

Abstract
Background
The receptor tyrosine kinase FLT3 with internal tandem duplications within the juxtamembrane domain (FLT3-ITD) is a poor prognostic factor; however, the prognostic significance of missense mutation in the tyrosine kinase domain (FLT3-TKD) is controversial. Furthermore, the accompanying mutations and fusion genes with FLT3 mutations are unclear in acute myeloid leukemia (AML).
Methods
We investigated FLT3 mutations and their correlation with other gene mutations and gene fusions through two RNA-seq based next-generation sequencing (NGS) method and prognostic impact in 207 de novo AML patients.
Results
FLT3-ITD mutations were positive in 58 patients (28%), and FLT3-TKD mutations were positive in 20 patients (9.7%). FLT3-ITD was associated with a higher white blood cell count (WBC, mean 72.9 × 109/L vs. 24.2 × 109/L, P = 0.000), higher bone marrow blasts (mean 65.9% vs. 56.0%, P = 0.024), and NK-AML (normal karyotype) (64.8% vs. 48.4%, P = 0.043). NPM1 and DNMT3A mutations were enriched in FLT3-ITD (53.5% vs. 15.3%, P = 0.000; 34.6% vs. 13%, P = 0.003). However, the mutations of CEBPA were excluded in FLT3-AML (3.8% vs. 0% vs. 19.8%, P = 0.005). Mutations of Ras and TP53 were unlikely associated with FLT3-ITD (1.9% vs. 20.6%, P = 0.006; 0% vs. 6.1%, P = 0.04). The common fusion genes (> 10%) in FLT3-ITD had MLL-rearrangement and NUP98-rearrangement, while the common fusion genes in FLT3-TKD had AML1-ETO and MLL-rearrangement. Two novel fusion genes PRDM16-SKI and EFAN2-ZNF238 were identified in FLT3-ITD patients. Gene fusions and NPM1 mutation were mutually excluded in FLT3-ITD and FLT3-TKD patients. Their patterns of mutual exclusivity and cooperation among mutated genes suggest that additional driver genetic alterations are required and reveal two evolutionary patterns of FLT3 pathogenesis. Patients with FLT3-ITD had a lower CR (complete remission) rate, lower 3-year OS (overall survival), DFS (disease-free survival), and EFS (event-free survival) compared to FLT3wtAML. NK-AML with FLT3-ITD had a lower 3-year OS, DFS, and EFS than those without, while FLT3-TKD did not influence the survival in whole cohort and NK-AML. Besides, we found that FLT3-ITD/TET2 bimutation defined a poor prognostic subgroup.
Conclusions
Our study offers deep insights into the molecular pathogenesis and biology of AML with FLT3-ITD and FLT3-TKD by providing the profiles of concurrent molecular alterations and the clinical impact of FLT3-ITD and FLT3-TKD on AML patients.
Keywords: Acute myeloid leukemia, FLT3-ITD, FLT3-TKD, TET2, Next-generation sequencing
Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy accompanied by complex molecular genetic abnormalities with an increasing incidence in the globe [1–3]. FLT3 mutation is one of the most common mutations in AML. FLT3 with internal tandem duplications within the juxtamembrane domain (FLT3-ITD) is present in 20–30% of AML patients, and a missense mutation in the tyrosine kinase domain (FLT3-TKD) accounts for about 10% of AML [4, 5]. FLT3-ITD alone does not trigger leukemia, indicating that other drivers are needed for pathogenies [6, 7]. FLT3-ITD with additional NPM1 mutation [8], AML1-ETO fusion gene [9], NUP98 fusion [10, 11], CBFβ-SMMHC fusion gene [12], and TET2 deletion [13, 14] can cause leukemia. The complex pathogenic mechanism and heterogeneous clinical features of FLT3-ITD necessitate comprehensive molecular profiling. Owing to the low incidence of FLT3-TKD mutation, neither the prognostic significance is clear nor the accompanying molecular alterations.
Gene fusion is a very important pathogenic mechanism, and each fusion gene has its unique clinical manifestations. BCR-ABL resulting from t(9;22) in chronic myelogenous leukemia (CML) is a classic example [15]. Since the discovery of BCR/ABL, CML entered the era of targeted treatment, significantly improving the survival [16, 17]. Fusion genes are effective targets for diagnosis, prognosis, therapy, and minimal residual disease (MRD) monitoring in hematological cancers [18]. The detection of common fusion genes with clinical significance has become a routine practice today. The next-generation sequencing (NGS) technique has much more advantages in detecting fusion genes. RNA-seq based NGS can provide information about the structure and transcript level of fusion genes. Its technical advance makes the global identification of fusion transcripts possible [19]. Targeted NGS sequencing for fusion genes in FLT3 mutant AML has not been reported before. In this study, coexisting gene mutations and fusion genes of FLT3-ITD and FLT3-TKD mutation in AML patients by NGS were analyzed to better understand this disease.
Patients and methods
Patients and study design
A total of 207 patients (older than 14 years) with newly diagnosed AML (non-M3) admitted to the hospital from August 2009 to October 2017 were analyzed. According to the cytogenetically defined MRC criteria, 23 patients of this cohort were assigned to the favorable-prognostic-risk group, 151 to the intermediate-prognostic-risk group, and 23 to the poor-prognostic-risk group. In detail, 103 patients had a normal karyotype; six patients had a complex aberrate karyotype; 23 patients had a t(8;21); eight patients had a 11q23 rearrangement; 57 patients had other aberrant karyotypes. Cytogenetics of 10 patients were not available because of analysis failure or missing information. The cohort included 115 male and 94 female patients. The median age was 45.4 years (ranging from 14 to 76 years). Incidence of FLT3 mutations and correlation with other recurrent mutations and fusions in AML were evaluated in this cohort.
Fifty-eight cases were FLT3-ITD positive (28%), and 20 cases were FLT3-TKD mutation-positive (9.7%), four of which carried both mutations. FLT3-ITD analysis was based on DNA capture sequencing. The filtered reads were compared to the reference genome sequence (HG19, NCBI Built 37) using Burrows–Wheeler alignment (BWA), and the insertion and deletion of FLT3 region were detected using Pindel (0.2.4) software to detect FLT3-ITD mutation. The variation was annotated using ANNOVAR. The reads were aligned using BWA tool to human genomic reference sequences (HG19, NCBI built 37). To identify SNPs and INDELs, GATK was performed with recommended parameters; Pindel (0.2.4) was performed to identify the FLT3-ITD. FLT3-ITD was simultaneously verified by Sanger sequencing. 52/58 FLT3-ITD patients were detected by two targeted NGS for mutations and fusions. Four FLT3-TKD patients co-occurring with FLT3-TKD were assigned to the FLT3-ITD group; the other 16 FLT3-TKD patients were also detected by NGS for mutations and fusion genes assigned as FLT3-TKD group. The other 133 FLT3 wild-type AML (FLT3wtAML) patients were detected by Sanger sequencing for molecular mutation analyses only. The study was designed following the Declaration of Helsinki and approved by the institutional review board of PLA general hospital.
