

Abstract
The modulation of the endocannabinoid system (ECS) has shown positive results in animal models of multiple sclerosis (MS) and immune and inflammatory disorders. However, chronic administration of CB1 receptor agonists and degrading enzyme inhibitors can lead to CB1 receptor desensitization and sedation. WOBE437 is the prototype of a new class of ECS modulators named selective endocannabinoid reuptake inhibitors (SERIs), which mildly and selectively increase central endocannabinoid levels with a self-limiting mode of action. In previous studies, WOBE437 demonstrated analgesic, anxiolytic, and anti-inflammatory effects. Here, we tested the therapeutic potential of WOBE437 in a clinically relevant mouse model of MS (experimental autoimmune encephalomyelitis). C57BL/6 mice were administered WOBE437 (10 mg/kg, 20 days) or vehicle using two therapeutic options: (1) starting the treatment at the disease onset or (2) before reaching the peak of the disease. In both strategies, WOBE437 significantly reduced disease severity and accelerated recovery through CB1 and CB2 receptor-dependent mechanisms. At the peak of the disease, WOBE437 increased endocannabinoid levels in the cerebellum, concurring with a reduction of central nervous system (CNS)-infiltrating immune cells and lower microglial proliferation. At the end of treatment, endocannabinoid levels were mildly increased in brain, cerebellum, and plasma of WOBE437-treated mice, without desensitization of CB1 receptor in the brain and cerebellum. In a mouse model of spasticity (Straub test), WOBE437 (10 mg/kg) induced significant muscle relaxation without eliciting the typical sedative effects associated with muscle relaxants or CB1 receptor agonists. Collectively, our results show that WOBE437 (and SERIs) may represent a novel therapeutic strategy for slowing MS progression and control major symptoms.
Keywords: 2-arachidonoylglycerol, anandamide, cannabinoid receptors, multiple sclerosis, spasticity, SERI
Multiple sclerosis (MS) is a chronic, neurodegenerative, and neuroinflammatory disease that affects approximately 2.8 million people worldwide, according to the 2020 report published in the Atlas of MS.1 MS is characterized by demyelination of the axons in the central nervous system (CNS) that gradually leads to neuronal dysfunction and is associated with a wide spectrum of symptoms, including muscle weakness, abnormal muscle spasms, difficulty in coordination and balance, cognitive impairment, and problems with speech, swallowing, and sight.2
Neuroinflammation, induced by migration of activated myelin-specific T lymphocytes across the blood–brain barrier (BBB) and the activation of innate immune cells (dendritic cell, macrophages, and microglia) plays a prominent role in the demyelination occurring in the CNS. This correlates with the production of proinflammatory mediators and the consequent damage to oligodendrocytes, loss of myelin production, axonal damage, synaptic alteration, and neuronal loss.3,4
The clinical course of MS is highly variable and follows two main paths: relapsing–remitting or progressive. Around 85% of the MS patients are initially diagnosed by the onset of recurring clinical symptoms followed by total or partial recovery, namely, the classic relapsing–-remitting form of MS (RRMS).1,5,6 Over time, around 10–15 years of disease, this pattern might become less frequent with gradual worsening of symptoms due to progressive deterioration, reaching the secondary progressive MS (SPMS) stage.5,6 However, about 12% of patients with MS are diagnosed with primary progressive MS (PPMS), where disease progression is relentless from the onset.1,5,6 Despite these distinctions, all clinical forms of MS appear to reflect the same underlying disease process.
Currently available treatments focus on speeding recovery from attacks, slowing disease progression, and palliative care of MS patients. Among the approved treatments for MS are monoclonal antibodies (such as ocrelizumab, rituximab, and natalizumab), interferons (such as IFN-β-1a), and corticosteroids for acute exacerbations.5 Symptomatic treatments include muscle relaxants for spasticity and anticonvulsants for neuropathic pain.7 Additionally, the use of cannabis preparations for the relief of some MS symptoms, such as spasticity and inflammatory pain, has become a new therapeutic option. Regulated cannabinoid preparations like Marinol (dronabinol, as active principle), Cesamet (nabilone, as active principle), and Sativex (Δ9-tetrahydrocannabinol [THC] and cannabidiol [CBD] in 1:1 ratio, as active principles) have shown positive response in clinical trials with MS patients by reducing symptoms of pain, spasticity, and bladder dysfunction.7−11 This has led to their approval in certain countries.
Research compassing different preclinical in vivo and ex vivo studies have demonstrated the important role of the endocannabinoid system (ECS) as therapeutic target in MS.12 THC and synthetic agonists of the two cannabinoid receptors (CB1 and CB2) have shown to ameliorate both tremor and spasticity in MS mouse models.13 Anti-inflammatory and neuroprotective roles of the ECS have been widely demonstrated, in which CB2 receptor activation mainly participates in the control of autoimmune inflammation and attenuation of disease progression.14,15 CB1 receptor controls spasticity16 and plays a neuroprotective role by regulating glutamate homeostasis, which is unbalanced in neuroinflammation.17,18 Importantly, altered levels in circulating endocannabinoid (eCB) levels have been reported in MS patients, increased or decreased compared to healthy volunteers depending in the clinical form of MS and given treatments;19−22 however, more studies are needed to understand the correlation between peripheral and central eCB levels.
Due to diverse CNS-related side effects associated with CB1 agonism (such as sedation and receptor desensitization), alternative pharmacological approaches to target CB1 and CB2 receptors have been envisaged in drug discovery and development, with the main goal being to increase eCB levels. Feasible strategies, with promising therapeutic effects in preclinical models, include the inhibition of the main eCB degrading enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). FAAH and MAGL inhibitors have been shown to reduce neuroinflammation, neurodegeneration, and spasticity in animal models of MS.23−25 Some of these inhibitors have been tested in early stage clinical trials for CNS and inflammatory diseases, but not MS.26
In 2017, WOBE437 was identified as the first potent selective eCB reuptake inhibitor (SERI).27In vitro and in vivo, WOBE437 induced a moderate but significant increase of the eCBs anandamide (AEA) and 2-arachydonoyl glycerol (2-AG), without targeting degrading enzymes, any known components of the ECS, or relevant CNS-associated proteins.27 Importantly, WOBE437 displayed beneficial pharmacological effects in animal models of acute pain, chronic inflammatory pain, anxiety, and endotoxemia, reaching bioactive concentrations in the brain around 15–30 min after intraperitoneal injection27 or gavage administration.28 WOBE437 exerts CB1-mediated antinociceptive effects after single oral dose, without significantly affecting the other parameters of the “classic” cannabinoid tetrad test (catalepsy, hypothermia, and motor coordination).27 Moreover, subchronic treatment for 7 days with WOBE437 led to a 1.5-fold increase of AEA and 2-AG levels, which was enough to elicit pharmacological effects without causing functional alterations (desensitization) of CB1 receptors, which is a typical feature of MAGL inhibitors that strongly increase 2-AG levels (>10-fold increase), the most abundant eCB in the CNS.27,29
The aim of this work was to evaluate the potential effect of WOBE437 in a relevant mouse model of MS, the experimental autoimmune encephalomyelitis (EAE) model, which is a CD4 T-cell-mediated neuroinflammatory disease that leads to some key pathological features of human MS: inflammation, demyelination, axonal loss, and gliosis.30 Given that previous studies already indicated the important roles of CB1 and CB2 receptors in the EAE pathophysiology, we aimed to understand whether the weak eCB modulation induced by SERIs would suffice to achieve therapeutic effects and reduce clinical symptoms. In order to understand the potential translational implications of this new eCB system modulation, two pharmacological approaches were considered to start the administrations: (1) on the individual day of disease onset and (2) close to the peak of the disease.
Results
WOBE437 Decreased Disease Severity in a Mouse Model of EAE
EAE was induced in female C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein (MOG) peptide (amino acids 35–55, emulsified in complete Freund’s adjuvant [CFA]) and pertussis toxin injections (see the Experimental Section). Mice showed the first clinical symptoms mainly between 10 and 16 days post-immunization (Figure S-1), and considering that the percentage of disease incidence variated between 50 and 100%, only mice showing clinical symptoms were included in the study. Therefore, administration of WOBE437 (10 mg/kg, intraperitoneally (i.p.)) or vehicle (dimethyl sulfoxide (DMSO), i.p.) was started individually at the day of onset of clinical disease for each mouse and continued for 20 days. For clarity, the time course of disease development (from immunization to the end of experimentation) was matched to the day of onset of each mouse; thus the time course of clinical symptoms is presented from the onset (day 1) to the end of treatment (day 20). Analysis of the time-course of clinical symptoms showed that WOBE437-treated mice developed a significantly milder disease (Figure 1) compared to vehicle-treated mice. The severity of symptoms was reduced by about 30–50% starting from 3 days post-treatment (p = 0.0712) and reaching statistical significance at 5 days post-treatment (p < 0.001), which was maintained until the last day of treatment (Figure 1A). WOBE437-treated mice showed an overall reduction of disease severity, analyzed using the area under the curve (AUC) of clinical scoring from day 0 to day 20 as compared to vehicle (Figure 1B) and exhibited lower occurrence of high clinical scores (>2; Figure 1C). In addition, the treatment with WOBE437 protected the animals from the strong weight loss observed in vehicle-treated mice (Figure 1D).
Figure 1.

Chronic treatment with WOBE437 reduced disease severity in EAE female C57BL/6 mice. (A) Time course of clinical score from day 0 to day 20; onset is represented as day 1 (red line). (B) AUC and (C) maximal clinical score observed from the time course of clinical score, from day 0 to day 20. (D) Time course of body weight changes from day −10 to day 20; onset is represented at day 1 (red line). Administration of vehicle (DMSO, n = 37) or WOBE437 (10 mg/kg, n = 29) started at the individual day of onset of each mouse; injections (20 μL) were done intraperitoneally once per day during 20 days. Data show (A, D) mean ± SEM; (B) median, percentile 25, percentile 75, minimum and maximum; or (C) cumulative frequency. (A–D) Data show the summary of four different cohorts, only mice showing symptoms were included in the study. Statistical differences were determined using (A, D) multiple t-tests corrected for multiple comparison with the Holm–Sidak method, (B) Mann–Whitney test, or (C) Chi-square test and Fisher’s exact test. *, p < 0.05 compared to vehicle group.
Role of CB1 and CB2 Receptors in the Pharmacological Effect of WOBE437
In order to test the role of CB1 and CB2 receptors in the pharmacological effect of WOBE437, we coadministered WOBE437 (10 mg/kg, i.p.) with the selective CB1 receptor antagonist rimonabant (3 mg/kg, i.p.) and the selective CB2 receptor antagonist AM630 (3 mg/kg, i.p.). As shown in Figure 2A, CB1 receptor blockade induced a partial reduction of WOBE437 effect with a slight decrease of clinical score during the peak of disease (4–8 days post-treatment; day 1 = onset) and full inhibition during the chronic/remission stage (10–20 days post-treatment; day 1 = onset). Importantly, animals administered with rimonabant (alone or in combination) exhibited a collective fatality ratio of 23%, while animal groups treated with vehicle and WOBE437 did not show any fatalities (data not shown). The number of fatalities was equally distributed in the cohort treated with rimonabant in monotherapy and in combination with WOBE437. The fatalities occurred between 1 (onset) and 11 days post-treatment. Due to the short period of administration, those cases were excluded from the time-course analysis but assigned with clinical score 3 (maximal clinical score) and included in the report of maximal clinical score (Figure 2C). Notably, in a previous trial, a slightly higher dose of rimonabant (5 mg/kg, i.p.) showed 55.6% fatality (10 out 18 mice) after 1 or 2 days of administration (data not shown).
