

Abstract

Inositol hexakisphosphate kinases (IP6Ks) catalyze pyrophosphorylation of inositol hexakisphosphate (IP6) into inositol 5-diphospho-1,2,3,4,6-pentakisphosphate (IP7), which is involved in numerous areas of cell physiology including glucose homeostasis, blood coagulation, and neurological development. Inhibition of IP6Ks may be effective for the treatment of Type II diabetes, obesity, metabolic complications, thrombosis, and psychiatric disorders. We performed a high-throughput screen (HTS) of 158 410 compounds for IP6K1 inhibitors using a previously developed ADP-Glo Max assay. Of these, 1206 compounds were found to inhibit IP6K1 kinase activity by more than 25%, representing a 0.8% hit rate. Structural clustering analysis of HTS-active compounds, which were confirmed in the dose–response testing using the same kinase assay, revealed diverse clusters that were feasible for future structure–activity relationship (SAR) optimization to potent IP6K inhibitors. Medicinal chemistry SAR efforts in three chemical series identified potent IP6K1 inhibitors which were further validated in an orthogonal LC-MS IP7 analysis. The effects of IP6K1 inhibitors on cellular IP7 levels were further confirmed and were found to correlate with cellular IP6K1 binding measured by a high-throughput cellular thermal shift assay (CETSA).
Keywords: inositol pyrophosphate, IP7, inositol hexakisphosphate kinases, high-throughput screen, cellular thermal shift assay (CETSA)
Inositol pyrophosphates, such as 5-diphosphoinositol pentakisphosphate (5PP-IP5, also abbreviated IP7), are involved in a diverse set of biological processes such as insulin secretion, vesicular trafficking, cell signaling, metabolism, telomere length regulation, apoptosis, and cell migration.1 Mammals express three isoforms of IP6K that convert inositol hexakisphosphate (IP6) to IP7. IP6K1 and IP6K2 are ubiquitously expressed, whereas IP6K3 is primarily expressed in the cerebellum and skeletal muscle.2 The physiological function and metabolism of IP7 have been primarily established using IP6K knockouts in mouse models and cell lines. IP6K1 knockout mice display an increased insulin sensitivity.3 These mice are resistant to a high-fat diet (HFD)-induced impaired glucose tolerance, insulin resistance, or hyperglycemia.3 In human clinical studies, muscle IP6K1 protein content is elevated after lean meat ingestion in obese adults but not in lean individuals.4 Therefore, dysregulation of IP6K1 in obese adults may contribute to the development of insulin resistance. IP6K1 knockout mice also display a decrease in polyphosphate production in platelets and are resistant to thromboembolism,5 suggesting that an IP6K1 inhibitor may provide thromboprotection. Furthermore, IP6K1 knockout mice display some social deficits and resistance to amphetamine-induced hyperlocomotion.6 Therefore, pharmacologic inhibition of IP6K may be of therapeutic benefit in the treatment of type II diabetes, obesity, metabolic complications, venous thrombosis, and psychiatric disorders.
IP6Ks can also function through catalytic-activity-independent mechanisms. IP6K1 regulates lipolysis through protein–protein interaction with PLIN1,7 whereas IP6K2 regulates nuclear factor kappa B (NF-κB) signaling through interaction with TRAF2.8 Therefore, the phenotype associated with a “knockout” of IP6K may reflect a loss of IP7 synthesis and/or loss of its scaffolding activities. A pharmacologic inhibitor of IP6K could be insightful in differentiating the role between catalytic activity and scaffolding functions of IP6K.
To date, only one IP6K inhibitor, N2-(m-(trifluoromethyl)benzyl)N6-(p-nitrobenzyl)purine (TNP), is routinely used as a chemical probe for IP6K function.9 However, TNP suffers significant limitations due to poor potency, low solubility, a growing list of off-target effects, no isoform selectivity, and an inability to penetrate the blood–brain barrier.10 Through a medicinal chemistry approach, we were able to improve some properties of TNP, including solubility and isoform selectivity.11 However, we were not able to significantly improve the potency against IP6K. We previously developed a high-through assay for IP6K1 inhibitor using an ADP-Glo Max assay and demonstrated the robustness of this assay by screening an annotated set of 1280 compounds, the Library of Pharmacologically Active Compounds (LOPAC).12 To identify chemically tractable inhibitors for IP6K1, we screened a collection of 158 410 compounds from the National Center for Advancing Translational Sciences (NCATS) and identified several structural clusters from which it should be feasible to develop potent IP6K-specific inhibitors. Medicinal chemistry efforts identified several potent IP6K1 inhibitors that were confirmed by an orthogonal LC-MS assay. The effect of these compounds on IP6K1 in cells were further confirmed by a high-throughput cellular thermal shift assay (CETSA) assay as well as changes in cellular IP7 levels.
Results and Discussion
High-Throughput Screening for IP6K1 Inhibitors
We had previously developed an ADP-Glo Max assay for IP6K1 in a 384-well format.12 To adapt it for screening the NCATS compound library, we miniaturized the assay into a 1536-well plate format by optimizing the assay into a 4 μL assay reaction volume, followed by the addition of 2 μL of ADP-Glo reagent and 4 μL of ADP-Glo substrate. We kept the final ATP concentration at 1 mM, which is similar to the Km for IP6K1, in order to maximize the chance for identifying non-ATP competitive inhibitors.12 One major difference from our previous assay in a 384-well format is that the 1536-well assay was performed at room temperature instead of 37 °C. Under these ambient conditions, the enzyme activity is much lower. Therefore, a much higher concentration (600 nM) of enzyme and a longer reaction time (2 h) were used. We first tested a pilot quantitative high-throughput screen (qHTS) on the LOPAC library at 7 different doses. The results are consistent with our prior screening using the LOPAC library.12 Notably, the top hits identified in this screening such as suramin sodium salt, aurintricarboxylic acid, myricetin, and Reactive Blue 2 were also the top hits identified from a previous screening using the LOPAC at 10 μM. The average signal-to-background ratio (S/B) for the screen is 13, and the average Z′ is 0.83, indicating that the qHTS format is robust for high-throughput screening.