Therapy
Forty-six FLT3-ITD patients, 14 FLT3-TKD patients, and 113 FLT3wt patients completed two cycles of induction, and they were evaluated for treatment response. TKI inhibitor was applied in the induction regimen for three patients with FLT3-ITD [Sunitinib + AA (n = 1) and Sorafenib + FLAG (n = 2)]. Consolidation therapy after complete remission (CR) was administered to 29 patients in the FLT3-ITD group, 10 patients in the FLT3-TKD group, and 93 FLT3wt patients. Sorafenib was administrated in consolidation chemotherapy for one patient with FLT3-ITD and after HSCT for one patient with FLT3-ITD to prevent relapse. In total, 13 patients with FLT3-ITD, 5 with FLT3-TKD, and 47 FLT3 wild type received SCT in CR1. Treatment options for chemotherapy and stem cell transplantation were not significantly different among the three groups (P = 0.865). The treatment flow diagram is shown in Additional file 1: Fig. S1. The data of two FLT3-ITD patients and one FLT3wt patient were cancelled for survival analysis due to the loss of follow-up.
Library preparation and NGS
The method and gene panel of NGS for mutation detection in AML are previously reported [20]. The NGS for fusion gene detection is based on targeted RNA-seq. In brief, RNA was extracted from patient samples using the Tempus Spin RNA Isolation Kit (Life) following the manufacturer’s instructions. The RNA quality [RNA integrity number (RIN)] was assessed using an Agilent 2100 Bioanalyzer and RNA 6000 Nano Kit and quantified using a Qubit® 3.0 fluorometer and Qubit RNA HS Assay Kit. Samples with a total of 1500 ng RNA and RIN ≥ 4.1 were used as the input for the next library preparation. Briefly, the first- and second-strand complementary DNA (cDNA) was synthesized using a PrimeScript Double Strand cDNA Synthesis Kit (Takara). Double-stranded cDNA was then cleaned with Agencourt AMPure XP beads (Beckman Coulter) and subjected to end-repair, adenylation, and ligation using a universal barcode adapter, subjected, and amplified by seven cycles to generate the mid-libraries. The target genes were captured with a specific panel from the mid-libraries, amplified, and then sequenced. Paired-end, 101 bp sequencing was performed using a HiSeq 2500 (Illumina) instrument in the Rapid Run mode. The sequence was aligned to the reference sequence using Hisat2 (2.0.3). FusionMap software was used to detect the fusion genes, and Blacklist filtering was used to remove the ribosomal genes, mitochondrial genes, and fusions of pseudogenes, as well as the fusions between gene families and homologous genes. The targeted fusion genes are shown in Additional file 4: Table S1.
Statistics method
The data were analyzed and processed using GraphPad 7.0 software. The measurement data conforming to normal distribution were compared using a Student’s t-test and variance analysis. The mean value of measurement data that did not conform to normal distribution was compared using a rank-sum test. The frequency of counting data was expressed in %, and the rates were compared by conducting a χ2 test. The survival curve was tested using the log-rank method. Overall survival (OS) was calculated from diagnosis to death. Disease-free survival (DFS) was calculated from the first CR to relapse or death, and patients who did not achieve CR were excluded. Event-free survival (EFS) was calculated from diagnosis to relapse or death of any cause. A statistical difference was considered at P < 0.05.
Results
Clinical associations
The frequency of FLT3-ITD and FLT3-TKD mutation was 28%, and 9.7%, respectively. The general characteristics of FLT3-ITD AML, FLT3-TKD AML, and FLT3wt AML are shown in Table 1. The count of white blood cell (WBC) and the proportion of blasts in the bone marrow of FLT3-ITD group was higher than that of FLT3wtAML group (P = 0.000 and P = 0.024, respectively). The count of WBC of FLT3-TKD group was also higher than that of FLT3wtAML group, P = 0.008. There was no significant difference in the proportion of bone marrow blasts between the FLT3-TKD group and FLT3wtAML group, P > 0.05. FLT3-ITD was associated with normal karyotype (64.8% vs. 48.4%, P = 0.043); in contrast, FLT3-TKD showed no difference in karyotype distribution compared to FLT3wt AML, (40% vs. 48.4%, P > 0.05).
Table 1.
Clinical, cytogenetics and molecular genetics characteristic of 207 analyzed AML patients
| Parameter |
FLT3-ITD (n = 58) |
FLT3-TKD (n = 16) |
FLT3wtAML (n = 133) |
P valuea |
|---|---|---|---|---|
| Male | 23 (39.7) | 12 (75) | 80 (60.2) | 0.009 |
| Age | 48 (14–73) | 41 (14–76) | 45 (15–76) | 0.367 |
| WBC at diagnosis, × 109/L | 72.9 (2.3–405.1) | 68.2 (1.8–251.1) | 24.2 (0.57–311.0) | 0.000 |
| Blasts in BM, % | 65.9 (22.0–95.6) | 55.5 (30.8–94.0) | 56.0 (14.4–94.5) | 0.040 |
| FAB subtype, n (%) | 0.983 | |||
| M0 | 0 | 0 | 0 | |
| M1 | 3 (5.2) | 1 (6.3) | 4 (3.0) | |
| M2 | 16 (27.6) | 5 (31.3) | 38 (28.6) | |
| M4 | 20 (34.5) | 6 (37.5) | 41 (30.8) | |
| M5 | 14 (24.1) | 4 (25.0) | 36 (27.1) | |
| M6 | 2 (34) | 0 | 5 (3.8) | |
| Unclassified | 1 (1.7) | 0 | 6 (4.5) | |
| Secondary-AML | 2 (3.4) | 0 | 3 (2.3) | |
| Cytogenetics, n (%) (n = 197) | ||||
| Normal karyotypes | 35 (64.8) | 6 (40.0) | 62 (48.4) | 0.079 |
| Aberrant karyotypes | 19 (35.2) | 9 (60.9) | 66 (51.6) | |
| Gene Mutationc, n (%) | ||||
| NPM1 | 28 (53.8)& | 4 (25) | 20 (15.3) | 0.000 |
| DNMT3A | 18 (34.6)& | 4 (25) | 17(13.0) | 0.003 |
| RUNX1 | 1 (1.7) | 1 (6.3) | 8 (6.1) | 0.492 |
| KIT | 3 (5.8) | 0 (0) | 6 (4.6) | 0.623 |
| RAS | 1 (1.9)& | 1 (6.3) | 27 (20.6) | 0.003 |
| PTPN11 | 5 (9.6) | 1 (6.3) | 6 (6.3) | 0.435 |
| TET2 | 6 (11.5) | 1 (6.3) | 10 (7.6) | 0.656 |
| IDH1/2 | 5 (19.6) | 3 (18.8) | 20 (15.3) | 0.522 |
| CEBPA | 2 (3.8)& | 0 (0) | 26 (19.8) | 0.005 |
| ASXL1 | 2 (3.8) | 2 (12.5) | 13 (9.9) | 0.348 |
| TP53 | 0 (0)& | 1/16 (6.3) | 8 (6.1) | 0.189 |
| Methylation-related genesb | 23 (44.2) | 6 (37.5) | 42 (32.1) | 0.297 |
