

Abstract
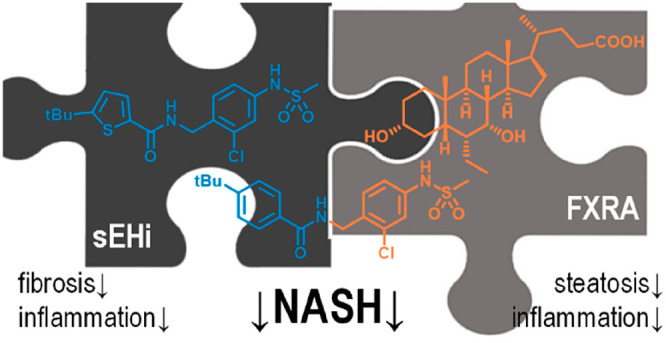
Nonalcoholic fatty liver disease (NAFLD) is an epidemic chronic liver disease and may progress over nonalcoholic steatohepatitis (NASH) to liver cirrhosis and hepatocellular carcinoma. The multiple metabolic, environmental, and genetic factors that are involved in NAFLD/NASH pathogenesis and progression suggest a need for multimechanistic interventions. We have developed and preliminarily characterized a concept of dual farnesoid X receptor (FXR) and soluble epoxide hydrolase (sEH) modulation as a promising polypharmacological strategy to counteract NASH. Here we report the profiling of FXR activation, sEH inhibition, and simultaneous FXR/sEH modulation as an interventional treatment in pre-established NASH in mice with diet-induced obesity (DIO). We found that full FXR activation was required to obtain antisteatosis effects but also worsened ballooning degeneration and fibrosis. In contrast, sEH inhibition and dual FXR/sEH modulation, despite a lack of antisteatosis activity, had anti-inflammatory effects and efficiently counteracted hepatic fibrosis. These results demonstrate great therapeutic potential of sEH inhibition to counteract hepatic fibrosis and validate the designed polypharmacology concept of dual FXR/sEH modulation as a potentially superior avenue for the effective treatment of the multifactorial condition NASH.
Keywords: farnesoid X receptor, soluble epoxide hydrolase, designed polypharmacology, nonalcoholic steatohepatitis
According to current understanding, nonalcoholic fatty liver disease (NAFLD) evolves from a complex interplay of multiple metabolic factors causing hepatic steatosis that progresses to nonalcoholic steatohepatitis (NASH) and liver fibrosis which can ultimately lead to liver cirrhosis and hepatocellular carcinoma.1,2 Obesity, elevated free fatty acid and cholesterol levels, insulin resistance, oxidative stress, and mitochondrial dysfunction are thought to be involved in the pathogenesis of NAFLD and NASH.1,2 In this context, the disease is considered as the hepatic manifestation of the metabolic syndrome and is closely associated with an unhealthy lifestyle.3 Recent estimations suggest NAFLD as an epidemic chronic liver disease affecting approximately one-quarter of the global adult population.3,4 Typical progression rates indicate that NASH develops in 6% of adults and progresses to cirrhosis in 2%.3 Importantly, NAFLD and its consequences are among the top reasons for liver transplantation and contribute to elevated incidence of primary liver cancer.3 The alarming global prevalence and serious consequences of NAFLD have fueled intensive research for pharmacological interventions, yet there is no approved drug for NASH treatment to date.