We then screened four libraries from the compound collection at NCATS, including two repurposing and mechanism-annotated collections, NIH Pharmaceutical Collection (NPC), NCATS Pharmacologically Active Chemical Toolbox (NPACT), and two diversity collections, the Sytravon and Genesis libraries, providing a total of 158 410 compounds tested at 58 and 19 μM. In total, we identified 1206 compounds with more than 25% inhibition at 58 μM, representing a hit rate of 0.76%. Of these, 645 compounds have more than 30% inhibition (Table 1). To further confirm the hits identified in the screen, we selected the 1206 compounds as well as compounds identified in the LOPAC screening and retested them as seven 3-fold dilutions starting at 58 μM. Of the compounds retested, 580 were confirmed to be active in this dose–response qHTS assay, representing a 43% confirmation rate. Importantly, almost all of these compounds were inactive in previous NCATS screens against PI4K2A and PI5P4Ka which also used the ADP-Glo format,13,14 suggesting that they are selective against other lipid kinases and not assay artifacts.
Table 1. Number of Hits Identified from Four Libraries of Compound Collection at NCATS.
| max response (%) | 100–80 | 80–50 | 50–30 | 30–25 | total (158 410) |
|---|---|---|---|---|---|
| NPC + NPACT | 2 | 11 | 65 | 31 | 1206 (>25% inhibition) |
| Sytravon | 11 | 20 | 226 | 329 | 645 (>30% inhibition) |
| Genesis | 7 | 68 | 235 | 201 |
Hit Confirmation by Dose–Response Assays
Among the set of confirmed hits, 306 compounds have a maximum inhibition of more than 50% at 58 μM. After eliminating compounds with PAINS substructures and performing a cluster analysis, we obtained 123 compounds from commercial (solid samples) or NCATS stock (in DMSO) and tested them in our 384-well format using a 10-point dose–response assay. The IC50’s of most of the compounds in the qHTS format are lower than those obtained by 384-well 10-point dose–response assay (Supplemental Table S1). There are two major differences between the qHTS and 384-well format assay: One is that the enzyme concentration used in the qHTS is much higher. The other is that qHTS was carried out at room temperature for 2 h, whereas the dose–response assay in the 384-well format was carried out at 37 °C for 30 min. Increasing the enzyme concentration 10-fold in the dose–response assays usually leads to an increase in IC50 for promiscuous inhibitors.15 Therefore, such a difference in enzyme concentration is unlikely to be the cause for the change in the IC50’s observed. The IP6K1 activity is much lower at room temperature than at 37 °C. It is possible that the enzyme may be in a less stable or less active stage, making it easier to be inhibited at room temperature. Of the 123 compounds selected for confirmation, 90 compounds are active in both assay formats. Structural analysis of these hits revealed several clusters (Figure 1), which were attractive for further development.
Figure 1.

IP6K1 inhibitory activity of selected HTS hits.
Hit Confirmation with an Orthogonal Assay
To further validate our hits for IP6K1 inhibition, we performed an orthogonal assay in which the effect of compounds on the production of IP7 was monitored by LC-MS. Separation of IP6 and IP7 by conventional reverse-phase chromatography with mass spectrometry friendly volatile buffers is challenging. By using octylamine as a lipophilic ion-pairing agent, EDTA to minimize nonspecific binding to metal tubing, and a PEEK-lined column, separation could be achieved (Figure 2A) as outlined in a recent patent application.16 Mass-labeled standards of 13C6–IP717 also enabled precise quantitation. We had previously identified myricetin as an IP6K1 inhibitor.12 Myricetin (M6760) inhibited IP7 production in a dose-dependent manner. Similarly, both LI-1753 and LI-1851 inhibited IP7 production in a dose-dependent manner (Figure 2B), confirming that these compounds are indeed be able to inhibit IP6K. The weaker hits LI-1776 and LI-1842 did not show a signal in this assay.
Figure 2.