| Number of mutations | 3.2 (1–7)& | 3.6 (1–6) | 2.7 (0–8) | 0.022 |
| CR after two cycles of induction | 29/46 (63) | 10/14 (71.4) | 100/113 (88.5) | 0.001 |
| Consolidation in CR1 | ||||
|
CT SCT |
16 (55.2) 13 (44.8) |
5 (50) 5 (50) |
6 (49.5) 47 (50.5) |
0.865 |
| Three-year OS (%) | 36 ± 9.1 | 65.6 ± 15.1 | 50.6 ± 7 | 0.020 |
| Three-year EFS (%) | 27.2 ± 8.1 | 55.9 ± 16.2 | 40.5 ± 6.5 | 0.005 |
Italic values indicate significance of P value (P < 0.05)
WBC white blood count, BM bone marrow, FAB French–America–British, CR complete remission, CT chemotherapy, SCT stem cell transplantation
aP-values for categorical variables are from chi-square test, P-values for continuous variables are from the ANOVA test
bMethylation related gene included DNMT3A, IDH1/2, and TET2
c52 FLT3-ITD, 16 FLT3-TKD and 131 FLT3 wildtype patients were analyzed for gene mutations
#p value for frequency of favorable, intermediate and unfavorable karyotype in three groups
&P value < 0.05 between the FLT3-ITD group and FLT3wt group
The CR rate after two cycles of induction of FLT3-ITD group was lower than that of FLT3wtAML group (63% and 88.5%, respectively, P = 0.000). The CR rate of FLT3-TKD patients was is 71.4%, not significantly different from that of the FLT3wtAML group, P = 0.077. The FLT3-ITD group had a lower three-year OS, DFS, and EFS than those of FLT3-TKD group and FLT3wtAML group (36% ± 9.1% vs. 65.6% ± 15.1% vs. 50.6% ± 4.6%, respectively, P = 0.02; 45.8% ± 10.8% vs. 70% ± 18.2% vs. 44.6% ± 7.4%, respectively, P = 0.052; 27.2% ± 8.1% vs. 55.9% ± 16.2% vs. 40.5% ± 6.5%, respectively, P = 0.005) (Fig. 1a–c). The three-year OS, DFS, and EFS of FLT3-ITD group, FLT3-TKD group, and FLT3-ITDwt group in normal karyotype (NK)-AML are shown in Fig. 1d–f. No difference was observed between FLT3-TKD group and FLT3wt group in three-year OS, DFS, and EFS in NK-AML (65.5% ± 20.9% vs. 54.4% ± 10.5%, P = 0.538; 53.3% ± 24.8% vs. 51.7% ± 8.4%, P = 0.43; 48.6% ± 22.7% vs. 47.9% ± 7.6%, P = 0.557). FLT3-ITD could stratify the outcomes of NK-AML patients (24% ± 19% vs. 54.4% ± 10.5%, P = 0.035; 0% vs. 51.7% ± 8.4%, P = 0.004; 0% vs. 47.9% ± 7.6%, P = 0.000). Furthermore, co-occurring TET2 mutation impaired the 3-year OS, DFS, and EFS of patients with FLT3-ITD (37.9% ± 10.3% vs. 25% ± 20.4%, P = 0.044; 48.9% ± 12.6% vs. 16.7% ± 15.2%, P = 0.002; 27.8% ± 9.2% vs. 16.7% ± 15.2%, P = 0.049) (Fig. 1g–i).
Fig. 1.

OS (a), DFS (b), and EFS (c) curve of FLT3-TKD (n = 16), FLT3-ITD (n = 56), and FLT3 wild type (n = 132) AML patients; OS (d), DFS (e), and EFS (f) curve of normal karyotype AML patients with (n = 18) or without (n = 74) FLT3-ITD mutation; OS (h), DFS (i), and EFS (g) curve of FLT3-ITD AML patients with (n = 6) or without (n = 44) TET2 mutation
Associations with fusion genes
Among the 52 patients with FLT3-ITD, 21 had fusion genes, and the incidence of fusion genes was 40.4% (Fig. 2a, b). The most common fusion genes of FLT3-ITD AML included seven MLL-rearranged (13.5%) (four MLL-PTD, two MLL-AF9, and one MLL-ELL) and seven NUP98-rearranged (13.5%) (four NUP98-NSD1 and three NUP98-HOX9A). Other recurrent fusions included three with AML1-ETO and two with DEK/CAN. One case with PRDM16-SKI and one case with EFAN2-ZNF238 fusion gene are reported for the first time.
Fig. 2.

Relationship between gene mutations and fusion genes of FLT3-ITD and FLT3-TKD AML. a, b Represent fusion genes by targeted NGS and its exclusive relationship with NPM1 mutation in FLT3-ITD positive AML (n = 60). c, d Represent fusion genes by targeted NGS and its exclusive relationship with NPM1 mutation in FLT3-TKD positive AML (n = 16)
Among the 16 FLT3-TKD mutant AML patients, 11 cases had fusion genes (four cases with AML1-ETO, two cases with MLL-AF9, one case with AML1-MDS1, one case with DEC-CAN, one case with BCR-ABL, one case with CBFB-MYH11, and one case with MLL-TMX2 -CTNND1) (Fig. 2c, d). The frequency of fusion genes of FLT3-TKD group was higher than that of FLT3-ITD group (68.75% vs. 40.4%, P = 0.014). Among patients with FLT3-TKD, 11 patients were associated with fusion genes, as described in the manuscript, four cases with AML1-ETO, two cases with MLL-AF9, one case with AML1-MDS1, one case with DEC-CAN, one case with BCR-ABL, one case with CBFB-MYH11, and one case with MLL-TMX2-CTNND1. Both patients with MLL rearrangement were refractory to induction therapy and died of disease progression. The prognosis of four patients with AML1-ETO was relatively good, among which two patients achieved long-term survival through chemotherapy and transplantation. Two patients with AML1-ETO relapsed and were salvaged by allo-HSCT, one of who relapsed and died after salvaged transplantation, and the other patient achieved long-term survival after salvaged transplantation. The patient with BCR/ABL1 fusion died during induction. The remaining patients with DEC-CAN, AML-MDS1, and CBFC-MYH11 achieved long-term survival. Among them, the patient with DEC-CAN received allogeneic transplantation, the patient with AML-MDS1 received autograft, and the patient with CBFB-MYH11 received chemotherapy as consolidation after remission.
Fusion genes and chromosome karyotype characteristics are shown in Table 2. NGS was efficient in the detection of gene fusions, especially in the rare fusions or MLL translocation partner genes. Cytogenetics analysis failed to detect all NUP98-NSD1 and four of five MLL fusions. Furthermore, one MLL/MLLT3 and one MLL/TMX2-CTNND1 were detected by only NGS. In normal karyotypes cases and negative cases by routine PCR, NGS identified seven fusion genes including four with NUP98-NSD1, four with AML/MDS1, one with PRDM16/SKI, one with EFCAB2/ZNF238, and one with MLL/MLLT3. One MLL fusion with rare translocation partner genes TMX2/CTNND1 was detected by NGS, while without providing information by karyotype analysis (Table 2).
Table 2.