Several therapeutic strategies for NASH have been studied in clinical trials and demonstrated promising results. The current NASH pipeline comprises of various nuclear receptor modulators, including the FXR agonists obeticholic acid (OCA), tropifexor and analogues, the PPAR agonists elafibranor (PPAR-α/δ), lanifibranor (pan-PPAR), seladelpar (PPARδ), and saroglitazar (PPARα/γ), as well as the THRβ agonist resmetirom.5−7 Until very recently, OCA and elafibranor had good prospects of satisfying the unmet clinical need of a pharmacological NASH treatment.8,9 Nevertheless, the FDA rejected an attempt for OCA approval,10 mainly because the anticipated benefit remains uncertain and does not sufficiently outweigh potential risks.10 Moreover, interim analysis of the RESOLVE-IT phase 3 clinical study of elafibranor in adults with NASH and fibrosis showed that the trial did not meet its primary end point of NASH resolution without worsening of fibrosis.11 Several approved antidiabetic agents such as licogliflozin (SGLT-2 inhibitor) and semaglutide (GLP-1 analogue), as well as FGF-21 analogues are also studied in advanced clinical trials and might benefit from these recent drawbacks.5−7 In addition, various further agents in clinical development with diverse targets and mechanisms such as the second-generation thiazolidinedione MSDC-0602K (a mitochondrial pyruvate carrier modulator),12 cenicriviroc (CCR2/CCR5 antagonist),13 and aramchol (SCD-1 inhibitor)14 illustrate strong efforts to develop a cure for NASH and an enormous diversity in experimental NASH treatment. However, the majority of advanced experimental drugs to counteract NASH to date suffers from limited efficacy. We therefore hypothesized that the multifactorial disease complex of NASH demands multiple modes of action for satisfactory therapeutic efficacy.
NASH can be characterized by three major factors: hepatic steatosis, fibrosis, and inflammation. We therefore proposed that a combination of antisteatosis, antifibrotic, and anti-inflammatory modes of action may provide superior efficacy in NASH. While FXR has been validated as a molecular target to treat NASH8,15 and counteracts steatosis, several lines of preclinical evidence also present inhibition of the soluble epoxide hydrolase (sEH) as a very promising strategy in NASH treatment with anti-inflammatory and antifibrotic activities.16−21 Among a variety of putative modes of action fulfilling these criteria, we thus chose FXR activation as validated antisteatosis contribution and soluble epoxide hydrolase (sEH) inhibition to obtain anti-inflammatory and antifibrotic effects. We reasoned that a simultaneous, polypharmacological modulation of both targets, FXR and sEH, by a single compound has a pharmacokinetic advantage over the combination of two selective modulators.22,23 Especially for achieving synergistic effects in liver tissue, a single compound appears preferable since two selective compounds with distinct pharmacokinetic profiles might result in unpredictable variations in concentrations and metabolic degradation.24,25 In addition to different distributions, two selective modulators might interact via cytochrome P450 enzymes in liver.25 Aiming to employ an effective tool to probe our multitarget hypothesis, we have developed the dual modulator, DM,26 that is an FXR activator (FXRA) and sEH inhibitor (sEHi) at low nanomolar concentrations. With this designed polypharmacology23 profile, FXRA/sEHi DM has previously revealed promising efficacy in two models of NASH in mice when used in a preventive fashion.27
In this study, we evaluated the dual FXRA/sEHi DM as a curative therapeutic intervention in mice with diet-induced obesity (DIO) and biopsy-confirmed NASH. After 12 weeks of treatment with the FXR agonist OCA, the FXRA/sEHi DM, or the sEH-selective analogue (sEHi) MH, we detected pronounced differences in the antisteatosis, antifibrotic, and anti-inflammatory effects in the treatment groups. OCA strongly reduced hepatic steatosis, while dual FXRA/sEHi DM and sEHi MH remarkably reversed hepatic fibrosis.
Results
Study Compounds
We have previously demonstrated appealing efficacy of the dual FXRA/sEHi DM in preventing disease development in toxin- and diet-induced NASH in mice.27 As the logical next step, we aimed to probe the potential of FXRA/sEHi treatment to reverse fully established NASH as a curative treatment in an interventional fashion and to capture the contributions of each molecular target to the attractive pharmacological effects of the dual modulator chemotype. We developed analogue MH with an altered activity profile as an sEH-selective control which possesses negligible FXR agonist activity while exhibiting equal potency as an sEH inhibitor despite its remarkable structural similarity (Table 1, Figure 1). With its weak, micromolar potency and low activation efficacy on FXR, as well as its low affinity to the recombinant FXR ligand binding domain (LBD), the FXR-mediated effects of MH can be neglected in vivo. Hence, MH was used as structurally related selective sEH inhibitor control for dual modulator DM.
Table 1. In Vitro Potencies of the Study Compounds FXRA/sEHi DM and sEHi MHa.