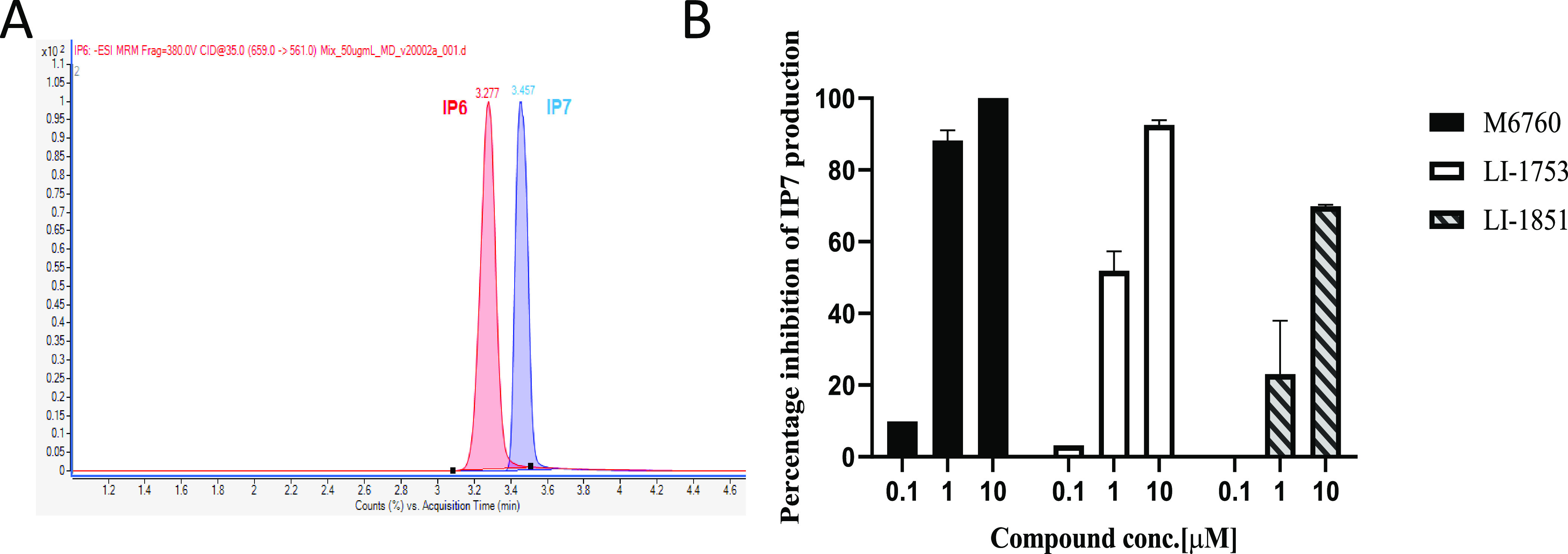
Orthogonal assay for IP6K1 inhibitors. (A) MRM chromatogram of 50 μg/mL IP6 and IP7. IP6 (red) and IP7 (blue) were separated by using a Triart PEEK-lined C18 column. LC/MS analysis was performed using a triple quad mass spectrometer (Agilent 6495B with 1290 UHPLC). (B) Percentage inhibition of IP7 measured by LC-MS. In vitro kinase assay was performed in the presence of DMSO or various concentration of different compounds. The concentration of IP7 was measured using LC-MS, and the percentage inhibition of IP7 was calculated.
Medicinal Chemistry Efforts Identified Potent IP6K1 Inhibitors
LI-1753 was an interesting initial hit as thiadiazolidindiones (TDZD) have been proposed to have a covalent mechanism of action toward other proteins.18 Compounds LI-1753 and LI-2355 have been studied as inhibitors of GSK3β, with LI-2355 (tideglusib) making it into clinical trials for Alzheimer’s disease.19,20 LI-2356 has been studied as an inhibitor of RGS proteins.21 To probe the SAR and potential covalent mechanism of action of the TDZD series, several analogs around this core were tested (Figure 3). Minimalized compound LI-2386 revealed that only the core TDZD structure is needed to achieve potency. We hypothesized that a protein nucleophile could attack the electrophilic sulfur on the TDZD and break the sulfur–nitrogen bond. When the sulfur was replaced with either a methylene (LI-2399) or an olefin (LI-2394), no activity was seen. When the nitrogen was replaced with an sp2 carbon (LI-2395), activity was also abolished. Finally, when the sulfur–nitrogen bond was replaced with a similarly electrophilic maleimide (LI-2406), the potency was restored. This SAR data implies a covalent mechanism for TDZD compounds with the sulfur–nitrogen bond acting as an electrophile. The TDZD series was not explored any further due to inactivity in cell-based assays and concerns of selectivity. Further exploration of LI-1851 (Figure 1) demonstrated the chemical instability of the core heterocycle, making assessment of the true active moiety challenging.
Figure 3.

IC50’s of TDZD compounds on IP6K1 inhibition.
We also pursued the spiro-oxindole piperidine lead, LI-1776 (Figure 4). Although its potency was modest, we were struck by the similarity of the scaffold to that of another spiro-oxindole, LI-2124, which is a known IP6K inhibitor.16 Addition of the important carboxylic acid functional group provided LI-2172, which displayed a dramatic improvement in IP6K1 potency, giving an IC50 of 20 nM for LI-2172. The initial hit LI-1842 also possesses a similar core structure, and inclusion of a carboxylic acid improved potency here as well, but more modestly, giving an IC50 of 1100 nM for LI-2240. Preliminary SAR studies around LI-2172 indicated that the hydrogens on both the carboxylic acid and the oxindole nitrogen are critical for IP6K1 inhibitory activity (Figure 5) as shown by a marked loss of potency for either methylated analogue. IP6K1 inhibitory activity is retained with a variety of substituents on the piperidine ring nitrogen of the tricyclic parent (Figure 6) which represents a vector for the modulation of physical properties. One example of this is the benzyl analogue LI-2178, which was 40-fold less potent than LI-2172, suggesting that benzoyl substitution is preferred over benzyl, at least for this particular matched pair of compounds. However, additional substitution of the aromatic ring similar to the pendant ring of LI-2124 is able to increase potency in the benzyl-substituted series as well as shown by LI-2242, with an IC50 of 31 nM.
Figure 4.

Comparison of IP6K1 inhibition of selected HTS hits to literature IP6K antagonist LI-2124 and results of the incorporation of a carboxyl acid functional group.
Figure 5.

SAR of methylation of the oxindole nitrogen and the carboxylic acid of LI-2172.
Figure 6.

SAR of amide and amine matched pairs of substituted (hetero)aryl analogues
To determine whether the lead compounds display any selelctivity against IP6K2, IP6K3 or inositol polyphosphate multikase (IPMK), we developed assays against these enzymes and performed dose-dependent inhibition of these kinases by six lead compounds (Table 2). All six compounds inhibit both IP6K2 and IP6K3 without displaying significant selectivity against IP6K isoforms. Both assays against IP6K1 and IPMK were performed using the concentration of their substrate and ATP at their respective Km; therefore, the results indicate that these compounds display 6- to 231-fold selectivity against IP6K1 compared to that against IPMK. The selectivities of LI-2172 and LI-2242 against other kinases were further profiled using KinaseProfiler service from Eurofins Discovery. None of the 58 kinases tested were inhibited by more than 30% by LI-2172 or LI-2242 at 10 μM. Particularly, these two compounds at 10 μM do not inhibit PI3K α, β, or γ isoforms (Supplemental Table 2), further supporting the selectivity of these compounds against other lipid kinases.
