permeabilized with 0.2% Triton X-100 (ThermoFisher, 85111) in dPBS for 20 min at room temperature without shaking. Plates were then blocked with antibody diluting buffer (ADB) containing 2% bovine serum albumin (BSA) in dPBS for 1 h at room temperature without shaking. Then, plates were incubated overnight at 4 °C with gentle shaking in ADB and a chicken anti-MAP2 antibody (1:10 000; EnCor, CPCA-MAP2), a guinea pig anti-vGLUT1 antibody (1:1000; Millipore, AB5905), and a mouse anti-PSD-95 antibody (1:500; Millipore, MABN68). The next day, plates were washed three times in dPBS and once in ADB. Plates were then incubated in ADB at room temperature containing an anti-chicken IgG secondary antibody conjugated to Alexa Fluor 488 (1:500; LifeTechnologies), an anti-guinea pig IgG secondary antibody conjugated to Cy3 (1:500; Jackson ImmunoResearch Inc., catalog no. 706-165-148), and an anti-mouse IgG secondary antibody conjugated to Alexa Fluor 647 (1:500; Jackson ImmunoResearch Inc., catalog no. 715-605-151) for 1 h. Next, plates were washed five times with dPBS, and after the final wash, 100 μL of dPBS was added to each well. All images were obtained using a Molecular Devices ImageXpress Micro XLS Widefield High-Content Analysis System at 9 sites per well using 40× magnification. Image analysis was performed using MetaXpress software. Analysis was completed by first using the MAP2 channel to establish a mask of the neuron using thresholds between 0 and 30 μm. Then, the mask of the neuron was expanded by 1 μm. The size of the objects for the presynaptic and postsynaptic fluorescent images were established, and only signals of 0–1.5 μm punctate were used to generate a mask. To measure synaptic density, the presynaptic and postsynaptic masks were overlaid using the logical operation “and” to retain only signal that colocalized to form the synapse mask. The number of events occurring in this synapse mask was quantified and normalized to the MAP2 channel mask area (number of counts per μm2). Normalized data were then tested for outliers using the ROUT method in Graphpad Prism (version 8) at Q = 1%. The outlier test was completed to remove artifacts in an unbiased manner. To prepare the representative images shown in Figure 3B, brightness/contrast was adjusted evenly across all images. After this, the vGLUT1 and PSD-95 channels were both processed using the “smooth” function in ImageJ (Fiji, Ver. 1.51v) followed by the plug-in “Image Calculator” with image1 corresponding to vGLUT1 and image2 corresponding to PSD-95. The operation used was “And”, and “Create new window” was checked. The resulting window contained all points of colocalization between the two images. This window was also processed through the “smooth” function once more. Next, the MAP2 channel was merged with the colocalization channel for a composite that was falsely colored to distinguish the two channels.
Acknowledgments
This work was supported by funds from the National Institutes of Health (NIH) (R01GM128997 to D.E.O.), an NIH training grant T32GM113770 (to C.L.), and a UC Davis Provost’s Undergraduate Fellowship (to A.C.G.). Several experiments utilized the Biological Analysis Core of the UC Davis MIND Institute Intellectual and Development Disabilities Research Center (U54 HD079125) or the Nikon high-content-analysis spinning-disk confocal microscope (1S10OD019980-01A1) located in the Light Microscopy Imaging Facility of the Department of Molecular and Cellular Biology at UC Davis. Lysergic acid diethylamide was generously provided by the NIDA Drug Supply Program.
Glossary
Abbreviations
- VEH
vehicle
- KET
ketamine
- LSD
lysergic acid diethylamide
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- TrkB
tyrosine kinase B
- mTOR
mammalian target of rapamycin
- BDNF
brain-derived neurotrophic factor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00065.
Effects of BDNF on neuronal growth; effects of LSD, ketamine, and 8-OH-DPAT on dendritic growth; KCl does not enhance psychoplastogen-induced growth (PDF)
Author Contributions
CL performed the majority of the dendritogeneis, spinogenesis, and synaptogenesis experiments with assistance from ACG, MVV, and WCD. ACGG and PJL supervised the high-content imaging experiments. DEO conceived the project and supervised experiments. DEO and CL wrote the manuscript with input from all authors.