Figure 2.

Evaluation of either CB1 or CB2 receptor antagonism in combination with WOBE437 treatment in EAE female C57BL/6 mice. (A, B) Time course of clinical score from day 0 to day 20 in mice administered with either a (A) CB1 antagonist or (B) CB2 antagonist, in combination with WOBE437 (10 mg/kg) or vehicle (DMSO); onset is represented at day 1 (red line). (C) Maximal clinical score observed during the time course of clinical score, from day 0 to day 20. Rimonabant (SR1, 3 mg/kg) was used as CB1 receptor antagonist/inverse agonist, and AM630 (3 mg/kg) was used as CB2 receptor antagonist/inverse agonist. Administration of vehicle and all compounds started at the individual day of onset (day 1) of each mouse; injections (20 μL) were done i.p. once per day during 20 days. Data show (A, B) mean ± SEM or (C) cumulative frequency; group sizes (mice per group) were as follows: vehicle n = 37, WOBE437, n = 29; SR1, n = 17; SR1 + WOBE437, n = 20; AM630, n = 16; AM630 + WOBE437, n = 21. Data show the summary of two or four different cohorts; only mice showing symptoms were included in the study. Statistical differences were determined using (A, B) multiple t-test corrected for multiple comparison with the Holm–Sidak method or (C) chi-square test and Fisher’s exact test. *, p < 0.05 comparing as indicated by brackets or comparing WOBE437 against SR1 + WOBE437 or against AM630 + WOBE437.
To evaluate the role of CB2 receptor, WOBE437 (10 mg/kg, i.p.) was coadministered with AM630 (3 mg/kg, i.p.), a selective CB2 receptor antagonist/inverse agonist. The combination fully prevented the pharmacological effect of WOBE437 during the whole duration of the treatment (Figure 2B). Importantly, administration of AM630 resulted in a fatality ratio of 32%, which occurred between 1 and 10 days after treatment started, with most cases occurring during the peak of disease severity (around days 3 and 5, day 1 = onset). Those data were handled as for rimonabant (see above).
Finally, animals treated with WOBE437 showed a significantly lower frequency of severe clinical scores, with no animals reaching score 3 (full paralysis and incontinence). On the contrary, animals treated with vehicle or coadministered with WOBE437 in combination with CB1 and CB2 receptors antagonists showed a significantly higher frequency of more severe clinical symptoms, including score 3 (Figure 2C). There were not significant differences in body weight among the groups (Figure S-2).
Central and Peripheral Increase in eCB levels after WOBE437 Treatment
WOBE437 is the first potent SERI which induces a significant increase in AEA and 2-AG levels in healthy mice, in plasma and brain, upon single or repeated (7 days) administrations.27,28 Here, we quantified using LC-MS/MS the levels of AEA and 2-AG in EAE mice treated with vehicle and WOBE437. To this end, brain, cerebellum, lumbar spinal cord (SC, L1–L6 segments), and plasma were collected at the peak of disease severity (4–6 days post-treatment; day 1 = onset) and at the end of treatment (20 days post-treatment; day 1= onset). Interestingly, at the peak of the disease, WOBE437 induced a significant increase of 2-AG (+32%) and AEA (+35%) levels in the cerebellum, compared to vehicle (Figure 3A,B). No significant changes were observed in brain, lumbar SC and plasma. However, at day 20, during the chronic/remission stage, WOBE437 induced a clear and significant increase of AEA and 2-AG levels in brain and cerebellum by 30–50% (Figure 3C,D). Notably, a 23% increase of 2-AG was observed in plasma (Figure 3C).
Figure 3.
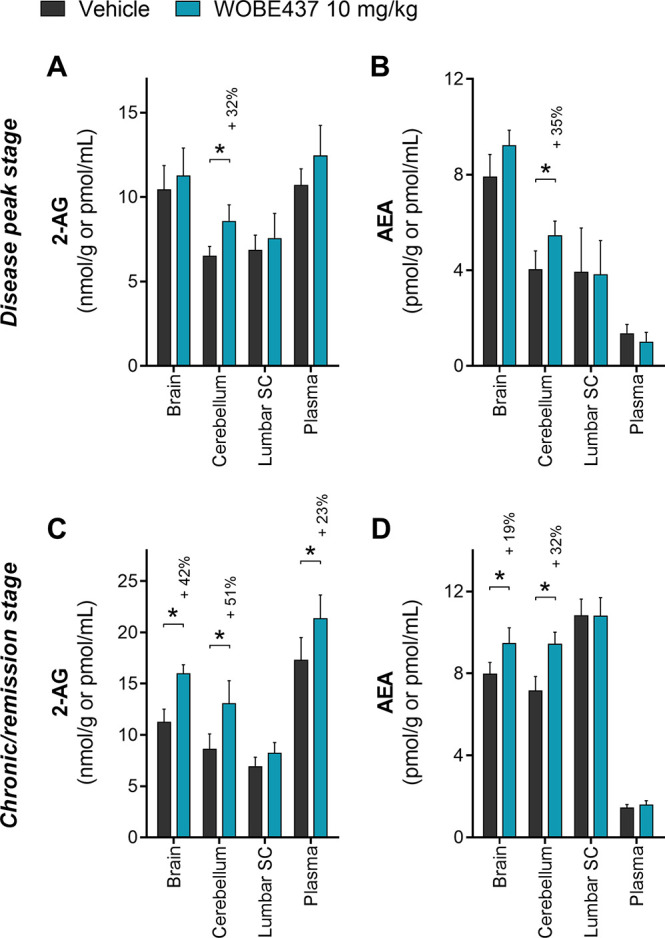
Quantification of endocannabinoid levels in EAE female C57BL/6 mice treated either with vehicle or WOBE437. (A) 2-AG and (B) AEA were quantified in brain, cerebellum, lumbar SC, and plasma at the peak of the disease (4–6 days after onset; average harvest day was 5 for both groups). (C) 2-AG and (D) AEA were quantified in brain, cerebellum, lumbar SC, and plasma at the chronic/remission stage (20 days after onset). 2-AG and AEA were quantified using LC-MS/MS, and values were normalized to the tissue amount (g) or volume (mL). Vehicle (DMSO) and WOBE437 were given i.p. (20 μL) starting at the disease onset (day 1). Sample size numbers (indicated as vehicle/WOBE437) were the following: (A, B) brain n = 8/9, cerebellum n = 8/9, lumbar SC n = 5/6, and plasma n = 8/9; (C) brain n = 21/17, cerebellum n = 20/15, lumbar SC n = 13/7, and plasma n = 21/17; (D) brain n = 21/17, cerebellum n = 20/15, lumbar SC n = 13/7, plasma n = 22/22. Data show mean ± SD. Statistical differences were determined using the Mann–Whitney test. *, p < 0.05 compare to vehicle group; if significant, then a percentage of increase is reported above.
CB1 Receptor Functional Activity in Brain after Chronic Treatment with WOBE437
In light of the increase of 2-AG and AEA concentrations in the brain and cerebellum of EAE mice after 20 days of treatment with WOBE437 (treatment started at onset), the functionality of CB1 receptors was assessed to exclude possible desensitization. [35S]GTPγS binding assay was conducted in brain membrane preparations obtained from EAE mice treated for 20 days with vehicle or WOBE437 (10 mg/kg, daily). After incubation with the CB1 receptor agonist CP-55940, both vehicle- and WOBE437-treated mouse brain preparations showed a significant activation of CB1 receptor by 146 and 157%, respectively, compared to control samples (Figure 4A). Similarly, CP-55940 induced a similar activation of CB1 receptor in the cerebellum (184% and 175% in vehicle and WOBE437 treated animals, respectively, Figure 4B). Co-incubation of CP-55940 with rimonabant fully abolished the increase of [35S]GTPγS binding, thus confirming the specificity of CB1 receptor activation in brain (Figure 4A) and cerebellum (Figure 4B). Finally, no statistical difference was observed between vehicle- and WOBE437-treated mouse brain and cerebellum preparations upon stimulation with CP-55940, rimonabant, and a combination of CP-55940 plus rimonabant (Figure 4A,B).
Figure 4.

Evaluation of CB1 receptor function in brains of EAE female C57BL/6 mice treated for 20 days with vehicle or WOBE437. (A) Brain or (B) cerebellum membrane preparations were evaluated using the [35S]GTPγS assay and incubation with vehicle (DMSO, control samples), 0.1 or 1 μM CP-55940 (CB1 agonist), 1 μM rimonabant (CB1 antagonist/inverse agonist), or the combination of both. The [35S]GTPγS bound to vehicle samples was used as a control to normalize the data. CB1 receptor activation was confirmed by coincubation with rimonabant. Daily treatments (i.p., 20 μL) in the EAE mice started individually on the day of disease onset. Sample size (vehicle/WOBE437) was as follows: (A) brain tissues n = 12/11 mice per group and (B) cerebellum n = 6/6 mice per group. Data show mean values ± SD. Statistical differences were determined using two-way ANOVA and Tukey’s multiple comparison test. ***, p < 0.001.
Evaluation of CNS Infiltrating Immune Cells during the Peak of Disease and WOBE437 Treatment
An important part of EAE pathogenesis involves disruption of the BBB and CNS infiltration of IFN-γ and IL-17 producing T cells, Th1 or Th17, respectively, which are autoreactive across myelin components.30 Immune cells were isolated from brain or spinal cord of vehicle- and WOBE437-treated mice during the peak of disease, 4–6 days post-treatment (day 1 = onset). Notably, each cell suspension sample was composed of isolated infiltrating cells pooled from two brain or spinal cord tissue samples, and each sample was resuspended in 1 mL of FACS buffer. After isolation, phenotyping of T cells was done after ex vivo stimulation with phorbol myristate acetate (PMA) and ionomycin, which promote cytokine production, along with a protein transport inhibitor to block cytokine release. Overall, WOBE437-treated mice showed a 55% decrease in the number of infiltrated CD45high cells in the brain, from 32.3 × 103 cells/mL in the vehicle group to 14.6 × 103 cells/mL in the WOBE437 group (Figure 5A). Further analysis of CD45high CNS infiltrating immune cells showed that both, CD4+IFNγ+IL-17+ and CD8+IFNγ+IL-17+ T cell subpopulations were significantly reduced in the brain of WOBE437-treated mice, compared to vehicle-treated mice by 50 and 71%, respectively (Figure 5B,C). Similarly, in the spinal cord we observed a tendency to reduce infiltration of CD45high infiltrating immune cells as well as CD4+IFNγ+IL-17+ and CD8+IFNγ+IL-17+ T cell subpopulations (Figure 5A-C). Additionally, CD4+ and CD8+ T cell subpopulations producing interleukin-10 (IL-10) or granulocyte–macrophage colony-stimulating factor (GM-CSF) were analyzed, observing an overall decreased of this subpopulations in the WOBE437 group (Figure S-3), in agreement with an overall decrease in the number of immune infiltrating cells.
Figure 5.