Table 2. Inhibition of Lead Compounds against IP6K1, IP6K2, IP6K3, and IPMK.
| LI-2124 | LI-2242 | LI-2260 | LI-2172 | LI-2180 | LI-2178 | |
|---|---|---|---|---|---|---|
| IP6K1 IC50 (nM) | 2.5 | 31 | 33 | 27 | 57 | 800 |
| IP6K2 IC50 (nM) | 3 | 42 | 31.7 | 25 | 58.5 | 1170 |
| IP6K3 IC50 (nM) | 1 | 8.7 | 9 | 8 | 135 | N/A |
| IPMK IC50 (nM) | 205 | 1944 | 1124 | 156 | 13163 | 5565 |
| IPMK/IP6K1 IC50 ratio | 82 | 63 | 34 | 6 | 231 | 7 |
IP6K1 Inhibitors Decrease IP7 Levels in HCT116 Cells
To determine whether the compounds identified above are able to inhibit IP7 production in the cell, we treated HCT116 cells with compounds, extracted inositol phosphates using TiO2, separated inositol phosphates by acrylamide gel, and monitored the change of cellular IP7.12 While we observed a dose-dependent decrease in IP7 levels, IP6 levels did not change significantly, suggesting that the decrease in IP7 level is not due to a cell toxicity effect (Figure 7). Consistent with this, we did not observe any cell toxicity when cells were treated with these three compounds at up to 100 μM for 30 min using CellTiter-Glo assay (data not shown). Unexpectedly, LI-2172 is less effective than LI-2178 in decreasing cellular IP7 even though LI-2172 is much more potent than LI-2178 in the cell-free enzymatic assay (Figure 7). Such a result suggests that other factors such as cell membrane penetration or nonspecific protein binding may also play important roles in affecting compound potency in the cells. The relative IP7 signal detected by Toluidine Blue staining decreases over time during destaining, and it is difficult to quantify the effect of compounds on cellular IP7 level using this method. In addition, the throughput of this assay is low because it is very time-consuming and requires a large number of cells (a confluence p150 plate for each condition). Therefore, we sought to develop alternative cell-based assays with a higher throughput to support a drug discovery project.
Figure 7.

Effect of IP6K1 inhibitors on cellular IP7 level in HCT116 cells. HCT116 cells were treated with various concentration of IP6K1 inhibitors. The inositol phosphates were extracted, separated by 30% acrylamide gel, and detected by Toluidine Blue staining. The first 3 lanes and the rest are on the same gels but were not adjacent to each other.
High-Throughput Cellular Thermal Shift Assay (CETSA) Revealed Different Target Engagement for Different Compounds
To confirm whether LI-2172 and LI-2178 are different in binding cellular IP6K1, we used CETSA to study the target engagement of these compounds in a cellular context. In the CETSA, unbound proteins denature and precipitate at elevated temperatures, whereas inhibitor-bound proteins remain in solution.22,23 IP6K1 shows temperature-dependent instability with a majority of the protein precipitating at 50 °C. A testing concentration of 100 μM LI-2124 completely blocks IP6K1 precipitation at 50 °C, but it loses its ability to stabilize IP6K1 at 55 °C (Figure 8A). Therefore, we compared the effect of various compounds in the CETSA assay at 50 °C. Neither LI-1776 nor LI-2172 increases the stability of IP6K1 at 50 °C as compared to DMSO (Figure 8B). In contrast, LI-2178 increases the thermal stability of IP6K1 although it is less potent than LI-2172 in the ADP-Glo IP6K1 inhibition assay. Therefore, LI-2172 is indeed less effective in binding to IP6K1 than is LI-2178 in a cellular environment, consistent with the results that LI-2172 is less effective than LI-2178 in decreasing IP7 levels in HCT116 cells (Figure 7).
Figure 8.

CETSA for IP6K1 inhibitors. (A) Amount of soluble IP6K1 detected by Western blot after cells treated with or without 100 μM LI-2124 followed by incubation at indicated temperature for 3 min. (B) Amount of soluble IP6K1 detected by Western blot after cells treated with DMSO or various compounds at 100 μM followed by incubation at 50 °C for 3 min.
