

Abstract

Lipophilicity is explored in the biodistribution (BD), pharmacokinetics (PK), radiation dosimetry (RD), and toxicity of an internally administered targeted alpha-particle therapy (TAT) under development for the treatment of metastatic melanoma. The TAT conjugate is comprised of the chelator DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate), conjugated to melanocortin receptor 1 specific peptidic ligand (MC1RL) using a linker moiety and chelation of the 225Ac radiometal. A set of conjugates were prepared with a range of lipophilicities (log D7.4 values) by varying the chemical properties of the linker. Reported are the observations that higher log D7.4 values are associated with decreased kidney uptake, decreased absorbed radiation dose, and decreased kidney toxicity of the TAT, and the inverse is observed for lower log D7.4 values. Animals administered TATs with lower lipophilicities exhibited acute nephropathy and death, whereas animals administered the highest activity TATs with higher lipophilicities lived for the duration of the 7 month study and exhibited chronic progressive nephropathy. Changes in TAT lipophilicity were not associated with changes in liver uptake, dose, or toxicity. Significant observations include that lipophilicity correlates with kidney BD, the kidney-to-liver BD ratio, and weight loss and that blood urea nitrogen (BUN) levels correlated with kidney uptake. Furthermore, BUN was identified as having higher sensitivity and specificity of detection of kidney pathology, and the liver enzyme alkaline phosphatase (ALKP) had high sensitivity and specificity for detection of liver damage associated with the TAT. These findings suggest that tuning radiopharmaceutical lipophilicity can effectively modulate the level of kidney uptake to reduce morbidity and improve both safety and efficacy.
Keywords: Biodistribution, Melanocortin receptor 1, Actinium-225, Lipophilicity, Radiotherapy, α-Particle emmiter
Drug discovery and development is a complex, labor-intensive, and lengthy process. This long road includes determining a clinical need, identification and validation of clinically relevant target(s), discovery of targeting moieties, preclinical testing of the drug candidates, and documentation of the efficacy and safety of the drug candidate in the clinical trial phase.1−3 On average, this process can take 12–15 years with a cost of hundreds of millions of dollars.4,5 Furthermore, roughly 10% of compounds that are selected for further clinical study make it to the marketplace as safe and effective drugs, demonstrating that the overall drug development process is formidable.6
The success of a new drug candidate is determined not only by its efficacy but also by proper pharmacokinetic behavior. Many promising drug candidates with appreciable activity in vitro fail to become marketable products, most often due to inappropriate pharmacokinetic behavior. Physicochemical properties can be used in the optimization of pharmacokinetics and the early prediction of absorption, distribution, metabolism, and excretion (ADME) can save time and money.7,8
Lipophilicity, which is the affinity of a molecule or a moiety for a lipophilic environment, is a key physicochemical property that plays a crucial role in ADME characteristics of drugs.9,10 It is one of the factors included in the rule of five formulated by Lipinski in 1997, a tool for medicinal chemists that is used for quick assessment of compounds during drug discovery.11 It also has considerable impact on solubility and membrane permeability,12,13 potency,14 selectivity,15 and promiscuity15 of drugs, therefore affecting their biodistribution (e.g., hepatic elimination vs renal excretion), pharmacodynamics, and toxicological profile.9,10,16 Lipophilicity is commonly described by the processes of partition between two phases, a nonpolar (organic phase) and a polar (mostly aqueous phase), and expressed as the logarithm of the n-octanol partition coefficient (log P or log D).17
Peptide–ligand drug conjugates can have high binding affinity and specificity for targets that are highly expressed on cancer cells but have low or nonexpression on normal cells of concern for toxicity. These properties have created significant interest in the use of peptide ligands in pharmaceutical research and development (R&D),18 as there are currently ∼140 therapeutics with peptide targeting ligands in clinical trials.2 The systemic clearance route of peptide-drug conjugates can be a significant factor in terms of dose-limiting toxicity (i.e., renal vs hepatic). Although several studies have correlated the route of clearance with the physicochemical properties of small molecules,9,19−21 to the best of our knowledge, these correlations are largely unexplored for peptide conjugates.
Because of the high linear energy transfer (LET) and short-range of α particles in tissue, there is significant interest in the development of targeted alpha-particle therapy (TAT) for the treatment of solid tumors (reviewed recently by our group).22 We recently published studies demonstrating the conjugation of a peptide based ligand (MC1RL) with high affinity and selectivity for the melanocortin 1 receptor (MC1R)23 to a metal chelator (dodecane tetraacetic acid, DOTA) using an aminohexanoic acid (Ahx) linker (DOTA-Ahx-MC1RL). This conjugate was radiolabeled with the α-particle-emitting radionuclide, 225Ac, for use as TAT for treatment of metastatic uveal melanoma.22 The melanocortin 1 receptor (MC1R) is expressed in 94% of uveal melanoma metastases.
Our preclinical studies demonstrated low toxicity, significantly prolonged survival, and decreased metastasis burden following treatment with 225Ac-DOTA-Ahx-MC1RL.22 We also observed that 225Ac-DOTA-Ahx-MC1RL was cleared more via the hepatic route, which differed from our previously reported near-infrared fluorescent dye conjugate (MC1RL-800) that has predominant renal clearance.24,25 We observed that the log D7.4 value of 225Ac-DOTA-Ahx-MC1RL was significantly higher than the value of MC1RL-800 and hypothesized that lipophilicity may have a role in determining the clearance route of these two conjugates. To test this hypothesis, we designed and synthesized a library of DOTA-linker-MC1RL compounds with diverse linkers and a predicted range of log D7.4 values. Herein, we report the synthesis and characterization of lipophilicity, biodistribution, clearance route, radiation dosimetry, and toxicity profile of this new library of compounds.
Materials and Methods
Synthesis of DOTA-linker-MC1RL Compound Library
MC1RL compounds23 with different linkers were synthesized on Rink Amide resin (initial loading: 0.568 mmol/g) using Nα-Fmoc-protecting amino acids and a HCTU/DIEA strategy. After the resin was swollen in DCM for 30 min, the Fmoc protecting group was removed with 2% DBU in DMF (2 × 5 min). The resin was washed with NMP and DCM three times each, and the first amino acid was coupled using HCTU and DIEA in NMP (4 equiv of Nα-Fmoc amino acid, 4 equiv of HCTU, and 8 equiv of DIEA). The double coupling was performed at all steps under the same coupling conditions due to the sequence deletion and the slower coupling rate in longer sequences. After coupling, the resin was washed with NMP (×3) and DCM (×3), and any unreacted free amine groups on the resin were treated using 50% acetic anhydride in pyridine for 5 min. After the resin was washed with NMP (×3) and DCM (×3), the same procedure was repeated for the next amino acid coupling until every residue in the sequence was coupled.
The Alloc protecting group of the C-terminal Lys was deprotected with piperidine (5–10 drops) and Pd(PPh3)4 (0.2 equiv) in anhydrous chloroform. Under nitrogen gas, the reaction mixture was stirred for 15 min and then repeated. After the resin was washed with chloroform, NMP, and DCM; DOTA (no-linker), aminohexanoic acid–DOTA, d-Lys-d-Lys–DOTA, or d-Lys-d-Glu–DOTA, were coupled to the free amine via HCTU coupling as described above.