| FXRA/sEHi (DM) | sEHi (MH) | |
|---|---|---|
| IC50 (sEH) | 0.004 μM | 0.002 μM |
| EC50 (FXR) | 0.02 μM | 1.4 μM |
| efficacy (FXR) | 35% | 14% |
| Kd (FXR LBD) | 0.13 μM | 1.4 μM |
Efficacy (FXR) refers to the maximum activation efficacy of the respective compound relative to 1 μM GW4064 which is defined as 100%.
Figure 1.

In vitro characteristics of the study compounds FXRA/sEHi DM and sEHi MH. (a) Effects of the study compounds on expression levels of selected FXR-regulated genes in human hepatocytes (HepG2). Data are mean ± S.E.M. fold mRNA induction compared to DMSO (0.1%) treated cells, n = 5. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 (t-test) vs DMSO (0.1%). CDCA vs CDCA+MH were not statistically different. (b) Selectivity profiles of the study compounds over relevant members of the nuclear receptor family. The heat map shows mean relative activation compared to the respective reference agonist, n = 5. (c) Microsomal stability of the study compounds. Data are mean ± S.E.M. remaining parent compound, n = 5.
Further characterization of DM and MH confirmed the different activity profiles on FXR. When we treated human hepatocytes (HepG2) with DM or MH and quantified expression of FXR-regulated genes at the mRNA level (Figure 1a), we observed an induction of bile salt export protein (BSEP) and small heterodimer partner (SHP) by DM with moderate efficacy compared to that of FXR agonist OCA. Congruently, the indirectly FXR-regulated cholesterol 7α hydroxylase (CYP7A1) and sterol regulatory element-binding protein 1c (SREBP-1c) were efficiently downregulated by DM. sEH-selective control MH only caused weak effects on BSEP and CYP7A1 mRNA expression at a high (10 μM) concentration and hardly affected the activity of the endogenous FXR agonist chenodeoxycholic acid (CDCA) on FXR-regulated gene expression when applied in a competitive fashion. In addition, DM and MH shared exceptional selectivity over related nuclear receptors (Figure 1b) and had equal stability against microsomal degradation, suggesting similar pharmacokinetics (PK).26 With these characteristics and its remarkable structural similarity, sEHi MH seemed suitable to capture therapeutic effects of the FXRA/sEHi DM that arise from sEH inhibition without the contributions of FXR activation.
Animal Model
To evaluate the potential of FXRA/sEHi to counteract fully established NASH, we selected the widely accepted disease model of NASH induced by a high-fat diet (40% fat, 20% fructose, 2% cholesterol) in DIO mice (Figure 2a,b). This model provides a robust NASH phenotype reflecting the human pathology28,29 and has served to evaluate several clinical candidates for NASH treatment.30−33 To establish NASH, C57BL/6JRj mice were fed the high-fat diet (Figure 2b) for a total of 53 weeks. Only mice with a NASH-positive biopsy in week 38 were included in the treatment phase (starting in week 42), stratified to ensure an equal baseline (Figure 2c) and randomized to receive DM (10 mg/kg, by mouth (per os, p.o.), twice per day (b.i.d.)), MH (10 mg/kg, p.o., b.i.d.), OCA (30 mg/kg, p.o., once per day (q.d.) + vehicle (p.o., q.d.)), or vehicle (p.o., b.i.d.) for 12 weeks with 16 animals per group (Figure 2d). The reference FXRA OCA was administered at 30 mg/kg based on previous studies which have shown therapeutic efficacy with this high OCA dose in similar settings.30,34 The healthy control group receiving normal chow and no intervention comprised of 10 mice. The study interventions were well-tolerated. One animal in the OCA group was found dead in week 2 of the treatment period due to a larger subcutaneous hemorrhage. All other animals completed the study. There were no differences in the daily food intake (Figure 3a) and body weight (Figure 3b) between the treatment groups throughout the study.
Figure 2.

Animal model outline. (a) Animal model outline and timing. (b) Key components of the NASH-inducing diet. (c) Prebiopsy PSR (%) at randomization. (d) Treatment groups.
Figure 3.