Fusion genes by NGS and PCR and Chromosome karyotype analysis in FLT3 mutant AML
| Fusion gene by NGS | Fusion gene by PCR | Chromosome karyotype | FLT3 mutation |
|---|---|---|---|
| AML1/ETO | AML1/ETO | 46, XY, t(8;21)(q22;q22)[20] | FLT3-ITD, FLT3-TKD |
| AML1/ETO | AML1/ETO | 46, X, -X,t(8;21)(q22;q22), del(9)(q22)[9]/46,XX,t(8;21)(q22;q22)[11] | FLT3-ITD |
| AML1/ETO | AML1/ETO | 45, X, -X,t(8;21)[20] | FLT3-ITD |
| MLL-PTD | MLL-PTD | 46, XY[20] | FLT3-ITD |
| MLL-PTD | MLL-PTD | 46, XY[20] | FLT3-ITD |
| MLL-PTD | MLL-PTD | 46, XX[20] | FLT3-ITD |
| MLL-PTD | MLL-PTD | 47, XY, + 8?[10]/46, XY[10] | FLT3-ITD |
| MLL/AF9 | MLL/AF9 | 46, XX,?der(2)(q11),inc[1] /46,XX[28]/hypodiploid [4] (44–45) | FLT3-ITD |
| MLL/AF9 | MLL/AF9 | 47, XY, + 8[7] | FLT3-ITD |
| MLL/ELL | MLL/ELL | 46, XX, t(11;19)(q23; q13)[10] | FLT3-ITD |
| NUP98/HOXA9 | NUP98/HOXA9 | NA | FLT3-ITD |
| NUP98/HOXA9 | NUP98/HOXA9 | 46, XX[20] | FLT3-ITD |
| NUP98/HOXA9 | NUP98/HOXA9 | NA | FLT3-ITD |
| NUP98-NSD1 | – | 46, XX[20] | FLT3-ITD |
| NUP98-NSD1 | – | 47, XX, + 6[14]/46, XX[6] | FLT3-ITD |
| NUP98-NSD1 | – | 46, XY[20] | FLT3-ITD |
| NUP98-NSD1 | – | 46, XY[25] | FLT3-ITD |
| DEK/CAN | DEK/CAN | 46, XX[20] | FLT3-ITD |
| DEK/CAN | DEK/CAN | 46, XY, ?t(6;9)(p23;34)[10]/46, XY,?t(6;9)(p23;q34),?del(8)(q21)[11]/46,XY[1] | FLT3-ITD |
| PRDM16-SKI | – | 46, XX[20] | FLT3-ITD |
| EFCAB2-ZNF238 | – | 46, XX [20] | FLT3-ITD |
| BCR/ABL | BCR/ABL | NA | FLT3-TKD |
| AML1/ETO | AML1/ETO | 45, X, -Y, t(8;21)(q22;q22)[22] | FLT3-TKD |
| AML1/ETO | AML1/ETO | 46, XY,t(8;21)(q22;q22)[26]/46,XY[1] | FLT3-TKD |
| AML1/ETO | AML1/ETO | 46, XX, t(8;21)(q22;q22)[20] | FLT3-TKD |
| AML1/ETO | AML1/ETO | 45,X,?Xq-,?8q-,-22[1]/43,X,?Xq-,-8,-10,-22[1]/45,X,-X[1]/47,XX, + mar[1]/40,-X,-X,-11,-21,-22, + mar[1]/46,XX[4] | FLT3-TKD |
| MLL/MLLT3 | – | 46, XX[20] | FLT3-TKD |
| DEK/CAN | DEK/CAN | 47, XY, chtb(4)(?q31),? + 9,-15,inc[1]/46, XY[27]/hypodiploid [2] (44–45) | FLT3-TKD |
| AML/MDS1 | – | 46, XY [20] | FLT3-TKD |
| SLC45A3/ELK4 | – | 47, XY, + 8[7] | FLT3-TKD |
| MLL/TMX2-CTNND1 | – | 42–47,XY, + 3,del(3)(p13),del(3)(q13),-4,?add(4)(q35),-8,-11,dic(11;?)(q25;?),-16,-17,-18,-19,-20, + r, + mar1, + mar2, + mar3,inc[cp22]/46,XY[1] | FLT3-TKD |
| CBFB/MYH11 | CBFB/MYH11 | 47, XY, + 22[2]/46, XY[23] | FLT3-TKD |
NGS, next generation sequencing; PCR, polymerase chain reaction
Associations with other molecular mutations
The mutation data were available in subcohorts as follows: 52 FLT3-ITD, 16 FLT3-TKD, and 131 FLT3wt patients. NPM1 and DNMT3A were concomitantly observed together with FLT3-ITD (Table 1; Figs. 3, 4). The frequency of NPM1 mutation was 53.8% in FLT3-ITD AML, higher than that of FLT3wtAML group (15.3%), P = 0.000. The second frequent mutation was DNMT3A, with a frequency of 34.6%, significantly higher than that of FLT3wtAML group, P = 0.001. However, the mutation in CEBPA and Ras were highly infrequent in FLT3-ITD AML (2/52 (3.8%) vs. 26/131 (19.8%), P = 0.007; 1/52(1.9%) vs. 27/131(20.6%), P = 0.002). RAS mutations in FLT3-ITD (n = 1) and FLT3-TKD (n = 1) were both NRAS mutation. RAS isoforms (n = 27) in FLT3wt patients were NRAS in 22 cases, KRAS in four cases, and both NRAS and KRAS in one case. Further, TP53 mutations were mutually exclusive of FLT3-ITD (0/58 vs. 8/131, P = 0.004). The average number of mutations in FLT3-ITD AML was 3.7, higher than that in FLT3wtAML (average number = 2.7, P = 0.011).
Fig. 3.

Distribution of somatic mutations and fusion genes in 82 AML patients with FLT3-ITD and FLT3-TKD. Each column displays an individual sample. White highlights in the top FAB subtype indicate that the information is not available (n.a.). Blue highlights indicate the presence of a gene mutation; grey highlights indicate wild-type status. CEBPA mutation is an allele double mutation in this panel. Mutated genes are clustered according to their pathways or family
Fig. 4.
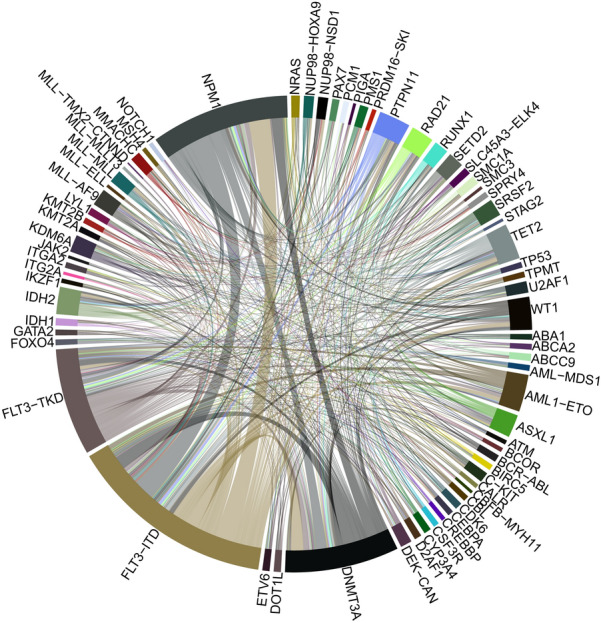
Circos of mutated genes and gene fusions in AML patients with FLT3 mutation. Ribbon widths are proportional to the frequency of a molecular event
In contrast to FLT3-ITD, no significant difference was observed in the incidence of NPM1, DNMT3A, and RAS mutations between FLT3-TKD group and FLT3wtAML group. Mutations of CEBPA were also excluded in FLT3-TKD mutant patients (0/16 vs. 26/131, P = 0.05) (Figs. 3, 4).
Clinical features of FLT3 AML with mutations and fusion genes
In AML patients with FLT3-ITD, NPM1 and DNMT3A were the most common mutations. FLT3-ITD with both NPM1 and DNMT3A mutations defines a poor prognosis. Three-year OS of FLT3-ITD patients with both NPM1 and DNMT3A mutations was 12.7% ± 11.5% (Additional file 2: Fig. S2A), though no significant difference was observed between the four subgroups in FLT3-ITD patients in DFS (Additional file 2: Fig. 2B).