CNS infiltrating CD45+ cells at the peak of disease in EAE female C57BL/6 mice treated with vehicle or WOBE437. Lymphocytes were gated considering their (A) CD45high expression, then gated in (B) CD4+IFNγ+IL-17+ and (C) CD8+IFNγ+IL-17+ T cell subpopulations. Microglia cells were estimated by gating a (D) CD45dim cell population. Infiltrating immune cells were isolated from brain or spinal cord collected at the peak of disease (4–6 days post-treatment, day 1 = onset). Daily treatment (i.p., 20 μL) with vehicle (DMSO) or WOBE437 (10 mg/kg) was started at the onset of disease. Data show mean ± SD of infiltrating immune cells isolated from the brain or spinal cord of n = 6 mice per group. Data is depicted as a summary of three independent experiments including n = 2 mice (pooled) per group. Isolated cells were resuspended in 1 mL of FACS buffer, and cellular count was assessed using the volumetric sample and sheath fluid delivery system of the Attune NxT cytometer. Statistical differences were determined using Mann–Whitney test. *, p < 0.05 compared to vehicle group.
Furthermore, microglial cells were estimated by gating the CD45dim cell population, since relative levels of CD45 can distinguish microglia (CD45dim) from CNS-infiltrating immune cells (CD45high).31,32 Interestingly, the number of CD45dim cells in brain and spinal cord of WOBE437-treated mice was significantly reduced by 67 and 64%, respectively, compared to vehicle-treated mice (Figure 5D).
To further explore the neuroinflammatory role of WOBE437 treatment in EAE mice, prostaglandin-E2 and -D2 levels were quantified in central and peripheral tissues collected either at the peak of disease or at the chronic/remission stage; however, no significant differences were observed compared to vehicle-treated mice (Figure S-4).
Potential of WOBE437 and SERIs as Disease-Modifying Agents
With the aim to evaluate the therapeutic potential of WOBE437 (and SERIs) in clinics, we conducted another experiment using a more clinically relevant treatment approach. In a new cohort of EAE mice, we started the daily administration of WOBE437 (10 mg/kg, i.p.), or vehicle, not at the onset of symptoms but when mice reached almost the peak of the disease (defined as clinical score 2, which is a limp tail and paraplegia of hind limbs), which occurred between 3 and 4 days after onset. Intriguingly, already at 1 day after starting administrations, WOBE437 induced a significant reduction of clinical score by 30%, which increased to 40% reduction after subsequent administrations and it was maintained throughout the remaining duration of the experiment (Figure 6A).
Figure 6.

WOBE437 as a disease-modifying agent in EAE female C57BL/6 mice. (A) Time course of clinical score from day 0 to day 20; onset is represented at day 1 (red line). The beginning of administration is represented with a blue line. (B) AUC and (C) maximal clinical score observed from the time course of disease severity from day 4 to day 20. (D) Time course of body weight change from day −10 to day 20; onset is represented at day 1. The beginning of administration is marked with a blue line. Administration of vehicle (DMSO, n = 18) or WOBE437 (10 mg/kg, n = 14) was started after disease onset, i.e., when the mouse showed a clinical score 2 or up to 4 days after onset; injections (20 μL) were done i.p. once per day during 15 days. Data show (A, D) mean ± SEM; (B) median, percentile 25, percentile 75, minimum, and maximum; or (C) cumulative frequency. (A–D) Data show the summary of two different cohorts; only mice showing symptoms were included in the study. Statistical differences were determined using (A, D) multiple t-test corrected for multiple comparison with the Holm-Sidak method; (B) Mann–Whitney test; or (C) chi-squared test and Fisher’s exact test. *, p < 0.05 compared to vehicle group.
In agreement, the overall disease severity calculated as AUC of the clinical score (4–20 days after onset) was significantly reduced by 35% in the WOBE437 group as compared to vehicle group (Figure 6B). Notably, only 30% of WOBE437-treated mice reached the clinical score 2, with no incidence of maximal clinical score (score = 3). In the control group around 60% of the mice reached score 2, and around 33% reached the maximal score of 3 (Figure 6C). Additionally, WOBE437-treated mice showed a faster recovery of body weight, showing significant improvements from day 9 after onset (5 days of treatment, Figure 6D) until the end of treatment. Importantly, starting from day 15 after onset, WOBE437-treated mice completely recovered from the disease-induced weight loss that reached its nadir (−3.7 g, ca. 20% reduction of initial body weight) 6 days after onset. On the contrary, at the end of the chronic-remitting phase (day 20 after onset), vehicle-treated mice only partially recovered from the weight loss, which was still significantly lower (−1.1 g) as compared to initial body weight.
WOBE437 Induces Muscle Relaxation without Sedative Effects
WOBE437 was administered to healthy mice to evaluate potential muscle relaxation properties using the well-established Straub tail test, which induces the elevation of the tail (Straub tail reaction) after morphine administration, as consequence of dopamine release in the CNS and the contraction of the sacro-coccygeus dorsalis muscle.33,34 We observed the maximum Straub tail response 15 min after administration of morphine (40 mg/kg, subcutaneous (s.c.)), and it lasted for more than 2 h. The elevation of the tail was evaluated with a 4-points scale score35 which is based on the angular degree of elevation from normal tail position. Baclofen (5 mg/kg, i.p., 30 min before the test), a GABAB receptor agonist used as muscle relaxant in human medicine, and diazepam (6 mg/kg, i.p., 30 min before the test), a GABAA-positive allosteric modulator approved to treat anxiety in humans with well-documented muscle relaxant properties, were used as positive controls in the Straub tail test. Both compounds significantly reduced the intensity of the Straub tail reaction, inducing a strong reduction (score 1–2) of tail elevation 15–60 min after morphine injection; for baclofen, this effect is lost 2 h after morphine injection (Figure 7A). The effects of diazepam were only followed for 1 h after morphine injection. Pretreatment with WOBE437 (10 mg/kg, i.p., 60 min before the Straub test) significantly reduced the intensity of the Straub reaction (score 2–3) as early as 15 min after morphine injection; notably, this effect lasted at least 2 h (Figure 7A). Additionally, we tested the nonselective CB1/CB2 full agonist WIN-55212-2 (3 mg/kg, i.p., 30 min before), which showed a stronger reduction of the Straub tail response for the evaluated time (1 h, Figure 7A).
Figure 7.

WOBE437 induced muscle relaxation without sedative effects in male C57BL/6 mice. (A) Muscle relaxation was evaluated using the Straub test after morphine administration (40 mg/kg, s.c.). (B) Spontaneous locomotion was evaluated in the open-field test (OFT) by placing each mouse in the arena and allowing it to freely move for 5 min. (C) The potential induction of catalepsy-like behavior was assessed in the bar test. Vehicle (DMSO, n = 10), WOBE437 (10 mg/kg, WOBE, n = 6), and WIN-55212-2 (3 mg/kg, WIN, n = 5) were injected (i.p.) 1 h before morphine injection. Baclofen (5 mg/kg, n = 7) and/or diazepam (6 mg/kg, DZP, n = 5) were injected (i.p.) 40 min before morphine injection. Importantly, the OFT and the bar test were conducted just before morphine administration: 8 and ∼2 min before, respectively. Data show (A) cumulative frequency or (B, C) median, percentile 25, percentile 75, minimum, and maximum. Statistical differences were determined using (A) the chi-square test and Fisher’s exact test or (B, C) the Kruskal–Wallis test followed by the Mann–Whitney test as post hoc analysis. *, p < 0.05 compared to vehicle group.
Importantly, unlike WIN-55212-2, baclofen, and diazepam, WOBE437 was the only muscle relaxant in this test that did not show the typical sedative effect of reduced locomotion (open field test) and catalepsy-like behavior (bar test) (Figure 7B,C).
Discussion
In the present study, we evaluated the therapeutic potential of WOBE437, the prototype of a new class of ECS modulators (SERIs), in a mouse model of MS. WOBE437 elicited a significant reduction of disease severity and promoted faster recovery, in a CB1- and CB2-receptor-dependent manner. Intriguingly, WOBE437 attenuated disease severity not only when the treatment started at the onset of the disease (day 1) but also when the treatment started just before the peak of disease severity (day 4). The latter represents a more clinically relevant therapeutic intervention to treat MS patients, who usually initiate pharmacotherapy after the symptoms become evident.
In line with its mode of action, WOBE437 induced a mild but significant increase of AEA and 2-AG levels by 30–50% in brain and cerebellum and an increase in circulating levels of 2-AG by 23%. Importantly, WOBE437 decreased the infiltration of immune cells into the CNS, in particular T cell subpopulations CD4+IFNγ+IL-17+ and CD8+IFNγ+IL-17+.
Activated Th1 cells and Th17 cells are thought to be primarily responsible for the pathological changes developed in EAE models and to participate during the pathology of human MS.36 Th1 are IFN-γ-producing T lymphocytes, and Th17 are IL-17-producing T lymphocytes, which are peripherally primed by dendritic cells and subsequently cross the BBB and encounter CNS-antigen-presenting cells. Th1 and Th17 cells produce an array of inflammatory factors and cytokines that lead to demyelination and axonal loss.37,38 Furthermore, Th1 and Th17 cells also activate resident microglia, which produce additional factors that attract additional inflammatory cells to the CNS and thereby perpetuate the inflammatory cascade.39,40 Notably, it was reported that T lymphocytes can produce cytokines from both linages, Th1 and Th17.41,42 Additionally, some key cell adhesion molecules are upregulated in the BBB, facilitating the entrance to the CNS of innate immune cells like monocytes that can become dendritic cells or macrophages; B cells also have been seen to infiltrate the CNS contributing to the inflammatory demyelination and neurodegeneration.30,43 Along with the reduction of CD4+IFNγ+IL-17+ and CD8+IFNγ+IL-17+ T cells infiltration, WOBE437 treatment reduced general infiltration of immune cells as seen by an overall reduction of CD45high cells. Th17 T cells also produce GM-CSF, which is also considered to be highly pathogenic during the development of EAE,44,45 and it was found reduced after WOBE437 treatment. Furthermore, CD45dim cells were also significantly reduced, which implied a reduction in the proliferation of microglial cells.32,46
Departing from anecdotal reports of the improvement of MS symptoms after the use of cannabis preparation in patients, a lot of knowledge has been gained in understanding the role of the ECS in the pathology of this complex neuroinflammatory disease. In animal models, either the deletion of CB1 receptor or its pharmacological inhibition were detrimental during the induction of EAE, resulting in an increase severity of the symptoms and fatality cases,17,47,48 which was also observed in our study. This has been associated with an increase in excitotoxicity, neuronal death due to toxic ion influxes (Ca2+), and caspase-3-mediated apoptosis.17,47 Furthermore, treatment of EAE mice with SR141716A (rimonabant) has led to a notable shift from Th2 (IL-4/IL-10) to Th1 (IFN-γ)/Th17 (IL-17) cytokines and increased level of chemokines during EAE.48 Consistently, local overexpression of CB1 receptor in the spinal cord exhibited neuroprotective effects in EAE, mainly suppressing inflammatory microenvironment and elevating neurotrophic factors, slightly declining IL-1β and IL-17 in the spleen, and increased IL-10 in the CNS.49 Notably, we found a significant increase of AEA (48%) and 2-AG (26%) levels in the spleen of WOBE437-treated mice compared to vehicle after the 20 days of treatment (chronic/remission stage; Figure S-5). This is in line with the previous finding that SERIs like WOBE437 act on immune cells.27 Additionally, CB2 receptor abundance was evaluated in spleen membrane preparations, and there were no differences between vehicle- and WOBE437-treated mice (Figure S-6).