The authors declare the following competing financial interest(s): D.E.O. is the president and chief scientific officer of Delix Therapeutics, Inc.
Supplementary Material
References
- James S. L.; et al. (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858. 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. (2014) Mental health: a world of depression. Nature 515, 180. 10.1038/515180a. [DOI] [PubMed] [Google Scholar]
- Cipriani A.; Furukawa T. A.; Salanti G.; Chaimani A.; Atkinson L. Z.; Ogawa Y.; Leucht S.; Ruhe H. G.; Turner E. H.; Higgins J. P. T.; Egger M.; Takeshima N.; Hayasaka Y.; Imai H.; Shinohara K.; Tajika A.; Ioannidis J. P. A.; Geddes J. R. (2018) Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta- analysis. Lancet 391, 1357–1366. 10.1016/S0140-6736(17)32802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush A. J.; Trivedi M. H.; Wisniewski S. R.; Nierenberg A. A.; Stewart J. W.; Warden D.; Niederehe G.; Thase M. E.; Lavori P. W.; Lebowitz B. D.; et al. (2006) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163, 1905–1917. 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Duman R. S.; Aghajanian G. K.; Sanacora G.; Krystal J. H. (2016) Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249. 10.1038/nm.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R. S.; Aghajanian G. K. (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72. 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry A. E.; Monteggia L. M. (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 64, 238–258. 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C.; Duman R. S. (2008) Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109. 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Olson D. E. (2018) Psychoplastogens: A Promising Class of Plasticity-Promoting Neurotherapeutics. J. Exp. Neurosci. 12, 1–4. 10.1177/1179069518800508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman R. M.; Cappiello A.; Anand A.; Oren D. A.; Heninger G. R.; Charney D. S.; Krystal J. H. (2000) Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354. 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Zarate C. A. Jr.; Singh J. B.; Carlson P. J.; Brutsche N. E.; Ameli R.; Luckenbaugh D. A.; Charney D. S.; Manji H. K. (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864. 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zarate C. A. Jr.; Brutsche N. E.; Ibrahim L.; Franco-Chaves J.; Diazgranados N.; Cravchik A.; Selter J.; Marquardt C. A.; Liberty V.; Luckenbaugh D. A. (2012) Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol. Psychiatry 71, 939–946. 10.1016/j.biopsych.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiazGranados N.; Ibrahim L. A.; Brutsche N. E.; Ameli R.; Henter I. D.; Luckenbaugh D. A.; Machado-Vieira R.; Zarate C. A. Jr. (2010) Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 71, 1605–1611. 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey M. L.; Drevets W. C. (2006) Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry 63, 1121–1129. 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets W. C.; Furey M. L. (2010) Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol. Psychiatry 67, 432–438. 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey M. L.; Khanna A.; Hoffman E. M.; Drevets W. C. (2010) Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology 35, 2479–2488. 10.1038/npp.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey M. L.; Nugent A. C.; Speer A. M.; Luckenbaugh D. A.; Hoffman E. M.; Frankel E.; Drevets W. C.; Zarate C. A. Jr. (2012) Baseline mood-state measures as predictors of antidepressant response to scopolamine. Psychiatry Res. 196, 62–67. 10.1016/j.psychres.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris R. L.; Goodwin G. M. (2017) The therapeutic potential of psychedelic drugs: past, present, and future. Neuropsychopharmacology 42, 2105–2113. 10.1038/npp.2017.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D. E.; Johnson M. W.; Nichols C. D. (2017) (2017). Psychedelics as medicines: an emerging new paradigm. Clin. Pharmacol. Ther. 101, 209–219. 10.1002/cpt.557. [DOI] [PubMed] [Google Scholar]