NPM1 mutation and fusion genes rarely occurred simultaneously in FLT3 mutant patients (Figs. 2, 5). Only two patients with FLT3-ITD are accompanied by both NPM1 mutation and fusion genes (NUP98/HOXA9 and PRDM16/SKI, respectively). The FLT3-ITD patient with NUP98/HOXA9 and NPM1 achieved CR after IA regimen and obtained continuous CR after HSCT. However, the other FLT3-ITD patient with PRDM16/SKI and NPM1 was refractory to induction and died of disease progress after four cycles of chemotherapy. The same trend was observed in FLT3-TKD group, with only one patient with both BCR/ABL and NPM1 mutation who died during induction due to disease progress. Interestingly, it was also rare to have neither an NPM1 mutation nor a fusion gene in FLT3 mutant AML. Only three FLT3-ITD patients had neither NPM1 mutation nor fusion genes, nor did two FLT3-TKD AML patients. The outcomes of these five patients were heterogeneous. One patient with FLT3-ITD did not receive chemotherapy. The other two patients with FLT3-ITD were both refractory to induction. One of the two patients with FLT3-TKD without NPM1 or fusions was refractory to induction, and the other one achieved long-term survival after chemotherapy.
Fig. 5.

Molecular heterogeneity of AML exemplified by mutational and fusion genes profiling in FLT3-AML. Each spoke radiating from the central FLT3-ITD or FLT3-TKD hub represents the molecular pattern of a single patient. Cooperating mutations are grouped into three tiers according to the function and color-coded according to the figure key, and white space indicates no mutation or fusion. Overall, based on molecular combination, patients are segregated into different subgroups
According to molecular alteration status, FLT3-ITD AML patients were divided into NPM1 + Fusion− group and Fusion + NPM1− group. The median age of NPM1 + Fusion− group was older than that of Fusion + NPM1− group, 52 and 36 years old, respectively, P = 0.006. NPM1 mutation was more likely to occur in older patients, while the fusion gene was more likely to be associated with younger age. The average number of mutations in NPM1 + Fusion− patients was 3.6, while the average number of mutations in Fusion + NPM1 − group was 2.2, indicating different molecular distributions. NPM1 mutation was associated with methylation-related mutations such as DNMT3A. No difference was observed in the CR rate and survival rate between the two groups (Additional file 3: Fig. S3). Different mutation distributions in NPM1 + Fusion − group and Fusion + NPM1 − group could be caused by different pathogenic mechanisms (Table 3; Fig. 6).
Table 3.
Clinical characteristic and outcomes of patients with NPM1 mutation or fusion genes in FLT3-ITD AML
| Characteristics |
FLT3-ITD + NPM1 + Fusion − |
FLT3-ITD + Fusion + NPM1 − |
P valuea |
|---|---|---|---|
| n | 31 | 24 | |
|
Age, years Median(range) |
52 (14–76) | 36 (12–65) | 0.007 |
|
Male n (%) |
12 (38.7) | 11 (45.8) | 0.254 |
|
WBC at diagnosis, × 109/L Median(range) |
31.4 (1.3–405.1) | 10.8 (0.7–306.9) | 0.278 |
|
Blast in BM, % Median(range) |
68.8 (21.2–96.4) | 56.4 (11.2–95.6) | 0.543 |
| FAB subtype | 0.671 | ||
| M0 | 0 (0) | 0 (0) | |
| M1 | 1 (3.3) | 1 (4.2) | |
| M2 | 8 (26.7) | 8 (33.3) | |
| M4 | 13 (43.3) | 9 (37.5) | |
| M5 | 6 (20.0) | 5 (20.8) | |
| M6 | 0 (0) | 1 (4.2) | |
| Unclassified | 0 (0) | 0 (0) | |
| Secondary-AML | 2 (6.7) | 0 (0) | |
| Karyotype | 0.015 | ||
| Favorable | 0 (0) | 7 (29.2) | |
| Intermediate | 27 (87.1) | 13 (54.2) | |
| Normal | 22 (71) | 9 (37.5) | 0.007 |
| Others | 5 (16.1) | 4 (16.7) | |
| Unfavorable | 2 (6.5) | 3 (12.5) | |
| Failed | 2 (6.5) | 1 (4.2) | |
| Immunophenotype | |||
| CD34 + | 18/25 (72.0) | 20/22 (90.9) | 0.203 |
| CD13 + | 25/26 (96.3) | 18/20 (90.0) | 0.814 |
| CD33 + | 26/26 (100) | 21/22 (95.5) | 0.458 |
| CD117 + | 23/25 (92) | 21/22 (95.5) | 1.000 |
| CD64 + | 10/24 (41.7) | 5/11 (45.5) | 1.000 |
| Mutations | |||
| Average num | 3.6 | 2.2 | 0.000 |
| DNMT3A | 13/28 (46.4) | 2/22 (9.1) | 0.004 |
| Methylation-related genesb | 16/28 (57.1) | 4/22 (18.2) | 0.005 |
| CR, n (%) | |||
| Yes | 14/25 (56) | 17/24 (70.8) | 0.282 |
| Relapse in 1 year | |||
| Yes | 16/22 (72.7) | 17/20 (85) | 0.460 |
Italic values indicate significance of P value (P < 0.05)
WBC white blood count, BM bone marrow, FAB French–America–British, CR complete remission
aP-values for categorical variables are from chi-square test, P-values for continuous variables are from the Mann–Whitney test and Fisher exact test
bMethylation-related genes included DNMT3A, IDH1/2, and TET2
Fig. 6.

Schematic model for the two paths of evolution of FLT3 mutant AML. The first step is the occurrence of mutations or fusions, and the second step is the hit of FLT3-ITD or FLT3-TKD mutations
Discussion
FLT3-ITD is a late acquired proliferative advantage in leukemogenesis. In addition to FLT3-ITD, other molecular alterations are necessary (Fig. 6). Because of the complex pathogenic mechanism, FLT3-ITD AML is heterogeneous. FLT3-ITD and FLT3-TKD mutants show distinct gain-of-function phenotypes with distinct differences in signaling properties and gene expression patterns. Whether FLT3-TKD has the same prognostic significance as FLT3-ITD is controversial. Comprehensive detection of molecular landscape of FLT3 mutant AML is significant to establish a risk classification in AML and guide therapy options. Here, we evaluated a cohort of 207 AML patients for mutations in FLT3 with two targeted sequencing approaches to obtain novel insights into the prognostic relevance of FLT3 mutations as well as their associations with other molecular markers.