Food intake and biochemical parameters in the different treatment groups. (a) Daily food intake during the treatment phase. Symbols are the mean; dotted lines are SD (b) Body weight development over the treatment period. Symbols are the mean; dotted lines are SD (c) Plasma biochemical parameters in the treatment groups at week 4 of the treatment period and at termination (week 12). Data are the mean ± SD. ***, p < 0.001 (Dunnett’s test) vs vehicle. (d) Blood glucose and plasma insulin levels in the treatment groups in week 11 of the treatment period. ***, p < 0.001 (Dunnett’s test). (e) Liver biochemical parameters in the treatment groups at termination (week 12). Data are the mean ± SD. *, p < 0.05; ***, p < 0.001 (Dunnett’s test) vs vehicle.
Biochemical Parameters
Biochemical parameters of liver health (plasma alanine aminotransferase, plasma ALT; plasma aspartate aminotransferase, plasma AST) and lipid homeostasis (plasma triglycerides, plasma TG; plasma total cholesterol, plasma TC) were determined after 4 weeks of treatment and at termination (Figure 3c). Plasma glucose and insulin levels were evaluated after 11 weeks of treatment (Figure 3d). No differences were observed between the treatment groups for plasma ALT, plasma AST, and plasma TG. Plasma TC was significantly reduced in the OCA group at week 4 and at termination. Plasma glucose levels were significantly increased in mice receiving OCA compared to vehicle treated animals. DM and MH did not affect glucose or insulin levels. At termination, OCA-treated animals had significantly lower liver weights, liver triglyceride, and liver cholesterol content compared to vehicle-treated mice, while DM and MH had no effect on these parameters (Figure 3e). Liver hydroxyproline (HP) content was not different between the treatment groups.
Histology and NAFLD Scores
After termination, livers of all animals were subjected to histopathological analysis using the NAFL activity score (NAS, Figure 4a) and immune-histochemistry. Representative histology images are shown in Figure 4b.
Figure 4.

NAFL-related scores and histology. (a) NAFL activity score (NAS) and individual scores for contributions by steatosis, fibrosis, lobular inflammation, and hepatocellular ballooning determined from all animals in the different treatment groups. Post-treatment scores were compared with pretreatment scores for each individual animal, and the change in the score is shown as improvement (lower score), no change, or worsening (higher score). (b) Representative immune-histochemistry images from post-treatment liver histology samples for the different treatment groups.
The NAS, which considers steatosis, fibrosis, lobular inflammation, and ballooning degeneration, was improved in the OCA group and showed a tendency to improvement in the FXRA/sEHi (DM) group. The individual parameters revealed that the pronounced effect of OCA on the NAS mainly resulted from strong antisteatosis effects and a slight improvement in lobular inflammation. In contrast, fibrosis was hardly affected, and ballooning degeneration was worsened by OCA compared to pretreatment and to vehicle. Treatment with FXRA/sEHi DM or sEHi MH had no effect on hepatic steatosis but improved fibrosis and reduced inflammation. Therein, FXRA/sEHi DM exhibited a stronger anti-inflammatory effect, while sEHi MH, interestingly, was more effective in reducing fibrosis.
OCA (8/15 animals) and FXRA/sEHi DM (9/16) decreased lobular inflammation with similar efficacy, while sEHi MH (5/16) was less active on inflammatory parameters (Figure 5a). Closer inspection of anti-inflammatory effects revealed similarly diminished numbers of liver inflammatory cells compared to vehicle for all treatment compounds (Figure 5b). Liver inflammatory foci were diminished by all treatments compared to vehicle with OCA having the strongest effect. OCA, the FXRA/sEHi DM, and the sEHi MH decreased the liver CD45-positive area as another measure of anti-inflammatory activity and OCA slightly reduced liver Galectin-3 (Gal3)-positive area.
Figure 5.

Inflammatory effects in the treatment groups. (a) Pre- and post-treatment lobular inflammation score in the different treatment groups. (b) Liver inflammatory parameters for the different treatment groups in post-treatment histopathological analysis. *, p < 0.05 (ANOVA with Dunnett’s test) vs vehicle.