- Sanches R. F.; de Lima Osó rio F.; Dos Santos R. G.; Macedo L. R.; Maia-de-Oliveira J. P.; Wichert-Ana L.; de Araujo D. B.; Riba J.; Crippa J. A.; Hallak J. E. (2016) Antidepressant effects of a single dose of ayahuasca in patients with recurrent depression: a SPECT study. J. Clin. Psychopharmacol. 36, 77–81. 10.1097/JCP.0000000000000436. [DOI] [PubMed] [Google Scholar]
- Osório F. de. L.; Sanches R. F.; Macedo L. R.; dos Santos R. G.; Maia-de-Oliveira J. P.; Wichert-Ana L.; de Araujo D. B.; Riba J.; Crippa J. A.; Hallak J. E. (2015) Antidepressant effects of a single dose of ayahuasca in patients with recurrent depression: a preliminary report. Rev. Bras. Psiquiatr. 37, 13–20. 10.1590/1516-4446-2014-1496. [DOI] [PubMed] [Google Scholar]
- Dos Santos R. G.; Osó rio F. L.; Crippa J. A.; Riba J.; Zuardi A. W.; Hallak J. E. (2016) Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther. Adv. Psychopharmacol. 6, 193–213. 10.1177/2045125316638008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris R. L.; Bolstridge M.; Rucker J.; Day C. M. J.; Erritzoe D.; Kaelen M.; Bloomfield M.; Rickard J. A.; Forbes B.; Feilding A.; Taylor D.; Pilling S.; Curran V. H.; Nutt D. J. (2016) Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psychiatry 3, 619–627. 10.1016/S2215-0366(16)30065-7. [DOI] [PubMed] [Google Scholar]
- Carhart-Harris R. L.; Roseman L.; Bolstridge M.; Demetriou L.; Pannekoek J. N.; Wall M. B.; Tanner M.; Kaelen M.; McGonigle J.; Murphy K.; et al. (2017) Psilocybin for treatment-resistant depression: fMRI-measured brain mechanisms. Sci. Rep. 7, 13187. 10.1038/s41598-017-13282-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker J. J.; Jelen L. A.; Flynn S.; Frowde K. D.; Young A. H. (2016) Psychedelics in the treatment of unipolar mood disorders: a systematic review. J. Psychopharmacol. 30, 1220–1229. 10.1177/0269881116679368. [DOI] [PubMed] [Google Scholar]
- Li N.; Lee B.; Liu R. J.; Banasr M.; Dwyer J. M.; Iwata M.; Li X. Y.; Aghajanian G.; Duman R. S. (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B.; Navarria A.; Liu R. J.; Banasr B.; Li N.; Terwilliger R.; Sanacora G.; Eid T.; Aghajanian G.; Duman R. S. (2013) Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol. Psychiatry 74, 742–749. 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly C.; Greb A. C.; Cameron L. P.; Wong J. M.; Barragan E. V.; Wilson P. C.; Burbach K. F.; Soltanzadeh Zarandi S.; Sood A.; Paddy M. R.; Duim W. C.; Dennis M. Y.; McAllister A. K.; Ori-McKenney K. M.; Gray J. A.; Olson D. E. (2018) Psychedelics Promote Structural and Functional Neural Plasticity. Cell Rep. 23, 3170–3182. 10.1016/j.celrep.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets W. C.; Zarate C. A. Jr.; Furey M. L. (2013) Antidepressant Effects of the Muscarinic Cholinergic Receptor Antagonist Scopolamine: A Review. Biol. Psychiatry 73, 1156–1163. 10.1016/j.biopsych.2012.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris R. L.; Bolstridge M.; Day C. M. J.; Rucker J.; Watts R.; Erritzoe D. E.; Kaelen M.; Giribaldi B.; Bloomfield M.; Pilling S. (2018) Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology 235, 399–408. 10.1007/s00213-017-4771-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough J. W.; Perez A. M.; Pillemer S.; Stern J.; Parides M. K.; aan het Rot M.; Collins K. A.; Mathew S. J.; Charney D. S.; Iosifescu D. V. (2013) Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol. Psychiatry 74, 250–256. 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu D. F.; Swee M. B.; Pavone K. J.; Taylor N.; Akeju O.; Baer L.; Nyer M.; Cassano P.; Mischoulon D.; Alpert J. E.; Brown E. N.; Nock M. K.; Fava M.; Cusin C. (2016) Rapid and Sustained Reductions in Current Suicidal Ideation Following Repeated Doses of Intravenous Ketamine: Secondary Analysis of an Open-Label Study. J. Clin. Psychiatry 77, e719–e725. 10.4088/JCP.15m10056. [DOI] [PubMed] [Google Scholar]