Generally, we observed an overall FLT3-ITD and FLT3-TKD mutation rate of 28% and 9.7% in 207 de novo AML, consistent with previous reports [21, 22]. Through novel targeted RNA-seq-based NGS, the profile of companying fusion genes with FLT3 mutations was revealed. The common fusion genes in FLT3-ITD had MLL-rearrangement and NUP98-rearrangement, while the common fusion genes in FLT3-TKD had AML1-ETO and MLL-rearrangement. Two novel fusion genes PRDM16-SKI and EFAN2-ZNF238 were identified in FLT3-ITD patients. Gene coexistence analysis revealed unbalanced gene mutation distributions in FLT3-ITD, FLT3-TKD, and FLT3wt AML. Gene fusions and NPM1 mutation are mutually excluded in FLT3-ITD and FLT3-TKD patients. Their patterns of mutual exclusivity and cooperation among mutated genes suggest that additional driver genetic alterations are required and reveal two evolutionary patterns of FLT3 pathogenesis. We observed unfavorable impact on CR and survival of FLT3-ITD in the whole cohort and NK-AML patients, consistent with other studies [21]. Besides, additional TET2 mutation further impaired the prognosis of patients with FLT3-ITD. In contrast to FLT3-ITD mutations, FLT3-TKD mutation did not affect the remission rate and survival of AML patients in this study. In patients with FLT3-TKD mutations in AML, the main prognostic factor still seems to be the concomitant fusion genes. MLL gene rearrangement is an identified adverse prognosis factor. The two patients with FLT3-TKD accompanied by MLL rearrangement were both primary refractory, while the patients with AML-ETO had a relatively good prognosis, 3/4 of them achieving long-term survival. Therefore, attention should be paid to the accompanying molecular abnormalities when stratifying risk in patients with FLT3-TKD mutations. FLT3-ITD mutation alone is insufficient to drive leukemogenesis, suggesting that additional mutations are necessary for full transformation. Genetic testing incorporating both molecular analysis and cytogenetic karyotyping is an integral part of definition and risk stratification of AML to guide therapy and monitor disease response/relapse [1, 23]. To further understand the pathogenic mechanism and prognostic effect of FLT3 mutations, gene mutations and gene fusions were examined using two targeted NGS methods in FLT3-mutant AML patients. NPM1 and DNMT3A were concomitantly observed together with FLT3-ITD. NPM1 mutation showed a strong correlation with FLT3-ITD in previous reports [8, 24, 25]. NPM1 is considered as one of the early cooperating mutations in leukemia leukemogenesis (Fig. 6) [26]. DNMT3A mutation is another common mutation in patients with FLT3-ITD. Epigenetics plays an important role in leukemogenesis. In Jifeng Yu et al.’s recent study, older AML patients (≥ 60 years) showed association with more incidence of DNA methylation compared with younger AML patients (87.7% vs. 75.4%, P = 0.0425) [27]. DNMT3A mutation functions, as an epigenetic regulator, are associated with aging [28]. This could explain the elder distributions of age of NPM1 mutation subgroup. We found that patients with comutation of FLT3-ITD and TET2 mutation had shorter survival compared to patients with FLT3-ITD mutation and wildtype TET2, identifying FLT3-ITD/TET2 bimutation as a high-risk AML subgroup. TET2 mutation and FLT3-ITD cooperatively remodeled DNA methylation and gene expression and triggered AML in vivo. Besides, the induced AML cell demonstrated refractory to standard AML chemotherapy and FLT3 targeted treatment [29]. We previously reported that TET2 mutation is an unfavorable prognostic factor in AML patients [30]. Furthermore, TET2 mutation with FLT3-ITD could further stratify AML patients with intermediate-risk cytogenetics. Interestingly, the mutations in Ras, CEBPA, and TP53 were found to be excluded in FLT3 mutant AML. CEBPA mutation was reported to be restricted in normal karyotype without FLT3-ITD and NPM1 mutation [31]. Ras also led to secondary events that occur later during leukemogenesis. Similar to Stirewalt’s study, the same negative association was observed between Ras mutation and FLT3 mutations in our study [32]. Most TP53 mutation is associated with abnormal cytogenetics, especially abnormalities in chromosomes 5 and 7, while FLT3-ITD is associated with normal karyotype [32]. These exclusive relationship between FLT3 mutation and mutations in Ras, CEBPA, and TP53 probably indicate that the use of a differential detection panel in genetic mutations may be convenient and economical [33].
The fusion genes are important pathogenic mechanism of leukemogenesis. It is considered as a potential therapeutic target and MRD monitoring marker. We found that MLL-rearrangement and NUP98-rearrangement are both recurrent fusion genes in FLT3-ITD, and their partner genes are multitudinous. In a Genome Atlas Research, 118 gene fusions were found in 178 de novo AML samples, including 74 reported recurring events and 57 novel gene fusions; most of them were not detected using cytogenetic studies [24]. The karyotype analysis relies on experts’ experience, and it is likely to be missed if the changes in chromosomal appearance after translocation are not easily discernible. Besides, the karyotype results may be inconsistent with the fusion gene expression under some conditions. In our study, 32 FLT3-mutant patients were identified with fusions, 28 of which were generated by translocations. However, only seven fusions showed consistent karyotype results.
PCR detection relies on targeted primer design; novel/rare fusion genes and common genes fusing at a rare site could be missed. On the other hand, even though chimeric RNA is almost the product of chromosomal rearrangement at the DNA level, it can also be generated from trans-splicing and cis-splicing between neighboring genes in some cases, which is only detectable at the RNA level than the DNA level [34–36]. Thus, RNA-based fusion gene detection is more comprehensive. The advantage of NGS is that it can detect atypical sites of classical fusions, identify fusions involving multiple fusion partner genes, and discover rare and unknown fusions [37]. In this study, 21 of 52 patients with FLT3-ITD had fusion genes detected by NGS, and the incidence of fusion genes is 40.4%. The most common fusions in FLT3-ITD included MLL-rearranged and NUP98-rearranged. The MLL fusion was associated with the fewest number of mutant genes in the newly diagnosed AML, indicating that the MLL gene alterations are very strong AML-initiating factors. Besides, NPM1 and DNMT3A gene mutations were exclusive in MLL fusions [24, 38]. MLL rearrangement accounts for about 10% of AML, and the prognosis is very poor. The median age of onset of leukemia in infants and young children closely related to MLL rearrangement is only six months, suggesting that MLL rearrangement is a very powerful pathogenic factor. A high expression of FLT3 is frequently observed in MLL-rearranged AML, but in vivo experiments showed that could induce AML independent of the FLT3 signaling pathway [39, 40]. A long-distance inverse PCR can be used to characterize MLL rearrangement; identification and distribution of MLL rearrangements of 579 AML samples including infants, pediatric, and adults were studied [41]. The most frequent fusion genes were MLL-MLLT3/AF9 (28.8%), MLL-MLLT10/AF10 (15.2%), MLL-ELL (11.4%), MLL-PTDs (11.4%), MLL-MLLT4/AF6 (9.5%), MLL-MLLT1/ENL (4.0%), MLL-SEPT6 (1.9%), and MLL-MLLT6/AF17 (1.6%). Adult AML patients were characterized by a higher frequency of MLL-PTD at 23.4% (64/272) compared to none and 1.9% in infants and pediatric, respectively. In this study, MLL-PTD was positive in four patients with FLT3-ITD, which is the second most frequent fusion in FLT3-ITD in our study. MLL-PTD is not sufficient to cause leukemia alone; an additional FLT3-ITD could trigger leukemia in mice [42]. Sun et al. identified the most frequent mutation in MLL-PTD, which was FLT3, and NPM1 was mutually exclusive with MLL-PTD, exhibiting the same trend as in this study [26]. Interestingly, NPM1 was wild type in these four MLL-PTD patients. MLL-PTD functioned as an early clonal driver mutation, while FLT3-ITD was acquired later.
NUP98-NSD1, created by the translocation of juxtaposition of Nucleoporin 98 (NUP98) and nuclear receptor binding SET-domain Protein 1 (NSD1) gene, is a common type of translocation in FLT3-ITD AML patients [10]. The incidence of translocation involving NUP98 in AML is very low, only 3% in adult AML [43]. It is reported that the frequency of NUP98-NSD1 in FLT3-ITD can reach 15%, and the frequency of NUP98-NSD1 combined with FLT3-ITD is 82% [44]. The incidence of NUP98 fusion accounted for 33.3% fusion genes in FLT3-ITD, and the incidence of NUP98-NSD1 in FLT3-ITD was 6.7% in our study, reflecting a strong synergistic effect. The lower incidence of NUP98-NSD1 in FLT3-ITD compared to the previous report is probably due to the insufficient number of cases. When NUP98-NSD1 and FLT3-ITD occur simultaneously, the CR rate is less than 30% in AML patients with concurrent NUP98-NSD1 and FLT3-ITD, and the patient’s prognosis is extremely poor [45].