A cell-specific involvement of the CB1 and CB2 receptors in the pathology of EAE/MS was previously shown, where CB1 receptor expression was important in the CNS but not in T cells, and the opposite was true for CB2 receptors.50 Maresz et al. also reported an increase in fatality cases (16%) in CB2 T cell conditional knockout mice that were subjected to an EAE model. In a CB2-deficient mouse model, induction of EAE resulted in a more severe disease with stronger axonal loss, exacerbated microglia activation and higher CD4+ T lymphocyte infiltration into the CNS; however, no fatality cases were reported.14 In agreement, our results showed an increase in disease severity after pharmacological inhibition of CB2 receptors with AM630, which was mainly reflected as fatality cases or euthanasia due to local law legislation. We also observed a tendency for a transitory mild reduction of disease severity with AM630 compared to vehicle at 4, 5, and 6 days after onset. In a few number of previous EAE studies, CB2 antagonist/inverse agonist did not show any significant difference in disease severity compared to vehicle.51,52 Bolognini et al.53 described AM630 acting as a protean agonist at the CB2 receptor in vitro. Protean agonists can behave as inverse agonist, neutral antagonist, or agonist, depending on to the proportion of constitutively activity in a receptor population. Salort et al.54 confirmed the protean agonism behavior of AM630 in mice upon repeated administrations. We may speculate that the partial and transitory improvement of disease severity observed in our study with AM630 could derive from an unbalanced distribution of constitutively active CB2 receptors, other compensatory mechanisms, intrinsic experimental variability, or a combination of those.
The equimolar combination of THC and CBD (Sativex) has been approved and successfully used in clinics for several years to control spasticity associated with MS.55,56 Although the Cannabinoid Use in Progressive Inflammatory brain Disease (CUPID) clinical trial failed to show a significant deceleration in MS progression over 3 years of treatment using the synthetic THC analogue dronabinol,57 recent investigations suggested that THC and CBD might exert complementary pharmacological actions which account for the control of MS-associated symptoms (humans and rodents) and slowing the disease progression (rodents).58 Therefore, CB receptors are considered potential pharmacological targets to reduce EAE/MS symptoms. Early experiments using nonselective CB1/CB2 receptor agonists such as WIN-55212-2 and either FAAH or MAGL inhibitors have shown a significant protection against EAE development in mice and rats, where those effects were mediated either through only one type of CB receptors (CB1 or CB2 receptor) or both.24,52,59−61
Control of spasticity symptoms has been reported using FAAH inhibitors such as CAY100400, CAY100402, and URB597 or MAGL inhibitors such as JZL184, using a mouse model where EAE was induced with spinal cord homogenate and Freund’s adjuvant.60 It was also reported that these FAAH inhibitors did not mediate typical cannabimimetic effects such as hypothermia and catalepsy. Notably, Pryce et al. also showed that FAAH deletion in mice induced a mild but significant reduction of CB1 receptor signaling activity (using [35S]GTPγS binding assay) in cerebellum and substantia nigra.60 Using the cuprizone-induced mouse model of T-cell-independent demyelination, it was reported that MAGL inactivation preserved myelin integrity and suppressed microglial activation. In additional in vitro experiments, the authors showed that JZL184, 2-AG and AEA, but not URB597, induced suppression of cell death in oligodendrocytes, and this effect was associated with protection from excitotoxicity, which was mediated by CB1 receptor activation and a reduction of AMPA-induced cytosolic calcium overload, mitochondrial membrane depolarization, and production of reactive oxygen species.62 In EAE mice, neuroprotective effects induced by CB1 receptor activation also have shown to involve the inhibition of TNFα effects (mainly release by activated microglia), which include abnormal expression and phosphorylation of AMPA receptors and increased frequency of excitatory postsynaptic currents (EPSC).18,63
Notably, irreversible MAGL inhibitors, such as JZL184 and KML29 induce a dramatic (>20- to 100-fold over basal levels) and sustained (up to 24 h) increase in 2-AG levels that can lead to CB1 receptor desensitization upon chronic treatment and possibly limiting the clinical development.29,64,65 Bernal-Chico et al. also showed that a relatively low dose (8 mg/kg, i.p.) of JZL184 did not lead to overall desensitization of CB1 receptors in the brain; however, it led to a region-specific desensitization, which was shown to be more pronounced in cerebellum and cortex after 21 days of daily administration of JZL184 in EAE mice.62 However, efforts to develop reversible MAGL inhibitors are ongoing. In this context, López-Rodríguez et al. reported a protective effect of the reversible MAGL inhibitor ⟨compound 21⟩ against EAE induction.24 ⟨Compound 21⟩ led to the reduction of leukocyte infiltration, microglia activation, prevention of axonal damage, and partial restoration of myelin morphology;24 however, no evaluation of receptor tolerance was carried out after the 21 days of treatment.24
It is well-known that CB1 receptor agonists induce sedation, undesired psychotropic effects, and development of receptor tolerance upon chronic administration. This limits the use of CB1 receptor agonist for therapeutic purposes. Alternative strategies to modulate the ECS aim to increasing the endogenous ligands (AEA and 2-AG) through the inhibition of their degradation. However, to date, FAAH and MAGL inhibitors have not been successful in large clinical trials, mainly due to a lack of efficacy or off-target effects.66−68 Additionally, some supposed AEA uptake inhibitors, which turned out to be nonselective compounds primarily inhibiting FAAH, have been tested in animal models of MS showing variable outcomes.52,69−71 However, these results are often biased due to the low potency of these compounds (micromolar), their metabolic instability and poor selectivity over other components of the ECS, as mentioned, in particular FAAH.72
Recently, we reported the development and biological characterization of WOBE437 as the first potent SERI.27 SERIs represent a new class of ECS modulators that mildly and selectively increase AEA and 2-AG levels, without targeting other components of the ECS like FAAH and MAGL.27 WOBE437 acts with a self-limiting mechanism of action which prevents an overflow of eCB beyond physiological concentrations (see refs (27) and (28) and current data); thus, it only moderately increases total eCB concentrations in the brain. Thanks to the selective increase of both AEA and 2-AG levels, SERIs can recapitulate the full spectrum of eCB biology, which is associated with a differential activation of CB receptors (partial vs full agonism) and other non-CB receptors.27,28,73,74
We previously showed that WOBE437 exerts anti-inflammatory effects in a LPS-induced endotoxemia mouse model, in the second phase of the formalin test, and in a mouse model of chronic inflammatory pain induced by monoarthritis.27,28 We also showed that WOBE437 shows analgesic/antinociceptive effects in different mouse models upon systemic and oral administration.27,28 In the CFA-induced monoarthritis mouse model, the effects of WOBE437 were mediated by both CB receptors but also involved PPARγ and TRPV1 receptors, indicating a clear multitarget mechanism.28 In the present study, WOBE437 administration in EAE mice elevates eCB levels in a time- and tissue-dependent manner, showing a localized increase of AEA and 2-AG only in the cerebellum during the disease peak and a less localized increase during the chronic/remission stage. Importantly, CB1 receptor tolerance was not observed upon the 20 days of subchronic daily administrations of WOBE437. A strong involvement of cerebellum during the development of EAE was shown previously, with gray matter atrophy, demyelination, inflammatory cell infiltration, and cell death of a major neuronal cell type within this structure.75 It is surprising that we could not see a significant change in eCB levels in lumbar SC upon WOBE437 treatment, since this tissue is also highly affected by EAE and has shown to be positively modulated after activation of the ECS.49,52 It is possible that in certain tissues, the weak increase of eCBs induced by WOBE437 may be difficult to detect analytically or that the amounts may only change in particular compartments rather than as net amounts. Nevertheless, WOBE437 treatment showed a significant reduction in infiltrating immune cells in both brain and spinal cord. Finally, in the context of spasticity, WOBE437 showed intriguing muscle relaxant properties in the Straub tail test (in healthy mice), and this effect lasted longer compared to the known and commercially available GABAB agonist baclofen. The dual CB1/CB2 agonist WIN-55212-2 showed more pronounced effects compare to both baclofen and WOBE437, suggesting a CB-receptor-dependent mechanism. However, baclofen and diazepam (used in human medicine also for muscle relaxation properties), as well as WIN-55212-2, induced typical side effects like catalepsy, hypolocomotion, and sedation. On the contrary, WOBE437 elicited muscle relaxation without inducing any of the above-mentioned side effects. Importantly, data obtained using the Straub tail test have shown a great correlation with the efficacy observed in humans.76,77
In conclusion, we show that WOBE437 treatment led to a significant improvement of clinical scores and significantly reduced the number of CNS-infiltrating immune cells in a EAE rodent model of MS. EAE is considered one of the most relevant preclinical models of MS because it resembles human pathology and symptoms in many ways.36
Notably, WOBE437 elicited a significant improvement of disease severity in two different treatment approaches: (1) preventing symptoms development with the treatment starting at the onset of the disease (day 1) and (2) reducing clinical score with the treatment starting before the peak of disease severity (4 days after onset). The latter represents a clinically relevant approach to treat MS patients, who initiate pharmacotherapy only after symptoms become evident.
Furthermore, acute (4–6 days, i.p.) and subchronic treatment (17–20 days, i.p.) with WOBE437 significantly increased brain levels of AEA and 2-AG, without leading to CB1 receptor desensitization in brain and cerebellum, therefore lacking tolerance after chronic use, which could have important translational implications.
Thanks to its combined pharmacological effects on regulating neurotransmission and reducing inflammation (see refs (28) and (78) and current data), SERIs like WOBE437 represent an innovative, safe, and effective therapeutic option for attenuating MS progression and tackling other unmet needs in the area of CNS and inflammatory disorders.
Experimental Section
Animals
Male or female C57BL/6J mice (7 weeks old; 15–18 g body weight) were supplied by Janvier Laboratories and kept under standard environmental conditions (24 ± 2 °C; light–dark cycle of 12:12 h) with food and water ad libitum. Mice were handled according Swiss federal legislation, and protocols were approved by the respective government authorities (Veterinäramt Kanton Bern, experimental license BE-07/17).
Induction of Active EAE
After 1 week of habituation, active EAE was induced in female C57BL/6J mice (8 weeks old) following a well-established protocol.79 In brief, mice were anesthetized under isoflurane flow and subcutaneously administered with 200 μg of myelin oligodendrocyte glycoprotein peptide (MOG, amino acids 35–55; 4 mg/mL in PBS), which was emulsified (1:1 ratio) with incomplete Freund’s adjuvant containing 4 mg/mL Mycobacterium tuberculosis; subcutaneous injections of the emulsion were done in both hips and tail root (100 μL final administration volume). Immediately after immunization, the mice were administered i.p. with 300 ng of Bordetella pertussis toxin in PBS. On the third day, 2 days post-immunization, a second administration of pertussis toxin was given.
Assessment of Clinical Disease and Treatments
Immediately after immunization and consecutively every day, each mouse was weighted and evaluated for the presence of any clinical symptom by using a previously described scale:80 0, healthy; 0.5, limp tail; 1, hind legs paraparesis; 2, hind legs paraplegia; and 3, hind legs paraplegia and incontinence. According to the animal welfare protocol of the Canton Bern, disease severity more intense than score 3 was considered as termination criteria.