Because the throughput of the above CETSA assay method is very low, we sought to develop a high-throughput CETSA (HT-CETSA). IP6K1 was fused with 11-amino acid HiBit-tag of nanoluc luciferase24 and expressed in HEK-293T cells, which allows for very sensitive detection of HiBit-IP6K1 expression. A linear signal from 3–12 500 cells can be detected with an R2 value of 0.9989 (Figure 9A). For indoleamine 2,3-dioxygenase (IDO) or the histone-lysine N-methyltransferase SMYD3 that was fused to the DiscoverX 42-amino acid ePL-tag, it had been demonstrated that the ePL-domain of the fusion proteins could not complement with the enzyme acceptor (EA) through enzyme fragment complementation (EFC) to form a functional beta-galactosidase if the fusion proteins were precipitated at elevated temperature.25 Similarly, we expected that thermally denatured HiBit-tagged IP6K1 may not complement LgBit nLuc to form active luciferase. Indeed, after treatment of cells at 51 °C for 3 min, the total luciferase signal detected by addition of lysis buffer containing LgBit and luciferase substrates was decreased by more than 90% (Figure 9B), suggesting that the HiBit-tagged IP6K1 in cells heated to 51 °C is not able to complement the LgBit fragment to form an active nLuc. In the presence of 10 μM LI-2124, 40% of the total luciferase signal was restored, consistent with the thermal stabilization of IP6K1 by the binding of IP6K1 inhibitor. Unlike the Western-blot-based CETSA, which requires separation of the precipitated IP6K1 from the soluble IP6K1, the HiBit-IP6K1-based HT-CETSA can be performed in a homogeneous-plate-based format without protein separation, thereby allowing us to analyze the effect of IP6K1 inhibitors with much higher throughput. Consistent with our Western-blot-based CETSA assay, we did not observe any effect of LI-2172 in this high-throughput CETSA (data not shown). Unlike the results from CETSA by Western blot showing that 100 μM LI-2124 could completely block the precipitation of IP6K1, the maximum recovery of total luciferase signal is only ∼50% with 100 μM LI-2124 (Figure 9B). To determine whether this effect is due to an inability of LI-2124 to block the precipitation of HiBit-IP6K1, we performed the Western-blot based CETSA assay for cells overexpressing HiBit-IP6K1. Consistent with what we observed for endogenous IP6K1 in Figure 7A, 100 μM LI-2124 also completely blocked the precipitation of HiBit-IP6K1 induced by heat treatment (Figure 9C). Therefore, heat treatment can lead not only to protein precipitation but also to a disordered HiBit domain. Such a disordered HiBit domain may be less effective in complementing with the LgBit fragment of nLuc, resulting a decrease in the total luciferase formation. Alternatively, such a disordered HiBit domain may be still able to complement with the LgBit fragment effectively and form the same amount of total luciferase, but these luciferases containing the disordered HiBit are less active. Either of these scenarios can lead to a lower luminescence signal. IP6K1 inhibitors may prevent the heat-induced precipitation of IP6K1 but not the heat-induced disorder of the HiBit fragment, resulting in a partial rescue of the total luminescence signal. It remains to be determined whether introducing a small peptide linker between the HiBit tag and IP6K1 can improve the recovery of HiBit-tagged IP6K1 detection upon heat treatment. Nevertheless, this homogeneous CETSA assay of HiBit-tagged IP6K1 allows us to measure the EC50 of compounds in restoring HiBit-IP6K1-dependent luciferase signal upon heat treatment (Figure 9D), providing a readout for the potency of compounds in cellular target engagement.
Figure 9.

Homogenous high-throughput CETSA for IP6K1 inhibitors using HiBit-tagged IP6K1. (A) Linear relationship between luminescence signal detected in varous amount of cells expressing HiBit-tagged IP6K1. (B) Effect of heat treatment with or without 100 μM LI-2124 on the total luminescence signal detected in 12 500 cells. Cells were treated with or without LI-2124 followed by incubation at 37 or 51 °C. Total luminesence signals from HiBit-IP6K1 in the cells were detected by addition of lysis buffer containing detection reagents. (C) Effect of LI-2124 on HiBit-tagged IP6K1 in CETSA assay. HEK293T cells were transfected with plasmid expressing HiBit-IP6K1 (+) or empty vector (−). Cells were treated with LI-2124 for 30 min followed by incubation at 37 or 51 °C. Soluble fraction of the cell lysates were probed with anti-IP6K1 antibody by Western blot. The HiBit-tag in IP6K1 migrates almost identical to the endogenous IP6K1 in this SDS-PAGE. (D) Dose–response assay for IP6K1 inhibitors using HT-CETSA.
In summary, we report the identification of multiple structurally diverse IP6K1 inhibitors through high-throughput screening. We further confirmed these hits by dose–response testing and an orthogonal LC-MS assay. Additional medicinal chemistry efforts took weak spirooxindole and benzimidazolinone scaffolds and produced very potent IP6K1 inhibitors. Further studies on the effect of IP6K1 inhibitors in inhibiting cellular IP7 level revealed that the potency is highly correlated with cellular target engagement measured by CETSA. We further developed a homogeneous HT-CETSA assay for a higher throughput demonstration of target engagement. This work opens an opportunity to further optimize IP6K1 inhibitors that could ultimately be advanced into the clinic for the treatment of type II diabetes, venous thrombosis, or psychiatric disorders.
Methods
Protein Expression and Purification
Full-length human IP6K1, IP6K2, and IP6K3 were purified as described previously.12 Several batches of IP6K1 protein were prepared and combined for the HTS. The purified proteins were concentrated to 1 mg/mL and stored at −80 °C.
Compound Screening
Compound collections from the National Center for Advancing Translational Science (NCATS), including NCATS Pharmaceutical Collection (NPC, 2816 compounds), the NCATS-NPAC Pharmacologically Active Chemical Toolbox (NPACT, 5632 compounds), the Sytravon library (45 000 compounds), and the Genesis library (105 000 compounds), were screened in 1536-well assay plates. A mixture of 1.3 mM ATP and 130 μM IP6 was prepared in assay buffer (25 mM Tris-HCl pH 6.9, 10 mM MgCl2, 2.5 mM DTT, and 0.02% Triton X-100), and 3 μL of the mixture were dispensed to the assay plates. A 23 nL aliquot of compounds of the top two doses (58 and 19 μM final concentration) of the libraries were dispensed into the mixture, followed by addition of 1 μL of 2.4 μM IP6K1. After incubation of the plates in room temperature for 2 h, 2 μL of ADP-Glo reagent was added to the wells. After 1 h of incubation at room temperature, 4 μL of ADP-Glo substrate was added, and the plates were incubated at room temperature for 45 min. The luminescence signals were then detected using ViewLux microplate imager (PerkinElmer).