The FLT3-TKD is positively correlated with normal karyotype, and the incidence of FLT3-TKD is 5–10% in normal karyotype AML [22, 46]. In the largest clinical study on FLT3-TKD, FLT3-TKD mutation alone did not affect prognosis [22]. Though no difference was observed in the incidence of NPM1 mutation in patients with FLT3-TKD and FLT3wt, NPM1 was observed to be one of the most common mutations in FLT3-TKD in this study. Expression of FLT3-TKD is insufficient to trigger leukemia in mice; however, a co-NPM1 mutation actively led to the onset of AML in mice. NPM1c altered the cellular localization of FLT3-TKD, leading to the aberrant activation of downstream STAT5 signaling pathway [47]. Interestingly, patients with FLT3-TKD and NPM1 comutation had a better prognosis than patients with FLT3-TKD or NPM1 mutation alone [22, 48]. FLT3-TKD had an unfavorable influence on prognosis in t(15;17)/PML-RARA and MLL-PTD/TKD double-mutated cases. Compared with FLT3-ITD, FLT3-TKD exhibits different molecular genetic profiles. The most frequent fusion gene in FLT3-TKD group was AML1-ETO. The high correlation between FLT3-TKD and AML1-ETO is probably one of the reasons why it has no adverse effect on prognosis. AML1-ETO fusion is one of the most common fusions in AML. In AML1-ETO AML patients, combined gene mutations are most frequently involved in the signal transduction pathway, including FLT3, KIT, and NRAS [49, 50]. The incidence of FLT3 mutation in core-binding factor (CBF) AML is 5–10%. FLT3 mutation combined with AML1-ETO gene fusion can lead to the onset of leukemia [9, 51]. In our previous study, 21 patients with AML1-ETO fusion-positive AML had a higher relapse rate and mortality with an FLT3 gene expression greater than 35% [52]. FLT3-ITD attenuates the good prognosis of AML1-ETO to some extent.
Because FLT3 mutation is insufficient to induce leukemia, additional gene aberration is necessary. We comprehensively examined the molecular genetic background of FLT3 mutant AML using two second-generation sequencing methods. In previous studies, FLT3 was found to be a late acquired genetic change; our results revealed two molecular collaborative patterns of FLT3 mutation in leukemia progression. Initiation of molecular alterations includes mutations and fusions. Initiation of cooperative gene mutation mainly includes NPM1 mutation and methylation-modified genes such as DNMT3A and TET2. In the other FLT3 mutant patients, fusions play an important role in leukemogenesis, especially the MLL-rearranged, NUP98 fusions, and AML1-ETO (Fig. 6).
Conclusions
In summary, this study elucidated the coexisting molecular landscape of AML with FLT3-ITD and FLT3-TKD mutations by NGS, revealing two patterns of two paths of evolution of FLT3 mutant AML. We confirmed the unfavorable prognostic effect of FLT3-ITD and no influence of FLT3-TKD on prognosis. Patients with FLT3-ITD/TET2 bimutation are a high-risk subgroup. Finally, this study provides further insight into the role that genetic alterations including fusion genes and mutations may eventually lead to the development of effective and precise targeted therapy in FLT3 mutated AML.
Supplementary Information
Additional file 1: Figure S1. Treatment flow diagram.
Additional file 2: Figure S2. Overall survival(A) and Disease-Free Survival(B) curves of FLT3-ITD patients divided into four subgroups according to NPM1 and DNMT3A status. The subscript wt and mut represents wildtype and mutant.
Additional file 3: Figure S3. Overall survival(A) and Disease-Free Survival(B) curves of FLT3-ITD AML patients with NPM1 mutation or fusion genes.
Additional file 4: Table S1. Gene penal list by next-generation sequencing.
Acknowledgements
The authors are grateful to the patients who consented to participate in this study and the families who supported them; to nurses and clinicians at the PLA general hospital for caring of patients, Wei Zhou at School of Medicine, Nankai University for sample processing, and Shengpu Niu at Gent University for data analysis.
Abbreviations
- ITD
Internal tandem duplications
- TKD
Tyrosine kinase domain
- AML
Acute myeloid leukemia
- NGS
Next-generation sequencing
- WBC
White blood count
- NK
Normal karyotype
- CR
Complete remission
- OS
Overall survival
- DFS
Disease-free survival
- EFS
Event-free survival
- CML
Chronic myelogenous leukemia
- MRD
Minimal residual disease
Authors’ contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by WG, LZ, and YL; EY, NL, and LF collected and analyzed patients’ data; YL, NF, FL, JZ, BW, and QL performed the NGS sequencing; YD and NW prepared clinical samples; MW analyzed data and drew figures; LW, YJ, and YL guided the study; LY provided funds support and supervised the study. The first draft of the manuscript was written by WG, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82070161, 81970151, 81870134, and 81800135), State Key Program of National Natural Science of China (82030076), Beijing Natural Science Foundation (Grant No. 7202186), and Major science and technology projects for major new drug creation (No. 2019ZX09201-002).
Availability of data and materials
The datasets generated and/or analyzed during this study are not publicly available due to privacy policy but are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All procedures performed in studies involving human participants followed the ethical standards of the Ethics Committee of Chinese PLA general hospital (S2016-076-01) and the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wei Guan, Lei Zhou and Yan Li contributed equally to this work
Supplementary Information
The online version contains supplementary material available at 10.1186/s40164-021-00207-4.