Treatment with testing compounds started individually at the disease onset of each mouse. To evaluate the effects of WOBE437, a dose of 10 mg/kg was i.p. administered once a day over 20 days; this dose was chosen considering our previous reports.27,28 To evaluate the involvement of CB1 and CB2 receptors, the mice were i.p. administered either 3 mg/kg rimonabant or 3 mg/kg AM630, which are inverse agonists or antagonists of CB1 and CB2 receptors, respectively; administration of rimonabant or AM630 was done 30 min before the administration of WOBE437. DMSO was used as vehicle for all the testing compounds in a final volume of 20 μL. Mice were assigned to each group using a simple random allocation strategy, in which each cage (n = 10–12 mice per cage) was randomly assigned to a treatment from the beginning.
To evaluate the potential of WOBE437 as a disease-modifying agent, two cohorts of mice were immunized and allowed to develop a maximum of clinical score 2 (hind legs paraplegia), marking the starting point of the daily administration of WOBE437 (10 mg/kg, i.p.) or vehicle. Considering the typical time-course of the clinical scores, the peak of severity occurs between 3 and 4 days after onset; therefore, day 4 was considered as a cutoff to start the treatment if a clinical score 2 was not seen before this day. Once the treatment started, daily i.p. injections were given until 20 days after onset. Mice were randomly assigned to each group on the day when each animal fulfilled one of the two possible selection criteria mention above; randomization was done using a combination of systemic allocation (day showing selection criteria to start the treatment) and stratified random allocation (cage sorting). Mice housed in the same cage were assigned to either of the groups in order to have a similar number of each treatment in each cage.
Quantification of eCBs and Prostaglandins by LC-MS/MS
Sample Preparation
eCBs were evaluated in central and peripheral tissues at the end of treatments (20 days after onset) or during the stage of highest severity of symptoms (4–6 days post-treatment, day 1 = onset). Mice were euthanized by decapitation, right after which brain, lumbar SC, and spleen were collected, rinsed in cold PBS, and snap frozen in liquid N2. Before freezing the brain, it was dissected to separate cerebellum and both hemispheres. Blood was also collected to obtain plasma. All samples were stored at −80 °C until extraction.
For the extraction of the interested molecules, our previously described protocol was used.27 Briefly, tissue samples were weighted while still frozen and transferred into 2 mL XXTuff reinforced microvials (Bio Spec Products Inc., Bartlesville, OK) with three chrome-steel beads (2.3 mm; Bio Spec Products Inc.Bartlesville, OK), and the corresponding volume of 0.1 M formic acid to reach 100 mg/mL. Samples were homogenized using a Mini-Beadbeater-24 (Bio Spec Products Inc., Bartlesville, OK). Right after homogenization, 0.25 mL of the tissue homogenate was extracted with 0.75 mL of a solution containing ethyl acetate plus hexane (9:1) and 0.1% formic acid. The internal standard mix solution (5 μL) was added to each sample. Samples were strongly vortexed for 30 s, sonicated in a cold bath for 5 min, and centrifuged at 1620g for 10 min at 4 °C. Samples were kept for 1 h at −20 °C to freeze the lower aqueous phase, and the upper organic phase was collected into 1.5 mL polypropylene tubes (Sarstedt, Germany). Samples were dried in a speed vacuum and reconstituted with 50 μL of acetonitrile plus 20% water. For the quantification in the LC-MS/MS system, 10 μL of the final sample were injected in the column.
LC-MS/MS Conditions
LC-MS/MS analysis was conducted using our previously described protocol,27 except for using a Shimadzu UFLC coupled to a TripleQuad 5500 QTRAP mass spectrometer (AB Sciex, Canada). AEA and 2-AG were analyzed using the Turbo-Ion Spray interface operated in positive mode. The LC column was a Reprosil-PUR C18 column (3 μm particle size; 2 × 50 mm; Dr. A. Maisch HPLC GmbH, Germany) maintained at 40 °C with a mobile phase flow rate of 0.35 mL/min. The mobile phase composition was a mixture of (A) 2 mM ammonium acetate plus 0.1% formic acid and (B) methanol plus 2 mM ammonium acetate. The MS parameters of the ESI source were as follows: curtain gas, 30 psig; Ion Spray voltage, 4.5 kV; temperature, 600 °C; ion source gas, 50 psig.
A gradient elution was used, starting with 85% phase A and 15% phase B until minute 0.5; phase B was then linearly increased to reach 70% at 3.5 min, followed by another slow linear increase reaching 99% B at 8 min. This 99% B was kept constant until 12 min. Finally, phase B was linearly decreased to 15% at 14 min and kept for 1 min for re-equilibration. The total analysis time was 15.0 min. The following MRM transitions were monitored: AEA, m/z 348.2 → 62.1 (AEA-d4, 352.2 → 66.0) and 2-AG, m/z 379.1 → 287.2 (2-AG-d5, 384.1 → 287.2). For quantification, an 11-point calibration curve was analyzed, determining the slope, intercept, and regression coefficient, and the concentration of each analyte in the samples was calculated applying the model previously described.27
Prostaglandin-E2 and -D2 were analyzed using the Turbo-Ion Spray interface operated in negative mode. The LC column was also a Reprosil-PUR C18 column (3 μm particle size; 2 × 50 mm) maintained at 40 °C with a mobile phase flow rate of 0.30 mL/min. The mobile phase composition was a mixture of (A) 2 mM ammonium acetate plus 0.1% formic acid and (B2) acetonitrile plus 0.1% formic acid. The MS parameters of the ESI source were as follows: curtain gas, 30 psig; Ion Spray voltage, 4.5 kV; temperature, 600 °C; ion source gas, 50 psig. A gradient elution was used starting with 95% phase A and linear increase of phase B2 reaching 40% at 3 min, then the linear increase rate was decreased to reach 65% B2 at 9 min. Another linear increase of phase B2 was made to reach 95% at 10 min, and this concentration was maintained for 4 min. Finally, phase B2 was linearly decreased to 5% at 15 min and held at this concentration for 2 min for re-equilibration. The total analysis time was 17.0 min. The following MRM transitions were monitored: PGE2, m/z 351.1 → 189.0 (PGE2-d4 355.0 → 319.0), PGD2 351.1 → 189.0 (PGE2-d4 355.0 → 319.0).
[35S]GTPγS Assay in Mouse Brain Membrane Preparation
As previously reported,27 brain and cerebellum membrane preparations were obtained from brains collected 20 days after the daily injection of vehicle or 10 mg/kg WOBE437 in active EAE mice. Tissues were homogenized in a glass dounce homogenizer with 1.5 mL of ice-cold lysis buffer (50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4). Homogenates were centrifuged twice at 800g for 10 min at 4 °C to remove debris. The resulting supernatants were transferred into 2 mL plastic tubes and centrifuged at 16 000g for 20 min at 4 °C. The harvested membranes were pooled and resuspended in 700 μL of 50 mM Tris-HCl pH 7.4. Membrane preparations were passed through a clean needle (27G), sonicated for 5 min in cold water, and stored at −80 °C. Total protein concentration was estimated with a BCA assay kit, which was conducted according to the manufacturer’s instructions.
[35S]GTPγS assay was conducted using 20 μg of brain membranes or 5 μg of cerebellum membranes, resuspended in 0.5% BSA binding buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4). The brain membrane preparation was preincubated at 37 °C for 30 min with 0.5 units/mL adenosine deaminase, 30 μM GDP, and 0.2 nM [35S]GTPγS, in a final volume of 200 μL. Afterward, preparations were kept on ice until binding was initiated by addition of CP-55940 (0.1 μM or 1 μM), rimonabant (1 μM), a combination of both ligands, or vehicle (DMSO). Nonspecific binding was determined with the addition of GTPγS (10 μM). The samples were incubated for 1 h at 30 °C and placed on ice right after incubation, until an aliquot (185 μL) of each was transferred into a GF/B glass fiber filter plate (presoaked with ice-cold 50 mM Tris-HCl plus 0.1% BSA). The filter plate was washed three times with 200 μL of 50 mM Tris-HCl plus 0.1% BSA and allowed to dry. Radioactivity associated with the filter was measured in a Microbeta Trilux counter, 15 min after the addition of 45 μL of liquid scintillation cocktail in each well. Data were normalized by subtracting the nonspecific [35S]GTPγS bound to the membranes and were expressed as percentage of the control (DMSO-treated membranes).
CB2 Receptor Binding in Spleen Tissues
Spleen membrane preparations were obtained from tissues collected 20 days after the daily injection of vehicle or 10 mg/kg WOBE437 in active EAE mice. Spleens were homogenized in a glass dounce homogenizer with 1.5 mL of ice-cold lysis buffer (50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, cocktail protease inhibitor [cOmplete ULTRA, Roche] pH 7.4). Homogenates were centrifuged at 800g for 10 min at 4 °C to remove debris, and the resulting pellets were resuspended in 1 mL of ice-cold lysis buffer and centrifuged again at 800g for 10 min at 4 °C. This step was repeated twice. The resulting supernatants were transferred into 2 mL plastic tubes and centrifuge at 16 000g for 20 min at 4 °C. The harvested membranes were pooled and resuspended in 500 μL of 50 mM Tris-HCl pH 7.4. Membrane preparations were passed through a clean needle (27G), sonicated for 5 min in cold water and stored at −80 °C. Total protein concentration was estimated with a BCA assay kit, which was conducted according to the manufacturer’s instructions.
For the binding assay, 50 μg of the membrane preparation was resuspended in 300 μL of assay buffer (50 mM Tris-HCl 2.5 mM EDTA 5 mM MgCl2 pH 7.5 with 0.5% BSA) and were transferred into silanized glass vials. Membranes were coincubated with vehicle (DMSO), 1 μM AM630, or 10 μM WIN-55212-2 (to determine nonspecific binding [NSB]) and 2 nM of [3H]CP-55940 (PerkinElmer, USA) for 90 min at 30 °C. After incubation, the samples were transferred into GF/B filter plate (previously soaked with 0.5% polyethylenimine for 1 h) and filtered through a vacuum pump. Each well was then washed 12 times with 100 μL of assay buffer and allowed to dry. Subsequently, 50 μL of MicroScint-20 were added to each well and incubated for 30 min at room temperature. Radioactivity was measured using TopCount scintillation counter (Packard, USA). NSB was substrated from the samples, and the counts per minute (CPM) were transform to disintegrations per minute (DPM) by considering the 85% efficiency of the instrument. The amount of [3H]CP-55940 bound to the membranes was then calculated considering its specific activity of 164.9 Ci/mmol. AM630 was used to confirm CB2 receptor activation.