- Barrett F. S.; Doss M. K.; Sepeda N. D.; Pekar J. J.; Griffiths R. R. (2020) Emotions and brain function are altered up to one month after a single high dose of psilocybin. Sci. Rep. 10, 2214. 10.1038/s41598-020-59282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aday J. S.; Mitzkovitz C. M.; Bloesch E. K.; Davoli C. C.; Davis A. K. (2020) Long-term effects of psychedelic drugs: A systematic review. Neurosci. Biobehav. Rev. 113, 179–189. 10.1016/j.neubiorev.2020.03.017. [DOI] [PubMed] [Google Scholar]
- Castrén E.; Antila H. (2017) Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 22, 1085–1095. 10.1038/mp.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori J.; Winkel F.; Didio G.; Llach Pou M.; Castrén E. (2018) iPlasticity: Induced juvenile-like plasticity in the adult brain as a mechanism of antidepressants. Psychiatry Clin. Neurosci. 72, 633–653. 10.1111/pcn.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantamäki T.; Hendolin P.; Kankaanpää A.; Mijatovic J.; Piepponen P.; Domenici E.; Chao M. V.; Männistö P. T.; Castrén E. (2007) Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology 32, 2152–2162. 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- Rantamäki T.; Vesa L.; Antila H.; Di Lieto A.; Tammela P.; Schmitt A.; Lesch K. P.; Rios M.; Castrén E. (2011) Antidepressant Drugs Transactivate TrkB Neurotrophin Receptors in the Adult Rodent Brain Independently of BDNF and Monoamine Transporter Blockade. PLoS One 6, e20567 10.1371/journal.pone.0020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fred S. M.; Laukkanen L.; Brunello C. A.; Vesa L.; Göös H.; Cardon I.; Moliner R.; Maritzen T.; Varjosalo M.; Casarotto P. C.; Castrén E. (2019) Pharmacologically diverse antidepressants facilitate TRKB receptor activation by disrupting its interaction with the endocytic adaptor complex AP-2. J. Biol. Chem. 294, 18150–18161. 10.1074/jbc.RA119.008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolder P. C.; Schmid Y.; Haschke M.; Rentsch K. M.; Liechti M. E. (2016) Pharmacokinetics and Concentration-Effect Relationship of Oral LSD in Humans. Int. J. Neuropsychopharmacol. 19, pyv072. 10.1093/ijnp/pyv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew S. J.; Shah A.; Lapidus K.; Clark C.; Jarun N.; Ostermeyer B.; Murrough J. W. (2012) Ketamine for Treatment-Resistant Unipolar Depression. CNS Drugs 26, 189–204. 10.2165/11599770-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai K.; Ido T.; Ishiwata K.; Hatazawa J.; Takahashi T.; Iwata R.; Matsuzawa T. (1986) In vivo kinetics and displacement study of a carbon-11-labeled hallucinogen, N,N- [11C]dimethyltryptamine. Eur. J. Nucl. Med. 12, 141–146. 10.1007/BF00276707. [DOI] [PubMed] [Google Scholar]
- Sitaram B. R.; Lockett L.; Talomsin R.; Blackman G. L.; McLeod W. R. (1987) In vivo metabolism of 5-methoxy-N,Ndimethyltryptamine and N,N-dimethyltryptamine in the rat. Biochem. Pharmacol. 36, 1509–1512. 10.1016/0006-2952(87)90118-3. [DOI] [PubMed] [Google Scholar]