IC50 Determination
The ADP-Glo Max assay was described previously.11,12 For the dose–response assays performed in the 1536-well plate format in NCATS, the assay condition is the same as that described in the HTS campaign except that seven serial 3-fold compound dilutions were used. For the dose–response assay performed in 384-well plate format in the Lieber Institute, 10 serial 3-fold compound dilutions were prepared in DMSO, and then each compound was diluted with assay buffer (50 mM Tris, 10 mM MgCl2, 2.5 mM DTT, and 0.02% Triton X-100, pH 6.9) to reach the desired inhibitor concentrations in 25% DMSO. Then, 1 μL of each dilution was added to a Corning low-volume 384-well white flat-bottomed polystyrene NBS microplate. A “kinase master mix” that contained IP6K1, IP6, and ATP was prepared on ice and mixed gently, and 4 μL of this was added to each compound resulting in a total reaction volume of 5 μL with 5% DMSO. The final concentrations for IP6K1, IP6, and ATP were 60 nM, 100 μM, and 1 mM, respectively. The reaction plate was quickly mixed on a plate shaker and centrifuged for 10 s at 1500 × g. The plate was then mixed and centrifuged a second time. The mixed reaction plate was placed into a 37 °C incubator and rotated on an orbital shaker at 300 rpm for 30 min. The reaction was terminated with 5 μL of Promega ADP-Glo reagent for 1 h followed by 10 μL of Promega Kinase Detection reagent for 1 h at room temperature in the dark. Luminescence generated was read on a Tecan Infinite M100 Pro plate reader. Samples were run in quadruplicate, and the data was analyzed using a four-point variable slope inhibition parameter on GraphPad Prism 9. IP6K2 and IP6K3 were tested as described above for IP6K1 with some variations. The IP6K2 assays used 7.5 nM enzyme, and the IP6K3 assays used 120 nM enzyme with a 2 h reaction time.
IC50 Determination for IPMK
Human recombinant IPMK was purchased from Origene (catalog no. TP309343). d-Myo-Iinositol-1,4,5,6-tetraphosphate (IP4) was purchased from Caymen Chem (catalog no. 10007783). The Km’s for IP4 and ATP were determined to be 5.6 and 10 μM, respectively. The optimal concentration for IPMK was determined to be 20 nM with the reaction time for 4 h at 37 °C. The ADP-Glo Max assays for dose-dependent compound inhibition were performed using the same format as that described in above for IP6K1 in 384-well format.
Orthogonal Assay for IP6K1 Activity by LC-MS Analysis
A 100 μL kinase assay containing 1 mM ATP, 100 μM IP6, and 60 nM IP6K1 in the presence of different concentrations of compounds was carried out at 37 °C for 30 min. The samples were analyzed following the protocol described in the patent of Takeda Phamaceutical with minor modification16 using an Agilent 6495 Triple Quadrupole with Jet Stream Electrospray Ionization Source (ESI) and Agilent 1290 Infinity II UHPLC. Solvents were 0.01% octylamine, 0.1% ammonia, and 10 μM ethylenediaminetetraacetic acid (EDTA) in water (A) and 0.02% octylamine and 0.01% ammonia in methanol (B). Chromatographic separation was achieved over 7.0 min using a YMC Triart PEEK-lined C18, 2.1 × 50 mm2 column with 1.9 μm particles. The flow rate was 600 μL min–1. Samples were kept at 4 °C during analysis. The LC gradient was as follows: 0–4.0 min, 20–95% B; 1.0–1.5 min; 50–70% B, 1.5–3.0 min, 70–95% B; 3.0–3.5 min, 95% B. Reaction samples were extracted with 25 μM 13C6–IP717,26 in 0.1% formic acid in acetonitrile. The extracts were centrifuged at 2650 rcf for 10 min at 4 °C. The supernatants were diluted 1:1 with mobile phase A, and 1 μL was injected. The mass spec acquisition was performed using MRM of IP6 (659–561 m/z), IP7 (739–641 m/z), and IP7-C13 (745–647 m/z) in negative ion mode with the following source conditions: drying gas at 200 °C and 18 L/min, sheath gas at 350 °C and 12 L/min; nebulizer −25 psig; capillary and nozzle voltages at 3000 and 1500 V, respectively. The high- and low-pressure RF were at 150 and 60 V, respectively. Analyte peak areas were determined from the extracted MRM chromatograms. Concentrations were calculated from the peak area ratios of IP7 and IP7-C13 by linear regression analysis using Agilent Masshunter Quantitative Analysis for QQQ Software (version 10.0).
Inhibition of IP7 in HCT116 Cells
HCT116 cells were grown in a 150 mL flask to confluence and treated with DMSO or compounds for 30 min. Cells were washed with ice cold PBS, and inositol polyphosphates were extracted using 1 M perchloric acid (Sigma). Titanium dioxide beads (Titansphere TiO 5 μm; GL Sciences) were used to pull down inositol phosphates and other phosphate-rich molecules from the extracts. These extracts were resolved using 33.3% PAGE gels as previously described12 and visualized by Toluidine Blue (Sigma) staining.
Determination of Apparent Melting Curve of Intracellular IP6K1 by CETSA
The apparent melting curve for intracellular IP6K1 was established according to the literature.27 In two individual T75 flasks, 293T cells were treated with LI-2124 at a final concentration of 100 μM or 1.2% DMSO as a negative control. The flasks were then incubated for 1 h in the CO2 incubator at 37 °C. The cells were collected from each flask and resuspended in 15 mL of PBS with protease inhibitor cocktail (Sigma-Aldrich, P8340) in individual conical tubes. Each cell suspension was then distributed into six different PCR tubes with 100 μL cell suspension in each tube, heated at their designated temperatures (40, 45, 50, 55, and 60 °C, respectively), followed by three freeze–thaw cycles in a cooling bath using dry ice and ethanol. The cell lysates were then centrifuged at 20,000 × g for 20 min at 4 °C. The resulting supernatants containing the soluble IP6K1 protein were separated by SDS-PAGE. Images of the Western blot using IP6K1 antibody (Sigma-Aldrich catalog no. HPA040825) were acquired with the LI-COR Odyssey CLx imaging system, and the amount of IP6K1 was quantified by Empiria Studio Software.