The peptide and every protecting group was cleaved using cleavage cocktail (88% TFA, 5% water, 5% phenol, and 2% triethylsilane) for 4 h. The crude peptide was isolated from the resin by filtration, and the filtrate was concentrated. The peptide was precipitated in ice cold diethyl ether, dissolved in water, and lyophilized. The off-white crude powder was purified by reverse-phase chromatography.
Complexation of the peptides with lanthanum (La3+) was accomplished by incubation (25 °C) of the purified peptide in 0.1 M of sodium acetate, pH 6 buffer solution with 3 equiv of lanthanum chloride heptahydrate. The complexation reaction was assayed for completion by monitoring the retention time shift of the ligands on a linear gradient of analytical RP-HPLC. After 16 h of incubation, the reaction was complete, and peptide ligand solutions were separated from the excess metal and buffer by semiprep RP-HPLC. Eu-DTPA-MC1RL was synthesized as described before.22
Cell Culture
HEK293/MC1R (HEK293 cells engineered to express MC1R)26 were grown in DMEM/F12 (Life Technologies) containing 10% FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin in 5% CO2 at 37 °C. All experiments were performed with cells of passage number less than 25. Cells were authenticated using short tandem repeat (STR) DNA typing according to ATCC’s guidelines by the Molecular Genomics Core Facility at Moffitt27 and monitored by microscopy to ensure maintenance of their original morphology. Cells were tested for mycoplasma contamination using the MycoAlert kit (Lonza).
Binding Assay
HEK293/MC1R cells28 were used to assess ligand binding in a lanthanide-based time-resolved fluorescence competitive binding assay as described previously.23 Briefly, the cells were plated on SigmaScreen poly-d-lysine coated plates (Sigma-Aldrich) at a density of 30 000 cells/well 1 day before the experiment. On the day of the experiment, the medium was aspirated, and 50 μL of nonlabeled competing ligand was added to each well in a series of decreasing concentrations (ranging from ∼1 μM to 0.1 nM), followed by 50 μL of Eu-DTPA-MC1RL at 10 nM. All ligands were diluted in binding medium (DMEM, 1 mM 1,10-phenanthroline, 200 mg/L bacitracin, 0.5 mg/L leupeptin, and 0.3% BSA). Cells were incubated with ligands for 30 min at 37 °C. Following incubation, cells were washed two times with PBS, and 100 μL of enhancement solution (PerkinElmer) was added to each well. Cells were incubated for an additional 30 min at 37 °C prior to reading. The plates were read on a PerkinElmer VICTORx4 2030 multilabel reader using the standard Eu time-resolved fluorescence (TRF) measurement (340 nm excitation, 400 μs delay, and emission collection for 400 μs at 615 nm). Competitive binding curves were analyzed with GraphPad Prism software using the sigmoidal dose–response (variable slope) classical equation for nonlinear regression analysis.
Lipophilicity
The lipophilicities of the unmetalated and La3+-chelated DOTA-linker-MC1RL compounds were determined by a miniaturized shake-flask method similar to a previous report.24 Stock solutions of the compounds (200 μM) in 10 mM, pH 7.4, phosphate buffer were prepared. Aliquots of the stock solutions were vortexed with three different ratios of n-octanol. The resulting emulsions were centrifugated and the PBS and n-octanol layers separated. Unknown compound concentrations in the aqueous and organic layers were measured by HPLC-triple quadrupole MRM mass spectroscopy. Triplicate measurements of compound concentration were used to calculate the log of partition between the lipid and aqueous phases at pH 7.4 (log D7.4).
Radiochemical Synthesis and Quality Control for 225Ac-DOTA-linker-MC1RL Compounds
Actinium-225 (225Ac: t1/2 = 10 days; Eαmax = 6–8 MeV) was purchased from Oak Ridge National Laboratories. The complexation of 225Ac was achieved for DOTA-Ahx-MC1RL as previously described35 and by reacting DOTA-MC1RL (no linker), DOTA-DLDL-MC1RL (d-Lys-d-Lys linker), or DOTA-DLDG-MC1RL (d-Lys-d-Glu linker) (5–10 μg in 5–10 μL water from 1.0 mg/mL solution) with 225Ac(NO3)3 (3.4 MBq) that was diluted in 100 μL of water containing 10 μL of 20% l-ascorbic acid. The pH of the resulting solution was adjusted to 5.5–6 using 1 M Tris buffer (10–12 μL) and then incubated at 60 °C for 1 h. Reaction progress and radiochemical purity were measured without further purification using ITLC with gamma counting, radio-TLC, and gamma counting of radio-HPLC fractions. Specific activity was calculated using a standard method.30 Analysis was performed 24 h after sample collection to ensure secular equilibrium was achieved between 225Ac and its daughter products. The proportion of chelated 225Ac in this preparation relative to the proportion of chelated La3+ is described in Supplemental Experimental Methods.
Animal Studies
All protocols were approved as University of South Florida IACUC protocol IS00004454.
Injected Activity Measurement
Syringes were prefilled with 225Ac radiopharmaceutical activity, and since α-particles from 225Ac cannot be directly measured in tissue due to the range,31 the activity was measured using the isomeric gamma spectra by the 4π well-type Atomlab 500 Wipe Test Gamma Counter (BioDex) and converted to 225Ac alpha activity using factors for γ ray abundance per alpha decay as described previously.22 The spectra were acquired using a full energy window to include gamma counts from 225Ac, with peak at 99.8 keV (abundance 1%), and its two γ-emitting daughters, 221Fr (4.9 min T1/2) with peak at 218.1 keV (abundance 11.4%) and 213Bi (46 min T1/2) with peak at 440.5 keV (abundance 25.9%).32 The alpha activity in 1 μCi of each radionuclide was determined by fitting each peak with a multi-Gaussian fit and integrating to determine the net number of counts while incorporating the acquisition time. The activity was injected into each mouse via tail-vein catheter. Activity remaining in the syringe and catheter postinjection were calculated and subtracted to determine net administered activity. Spectra were acquired at ≥24 h post-radiosynthesis or tissue rendering ensuring that 225Ac and daughters were in secular equilibrium.33
Biodistribution (BD)
Male and female BALB/c mice (10–12 weeks, 18–22 g, Charles River) were used for the BD studies. Tail vein catheters were used for agent administration to mice. All mice were intravenously administered 148 kBq ± 10% of 225Ac alpha activity in the syringe. The animals were euthanized at multiple time-points between 24 h and 1 week post-injection (p.i.) (n = 5 mice per time point), and tissues were removed and weighed. The isomeric gamma spectra were measured for each tissue sample as described above. Using the net administered activity for 225Ac, 221Fr, and 213Bi, the percent injected activity per gram (%IA/g) were then calculated and compared to a weighed, counted standard for all groups.