The most pronounced effects of the dual modulator FXRA/sEHi DM and the sEHi MH were evident in fibrosis (Figure 6). While fibrosis stage worsened in the OCA group over the treatment period and was not improved compared to vehicle, DM and MH led to a reversal of fibrosis as observed by improvements in the fibrosis score (Figure 6a) and a markedly decreased liver Picrosirius Red (PSR)-positive area in the post-treatment histology (Figure 6b). This pronounced antifibrotic effect is also illustrated in the comparison of pre- and post-treatment histology of individual animals treated with DM or MH (Figure 7), and interestingly, the sEHi MH reversed fibrosis slightly more efficiently than did the FXRA/sEHi DM. No reduction of fibrosis stage (Figure 6a) or PSR (Figures 6b and 7) was seen with OCA, but the FXRA slightly reduced the Gal3-positive area (Figure 5b) potentially suggesting a mild improvement in fibrosis despite no apparent change in PSR upon OCA treatment. The weak antifibrotic effect of OCA is in contrast to the markedly improved NAS in OCA-treated animals further underlining the pronounced antisteatosis activity of FXR activation. Notably, OCA had detrimental effects on hepatocellular ballooning as observed by a worsening of the respective score (Figure 6c) and a significant increase in ballooning cells in liver (Figure 6d). DM and MH hardly affected ballooning degeneration compared to vehicle.
Figure 6.

Effects of the study compounds on fibrosis and hepatocellular ballooning. (a) Changes in fibrosis stage from pre- to post-treatment in the different treatment groups. *, p < 0.05 (one-sided Fisher’s exact test with Bonferroni correction). (b) Changes in liver PSR positive area from pre- to post-treatment in the different treatment groups. *, p < 0.05; **, p < 0.01 (paired t test). (c) Changes in ballooning degeneration from pre- to post-treatment in the different treatment groups. (d) Number (1/mm2) of ballooning cells in liver after the treatment in the different treatment groups. *, p < 0.05 (ANOVA with Dunnett’s test) vs vehicle.
Figure 7.

Pre- and post-treatment histology images for DM-, MH-, and OCA-treated animals. Each representative set of images (pre- and post-treatment) originates from the same animal.
Discussion
NAFLD evolves from an interplay of several factors involving obesity, elevated levels of free fatty acids and cholesterol, insulin resistance, and adipose tissue dysfunction leading to hepatic fat accumulation. Hepatic steatosis and these factors trigger oxidative and endoplasmic reticulum stress, mitochondrial dysfunction, and inflammation in the liver which in turn eventually cause hepatocellular damage and liver fibrosis.1,2 In addition, the disease is closely linked to other manifestations of the metabolic syndrome which promote each other.1,2 Considering these multiple processes, strong therapeutic efficacy in this complex pathology likely requires multiple modes of action, which has recently prompted us to develop a class of potent dual agents that simultaneously activate FXR and inhibit sEH.26,27 Previous studies have demonstrated superior effects for combination therapies in counteracting NASH compared to single-target pharmacologies, for example, with simultaneous FXR and PPAR activation30 or FXR and glucagon-like peptide-1 receptor agonism.32 In toxin- and diet-induced models of NASH, the FXRA/sEHi DM already proved effective in preventing disease development.27 In this study, we evaluated the potential of the dual modulator concept as an interventional treatment of pre-established NASH and probed the contributions of the underlying modes of action with selective control treatments.
In treating mice with pre-established, biopsy-confirmed NASH with the FXRA OCA, the FXRA/sEHi DM, or the sEHi MH, we observed remarkable differences in the therapeutic effects of these interventions. The selective full FXR agonist OCA was clearly superior in reducing hepatic steatosis, while weak activation (FXRA/sEHi DM) or lack of FXR activation (sEHi MH) were insufficient to achieve strong antisteatosis efficacy. This in turn demonstrates that high FXR activation efficacy is required to counteract established hepatic steatosis in NASH while partial FXR agonism and sEH inhibition do not efficiently exhibit such activity.