HiBit-IP6K1 Plasmid Construct
Oligos encoding HiBit peptide 5′-gatccatgGTGAGCGGCTGGCGGCTGTTCAAGAAGATT AGCgg-3′ and 5′-aattccGCTAATCTTCTTGAACAGCCGCCAGCCGCTCACcatg-3′ were annealed and cloned into the BamHI and EcoRI sites of the GEX-IP6KI plasmid to form the GEX-HiBit-IP6K1 plasmid. The sequence encoding HiBit-IP6K1 fusion protein between the BamHI and NotI sites in the GEX-HiBit-IP6K1 plasmid was then subcloned into the pcDNA(+)3.1 plasmid to form the pcDNA3.1-HiBit-IP6K1 plasmid. Both clones were confirmed by DNA sequencing.
Determination of Effect of Compounds on Intracellular IP6K1 by CETSA
293T cells were collected from a T75 flask and transferred to 12-well plates at about 2 × 106 cells per well. The cells were treated with or without compounds at 100 μM, followed by 30 min of incubation at 37 °C in a CO2 incubator. Cell suspensions in individual wells were transferred into individual microcentrifuge tubes, washed with 1 mL of PBS, resuspended in 100 μL of PBS with protease inhibitor cocktail, and transferred into individual PCR tubes. Cells were heated at 50 °C for 3 min in a PCR machine. Cells were then lysed, and soluble IP6K1 was detected as described in the method for determination of apparent of melting curve of intracellular IP6K1 by CETSA.
HT-CETSA
HEK293T cells (6 ×106 cells) were prepared in 12 mL of DMEM with 10% FBS, plated onto 75 cm flask, and incubated overnight at 37 °C with 5% CO2. Cells were then transfected with 15 μg of pcDNA3.1-HiBit-IP6K1 plasmid by Lipofectamine 3000 (ThermoFish catalog no. L3000015). Cells were harvested at 24 h post-transfection (2.5 × 106 cells/mL) and aliquoted into a 96-well plate at 100 μL/well. DMSO or 3-fold serially diluted compounds were added to cells, and the plates were incubated at 37 °C for 30 min with shaking at 300 rpm. Then, 30 μL of each cell mixture was then transferred into a 96-well PCR plate and heated at 51 °C for 3 min in a PCR machine. Next, 30 μL of Hibit lytic reagent (Promega catalog no. N3040) was added to each well. After thoroughly mixing, the cell–lysis solution mixtures were transferred to a 384-well white assay plate (Corning ref no. 3824BC) at 15 μL per well in triplicate, and the luminescence signals were detected via TECAN infinite M1000Pro.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00218.
Procedures, compound synthesis and characterization data, summary of IC50’s for 123 hits in 10-point DRCs, inhibition against 58 kinases by LI-2172 and LI-2242 (PDF)
Author Present Address
∇ H.W.: Department of Neurology, pharmacology and Johns Hopkins Drug Discovery, Johns Hopkins School of Medicine, Baltimore, MD 21205.
Author Present Address
◆ M.W.: John Hopkins Technology Ventures, John Hopkins University, Baltimore MD 21205.
Author Contributions
¶ G.L. and W.Y. contributed equally to these work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was fund by the Lieber Institute for Brain Development and U01 MH112658–01A1 from NIH/NIMH and the NIH Intramural Research Program to NCATS.
The authors declare no competing financial interest.
Supplementary Material
References
- Chakraborty A. (2018) The inositol pyrophosphate pathway in health and diseases. Biol. Rev. Camb Philos. Soc. 93, 1203–1227. 10.1111/brv.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritoh Y.; Oka M.; Yasuhara Y.; Hozumi H.; Iwachidow K.; Fuse H.; Tozawa R. (2016) Inositol Hexakisphosphate Kinase 3 Regulates Metabolism and Lifespan in Mice. Sci. Rep. 6, 32072. 10.1038/srep32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A.; Koldobskiy M. A.; Bello N. T.; Maxwell M.; Potter J. J.; Juluri K. R.; Maag D.; Kim S.; Huang A. S.; Dailey M. J.; Saleh M.; Snowman A. M.; Moran T. H.; Mezey E.; Snyder S. H. (2010) Inositol Pyrophosphates Inhibit Akt Signaling, Thereby Regulating Insulin Sensitivity and Weight Gain. Cell 143, 897–910. 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay R. D.; Beals J. W.; Drnevich J.; Imai B. S.; Yau P. M.; Ulanov A. V.; Tillin N. A.; Villegas-Montes M.; Paluska S. A.; Watt P. W.; De Lisio M.; Burd N. A.; Mackenzie R. W. (2020) Ingestion of lean meat elevates muscle inositol hexakisphosphate kinase 1 protein content independent of a distinct post-prandial circulating proteome in young adults with obesity. Metab., Clin. Exp. 102, 153996. 10.1016/j.metabol.2019.153996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Shukla D.; Suman K.; Lakshmi B. J.; Manorama R.; Kumar S.; Bhandari R. (2013) Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood 122, 1478–1486. 10.1182/blood-2013-01-481549. [DOI] [PubMed] [Google Scholar]
- Chakraborty A.; Latapy C.; Xu J.; Snyder S. H.; Beaulieu J. M. (2014) Inositol hexakisphosphate kinase-1 regulates behavioral responses via GSK3 signaling pathways. Mol. Psychiatry 19, 284–293. 10.1038/mp.2013.21. [DOI] [PubMed] [Google Scholar]