In-house-designed Matlab (MathWorks) code was used to calculate the pharmacokinetics (PK) rates of uptake and clearance for each compound. Data were fit using the nonlinear least-squares approach and the logarithmic equation for uptake and two-exponential decay equation for clearance to find relevant parameters (e.g., rates of uptake and clearance).34 The following equation was used for uptake:
where y [%IA/g] is the percentage of measured radioactivity per gram of tissue, A [%IA/g] is the peak percentage of radioactivity per gram of tissue, and λ [%IA/g/h] is the rate of uptake.
A two-compartment model was used where rapid clearance of radiopharmaceutical circulating in the blood and a slower clearance of radiopharmaceutical from tissues are observed. Hence, the following two-exponential equation was used for clearance:
where y [%IA/g] is the percentage of measured radioactivity per gram of tissue, A1 [%IA/g] and A2 [%IA/g] are the initial percentages of radioactivity per gram of tissue at two different phases of clearance, and λ1 [%IAg/h] and λ2 [%IAg/h] are the rates of clearance for two different phases.
Radiation Dosimetry (RD)
Dosimetry calculations were performed as previously described.35 As described above for BD studies, isomeric γ-emission spectra of 225Ac and the two radionuclides in the decay chain with detectable emissions, 221Fr and 213Bi, were acquired in tissues rendered at different p.i. time points and used for these calculations. Because of their short half-lives, 217At (32.2 ms) and 213Po (4.2 μs) were assumed to decay at the site of their parent radionuclides, 221Fr and 213Bi, respectively. The β decay branching ratio for 217At to 217Rn is only 0.01%; therefore, it was assumed that all decays of 217At progressed by α emission to 213Bi. The branching ratios for decay of 213Bi are 98% to 213Po and 2% to 209Tl and were accounted for in the calculation. Due to the relatively low LET and the small dimensions of the target tissues, the β emissions from 217At, 213Bi, 209Tl, and 209Pb were assumed to be negligible and are not included in the calculations.36
Organ and tumor absorbed doses from 225Ac, 221Fr, 217At, 213Bi, and 213Po were determined using acquired BD data. Dosimetry calculations were performed using the generalized internal dosimetry schema of the MIRD Committee for α-particle emitters.36,37 According to MIRD #21, the absorbed radiation dose due to particle type x, Dx(rT, TD), is the mean energy imparted to target tissue rT per unit tissue mass, defined by eq 1:
| 1 |
where Ã(rS, TD) is the total number of nuclear transitions in the target region (accumulated activity), Δix is the mean energy emitted per nuclear transition, φ(rT ← rS; Ei) is the fraction of energy emitted per nuclear transition in the source region that is absorbed in the target region by the ith emission that is emitted with initial energy E, k is a conversion factor, and M(rT) is the mass of the target tissue. Time–activity curves were generated for each organ and fit with an exponential decay nonlinear regression. Accumulated activity in each organ/tumor was determined by analytically integrating the resulting equation of fit. Several assumptions were made in the calculation of absorbed dose. It was assumed that the α-particle activity was uniformly distributed in each organ/tumor volume, and as a result of its short-range in tissue, no α particles escaped from its source organ/tumor. Electron and photon contributions were assumed to be negligible compared to α-particle energy deposition.38 After these assumptions, eq 1 simplifies to
| 2 |
It was assumed that α particles from 221Fr (4.9 min T1/2), 217At (32.2 ms T1/2), 213Bi (46 min T1/2), and 213Po (4.2 μs T1/2) were deposited in the same location as 225Ac (10 day T1/2) due to the relatively shorter half-lives of these daughter isotopes. Although 217At and 213Po do not have detectable γ emissions, under the assumption that the decay chain had reached secular equilibrium, the accumulated activity of these two daughters would equal that of 221Fr and 213Bi, respectively. The total absorbed α-particle dose was calculated from the summation of doses from 225Ac, 221Fr, 217At, 213Bi, and 213Po.
Toxicity
Toxicity studies were conducted using cohorts of normal BALB/c mice. After a single intravenous injection of each 225Ac conjugate ranging from 3 to 314 kBq 225Ac activity or saline, cohorts (n = 6 mice/cohort) were weighed 2 times per week and monitored for 7 months for signs of distressed behavior. At the end of that period, animals were euthanized, and serum was collected by centrifugation at 1500 × g. Blood urea nitrogen (BUN), creatinine, alkaline phosphatase (ALKP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) content in serum were analyzed using the Catalyst DX automated chemistry analyzer and CLIP (IDEXX, ME). Liver, kidney, spleen, pancreas, cecum, small intestine, lymph nodes, bone marrow, muscle, salivary gland, and bone were harvested and fixed in 10% formalin. Bone was decalcified in 14% ethylene diaminetetraacetic acid (EDTA) solution after fixation in formalin. All tissues were embedded in paraffin, sectioned (4–6 μm thickness), and stained with hematoxylin and eosin. The slides were scanned in the Moffitt Analytical Microscopy Core Facility using a ScanScope XT digital slide scanner (Aperio, CA) and examined by the veterinary pathologist (R.E.), in a blind manner.
Statistical Analysis
Analysis of variance (ANOVA) was used to assess the relationship of BD (%IA/g) with the log D value, with log D and time as the independent variables. These analyses were modeled on liver BD, kidney BD, and the kidney/liver BD ratio (i.e., three separate models). An interaction effect with time and BD (either kidney or liver or the ratio) was included too.
Logistic regression analyses were used to assess the toxicities for each compound and injected activity group using standard reference ranges (adjusted for control levels if necessary) based on the injected activity. Weight change for each compound and activity was evaluated relative to the control group’s weight change. A predicted probability of toxicity was then generated across the range of injected activity levels to generate curves for each of compound group.
Using the Spearman’s correlation coefficient, the correlation of log D values with uptake values for both kidney and liver were estimated for each radiopharmaceutical. Also, for each derived toxicity measure for either kidney or liver, we calculated the sensitivity and specificity of toxicity using pathology as the standard.
Results
Synthesis and Characterization of DOTA-linker-MC1RL Compounds
The library of DOTA-linker-MC1RL compounds were synthesized using the synthetic route shown in Scheme 1. Synthesis of the DOTA-Ahx-MC1RL version was previously described.35 Each compound was chelated with La3+ to facilitate in vitro characterizations. La3+ has been reported as a useful surrogate for compounds being developed with Ac, since both exist as trivalent ions in solution.28 See Figures S1–S8 for HPLC chromatograms and MALDI-TOF spectra of each compound, with and without La3+ chelation.
Scheme 1. Synthetic Route of DOTA-Linker-MC1RL Compounds.

Via competitive binding assay, we determined that all of the La3+-DOTA-linker-MC1RL compounds have high binding affinity for MC1R (i.e., 0.08, 0.20, 0.06, and 0.23 nM Ki) for versions with the following linkers: no linker, Ahx, DLDL, and DLDG, respectively (Figure 1).
Figure 1.
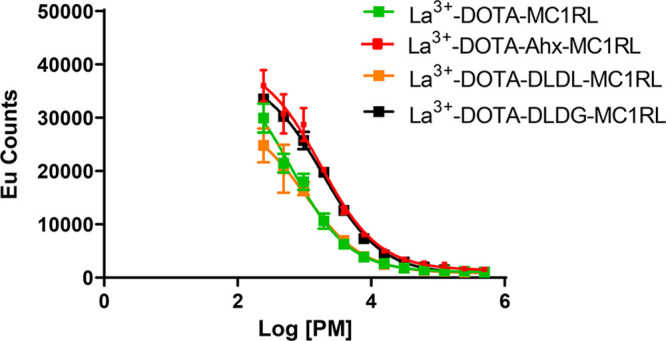
Competitive binding assay for La3+-DOTA-linker-MC1RL compounds.