- González-Burgos I.; López-Vázquez M. A.; Beas-Zárate C. (2004) Density, but not shape, of hippocampal dendritic spines varies after a seizure-inducing acute dose of monosodium glutamate in rats. Neurosci. Lett. 363, 22–24. 10.1016/j.neulet.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Ly C.; Shimizu A. J.; Vargas M. V.; Duim W. C.; Wender P. A.; Olson D. E. (2020) Bryostatin 1 Promotes Synaptogenesis and Reduces Dendritic Spine Density in Cortical Cultures Through a PKC-Dependent Mechanism. ACS Chem. Neurosci. 11, 1545–1554. 10.1021/acschemneuro.0c00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R. S. (2018) Ketamine and rapid-acting antidepressants: a new era in the battle against depression and suicide. F1000Research 7, 659. 10.12688/f1000research.14344.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb E. S.; Gerhard D.; Thomas A.; Duman R. S. (2016) Molecular and Cellular Mechanisms of Rapid-Acting Antidepressants Ketamine and Scopolamine. Curr. Neuropharmacol. 15, 11–20. 10.2174/1570159X14666160309114549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian G. K.; Marek G. J. (1999) Serotonin, via 5-HT2A receptors, increases EPSCs in layer V pyramidal cells of prefrontal cortex by an asynchronous mode of glutamate release. Brain Res. 825, 161–171. 10.1016/S0006-8993(99)01224-X. [DOI] [PubMed] [Google Scholar]
- Jiang C.; Lin W. J.; Salton S. R. (2019) Role of a VGF/BDNF/TrkB Autoregulatory Feedback Loop in Rapid-Acting Antidepressant Efficacy. J. Mol. Neurosci. 68, 504–509. 10.1007/s12031-018-1124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvald E. E.; Tuvikene J.; Sirp A.; Patil S.; Bramham C. R.; Timmusk T. (2020) CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J. Neurosci. 40, 1405–1426. 10.1523/JNEUROSCI.0367-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvikene J.; Pruunsild P.; Orav E.; Esvald E. E.; Timmusk T. (2016) AP-1 Transcription Factors Mediate BDNF-Positive Feedback Loop in Cortical Neurons. J. Neurosci. 36, 1290–1305. 10.1523/JNEUROSCI.3360-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambah-Mukku D.; Travaglia A.; Chen D. Y.; Pollonini G.; Alberini C. M. (2014) A Positive Autoregulatory BDNF Feedback Loop via C/EBPβ Mediates Hippocampal Memory Consolidation. J. Neurosci. 34, 12547–12559. 10.1523/JNEUROSCI.0324-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M.; Prémont J.; Mann A.; Girard N.; Kellendonk C.; Rognan D. (2011) Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest. 121, 1846–1857. 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer C. A.; Klann E. (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingamaneni R.; Hemmings H. C. Jr. (1999) Effects of anticonvulsants on veratridine- and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci. Lett. 276, 127–130. 10.1016/S0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- Koike H.; Iijima M.; Chaki S. (2011) Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res. 224, 107–111. 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Wang N.; Yang C.; Li X. M.; Zhou Z. Q.; Yang J. J. (2014) Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 29, 419–423. 10.1016/j.eurpsy.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Koike H.; Chaki S. (2014) Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 271, 111–115. 10.1016/j.bbr.2014.05.065. [DOI] [PubMed] [Google Scholar]
- Li S.; Luo X.; Hua D.; Wang Y.; Zhan G.; Huang N.; Jiang R.; Yang L.; Zhu B.; Yuan X.; et al. (2020) Ketamine Alleviates Postoperative Depression-Like Symptoms in Susceptible Mice: The Role of BDNF-TrkB Signaling. Front. Pharmacol. 10, 1702. 10.3389/fphar.2019.01702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K.; Fogaça M. V.; Liu R.; Duman C.; Kato T.; Li X.; Duman R. S. (2019) Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)- hydroxynorketamine. Proc. Natl. Acad. Sci. U. S. A. 116, 297–302. 10.1073/pnas.1814709116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal S.; Bang E.; Yue W.; Hare B. D.; Lepack A. E.; Girgenti M. J.; Duman R. S. (2018) Activity-Dependent Brain-Derived Neurotrophic Factor Release Is Required for the Rapid Antidepressant Actions of Scopolamine. Biol. Psychiatry 83, 29–37. 10.1016/j.biopsych.