Isolation of CNS-Infiltrating Immune Cells and Sorting by Flow Cytometry
During the stage of highest severity of EAE symptoms (clinical score 2 or 4–6 days after onset), inflammatory cells were isolated from the CNS as previously described.81 Briefly, anesthetized mice were perfused with 15–20 mL of cold PBS, and brain and spinal cord tissues were carefully isolated and transferred into washing buffer (DMEM supplemented with 5% calf serum and 25 mM HEPES). Importantly, two brains or two spinal cords were pooled per sample. The tissues were homogenized between two glass slides and digested with collagenase VIII (0.25 mg/mL; Sigma-Aldrich) at 37 °C for 30 min in the presence of DNase I (1 U/mL; Sigma-Aldrich). Subsequently, samples were filtered through a prewet 100 μm nylon mesh and spin-down for 10 min at 4 °C and 330g. For isolation of inflammatory cells, cell pellets were resuspended in Percoll (GE Healthcare Life Sciences) gradient (50 and 30% isotonic Percoll) and centrifuged for 30 min at 4 °C and 1300g. Inflammatory cells are collected in washing buffer and washed twice (centrifugation for 10 min at 4 °C, and 330g). On a Petri dish (60 × 15 mm2), isolated cells were stimulated with 50 ng/mL PMA (Alexix Biochemicals), 1 μg/mL ionomycin (BioVision, Inc.), and 3.3 μL/10 mL GolgiStop (BD Biosciences) for 5 h at 37 °C in 5 mL of restimulation medium (RPMI1640 [Gibco] supplemented with 10% FBS, 2% l-glutamine [Gibco], 1% NEAA [Gibco], 1% sodium pyruvate [Gibco], 1% penicillin–streptavidin [Gibco], and 0.4% 14.3 mM β-mercaptoethanol [Merck]). After stimulation, cells were spun down and resuspended in 1 mL of FACS buffer (DPBS, 2.5% FBS, and 0.1% NaN3) and stained for intracellular cytokines (IFN-γ, IL-17, IL-10, and GM-CSF) and surface markers (CD45, CD4, and CD8). After a washing step, cells were fixed and permeabilized for 20 min on ice (BD Biosciences; Cytofix/CytopermTM). Cells were washed and incubated with primary antibody mixes (Table S-1) in Perm/WashTM solution (BD Biosciences) for 30 min on ice. Cells were washed twice after the staining and resuspended in FACS buffer. Flow cytometry analysis was conducted using an Attune NxT flow cytometer (Thermofisher scientific), which uses a positive displacement syringe pump to control sample volume and allows measurement of volumetric cell counts in a known volume (gated and total events).82 Data analysis was conducted with FlowJo software.
Straub Test
Morphine induces a characteristic and reproducible elevation of the tail in mice, commonly called “Straub tail response”. This response is used as an animal model of muscle spasticity and to evaluate the induction of muscle relaxation by new drugs.76,77 For the test, vehicle (DMSO, 20 μL) or 10 mg/kg WOBE437 were administered i.p. in male C57BL/6J mice (8–9 weeks old) 60 min before subcutaneous injection of 40 mg/kg morphine (10 mg/mL). Baclofen (5 mg/kg in 100 μL of PBS) and diazepam (6 mg/kg in 20 μL of DMSO) were used as positive control and given i.p. 40 min before morphine injection. For comparison, WIN-55212-2 (3 mg/kg in 20 μL of DMSO) was used as a control for CB1 receptor agonism. The intensity of the tail elevation was evaluated 15, 60, and 120 min after morphine injection, using a previously reported score scale:35 0, lower than 30°; 1, 30–45°; 2, 46–90°; 3, higher than 90°; and 4, “S” shaped tail. Notably, there have not been reports of differences in this test when comparing male and female mice.83
Open-Field Test
Spontaneous locomotion was evaluated in an open-field box (40 × 40 × 40 cm, Noldus). The mice were individually placed in south right corner of the field and allowed to move freely for 5 min. The activity was recorded, and distance traveled was analyzed with EthoVision XT version 13 (Noldus, Netherlands). After every experiment, the box was cleaned with 70% ethanol to remove odors.
Bar Test
Catalepsy-like behavior was measured using the bar test, where the mouse was retained in an imposed position with forelimbs resting on a bar of 4 cm height. Latency in this position was measured until both front limbs were removed or after a cutoff time of 120 s.
Statistical Analysis
For clarity, data is presented in different ways, according to the type of analysis and parameters evaluated. Considering data distribution or sample size, statistical analysis was evaluated by either parametric or nonparametric approach. In brief, the time course data shown in Figures 1A,D, 2A,B, and 6A,D are presented as mean ± SEM and were analyzed using multiple t-tests corrected for multiple comparison with the Holm–Sidak method. Data shown in Figures 1B, 6B, and 7B,C are presented as box plot highlighting median, 25th percentile, 75th percentile, minimum, and maximum; data in Figures 1B and 6B were analyzed using the Mann–Whitney test. Data in Figure 7B,C were analyzed using the Kruskal–Wallis test followed by the Mann–Whitney test as post hoc analysis. Data in Figures 1C, 2C, 6C, and 7A are shown as cumulative frequency in percentage and were analyzed with the chi-square and Fisher’s exact tests. Data in Figures 3A–F and 5A–D are shown as mean ± SD and were analyzed using the Mann–Whitney test. Data in Figure 4 shows mean values ± SD and was analyzed using two-way ANOVA and Tukey’s multiple comparison test. Data representation style and statistical analysis information are given in all the figure legends. A confidence level of p < 0.05 was considered statistically significant. Analyses were carried out using the GraphPad Prism software version v8.0 (La Jolla, CA); except for the chi-square and Fisher’s exact tests, which were carried out using IBM SPSS software v22.0 (Hamlet, NY).
Acknowledgments
The authors thank Dr. Roch-Philippe Charles for the support on animal experimentation and veterinary license.
Glossary
Abbreviations
- 2-AG
2-arachidonoyl glycerol
- AEA
anandamide
- AUC
area under the curve
- CB1
cannabinoid type 1 receptor
- CB2
cannabinoid type 2 receptor
- CBD
cannabidiol
- CFA
complete Freund’s adjuvant
- DMSO
dimethyl sulfoxide
- EAE
experimental autoimmune encephalomyelitis
- ECS
endocannabinoid system
- eCB
endocannabinoid
- FAAH
fatty acid amide hydrolase
- i.p.
intraperitoneally
- MAGL
monoacylglycerol lipase
- MS
multiple sclerosis
- MOG
myelin oligodendrocyte glycoprotein
- SERI
selective endocannabinoid reuptake inhibitor
- THC
(−)-trans-Δ9-tetrahydrocannabinol
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00214.
Flow cytometry antibodies, distribution of the day of onset showed in all the mice included in the study, evaluation of body weight changes in groups treated in combination with CB1 and CB2 receptor antagonists, evaluation of IL-10- or GM-CSF-producing CD4+ and CD8+ T cells isolated from brain and spinal cord of EAE female C57BL/6 mice treated with vehicle or WOBE437, quantification of prostaglandin levels in central and peripheral tissues, endocannabinoid levels in spleen samples collected from EAE female C57BL/6 mice treated with either vehicle or WOBE437, and evaluation of CB2 receptor abundance in spleen membrane preparations from EAE female C57BL/6 mice treated either with vehicle or WOBE437 (PDF)
Author Contributions
A.C. conceived the idea. A.C., B.E., S.T., J.G., and I.R. discussed and designed the experiments. A.C. and I.R. led the writing of the manuscript. I.R. and S.T. conducted the animal experiments. S.T. conducted the isolation of CNS infiltrating immune cells and flow cytometry measurements. I.R. and E.V. conducted LC-MS/MS measurements and CB1 receptor functional assays. I.R. and A.C. led the statistical analyses. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
A.C., B.E., and J.G. thank the Swiss Multiple Sclerosis Society for a grant. J.G. acknowledges the NCCR TransCure for continuous funding.
The authors declare the following competing financial interest(s): A.C. and J.G. are co-founders of Synendos Therapeutics, a spin-off company dedicated to developing selective endocannabinoid reuptake inhibitors for human medicine. A.C. has recently joined the Synendos Therapeutics team.
Supplementary Material
References
- The Multiple Sclerosis International Federation . (2020) Mapping multiple sclerosis around the world key epidemiology findings, in Atlas of MS, 3rd ed., The Multiple Sclerosis International Federation. [Google Scholar]
- Brownlee W. J.; Hardy T. A.; Fazekas F.; Miller D. H. (2017) Diagnosis of multiple sclerosis: progress and challenges. Lancet 389, 1336–1346. 10.1016/S0140-6736(16)30959-X. [DOI] [PubMed] [Google Scholar]
- Nafee T., Watanabe R., and Fregni F. (2018) Multiple Sclerosis, in Neuromethods, pp 263–295, Elsevier Ltd. [Google Scholar]
- Høglund R. A. (2014) Multiple sclerosis and the role of immune cells. World J. Exp. Med. 4, 27. 10.5493/wjem.v4.i3.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser S. L.; Cree B. A. C. (2020) Treatment of Multiple Sclerosis: A Review. Am. J. Med. 133, 1380. 10.1016/j.amjmed.2020.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin F. D.; Reingold S. C.; Cohen J. A.; Cutter G. R.; Sorensen P. S.; Thompson A. J.; Wolinsky J. S.; Balcer L. J.; Banwell B.; Barkhof F.; Bebo B.; Calabresi P. A.; Clanet M.; Comi G.; Fox R. J.; Freedman M. S.; Goodman A. D.; Inglese M.; Kappos L.; Kieseier B. C.; Lincoln J. A.; Lubetzki C.; Miller A. E.; Montalban X.; O’Connor P. W.; Petkau J.; Pozzilli C.; Rudick R. A.; Sormani M. P.; Stuve O.; Waubant E.; Polman C. H. (2014) Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 83, 278–286. 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero-Romero S.; Sastre-Garriga J.; Comi G.; Hartung H.-P.; Soelberg Sørensen P.; Thompson A. J.; Vermersch P.; Gold R.; Montalban X. (2016) Pharmacological management of spasticity in multiple sclerosis: Systematic review and consensus paper. Mult. Scler. J. 22, 1386–1396. 10.1177/1352458516643600. [DOI] [PubMed] [Google Scholar]
- Martyn C.; Illis L.; Thom J. (1995) Nabilone in the treatment of multiple sclerosis. Lancet 345, 579. 10.1016/S0140-6736(95)90485-9. [DOI] [PubMed] [Google Scholar]
- Wissel J.; Haydn T.; Müller J.; Brenneis C.; Berger T.; Poewe W.; Schelosky L. D. (2006) Low dose treatment with the synthetic cannabinoid Nabilone significantly reduces spasticity-related pain. J. Neurol. 253, 1337–1341. 10.1007/s00415-006-0218-8. [DOI] [PubMed] [Google Scholar]