Log D7.4, which can influence the tissue distribution of a compound and is an indicator of lipophilicity or solubility, was determined for the unmetalated and La3+-chelated compounds (Table S1). Complexation with La3+ reduced the log D7.4 for each compound. These values suggest that the compounds are soluble in aqueous buffer, but with variable lipophilicities (ranging from −1.79 to −2.75 for the unmetalated compounds and from −2.14 to −3.88 for the compounds chelated with La3+).
Radiosynthesis and Characterization of 225Ac-DOTA-linker-MC1RL Radiopharmaceuticals
Radiosynthesis (Scheme 2) for all 225Ac-DOTA-linker-MC1RL radiopharmaceuticals provided a greater than 95% yield with high radiochemical purity (≥99.0%) as determined by radio-TLC and radio-HPLC (Figures S9–S12).
Scheme 2. Radiochemical Synthesis of 225Ac-DOTA-linker-MC1RL Compounds.

Biodistribution and Pharmacokinetics
Tissue BD of the 225Ac-DOTA-linker-MC1RL radiopharmaceuticals were determined in BALB/c mice. Following intravenous injection, 225Ac activity was observed primarily in clearance tissues (i.e., liver, kidneys, and intestine), and clearance occurred over a period of weeks (Figure 2). Using the BD data, the pharmacokinetics of kidney and liver uptake and clearance rates, and blood clearance rates were calculated for each radiopharmaceutical (Table 1). The uptake data were best fit using the logarithmic uptake equation. A two-compartment model for clearance was assumed, and clearance data were fitted using the two-exponential clearance equation.
Figure 2.

BD results for (A) 225Ac-DOTA-MC1RL, (B) 225Ac-DOTA-Ahx-MC1RL, (C) 225Ac-DOTA-DLDL-MC1RL, and (D) 225Ac-DOTA-DLDG-MC1RL. Activities were calculated for tissues rendered from BALB/c mice (n = 6 per time point). The time courses vary among the different compounds due to differences in animal survival and liver clearance.
Table 1. Pharmacokinetics of Uptake and Clearance Rate Constants for Each Compound.
| compound | organ | uptake rate constant, λ (%IA/g/h) | fast clearance rate constant, λ1 (%IA/g/h) | slow clearance rate constant, λ2 (%IA/g/h) |
|---|---|---|---|---|
| 225Ac-DOTA-MC1RL | blood | 0.00756 | 1.89 | |
| kidney | 3.98 | 0.0361 | 0.0275 | |
| liver | 30.1 | 63.8 | 0.00737 | |
| 225Ac-DOTA-Ahx-MC1RL | blood | 0.507 | 0.0125 | |
| kidney | 5.63 | 0.0605 | 0.00514 | |
| liver | 17.3 | 0.0662 | 0.00319 | |
| 225Ac-DOTA-DLDL-MC1RL | blood | 1.87 | 0.00577 | |
| kidney | 179 | 0.0229 | –0.0109 | |
| liver | 55.9 | 0.00511 | –0.0323 | |
| 225Ac-DOTA-DGDL-MC1RL | blood | 0.0198 | 2.00 | |
| kidney | 44.1 | 0.0117 | –0.0118 | |
| liver | 8.97 | 0.0412 | 0.00360 |
Radiation Dosimetry
Radiation dosimetry (RD) calculations were based on the data obtained from the BD studies. RD for targeted radiotherapy is the determination of the absorbed energy deposited per unit mass by ionizing radiation in the different tissue compartments within the body. As described in the Materials and Methods, the α-particle dose from 225Ac and each of its α-emitting daughters was calculated using γ spectroscopy of 225Ac, 221Fr, and 213Bi (Figure S13).
BD data for the different tissues were fitted using an exponential decay nonlinear regression, allowing the estimation of clearance kinetics, tissue biological half-life, accumulated activity, and absorbed dose/injected activity (Gy/kBq) for each radionuclide in each tissue. See Table S2A–C for 225Ac-DOTA-MC1RL, 225Ac-DOTA-DLDL-MC1RL, and 225Ac-DOTA-DLDG-MC1RL. RD for 225Ac-DOTA-Ahx-MC1RL was previously reported.35 The total absorbed dose is the summation of the values for the five α-emitting radionuclides. The calculated total absorbed dose for each of the 225Ac-labeled compounds was minimal in all tissues except clearance organs (kidneys and liver). The total absorbed dose of 225Ac and daughters was greater in the liver relative to the kidneys for 225Ac-DOTA-MC1RL (no linker) and 225Ac-DOTA-Ahx-MC1RL with 0.56 and 0.24 Gy/kBq for kidneys and 0.79 and 0.70 Gy/kBq for liver, respectively. The total absorbed dose of 225Ac and daughters was greater in the kidneys (1.59 and 0.92 Gy/kBq) relative to the liver (1.36 and 0.45 Gy/kBq) for 225Ac-DOTA-DLDL-MC1RL and 225Ac-DOTA-DLDG-MC1RL, respectively. Figure 3 presents graphs of the absorbed dose from each radionuclide per tissue for all four radiopharmaceuticals.
Figure 3.

Radiation dosimetry of 225Ac and daughters following administration of (A) 225Ac-DOTA-MC1RL (no linker), (B) 225Ac-DOTA-Ahx-MC1RL, (C) 225Ac-DOTA-DLDL-MC1RL, and (D) 225Ac-DOTA-DLDG-MC1RLin BALB/c mice.
It is noted that in tissues with significant uptake, the effective decay half-lives (Teff) calculated for 225Ac were shorter than the radiodecay half-life of 225Ac (10 days) for all the compounds, indicating biological clearance. For example, the calculated Teff in kidneys and liver for 225Ac–no linker was 4.8 and 5.7 days, respectively (Table S2A). Hence, Teff is a composite of radiodecay and active biological clearance. The Teff was only calculated to be longer in some tissues with minimal uptake where instrument background likely interfered with the accuracy of measurement.
Toxicity
Cohorts of mice received a single intravenous injection of a range of 225Ac activities from 3 to 314 kBq of each radiopharmaceutical. Animals were weighed twice per week and monitored for 7 months for signs of discomfort or declining condition. At the completion of the study, serum was collected for renal and hepatic toxicity assays, and tissues were collected for histological staining and pathology examination. See Table S3 for the histopathology scoring of all tissues in the study.
Death due to acute renal failure was observed in some cohorts. There were no deaths observed in mice administered 225Ac-DOTA-MC1RL (no linker) or 225Ac-DOTA-Ahx-MC1RL at any injected activity. All mice treated with ≥77.6 kBq of 225Ac-DOTA-DLDL-MC1RL and a single mouse injected with 44.0 kBq of 225Ac-DOTA-DLDG-MC1RL died within 1–2 weeks p.i., and at necropsy, kidneys appeared grossly pale, pitted, small, and irregular in shape (Figure 4A). Histologically, tubular cell necrosis was characterized by intense cytoplasmic eosinophilia with pyknotic nuclei, while other tubules appeared regenerative with cytoplasmic basophilia and nuclear crowding (Figure 4C).