References
- 1.Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29–41. doi: 10.1182/blood-2015-07-604496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wakita S, Yamaguchi H, Ueki T, Usuki K, Kurosawa S, Kobayashi Y, et al. Complex molecular genetic abnormalities involving three or more genetic mutations are important prognostic factors for acute myeloid leukemia. Leukemia. 2016;30(3):545–554. doi: 10.1038/leu.2015.288. [DOI] [PubMed] [Google Scholar]
- 3.Ming Y, Anping L, Linghui Z, Qian C, Yongping S, Kongming W. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017. J Hematol Oncol. 2020;13(1):72. doi: 10.1186/s13045-020-00908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010. doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- 5.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kresinsky A, Schnöder TM, Jacobsen ID, Rauner M, Hofbauer LC, Ast VKR, et al. Lack of CD45 in FLT3-ITD mice results in a myeloproliferative phenotype, cortical porosity, and ectopic bone formation. Oncogene. 2019;38(24):4773–4787. doi: 10.1038/s41388-019-0757-y. [DOI] [PubMed] [Google Scholar]
- 7.Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Haridas Keloth S, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126(22):2491–2501. doi: 10.1182/blood-2015-05-646240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dovey OM, Cooper JL, Mupo A, Grove CS, Lynn C, Conte N, et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood. 2017;130(17):1911–1922. doi: 10.1182/blood-2017-01-760595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005;115(8):2159–2168. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thanasopoulou A, Tzankov A, Schwaller J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica. 2014;99(9):1465–1471. doi: 10.3324/haematol.2013.100917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michmerhuizen NL, Klco JM, Mullighan CG. Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood. 2020;136(20):2275–2289. doi: 10.1182/blood.2020007093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HG, Kojima K, Swindle CS, Cotta CV, Huo Y, Reddy V, et al. FLT3-ITD cooperates with inv(16) to promote progression to acute myeloid leukemia. Blood. 2008;111(3):1567–1574. doi: 10.1182/blood-2006-06-030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ah S, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502–515. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunimoto H, Nakajima H. TET2: a cornerstone in normal and malignant hematopoiesis. Cancer Sci. 2020;112(1):31–40. doi: 10.1111/cas.14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tkachuk DC, Westbrook CA, Andreeff M, Donlon TA, Cleary ML, Suryanarayan K, et al. Detection of bcr-abl fusion in chronic myelogeneous leukemia by in situ hybridization. Science. 1990;250(4980):559–562. doi: 10.1126/science.2237408. [DOI] [PubMed] [Google Scholar]
- 16.Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 17.Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–1054. doi: 10.1038/leu.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Handschuh L. Not only mutations matter: molecular picture of acute myeloid leukemia emerging from transcriptome studies. J Oncol. 2019;2019:7239206. doi: 10.1155/2019/7239206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacher U, Shumilov E, Flach J, Porret N, Joncourt R, Wiedemann G, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018;8(11):113. doi: 10.1038/s41408-018-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, Liu Y, Hou G, Wang L, Lv N, Xu Y, et al. Mutational spectrum and risk stratification of intermediate-risk acute myeloid leukemia patients based on next-generation sequencing. Oncotarget. 2016;31(7):32065–32078. doi: 10.18632/oncotarget.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute lyelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi: 10.1182/blood.V99.12.4326. [DOI] [PubMed] [Google Scholar]
- 22.Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111(5):2527–2537. doi: 10.1182/blood-2007-05-091215. [DOI] [PubMed] [Google Scholar]
- 23.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Döhner K, Thiede C, Jahn N, Panina E, Gambietz A, Larson RA, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood. 2020;135(5):371–380. doi: 10.1182/blood.2019002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun QY, Ding LW, Tan KT, Chien W, Mayakonda A, Lin DC, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD) Leukemia. 2017;31(1):1–10. doi: 10.1038/leu.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Li Y, Li T, Li Y, Xing H, Sun H, et al. Gene mutational analysis by NGS and its clinical significance in patients with myelodysplastic syndrome and acute myeloid leukemia. Exp Hematol Oncol. 2020;9:2. doi: 10.1186/s40164-019-0158-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502–515. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R, Gao X, Yu L. The prognostic impact of tet oncogene family member 2 mutations in patients with acute myeloid leukemia: a systematic-review and meta-analysis. BMC Cancer. 2019;19(1):389. doi: 10.1186/s12885-019-5602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konstandin NP, Pastore F, Herold T, Dufour A, Rothenberg-Thurley M, Hinrichsen T, et al. Genetic heterogeneity of cytogenetically normal AML with mutations of CEBPA. Blood Adv. 2018;2(20):2724–2731. doi: 10.1182/bloodadvances.2018016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97(11):3589–3595. doi: 10.1182/blood.V97.11.3589. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Li Y, Zhang D, Wan D, Jiang Z. Clinical implications of recurrent gene mutations in acute myeloid leukemia. Exp Hematol Oncol. 2020;9. [DOI] [PMC free article] [PubMed]
- 34.Gingeras TR. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461(7261):206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Wang J, Ma X, Sklar J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle. 2009;8(2):218–222. doi: 10.4161/cc.8.2.7358. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012;2(7):598–607. doi: 10.1158/2159-8290.CD-12-0042. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109(3):513–522. doi: 10.1111/cas.13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albouhair S, Morgado E, Lavau C. Flt3 does not play a critical role in murine myeloid leukemias induced by MLL fusion genes. PLoS ONE. 2013;8(8):e72261. doi: 10.1371/journal.pone.0072261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamezaki K, Luchsinger LL, Hans-Willem WS. Differential requirement for wild-type Flt3 in leukemia initiation among mouse models of human leukemia. Exp Hematol. 2014;42(3):192–203. doi: 10.1016/j.exphem.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27(11):2165–2176. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood. 2012;120(3):1130–1136. doi: 10.1182/blood-2012-03-415067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohanty S, Jyotsana N, Sharma A, Kloos A, Gabdoulline R, Othman B, et al. Targeted inhibition of the NUP98-NSD1 fusion oncogene in acute myeloid leukemia. Cancers (Basel) 2020;12(10):2766. doi: 10.3390/cancers12102766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostronoff F, Othus M, Gerbing RB, Loken MR, Raimondi SC, Hirsch BA, et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood. 2014;124(15):2400–2407. doi: 10.1182/blood-2014-04-570929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimada A, Iijima-Yamashita Y, Tawa A, Tomizawa D, Yamada M, Norio S, et al. Risk-stratified therapy for children with FLT3-ITD-positive acute myeloid leukemia: results from the JPLSG AML-05 study. Int J Hematol. 2018;107(5):586–595. doi: 10.1007/s12185-017-2395-x. [DOI] [PubMed] [Google Scholar]
- 46.Sakaguchi M, Yamaguchi H, Kuboyama M, Najima Y, Usuki K, Ueki T, et al. Significance of FLT3-tyrosine kinase domain mutation as a prognostic factor for acute myeloid leukemia. Int J Hematol. 2019;110(5):566–574. doi: 10.1007/s12185-019-02720-z. [DOI] [PubMed] [Google Scholar]
- 47.Rudorf A, Müller TA, Klingeberg C, Kreutmair S, Poggio T, Gorantla SP, et al. NPM1c alters FLT3-D835Y localization and signaling in acute myeloid leukemia. Blood. 2019;134(4):383–388. doi: 10.1182/blood.2018883140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boddu P, Kantarjian H, Borthakur G, Kadia T, Daver N, Pierce S, et al. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv. 2017;1(19):1546–1550. doi: 10.1182/bloodadvances.2017009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci USA. 2001;98(18):10398–10403. doi: 10.1073/pnas.171321298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen G, Liu A, Xu Y, Gao L, Jiang M, Li Y, et al. The RUNX1-ETO fusion protein trans-activates c-KIT expression by recruiting histone acetyltransferase P300 on its promoter. FEBS J. 2019;286(5):901–912. doi: 10.1111/febs.14751. [DOI] [PubMed] [Google Scholar]
- 51.Sun XJ, Wang Z, Wang L, Jiang Y, Kost N, Soong TD, et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature. 2013;500(7460):93–97. doi: 10.1038/nature12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie HM, Gao L, Wang N, Xu YY, Shi JL, Yu L, et al. FLT3 gene overexpression and its clinical significance in acute myeloid leukemia with AML1/ETO fusion gene positive. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014;22(5):1199–1205. doi: 10.7534/j.issn.1009-2137.2014.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Treatment flow diagram.
Additional file 2: Figure S2. Overall survival(A) and Disease-Free Survival(B) curves of FLT3-ITD patients divided into four subgroups according to NPM1 and DNMT3A status. The subscript wt and mut represents wildtype and mutant.
Additional file 3: Figure S3. Overall survival(A) and Disease-Free Survival(B) curves of FLT3-ITD AML patients with NPM1 mutation or fusion genes.
Additional file 4: Table S1. Gene penal list by next-generation sequencing.
Data Availability Statement
The datasets generated and/or analyzed during this study are not publicly available due to privacy policy but are available from the corresponding author on reasonable request.