- Riva N.; Mora G.; Sorarù G.; Lunetta C.; Ferraro O. E.; Falzone Y.; Leocani L.; Fazio R.; Comola M.; Comi G.; Formaglio F.; Rossi P.; Clerici M.; Falzone Y. M.; Pozzi L.; Martinelli D.; Cerri F.; Lopez I. D.; Martinelli-Boneschi F.; Quattrini A.; Pieri E.; Marinou K.; Querin G.; Sansone V.; Maestri E.; Calvo A.; Chio A. (2019) Safety and efficacy of nabiximols on spasticity symptoms in patients with motor neuron disease (CANALS): a multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 18, 155–164. 10.1016/S1474-4422(18)30406-X. [DOI] [PubMed] [Google Scholar]
- Rice J.; Hugos C.; Hildebrand A.; Cameron M. (2020) Cannabis use in people with multiple sclerosis and spasticity: A cross-sectional analysis. Mult. Scler. Relat. Disord. 41, 102009. 10.1016/j.msard.2020.102009. [DOI] [PubMed] [Google Scholar]
- Chiurchiù V.; van der Stelt M.; Centonze D.; Maccarrone M. (2018) The endocannabinoid system and its therapeutic exploitation in multiple sclerosis: Clues for other neuroinflammatory diseases. Prog. Neurobiol. 160, 82–100. 10.1016/j.pneurobio.2017.10.007. [DOI] [PubMed] [Google Scholar]
- Kubajewska I.; Constantinescu C. S. (2010) Cannabinoids and experimental models of multiple sclerosis. Immunobiology 215, 647–657. 10.1016/j.imbio.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Palazuelos J.; Davoust N.; Julien B.; Hatterer E.; Aguado T.; Mechoulam R.; Benito C.; Romero J.; Silva A.; Guzmán M.; Nataf S.; Galve-Roperh I. (2008) The CB 2 Cannabinoid Receptor Controls Myeloid Progenitor Trafficking. J. Biol. Chem. 283, 13320–13329. 10.1074/jbc.M707960200. [DOI] [PubMed] [Google Scholar]
- Kong W.; Li H.; Tuma R. F.; Ganea D. (2014) Selective CB2 receptor activation ameliorates EAE by reducing Th17 differentiation and immune cell accumulation in the CNS. Cell. Immunol. 287, 1–17. 10.1016/j.cellimm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce G.; Baker D. (2007) Control of Spasticity in a Multiple Sclerosis Model is mediated by CB 1, not CB 2, Cannabinoid Receptors. Br. J. Pharmacol. 150, 519–525. 10.1038/sj.bjp.0707003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musella A.; Sepman H.; Mandolesi G.; Gentile A.; Fresegna D.; Haji N.; Conrad A.; Lutz B.; Maccarrone M.; Centonze D. (2014) Pre- and postsynaptic type-1 cannabinoid receptors control the alterations of glutamate transmission in experimental autoimmune encephalomyelitis. Neuropharmacology 79, 567–572. 10.1016/j.neuropharm.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Rossi S.; Furlan R.; Chiara V. De; Muzio L.; Musella A.; Motta C.; Studer V.; Cavasinni F.; Bernardi G.; Martino G.; Cravatt B. F.; Lutz B.; Maccarrone M.; Centonze D. (2011) Cannabinoid CB1 receptors regulate neuronal TNF-α effects in experimental autoimmune encephalomyelitis. Brain, Behav., Immun. 25, 1242–1248. 10.1016/j.bbi.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Jean-Gilles L.; Feng S.; Tench C. R.; Chapman V.; Kendall D. A.; Barrett D. A.; Constantinescu C. S. (2009) Plasma endocannabinoid levels in multiple sclerosis. J. Neurol. Sci. 287, 212–215. 10.1016/j.jns.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Di Filippo M.; Pini L. A.; Pelliccioli G. P.; Calabresi P.; Sarchielli P. (2008) Abnormalities in the cerebrospinal fluid levels of endocannabinoids in multiple sclerosis. J. Neurol., Neurosurg. Psychiatry 79, 1224–1229. 10.1136/jnnp.2007.139071. [DOI] [PubMed] [Google Scholar]
- Centonze D.; Bari M.; Rossi S.; Prosperetti C.; Furlan R.; Fezza F.; De Chiara V.; Battistini L.; Bernardi G.; Bernardini S.; Martino G.; Maccarrone M. (2007) The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 130, 2543–2553. 10.1093/brain/awm160. [DOI] [PubMed] [Google Scholar]
- Sánchez López A. J.; Román-Vega L.; Ramil Tojeiro E.; Giuffrida A.; García-Merino A. (2015) Regulation of cannabinoid receptor gene expression and endocannabinoid levels in lymphocyte subsets by interferon-β: a longitudinal study in multiple sclerosis patients. Clin. Exp. Immunol. 179, 119–127. 10.1111/cei.12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Gutiérrez S.; Molina-Holgado E.; Arévalo-Martín Á.; Correa F.; Viso A.; López-Rodríguez M. L.; Di Marzo V.; Guaza C. (2005) Activation of the endocannabinoid system as a therapeutic approach in a murine model of multiple sclerosis. FASEB J. 19, 1338–1340. 10.1096/fj.04-2464fje. [DOI] [PubMed] [Google Scholar]
- Hernandez-Torres G.; Cipriano M.; Heden E.; Bjorklund E.; Canales A.; Zian D.; Feliu A.; Mecha M.; Guaza C.; Fowler C. J.; Ortega-Gutierrez S.; Lopez-Rodriguez M. L. (2014) A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew. Chem., Int. Ed. 53, 13765–13770. 10.1002/anie.201407807. [DOI] [PubMed] [Google Scholar]
- Baker D.; Pryce G.; Croxford J. L.; Brown P.; Pertwee R. G.; Makriyannis A.; Khanolkar A.; Layward L.; Fezza F.; Bisogno T.; Di Marzo V. (2001) Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 15, 300–302. 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov.
- Chicca A.; Nicolussi S.; Bartholomäus R.; Blunder M.; Aparisi Rey A.; Petrucci V.; Reynoso-Moreno I. D. C.; Viveros-Paredes J. M.; Dalghi Gens M.; Lutz B.; et al. (2017) Chemical probes to potently and selectively inhibit endocannabinoid cellular reuptake. Proc. Natl. Acad. Sci. U. S. A. 114, E5006–E5015. 10.1073/pnas.1704065114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso-Moreno I.; Chicca A.; Flores-Soto M. E.; Viveros-Paredes J. M.; Gertsch J. (2018) The Endocannabinoid Reuptake Inhibitor WOBE437 Is Orally Bioavailable and Exerts Indirect Polypharmacological Effects via Different Endocannabinoid Receptors. Front. Mol. Neurosci. 11, 180. 10.3389/fnmol.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg J. E.; Blankman J. L.; Long J. Z.; Nomura D. K.; Pan B.; Kinsey S. G.; Nguyen P. T.; Ramesh D.; Booker L.; Burston J. J.; Thomas E. A.; Selley D. E.; Sim-Selley L. J.; Liu Q. S.; Lichtman A. H.; Cravatt B. F. (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 13, 1113–1119. 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu C. S.; Farooqi N.; O’Brien K.; Gran B. (2011) Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 164, 1079–1106. 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford A. L.; Goodsall A. L.; Hickey W. F.; Sedgwick J. D. (1995) Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 154, 4309–4321. [PubMed] [Google Scholar]
- Remington L. T.; Babcock A. A.; Zehntner S. P.; Owens T. (2007) Microglial Recruitment, Activation, and Proliferation in Response to Primary Demyelination. Am. J. Pathol. 170, 1713–1724. 10.2353/ajpath.2007.060783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbey D. L. J.; Salem H.; Grossman M. H. (1960) The Anatomical Basis Of The Straub Phenomenon. Br. J. Pharmacol. Chemother. 15, 540–543. 10.1111/j.1476-5381.1960.tb00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M. L.; Nath R.; Gupta T. K.; Gupta G. P. (1988) A study of central neurotransmitter mechanisms in morphine-induced “Straub reaction” in mice: Role of central dopamine receptors. Clin. Exp. Pharmacol. Physiol. 15, 727–732. 10.1111/j.1440-1681.1988.tb01012.x. [DOI] [PubMed] [Google Scholar]
- Kameyama T.; Ukai M.; Nabeshima T. (1978) Effects of catecholaminergic or tryptaminergic agents on the morphine-induced Straub tail reaction. Jpn. J. Pharmacol. 28, 249–257. 10.1254/jjp.28.249. [DOI] [PubMed] [Google Scholar]
- Robinson A. P., Harp C. T., Noronha A., and Miller S. D. (2014) The experimental autoimmune encephalomyelitis (EAE) model of MS. utility for understanding disease pathophysiology and treatment, in Handbook of Clinical Neurology, pp 173–189, Elsevier B.V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia M. K.; Paintlia A. S.; Singh A. K.; Singh I. (2011) Synergistic activity of interleukin-17 and tumor necrosis factor-α enhances oxidative stress-mediated oligodendrocyte apoptosis. J. Neurochem. 116, 508–521. 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami A.; Ciric B. (2013) Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 333, 76–87. 10.1016/j.jns.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy Á. C.; Lalor S. J.; Lynch M. A.; Mills K. H. G. (2010) Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain, Behav., Immun. 24, 641–651. 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Prajeeth C. K.; Kronisch J.; Khorooshi R.; Knier B.; Toft-Hansen H.; Gudi V.; Floess S.; Huehn J.; Owens T.; Korn T.; Stangel M. (2017) Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflammation 14, 204. 10.1186/s12974-017-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K.; Duarte J. H.; Veldhoen M.; Hornsby E.; Li Y.; Cua D. J.; Ahlfors H.; Wilhelm C.; Tolaini M.; Menzel U.; Garefalaki A.; Potocnik A. J.; Stockinger B. (2011) Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263. 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato F.; Cosmi L.; Santarlasci V.; Maggi L.; Liotta F.; Mazzinghi B.; Parente E.; Filì L.; Ferri S.; Frosali F.; Giudici F.; Romagnani P.; Parronchi P.; Tonelli F.; Maggi E.; Romagnani S. (2007) Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861. 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland H. F.; Martin R. (2007) Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 8, 913–919. 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- Luchtman D. W.; Ellwardt E.; Larochelle C.; Zipp F. (2014) IL-17 and related cytokines involved in the pathology and immunotherapy of multiple sclerosis: Current and future developments. Cytokine Growth Factor Rev. 25, 403–413. 10.1016/j.cytogfr.2014.07.013. [DOI] [PubMed] [Google Scholar]
- El-Behi M.; Ciric B.; Dai H.; Yan Y.; Cullimore M.; Safavi F.; Zhang G.-X.; Dittel B. N.; Rostami A. (2011) The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 12, 568–575. 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran M.; Marino M. E.; Busch R.; Keim C.; King C.; Lee J.; Killion S.; Awada M.; Hellerstein M. K. (2007) Measurement of brain microglial proliferation rates in vivo in response to neuroinflammatory stimuli: Application to drug discovery. J. Neurosci. Res. 85, 2374–2384. 10.1002/jnr.21389. [DOI] [PubMed] [Google Scholar]
- Pryce G.; Ahmed Z.; Hankey D. J. R.; Jackson S. J.; Croxford J. L.; Pocock J. M.; Ledent C.; Petzold A.; Thompson A. J.; Giovannoni G.; Cuzner M. L.; Baker D. (2003) Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 126, 2191–2202. 10.1093/brain/awg224. [DOI] [PubMed] [Google Scholar]
- Lou Z. Y.; Zhao C. B.; Xiao B. G. (2012) Immunoregulation of experimental autoimmune encephalomyelitis by the selective CB1 receptor antagonist. J. Neurosci. Res. 90, 84–95. 10.1002/jnr.22721. [DOI] [PubMed] [Google Scholar]