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry A. E.; Adachi M.; Nosyreva E.; Na E. S.; Los M. F.; Cheng P.; Kavalali E. T.; Monteggia L. M. (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack A. E.; Fuchikami M.; Dwyer J. M.; Banasr M.; Duman R. S. (2015) BDNF Release Is Required for the Behavioral Actions of Ketamine. Int. J. Neuropsychopharmacol. 18, pyu033. 10.1093/ijnp/pyu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm C.; Monteggia L. M. (2016) BDNF — a key transducer of antidepressant effects. Neuropharmacology 102, 72–79. 10.1016/j.neuropharm.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moda-Sava R. N.; Murdock M. H.; Parekh P. K.; Fetcho R. N.; Huang B. S.; Huynh T. N.; Witztum J.; Shaver D. C.; Rosenthal D. L.; Alway E. J.; et al. (2019) Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, eaat8078 10.1126/science.aat8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Cui Y.; Sang K.; Dong Y.; Ni Z.; Ma S.; Hu H. (2018) Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322. 10.1038/nature25509. [DOI] [PubMed] [Google Scholar]
- Cohen I.; Vogel W. H. (1972) Determination and physiological disposition of dimethyltryptamine and diethyltryptamine in rat brain, liver and plasma. Biochem. Pharmacol. 21, 1214–1216. 10.1016/0006-2952(72)90119-0. [DOI] [PubMed] [Google Scholar]
- Jourdi H.; Hsu Y. T.; Zhou M.; Qin Q.; Bi X.; Baudry M. (2009) Positive AMPA Receptor Modulation Rapidly Stimulates BDNF Release and Increases Dendritic mRNA Translation. J. Neurosci. 29, 8688–8697. 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N.; Inamura N.; Kawamura M.; Namba H.; Hara K.; Yonezawa K.; Nawa H. (2004) Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 24, 9760–9769. 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N.; Numakawa T.; Kozaki S.; Sakai N.; Endo Y.; Takahashi M.; Hatanaka H. (1998) Brain-derived neurotrophic factor induces rapid and transient release of glutamate through the non-exocytotic pathway from cortical neurons. J. Biol. Chem. 273, 27620–27624. 10.1074/jbc.273.42.27620. [DOI] [PubMed] [Google Scholar]
- Fukumoto K.; Iijima M.; Funakoshi T.; Chaki S. (2018) Role of 5-HT1A Receptor Stimulation in the Medial Prefrontal Cortex in the Sustained Antidepressant Effects of Ketamine. Int. J. Neuropsychopharmacol. 21, 371–381. 10.1093/ijnp/pyx116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoumthipphavong V.; Barthas F.; Hassett S.; Kwan A. C. (2016) Longitudinal Effects of Ketamine on Dendritic Architecture In Vivo in the Mouse Medial Frontal Cortex. eNeuro 3, ENEURO.0133-15.2016 10.1523/ENEURO.0133-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibicke M.; Landry A. N.; Kramer H. M.; Talman Z. K.; Nichols C. D. (2020) Psychedelics, but Not Ketamine, Produce Persistent Antidepressant-like Effects in a Rodent Experimental System for the Study of Depression. ACS Chem. Neurosci. 11, 864–871. 10.1021/acschemneuro.9b00493. [DOI] [PubMed] [Google Scholar]
- Cameron L. P.; Olson D. E. (2018) Dark Classics in Chemical Neuroscience: N,N-Dimethyltryptamine (DMT). ACS Chem. Neurosci. 9, 2344–2357. 10.1021/acschemneuro.8b00101. [DOI] [PubMed] [Google Scholar]
- Nutt D.; Erritzoe D.; Carhart-Harris R. (2020) Psychedelic Psychiatry’s Brave New World. Cell 181, 24–28. 10.1016/j.cell.2020.03.020. [DOI] [PubMed] [Google Scholar]
- Dunlap L. E.; Azinfar A.; Ly C.; Cameron L. P.; Viswanathan J.; Tombari R. J.; Myers-Turnbull D.; Taylor J. C.; Grodzki A. C.; Lein P. J.; Kokel D.; Olson D. E. (2020) Identification of Psychoplastogenic N,N-Dimethylaminoisotryptamine (isoDMT) Analogs Through Structure-Activity Relationship Studies. J. Med. Chem. 63, 1142–1155. 10.1021/acs.jmedchem.9b01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets analyzed as part of this study are available via Mendeley Data: https://data.mendeley.com/datasets/6f5wf65fbf/draft?a=5311108e-7332-4df8-9d4c-86dcdd023cd0.