- Ghoshal S.; Tyagi R.; Zhu Q.; Chakraborty A. (2016) Inositol hexakisphosphate kinase-1 interacts with perilipin1 to modulate lipolysis. Int. J. Biochem. Cell Biol. 78, 149–155. 10.1016/j.biocel.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B. H.; Bauer J. A.; Lupica J. A.; Tang Z.; Schmidt H.; DiDonato J. A.; Lindner D. J. (2007) Effect of inositol hexakisphosphate kinase 2 on transforming growth factor beta-activated kinase 1 and NF-kappaB activation. J. Biol. Chem. 282, 15349–15356. 10.1074/jbc.M700156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal S.; Zhu Q.; Asteian A.; Lin H.; Xu H.; Ernst G.; Barrow J. C.; Xu B.; Cameron M. D.; Kamenecka T. M.; Chakraborty A. (2016) TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol. Metab. 5, 903–917. 10.1016/j.molmet.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C.; Stashko M. A.; Puhl-Rubio A. C.; Chakraborty M.; Chakraborty A.; Frye S. V.; Pearce K. H.; Wang X.; Shears S. B.; Wang H. (2019) Inhibition of Inositol Polyphosphate Kinases by Quercetin and Related Flavonoids: A Structure-Activity Analysis. J. Med. Chem. 62, 1443–1454. 10.1021/acs.jmedchem.8b01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormald M. M.; Ernst G.; Wei H.; Barrow J. C. (2019) Synthesis and characterization of novel isoform-selective IP6K1 inhibitors. Bioorg. Med. Chem. Lett. 29, 126628. 10.1016/j.bmcl.2019.126628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormald M.; Liao G.; Kimos M.; Barrow J.; Wei H. (2017) Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS One 12, e0188852 10.1371/journal.pone.0188852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta N.; Jovic M.; Barnaeva E.; Kim D. W.; Hu X.; Southall N.; Dejmek M.; Mejdrova I.; Nencka R.; Baumlova A.; Chalupska D.; Boura E.; Ferrer M.; Marugan J.; Balla T. (2019) A large scale high-throughput screen identifies chemical inhibitors of phosphatidylinositol 4-kinase type II alpha. J. Lipid Res. 60, 683–693. 10.1194/jlr.D090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz T. D.; Sivakumaren S. C.; Ferguson F. M.; Zhang T.; Yasgar A.; Seo H. S.; Ficarro S. B.; Card J. D.; Shim H.; Miduturu C. V.; Simeonov A.; Shen M.; Marto J. A.; Dhe-Paganon S.; Hall M. D.; Cantley L. C.; Gray N. S. (2020) Discovery and Structure-Activity Relationship Study of (Z)-5-Methylenethiazolidin-4-one Derivatives as Potent and Selective Pan-phosphatidylinositol 5-Phosphate 4-Kinase Inhibitors. J. Med. Chem. 63, 4880–4895. 10.1021/acs.jmedchem.0c00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 45, 1712–1722. 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- Terao Y., Takahashi M., Hara R., Hidaka K., Furukawa H., Yamasaki T., and Kasai S. (2018) Ip6k inhibitors, WO2018182051 A1.
- Puschmann R.; Harmel R. K.; Fiedler D. (2019) Scalable Chemoenzymatic Synthesis of Inositol Pyrophosphates. Biochemistry 58, 3927–3932. 10.1021/acs.biochem.9b00587. [DOI] [PubMed] [Google Scholar]
- Dominguez J. M.; Fuertes A.; Orozco L.; del Monte-Millan M.; Delgado E.; Medina M. (2012) Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib. J. Biol. Chem. 287, 893–904. 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A.; Alonso M.; Castro A.; Pérez C.; Moreno F. J. (2002) First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299. 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- Matsunaga S.; Fujishiro H.; Takechi H. (2019) Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer's Dis. 69, 1031–1039. 10.3233/JAD-190256. [DOI] [PubMed] [Google Scholar]
- Blazer L. L.; Zhang H.; Casey E. M.; Husbands S. M.; Neubig R. R. (2011) A nanomolar-potency small molecule inhibitor of regulator of G-protein signaling proteins. Biochemistry 50, 3181–3192. 10.1021/bi1019622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundback T.; Nordlund P.; Molina D. M. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Sasaki M.; Anindita P. D.; Phongphaew W.; Carr M.; Kobayashi S.; Orba Y.; Sawa H. (2018) Development of a rapid and quantitative method for the analysis of viral entry and release using a NanoLuc luciferase complementation assay. Virus Res. 243, 69–74. 10.1016/j.virusres.2017.10.015. [DOI] [PubMed] [Google Scholar]
- McNulty D. E.; Bonnette W. G.; Qi H.; Wang L.; Ho T. F.; Waszkiewicz A.; Kallal L. A.; Nagarajan R. P.; Stern M.; Quinn A. M.; Creasy C. L.; Su D. S.; Graves A. P.; Annan R. S.; Sweitzer S. M.; Holbert M. A. (2018) A High-Throughput Dose-Response Cellular Thermal Shift Assay for Rapid Screening of Drug Target Engagement in Living Cells, Exemplified Using SMYD3 and IDO1. SLAS Discov 23, 34–46. 10.1177/2472555217732014. [DOI] [PubMed] [Google Scholar]
- Harmel R. K.; Puschmann R.; Nguyen Trung M.; Saiardi A.; Schmieder P.; Fiedler D. (2019) Harnessing (13)C-labeled myo-inositol to interrogate inositol phosphate messengers by NMR. Chem. Sci. 10, 5267–5274. 10.1039/C9SC00151D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundbäck T.; Nordlund P.; Molina D. M. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.