- Lou Z.-Y. Y.; Yu W.-B. B.; Chen J.; Li L.; Jiang L.-S. S.; Xiao B.-G. G.; Liu Z.-G. G. (2016) Neuroprotective Effect Is Driven Through the Upregulation of CB1 Receptor in Experimental Autoimmune Encephalomyelitis. J. Mol. Neurosci. 58, 193–200. 10.1007/s12031-015-0656-9. [DOI] [PubMed] [Google Scholar]
- Maresz K.; Pryce G.; Ponomarev E. D.; Marsicano G.; Croxford J. L.; Shriver L. P.; Ledent C.; Cheng X.; Carrier E. J.; Mann M. K.; Giovannoni G.; Pertwee R. G.; Yamamura T.; Buckley N. E.; Hillard C. J.; Lutz B.; Baker D.; Dittel B. N. (2007) Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med. 13, 492–497. 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- Wen J.; Ribeiro R.; Tanaka M.; Zhang Y. (2015) Activation of CB2 receptor is required for the therapeutic effect of ABHD6 inhibition in experimental autoimmune encephalomyelitis. Neuropharmacology 99, 196–209. 10.1016/j.neuropharm.2015.07.010. [DOI] [PubMed] [Google Scholar]
- de Lago E.; Moreno-Martet M.; Cabranes A.; Ramos J. A.; Fernández-Ruiz J. (2012) Cannabinoids ameliorate disease progression in a model of multiple sclerosis in mice, acting preferentially through CB1 receptor-mediated anti-inflammatory effects. Neuropharmacology 62, 2299–2308. 10.1016/j.neuropharm.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Bolognini D.; Cascio M. G.; Parolaro D.; Pertwee R. G. (2012) AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br. J. Pharmacol. 165, 2561–2574. 10.1111/j.1476-5381.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salort G.; Álvaro-Bartolomé M.; García-Sevilla J. A. (2017) Regulation of cannabinoid CB2 receptor constitutive activity in vivo: repeated treatments with inverse agonists reverse the acute activation of JNK and associated apoptotic signaling in mouse brain. Psychopharmacology (Berl). 234, 925–941. 10.1007/s00213-017-4537-5. [DOI] [PubMed] [Google Scholar]
- Giacoppo S.; Bramanti P.; Mazzon E. (2017) Sativex in the management of multiple sclerosis-related spasticity: An overview of the last decade of clinical evaluation. Mult. Scler. Relat. Disord. 17, 22–31. 10.1016/j.msard.2017.06.015. [DOI] [PubMed] [Google Scholar]
- Novotna A.; Mares J.; Ratcliffe S.; Novakova I.; Vachova M.; Zapletalova O.; Gasperini C.; Pozzilli C.; Cefaro L.; Comi G.; Rossi P.; Ambler Z.; Stelmasiak Z.; Erdmann A.; Montalban X.; Klimek A.; Davies P. (2011) A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex ®), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur. J. Neurol. 18, 1122–1131. 10.1111/j.1468-1331.2010.03328.x. [DOI] [PubMed] [Google Scholar]
- Zajicek J.; Ball S.; Wright D.; Vickery J.; Nunn A.; Miller D.; Cano M. G.; McManus D.; Mallik S.; Hobart J. (2013) Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 12, 857–865. 10.1016/S1474-4422(13)70159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce G., and Baker D. (2015) Endocannabinoids in multiple sclerosis and amyotrophic lateral sclerosis, in Endocannabinoids, pp 213–231, Springer International Publishing. [DOI] [PubMed] [Google Scholar]
- Hasseldam H.; Fryd Johansen F. (2011) Cannabinoid Treatment Renders Neurons Less Vulnerable Than Oligodendrocytes in Experimental Autoimmune Encephalomyelitis. Int. J. Neurosci. 121, 510–520. 10.3109/00207454.2011.582237. [DOI] [PubMed] [Google Scholar]
- Pryce G.; Cabranes A.; Fernández-Ruiz J.; Bisogno T.; Di Marzo V.; Long J.; Cravatt B.; Giovannoni G.; Baker D. (2013) Control of experimental spasticity by targeting the degradation of endocannabinoids using selective fatty acid amide hydrolase inhibitors. Mult. Scler. J. 19, 1896–1904. 10.1177/1352458513485982. [DOI] [PubMed] [Google Scholar]
- Arena C.; Gado F.; Di Cesare Mannelli L.; Cervetto C.; Carpi S.; Reynoso-Moreno I.; Polini B.; Vallini E.; Chicca S.; Lucarini E.; Bertini S.; D’Andrea F.; Digiacomo M.; Poli G.; Tuccinardi T.; Macchia M.; Gertsch J.; Marcoli M.; Nieri P.; Ghelardini C.; Chicca A.; Manera C. (2020) The endocannabinoid system dual-target ligand N-cycloheptyl-1,2-dihydro-5-bromo-1-(4-fluorobenzyl)-6-methyl-2-oxo-pyridine-3-carboxamide improves disease severity in a mouse model of multiple sclerosis. Eur. J. Med. Chem. 208, 112858. 10.1016/j.ejmech.2020.112858. [DOI] [PubMed] [Google Scholar]
- Bernal-Chico A.; Canedo M.; Manterola A.; Victoria Sánchez-Gómez M.; Pérez-Samartín A.; Rodríguez-Puertas R.; Matute C.; Mato S. (2015) Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia 63, 163–176. 10.1002/glia.22742. [DOI] [PubMed] [Google Scholar]
- Centonze D.; Muzio L.; Rossi S.; Cavasinni F.; De Chiara V.; Bergami A.; Musella A.; D’Amelio M.; Cavallucci V.; Martorana A.; Bergamaschi A.; Cencioni M. T.; Diamantini A.; Butti E.; Comi G.; Bernardi G.; Cecconi F.; Battistini L.; Furlan R.; Martino G. (2009) Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 29, 3442–3452. 10.1523/JNEUROSCI.5804-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B. M.; Ghosh S.; Crowe M. S.; Kinsey S. G.; Niphakis M. J.; Abdullah R. A.; Tao Q.; O’ Neal S. T.; Walentiny D. M.; Wiley J. L.; Cravatt B. F.; Lichtman A. H. (2014) In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 171, 1392–1407. 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg J. E.; Kinsey S. G.; Ignatowska-Jankowska B.; Ramesh D.; Abdullah R. A.; Tao Q.; Booker L.; Long J. Z.; Selley D. E.; Cravatt B. F.; Lichtman A. H. (2014) Prolonged Monoacylglycerol Lipase Blockade Causes Equivalent Cannabinoid Receptor Type 1 Receptor–Mediated Adaptations in Fatty Acid Amide Hydrolase Wild-Type and Knockout Mice. J. Pharmacol. Exp. Ther. 350, 196–204. 10.1124/jpet.114.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi R. K. P. (2020) A perspective review on fatty acid amide hydrolase (FAAH) inhibitors as potential therapeutic agents. Eur. J. Med. Chem. 188, 111953. 10.1016/j.ejmech.2019.111953. [DOI] [PubMed] [Google Scholar]
- Toczek M.; Malinowska B. (2018) Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 204, 20–45. 10.1016/j.lfs.2018.04.054. [DOI] [PubMed] [Google Scholar]
- H. Lundbeck A/S . (2020) Lundbeck announces phase IIa study results of Lu AG06466 in adults with Tourette Syndrome, https://investor.lundbeck.com/news-releases/news-release-details/lundbeck-announces-phase-iia-study-results-lu-ag06466-adults.
- de Lago E.; Ligresti A.; Ortar G.; Morera E.; Cabranes A.; Pryce G.; Bifulco M.; Baker D.; Fernandez-Ruiz J.; Di Marzo V. (2004) In vivo pharmacological actions of two novel inhibitors of anandamide cellular uptake. Eur. J. Pharmacol. 484, 249–257. 10.1016/j.ejphar.2003.11.027. [DOI] [PubMed] [Google Scholar]
- de Lago E.; Fernández-Ruiz J.; Ortega-Gutiérrez S.; Cabranes A.; Pryce G.; Baker D.; López-Rodríguez M.; Ramos J. A. (2006) UCM707, an inhibitor of the anandamide uptake, behaves as a symptom control agent in models of Huntington’s disease and multiple sclerosis, but fails to delay/arrest the progression of different motor-related disorders. Eur. Neuropsychopharmacol. 16, 7–18. 10.1016/j.euroneuro.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Loría F.; Petrosino S.; Hernangómez M.; Mestre L.; Spagnolo A.; Correa F.; Di Marzo V.; Docagne F.; Guaza C. (2010) An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol. Dis. 37, 166–176. 10.1016/j.nbd.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Nicolussi S.; Gertsch J. (2015) Endocannabinoid transport revisited. Vitam. Horm. 98, 441–485. 10.1016/bs.vh.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. (2018) New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discovery 17, 623–639. 10.1038/nrd.2018.115. [DOI] [PubMed] [Google Scholar]
- Cristino L.; Bisogno T.; Di Marzo V. (2020) Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 16, 9–29. 10.1038/s41582-019-0284-z. [DOI] [PubMed] [Google Scholar]
- MacKenzie-Graham A.; Tiwari-Woodruff S. K.; Sharma G.; Aguilar C.; Vo K. T.; Strickland L. V.; Morales L.; Fubara B.; Martin M.; Jacobs R. E.; Johnson G. A.; Toga A. W.; Voskuhl R. R. (2009) Purkinje cell loss in experimental autoimmune encephalomyelitis. NeuroImage 48, 637–651. 10.1016/j.neuroimage.2009.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack G. D.; Zwolshen J. M. (1983) Predictive value of muscle relaxant models in rats and cats. J. Pharmacol. Methods 10, 175–183. 10.1016/0160-5402(83)90028-1. [DOI] [PubMed] [Google Scholar]
- Belozertseva I. V.; Dravolina O. A.; Tur M. A.; Semina M. G.; Zvartau E. E.; Bespalov A. Y. (2016) Morphine-induced Straub tail reaction in mice treated with serotonergic compounds. Eur. J. Pharmacol. 791, 1–7. 10.1016/j.ejphar.2016.08.014. [DOI] [PubMed] [Google Scholar]
- Soethoudt M.; Grether U.; Fingerle J.; Grim T. W.; Fezza F.; De Petrocellis L.; Ullmer C.; Rothenhäusler B.; Perret C.; Van Gils N.; Finlay D.; Macdonald C.; Chicca A.; Gens M. D.; Stuart J.; De Vries H.; Mastrangelo N.; Xia L.; Alachouzos G.; Baggelaar M. P.; Martella A.; Mock E. D.; Deng H.; Heitman L. H.; Connor M.; Di Marzo V.; Gertsch J.; Lichtman A. H.; Maccarrone M.; Pacher P.; Glass M.; Van Der Stelt M. (2017) Cannabinoid CB2receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 8, 13958. 10.1038/ncomms13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz S. M.; Engelhardt B. (2019) Visualizing Impairment of the Endothelial and Glial Barriers of the Neurovascular Unit during Experimental Autoimmune Encephalomyelitis In Vivo. J. Visualized Exp. 10.3791/59249. [DOI] [PubMed] [Google Scholar]
- Tietz S. M.; Zwahlen M.; Haghayegh Jahromi N.; Baden P.; Lazarevic I.; Enzmann G.; Engelhardt B. (2016) Refined clinical scoring in comparative EAE studies does not enhance the chance to observe statistically significant differences. Eur. J. Immunol. 46, 2481–2483. 10.1002/eji.201546272. [DOI] [PubMed] [Google Scholar]
- Döring A.; Wild M.; Vestweber D.; Deutsch U.; Engelhardt B. (2007) E- and P-Selectin Are Not Required for the Development of Experimental Autoimmune Encephalomyelitis in C57BL/6 and SJL Mice. J. Immunol. 179, 8470–8479. 10.4049/jimmunol.179.12.8470. [DOI] [PubMed] [Google Scholar]
- Attune NxT Flow Cytometer Features, https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometers/attune-nxt-flow-cytometer/features.html. [Google Scholar]
- Daniela B.; Jiang L.; Goto S.; Gantois I.; Schutz G.; Lawrence A. J.; Waddington J. L.; Drago J. (2013) Behavioural and anatomical characterization of mutant mice with targeted deletion of D1 dopamine receptor-expressing cells: Response to acute morphine. J. Pharmacol. Sci. 121, 39–47. 10.1254/jphs.12214FP. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.