Figure 4.

Gross appearance of kidneys (A, B) and histological appearance of kidney (C–F) and liver (G–J) from mice treated with different peptides in the toxicity study. (A, B) Gross appearance of kidneys from mice injected with 79.4 kBq of 225Ac-DOTA-DLDL-MC1RL and 305.4 kBq 225Ac-DOTA-MC1RL. (C) Kidney histology of a 135.4 kBq of 225Ac-DOTA-DLDL-MC1RL administered mouse, with acute tubular necrosis resulting in death at 11 days after administration. At necropsy, kidneys appeared grossly pale, pitted, small, and irregular in shape. Histologically, tubular cell necrosis was characterized by intense cytoplasmic eosinophilia with pyknotic nuclei (arrows), while other tubules appeared regenerative with cytoplasmic basophilia and nuclear crowding (arrowheads). (D) Kidney of a 293.4 kBq of the 225Ac–No linker administered mouse with nephropathy comprised of tubular epithelial cell degeneration with cytoplasmic vacuolization, necrosis, and regeneration with cytoplasmic basophilia and nuclear crowding, hypercellularity of glomerular tufts, and focal infiltration of mononuclear cells (arrow). (E) Kidney histology of a 84.2 kBq of 225Ac-DOTA-DLDG-MC1RL administered mouse euthanized 7 months after administration, with chronic progressive nephropathy. Little normal renal parenchyma remains due to extensive tubular cell necrosis (arrow), epithelial sloughing and cast formation, extensive tubular cell regeneration (arrowheads), diffuse interstitial edema and fibrosis with mild mononuclear inflammatory cell infiltrates, and hypercellular glomerular tufts. (F) Kidney of saline-treated mouse with normal eosinophilic cuboidal tubular epithelium, and normal glomerluli. (G) Liver of 292.2 kBq of the 225Ac-DOTA-Ahx-MC1RL administered mouse with hepatocellular eosinophillic cytoplasmic swelling, single hepatocellular apoptosis (arrows) with hypereosinophillic cytoplasm and pyknotic nuclei, and single hepatocellular necrosis (arrowheads) with pale eosinophilic cytoplasm and karyolysis. (H) Liver histology of a 135.4 kBq of 225Ac-DOTA-DLDL-MC1RL administered mouse, with focal hepatocellular eosinophillic cytoplasmic swelling, single hepatocellular apoptosis (arrows) with hypereosinophillic cytoplasm and pyknotic nuclei, and single hepatocellular necrosis (arrowhead) with pale eosinophilic cytoplasm and karyolysis. (I) Liver histology of a 36.0 kBq of 225Ac-DOTA-DLDL-MC1RL administered mouse, with hepatocellular eosinophillic cytoplasmic swelling, hepatocellular apoptosis (arrows), and hepatocellular necrosis (arrowhead). (J) Liver of saline administered mouse with normal, well-delineated hepatic cords and sinusoids, a portal triad (left), and a central vein (right).
By the end of the study, mice injected with 225Ac-DOTA-MC1RL, 225Ac-DOTA-Ahx-MC1RL, 225Ac-DOTA-DLDL-MC1RL, and 225Ac-DOTA-DLDG-MC1RL at ≥268.9, 181.5, 11.5, and 50.2 kBq of 225Ac activity, respectively, had chronic progressive nephropathy. Histologically, this pathophysiological entity was characterized by little normal renal parenchyma remains due to extensive tubular cell necrosis, epithelial sloughing and cast formation, extensive tubular cell regeneration, diffuse interstitial edema and fibrosis with mild mononuclear inflammatory cell infiltrates, and hypercellular glomerular tufts (Figure 4D,E). Animals injected with less than the above activities survived without abnormal kidney pathology.
None of the animals injected with 225Ac-DOTA-MC1RL or 225Ac-DOTA-DGDL-MC1RL had histopathological liver damage. By the end of the study, mice injected with greater than >317.8 kBq of 225Ac-DOTA-Ahx-MC1RL had treatment-related liver damage such as bile duct hyperplasia and hepatocyte apoptosis (Figure 4G), and mice injected with >27.0 kBq of 225Ac-DOTA-DLDL-MC1RL had treatment related hepatocyte granular degeneration, apoptosis, and necrosis (Figure 4H,I). Incidental hepatocyte glycogen accumulation and mild, focal, hepatocellular fatty changes were observed in the livers of some treated and untreated control mice.
The spleen of some animals injected with >125.8 kBq of 225Ac-DOTA-DLDL-MC1RL had moderate lymphoid depletion. A mild multifocal lymphoplasmacytic infiltration of salivary glands was observed in some treated mice.
The animal weights for each radiopharmaceutical and administered activity are plotted in Figures S14–S17. By the end of the study, mice injected with 225Ac-DOTA-MC1RL, 225Ac-DOTA-DLDL-MC1RL, and 225Ac-DOTA-DGDL-MC1RL at ≥268.9, 25.5, and 44.4 kBq of 225Ac activity, respectively, decreased in weight over the course of the study. Mice injected with ≥192.9 kBq of 225Ac-DOTA-Ahx-MC1RL lost weight, but three mice injected with 200, 212, and 315 kBq of activity gained weight. Mice administered lower activities gained weight over the course of the study.
Serum specimens were tested for BUN and creatinine levels as indicators of renal toxicity, and ALKP, ALT, and AST levels as indicators of hepatic toxicity (Table S4).39 Elevated BUN levels were observed in some mice injected with 225Ac-DOTA-MC1RL, 225Ac-DOTA-Ahx-MC1RL, 225Ac-DOTA-DLDL-MC1RL, and 225Ac-DOTA-DLDG-MC1RL at ≥193.6, 200.1, 11.5, and 19.36 kBq of 225Ac activity, respectively.
Since histopathology is considered the gold standard of determining toxicity of exposure to ionizing radiation and since weight loss and serum biomarkers are indirect indicators of damage due to toxicity, the sensitivity and specificity of these indirect measures were evaluated relative to pathology (Table 2). Weight loss was shown to have high sensitivity and specificity for tissue damage to either kidney or liver. BUN levels had high sensitivity and specificity for kidney damage, and ALKP levels had high sensitivity and specificity for liver damage. The probabilities of kidney or liver damage by pathology, elevated BUN or ALKP levels, or weight loss per injected activity for each radiopharmaceutical are shown in Figure 5A–E.
Table 2. Determination of Sensitivity and Specificity of the Indirect Indicators of Toxicity Incomparison to Pathology.
| parameter | % sensitivity | % specificity |
|---|---|---|
| Kidney Pathology | ||
| weight loss | 87 | 95 |
| BUN | 74 | 97 |
| Liver Pathology | ||
| weight loss | 85 | 74 |
| AST | 14 | 90 |
| ALKP | 85 | 86 |
| ALT | 28 | 95 |
Figure 5.

Toxicity probability curves per injected activity for each radiopharmaceutical per (A) kidney pathology, (B) liver pathology [no toxicities were observed for the other two radiopharmaceuticals], (C) BUN, (D) ALKP, and (E) weight loss.
Relationship of BD, PK, RD, and Toxicity with Compound Lipophilicity
Using the BD data for all four compounds, a significant relationship was determined between log D7.4 and kidney BD, p < 0.05, and between log D7.4 and the kidney-to-liver BD ratio, p < 0.001 (Table S5). It is noted that no relationship was observed between log D7.4 and liver BD. Since BD is a composite of agent uptake and clearance, the correlations among log D7.4 and kidney and liver uptake, and clearance rates and toxicity indicators (weight loss, pathology, BUN and ALKP levels) were examined (Table S6). The statistical analysis showed that log D7.4 is significantly associated with weight loss, p < 0.0001, and that both log D7.4 and weight loss are significantly associated with kidney clearance rates, p < 0.0001. Furthermore, BUN levels are significantly associated with kidney uptake rates, p < 0.0001, and liver clearance rates, p < 0.0001.
Discussion
Lipophilicity is a major determinant of route of clearance of small molecule drugs due to the dependence of renal clearance on membrane permeability.40 However, this observation has not been fully substantiated for peptide conjugates.41 Mass can influence the route of clearance (i.e., molecules <50 kDa are removed from circulation via renal glomeruli, while larger macromolecules clear via the splenic and hepatic routes).41 With the goal of characterizing the relationship between lipophilicity and clearance route, we designed and synthesized a set of peptide-conjugate based radiopharmaceuticals for TAT with a range of lipophilicities (log D7.4 values) and comparable mass values ranging from 1361 to 1618 MW without metal chelation. Also, unnatural amino acids are incorporated into the peptidic component of these compounds, and high radiochemical stability has been previously demonstrated for the 225Ac-DOTA-Ahx-MC1RL radiopharmaceutical.35 Hence, peptide metabolism is likely a minor factor in determining the route of clearance. Herein, we report characterization of the BD, PK, RD, and toxicity of these compounds in mice and the relationships of log D7.4 values with the route and PK of uptake and clearance of the administered TAT.
Since the lipophilicities of the compounds were adjusted by inserting linkers that have different structures and chemical properties between the peptide targeting moiety and the metal chelator, quality control characterizations were also performed. As a nonradioactive surrogate of 225Ac, La3+ chelates were used for these assays. The high binding affinity (low nM Ki) of the MC1RL peptide for the MC1R receptor and log D7.4 values were confirmed for each compound.
For BD, PK, and RD determinations, each compound was labeled with 225Ac with high radiochemical yield and purity. When evaluating the BD, PK, and RD data, general observations can be made that the metalated compounds with log D7.4 values above −2.6 have lower kidney uptake rates, are cleared more by the hepatic route than renal, and have a lower radiation dose to the kidney when compared to the compounds with log D7.4 values below −3.4, which are cleared more by the renal route, have higher kidney uptake rates, and have a higher radiation dose to the kidney. This negative correlation of decreased kidney uptake and decreased radiation dose with increasing lipophilicity can be explained as a result of an increase in passive reabsorption in the kidney tubule, leading to less renal uptake.21,40
Radiation-induced renal toxicity is a major concern in the therapeutic application of peptides labeled with radiometals.42 In this study, we observed two distinct pathological features: acute kidney damage and chronic kidney damage. Acute kidney failure was observed in animals that were administered compounds with log D7.4 values below −3.4 and was grossly characterized by pale, pitted, small, and irregular shaped kidneys at necropsy and by acute nephropathy via pathology examination. Acute kidney damage resulted in a rapid loss of kidney function and death within the week following treatment. Animals administered compounds with log D7.4 values above −2.6 lived for the duration of the 7 month study and exhibited chronic pathologies of the kidney and liver at high administered activities. The relative probabilities of pathology in the kidney and liver by administered activity were calculated for all cohorts. Weight loss and serum biomarkers of kidney and liver toxicity were also quantified, and weight loss was shown to have high sensitivity and specificity for detection of both kidney and liver pathology. While creatinine and BUN levels are typically used as biomarkers of kidney damage,43 BUN was identified in our study as having higher sensitivity and specificity of detection of kidney pathology. Of all liver enzymes tested, ALKP levels had both high sensitivity and high specificity for detection of liver damage. Statistical analyses identified significant relationships between log D7.4 and kidney BD, the kidney-to-liver BD ratio, weight loss, and kidney clearance rates. Weight loss also significantly correlated with kidney clearance rates. BUN levels were significantly correlated with kidney uptake and liver clearance rates.
Table 3 was prepared to highlight the relevant relationships among the log D7.4 values of the La3+-chelated compounds and the kidney uptake, kidney total absorbed dose, and kidney toxicity values of the corresponding 225Ac-chelated compounds. It is notable that the compounds with log D7.4 values above −2.6 have kidney uptake rates, total absorbed doses, and probabilities of toxicity that range from 3- to 100-fold lower than the compounds with log D7.4 values below −3.4.
Table 3. Relationship of log D7.4 with Kidney Uptake, Radiation Doses, and Toxicity.
| kidneyb |
||||||
|---|---|---|---|---|---|---|
| probability
of toxicity at 148 kBq injected activity |
||||||
| linker | log D7.4a | uptake rate constant, λ (%IA/g/h) | total absorbed dose (Gy/kBq) | pathology | BUN | weight loss |
| no linker | –2.41 ± 0.26 | 3.985 | 0.5628 | 0.00 | 0.04 | 0.01 |
| Ahx | –2.59 ± 0.20 | 5.625 | 0.1366 | 0.23 | 0.10 | 0.10 |
| DLDL | –3.49 ± 0.27 | 178.5 | 1.5980 | 0.95 | 1.00 | 1.00 |
| DGDL | –3.88 ± 0.17 | 44.08 | 0.9200 | 0.95 | 0.94 | 0.99 |
Determined using the La3+ chelate.
Determined using the 225Ac chelate.
The arginine side chain in the MC1R targeting ligand is likely to be positively charged in all of the peptide conjugates at pH 7.4. This likely gives an overall plus one charge to the metalated no-linker, Ahx linker, and DLDG analogs and no overall charge for those same unmetalated analogs. The +3 metals, La and 225Ac, are balanced by the three carboxylates giving no overall charge for the DOTA-metal-chelated portion of the conjugates. The free DOTA-MC1RL species have three negative charges from the DOTA carboxylates and two positive charges from the protonation of two of the four tertiary amines in the 1,4,7,10-tetrazadodecane ring system. The metalated and unmetalated DLDL MC1RL analogs are expected to have overall +3 or +4 charge, respectively, but they are less polar than the relevant DLDG MC1RL analogs, which suggests significantly reduced basicity for the Lys side chains at physiological pH due to the overall charge density of DLDL analogs.
All of the linkers used for conjugation are expected to be stable to metabolic changes and all of the conjugates have epsilon lysine-amide linkages which are unlikely to be substrates of proteolytic enzymes. d-Amino acids were used in the polar linkers to inhibit possible proteolytic cleavage between the first and second amino acids in the linkers themselves.
In conclusion, by using different linker chemistry to increase the lipophilicity of a TAT radiopharmaceutical, we demonstrated that the kidney uptake rate can be significantly decreased, which also decreases the total absorbed dose and probability of toxicity to the kidney. Liver uptake, dose, and toxicity were not significantly influenced by the increase in lipophilicity. Hence, there is significant potential to decrease the kidney toxicity of radiopharmaceuticals via medicinal chemistry approaches that adjust lipophilicity.
Acknowledgments
Animal studies were conducted in the Moffitt Vivarium administered by the University of South Florida Comparative Medicine Department. The actinium-225 isotope used in this research was supplied by the U.S. Department of Energy Office of Science by the Isotope Program in the Office of Nuclear Physics. Funding was provided by an NIH/NCI SBIR Phase 1 Contract to Modulation Therapeutics, Inc. (PI: N.K.T.), a Melanoma Research Alliance Team Science Award (PI: D.L.M.), and a Moffitt Radiology Pilot Award (K.L.G.). This work was supported by the Analytic Microscopy, Bioinformatics and Biostatistics, and Tissue and Small Animal Imaging Laboratory cores at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
Glossary
Abbreviations
- MC1RL
Melanocortin receptor 1 ligand
- Ahx
6-Aminohexanoic acid linker
- BD
Biodistribution
- BUN
Blood urea nitrogen
- Fmoc
Fluorenylmethyloxycarbonyl
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIEA
Diisopropylethylamine
- DCM
Dichloromethane
- DBU
1,8-Diazabicyclo(5.4.0)undec-7-ene
- DMF
Dimethylformamide
- NMP
N-methyl-2-pyrrolidone
- Alloc
allyloxycarbonyl
- Pd(PPh3)4
Tetrakis(triphenylphosphine)palladium(0)
- DOTA
1,4,7,10-Tetraazacyclododecane-1,4,7-tris-tert-butyl acetate-10-acetic acid
- d-Glu
Fmoc-d-glutamic acid
- d-Lys
Fmoc-d-lysine
- TFA
Trifluoroacetic acid
- RT-HPLC
Reverse-phase high-performance liquid chromatography
- Ahx
Aminohexanoic acid
- DLDL
d-Lys-d-Lys
- DLDG
d-Lys-d-Glu
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00035.
Lipophilicty (log D7.4) for unmetalated and La3+ chelated compounds, radiation dosimetry and clearance kinetics parameters in BALB/c mice; relationship between log D and kidney, liver and kidney/liver biodistrbution (BD): statistical analysis, correlation of log D7.4, and kidney and liver uptake and clearance with indicators of toxicity (weight change, pathology, BUN and ALKP); analytical HPLC chromatograms and MALDI-TOF spectra; radiochemical purities of 225Ac-DOTA-MC1RL, 225Ac-DOTA-Ahx-MC1RL, 225Ac-DOTA-DLDL-MC1RL, and 225Ac-MC1RL-DLDG-DOTA; isomeric gamma spectrum of mouse kidneys for 225Ac-DOTA-MC1RL (no linker) at 24 h postinjection; weights over time of the animals in this study injected with different activities of radiopharmaceutical or saline (PDF)
Histopathology scoring for all tissues in the study (XLSX)
Toxicity indicators: weight change, pathology and serum markers (XLSX)
The authors declare the following competing financial interest(s): D.L.M. and N.K.T. are co-inventors of an awarded patent. D.L.M., T.J.W., M.L.M., H.K., and N.K.T. are co-inventors on a pending patent application. Modulation Therapeutics, Inc., has licensed related intellectual property, and M.L.M. is a co-founder of that company.
Supplementary Material
References
- Campbell I. B.; Macdonald S. J. F.; Procopiou P. A. (2018) Medicinal chemistry in drug discovery in big pharma: past, present and future. Drug Discovery Today 23 (2), 219–234. 10.1016/j.drudis.2017.10.007. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. (2015) Peptide therapeutics: current status and future directions. Drug Discovery Today 20 (1), 122–8. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Speck-Planche A.; Cordeiro M. N. (2015) Multitasking models for quantitative structure-biological effect relationships: current status and future perspectives to speed up drug discovery. Expert Opin. Drug Discovery 10 (3), 245–56. 10.1517/17460441.2015.1006195. [DOI] [PubMed] [Google Scholar]
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. (2016) Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ 47, 20–33. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Prasad V.; Mailankody S. (2017) Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Intern Med. 177 (11), 1569–1575. 10.1001/jamainternmed.2017.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S. M.; Mytelka D. S.; Dunwiddie C. T.; Persinger C. C.; Munos B. H.; Lindborg S. R.; Schacht A. L. (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discovery 9 (3), 203–14. 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Kola I.; Landis J. (2004) Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discovery 3 (8), 711–5. 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Zhivkova Z. D. (2017) Quantitative Structure - Pharmacokinetic Relationships for Plasma Clearance of Basic Drugs with Consideration of the Major Elimination Pathway. J. Pharm. Pharm. Sci. 20 (0), 135–147. 10.18433/J3MG71. [DOI] [PubMed] [Google Scholar]
- Arnott J. A.; Planey S. L. (2012) The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discovery 7 (10), 863–75. 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- Rutkowska E.; Pajak K.; Jozwiak K. (2013) Lipophilicity--methods of determination and its role in medicinal chemistry. Acta Polym. Pharm. 70 (1), 3–18. [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46 (1–3), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu X.; Testa B.; Fahr A. (2011) Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 28 (5), 962–77. 10.1007/s11095-010-0303-7. [DOI] [PubMed] [Google Scholar]
- Waring M. J. (2009) Defining optimum lipophilicity and molecular weight ranges for drug candidates-Molecular weight dependent lower log D limits based on permeability. Bioorg. Med. Chem. Lett. 19 (10), 2844–51. 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. (2011) Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discovery 10 (3), 197–208. 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 6 (11), 881–90. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; Decrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; Greene N.; Huang E.; Krieger-Burke T.; Loesel J.; Wager T.; Whiteley L.; Zhang Y. (2008) Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 18 (17), 4872–5. 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Kwon Y. (2001) Handbook of Essential Pharmacokinetics, Pharmacodynamics, and Drug Metabolism for Industrial Scientists, p xix, Kluwer Academic/Plenum Publishers, New York. [Google Scholar]
- Aina O. H.; Sroka T. C.; Chen M. L.; Lam K. S. (2002) Therapeutic cancer targeting peptides. Biopolymers 66 (3), 184–99. 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. (2008) Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 51 (4), 817–34. 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Obach R. S.; Lombardo F.; Waters N. J. (2008) Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos. 36 (7), 1385–405. 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- Varma M. V.; Feng B.; Obach R. S.; Troutman M. D.; Chupka J.; Miller H. R.; El-Kattan A. (2009) Physicochemical determinants of human renal clearance. J. Med. Chem. 52 (15), 4844–52. 10.1021/jm900403j. [DOI] [PubMed] [Google Scholar]
- Tafreshi N. K.; Doligalski M. L.; Tichacek C. J.; Pandya D. N.; Budzevich M. M.; El-Haddad G.; Khushalani N. I.; Moros E. G.; McLaughlin M. L.; Wadas T. J.; Morse D. L. (2019) Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules 24 (23), 4314. 10.3390/molecules24234314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkey N. M.; Tafreshi N. K.; Josan J. S.; De Silva C. R.; Sill K. N.; Hruby V. J.; Gillies R. J.; Morse D. L.; Vagner J. (2011) Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. J. Med. Chem. 54 (23), 8078–84. 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafreshi N. K.; Huang X.; Moberg V. E.; Barkey N. M.; Sondak V. K.; Tian H.; Morse D. L.; Vagner J. (2012) Synthesis and characterization of a melanoma-targeted fluorescence imaging probe by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Bioconjugate Chem. 23 (12), 2451–9. 10.1021/bc300549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafreshi N. K.; Silva A.; Estrella V. C.; McCardle T. W.; Chen T.; Jeune-Smith Y.; Lloyd M. C.; Enkemann S. A.; Smalley K. S.; Sondak V. K.; Vagner J.; Morse D. L. (2013) In vivo and in silico pharmacokinetics and biodistribution of a melanocortin receptor 1 targeted agent in preclinical models of melanoma. Mol. Pharmaceutics 10 (8), 3175–85. 10.1021/mp400222j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handl H. L.; Vagner J.; Yamamura H. I.; Hruby V. J.; Gillies R. J. (2004) Lanthanide-based time-resolved fluorescence of in cyto ligand-receptor interactions. Anal. Biochem. 330 (2), 242–50. 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Reid Y., Storts D., Riss T., and Minor L. (2004) Authentication of Human Cell Lines by STR DNA Profiling Analysis, in Assay Guidance Manual (Sittampalam G. S., Coussens N. P., Brimacombe K., Grossman A., Arkin M., Auld D., Austin C., Baell J., Bejcek B., and Chung T. D. Y., et al. , Eds.), Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD. [Google Scholar]
- Morss L. R., Edelstein N., Fuger J., and Katz J. J. (2011) The Chemistry of the Actinide and Transactinide Elements (Morss L. R., Edelstein N., Fuger J., and Katz J. J., Eds.) 4th ed., Vol. 1–6, Springer Science and Business Media. [Google Scholar]
- Tafreshi N. K.; Tichacek C. J.; Pandya D. N.; Doligalski M. L.; Budzevich M. M.; Kil H.; Bhatt N. B.; Kock N. D.; Messina J. L.; Ruiz E. E.; Delva N. C.; Weaver A.; Gibbons W. R.; Boulware D. C.; Khushalani N. I.; El-Haddad G.; Triozzi P. L.; Moros E. G.; McLaughlin M. L.; Wadas T. J.; Morse D. L. (2019) Melanocortin 1 Receptor-Targeted alpha-Particle Therapy for Metastatic Uveal Melanoma. J. Nucl. Med. 60 (8), 1124–1133. 10.2967/jnumed.118.217240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonardi M. L.; de Goeij J. J. M. (2005) How do we ascertain specific activities in no-carrier-added radionuclide preparations?. J. Radioanal. Nucl. Chem. 263 (1), 87–92. 10.1007/s10967-005-0017-1. [DOI] [Google Scholar]
- Ma D.; McDevitt M. R.; Finn R. D.; Scheinberg D. A. (2001) Breakthrough of 225Ac and its radionuclide daughters from an 225Ac/213 Bi generator: development of new methods, quantitative characterization, and implications for clinical use. Appl. Radiat. Isot. 55 (5), 667–78. 10.1016/S0969-8043(01)00062-8. [DOI] [PubMed] [Google Scholar]
- Apostolidis C.; Molinet R.; Rasmussen G.; Morgenstern A. (2005) Production of Ac-225 from Th-229 for targeted alpha therapy. Anal. Chem. 77 (19), 6288–91. 10.1021/ac0580114. [DOI] [PubMed] [Google Scholar]
- Robertson A. K. H.; Ramogida C. F.; Rodriguez-Rodriguez C.; Blinder S.; Kunz P.; Sossi V.; Schaffer P. (2017) Multi-isotope SPECT imaging of the (225)Ac decay chain: feasibility studies. Phys. Med. Biol. 62 (11), 4406–4420. 10.1088/1361-6560/aa6a99. [DOI] [PubMed] [Google Scholar]
- Dennis J. E. (1977) Nonlinear least-squares, in State of the Art in Numerical Analysis (Jacobs D., Ed.), pp 269–312, Academic Press. [Google Scholar]
- Bolch W. E.; Eckerman K. F.; Sgouros G.; Thomas S. R. (2009) MIRD pamphlet No. 21: a generalized schema for radiopharmaceutical dosimetry--standardization of nomenclature. J. Nucl. Med. 50 (3), 477–84. 10.2967/jnumed.108.056036. [DOI] [PubMed] [Google Scholar]
- Song H.; Hobbs R. F.; Vajravelu R.; Huso D. L.; Esaias C.; Apostolidis C.; Morgenstern A.; Sgouros G. (2009) Radioimmunotherapy of breast cancer metastases with alpha-particle emitter 225Ac: comparing efficacy with 213 Bi and 90Y. Cancer Res. 69 (23), 8941–8. 10.1158/0008-5472.CAN-09-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil C.; Bruchertseifer F.; Rathke H.; Bronzel M.; Apostolidis C.; Weichert W.; Haberkorn U.; Giesel F. L.; Morgenstern A. (2017) Targeted Alpha Therapy of mCRPC with 225Actinium-PSMA-617: Dosimetry estimate and empirical dose finding. J. Nucl. Med. 58, 1624–1631. 10.2967/jnumed.117.191395. [DOI] [PubMed] [Google Scholar]
- Choi S. I.; Kim J. E.; Hwang I. S.; Lee H. R.; Lee Y. J.; Kwak M. H.; Son H. J.; Lee H. S.; Lee J. S.; Kang B. C.; Hwang D. Y. (2012) Toxicity of red Liriope platyphylla manufactured by steaming process on liver and kidney organs of ICR mice. Lab Anim Res. 28 (4), 229–38. 10.5625/lar.2012.28.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van De Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. (2001) Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 44 (9), 1313–33. 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- Datta-Mannan A. (2019) Mechanisms Influencing the Pharmacokinetics and Disposition of Monoclonal Antibodies and Peptides. Drug Metab. Dispos. 47 (10), 1100–1110. 10.1124/dmd.119.086488. [DOI] [PubMed] [Google Scholar]
- Behr T. M.; Sharkey R. M.; Sgouros G.; Blumenthal R. D.; Dunn R. M.; Kolbert K.; Griffiths G. L.; Siegel J. A.; Becker W. S.; Goldenberg D. M. (1997) Overcoming the nephrotoxicity of radiometal-labeled immunoconjugates: improved cancer therapy administered to a nude mouse model in relation to the internal radiation dosimetry. Cancer 80, 2591–610. 10.1002/(SICI)1097-0142(19971215)80:12+<2591::AID-CNCR35>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Bonventre J. V.; Vaidya V. S.; Schmouder R.; Feig P.; Dieterle F. (2010) Next-generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 28 (5), 436–40. 10.1038/nbt0510-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.