Regarding hepatic inflammation as the second major factor of NASH, the three interventions revealed comparable therapeutic activity. The FXRA OCA, the FXRA/sEHi DM, and the sEHi MH reduced the lobular inflammation score and diminished liver inflammatory cells and markers. Despite only small differences between the treatments, OCA was slightly more active in reducing liver inflammatory foci and Gal3-positive area in the post-treatment biopsies, while the FXRA/sEHi DM demonstrated the strongest effect on the overall lobular inflammation score. The selective sEHi MH exhibited the least overall anti-inflammatory activity. The observation that the FXRA/sEHi DM achieved slightly higher anti-inflammatory effects further confirms our hypothesis that both modes of action contribute to counteracting hepatic inflammation in NASH and supports the dual modulator concept.
The most intriguing finding of this study, however, was the remarkable fibrosis-reversing activity of the FXRA/sEHi DM and the sEHi MH. Both interventions decreased the fibrosis stage and reduced the post-treatment liver PSR-positive area compared to pretreatment levels. Therein, the selective sEHi MH exhibited a stronger efficacy by causing a reduction of the fibrosis stage in >50% of animals and no fibrosis worsening in the remaining mice. The FXR agonist OCA, in contrast, failed to achieve beneficial effects on fibrosis compared to pretreatment or compared to vehicle but caused fibrosis worsening in >25% of animals. In line with this, OCA also induced a notable increase in the number of ballooning cells in the liver and worsened the ballooning degeneration score in >25% of animals. The FXRA/sEHi DM and the sEHi MH exhibited less unfavorable effects on hepatocellular ballooning, suggesting preferable effects on liver health compared to those of OCA. Importantly, fibrosis presents as the major prognostic marker for NASH progression3,4 which gives the interventional activity of the FXRA/sEHi DM and the sEHi MH considerable relevance. The fact that both the dual modulator DM and the selective sEHi MH efficiently counteracted fibrosis points to a major contribution of sEHi to this activity and to the great potential of sEHi in NASH treatment. It may also be speculated that this attractive reduction of fibrosis by sEHi without worsening of ballooning degeneration can ameliorate the negative effects on hepatocellular ballooning that we observed for the FXRA OCA. Thereby, the polypharmacological approach of FXRA/sEHi not only combines the beneficial activities of the two mechanisms but also complements FXRA with sEHi, compensating for adverse effects.
In summary, our observations confirm FXR as a valuable antisteatosis target with additional anti-inflammatory effects in the liver but also demonstrate a lack of antifibrotic effects and an unfavorable progression of ballooning degeneration associated with strong FXR activation. In this context, inhibition of sEH evolves as a promising antifibrotic strategy since both sEH inhibitors in our study revealed pronounced fibrosis-reversing activity. Additionally, combined partial FXR agonism and sEH inhibition resulted in slightly higher improvements in lobular inflammation. According to our results, both modes of action, FXR activation and sEH inhibition, thus present as suitable therapeutic strategies to counteract NASH but exhibit effects against different aspects of the multifactorial disease complex. The antisteatosis activity of FXR activation antagonized hepatic fat accumulation as major underlying pathogenic factor of NAFLD and NASH, whereas sEH inhibition reversed fibrosis which is the deleterious consequence of prolonged liver stress and damage in NASH. While both individual effects alone likely provide therapeutic benefit, they also complement each other, and their combination may strengthen therapeutic efficacy in NASH treatment. However, an optimal balance of sufficient FXR activation efficacy for antisteatosis effects and FXR overactivation leading to potential liver damage remains to be determined.
Overall, our results provide another example of remarkable antisteatosis efficacy of the FXR agonist OCA that also translated into strong efficacy of the semisynthetic bile acid in improving the NAS. Other factors of NASH, however, failed to confirm this pronounced therapeutic activity of OCA. Particularly, the beneficial effect of OCA on liver fibrosis was weak, and ballooning degeneration was worsened by OCA treatment which is in line with the FDA rejection of the drug for NASH treatment.10 In light of these recent the drawbacks of OCA and other leading anti-NASH drugs, the dual FXR/sEH modulator concept is particularly appealing. The observed fibrosis reversal counteracts a major factor for disease progression of NASH toward liver cirrhosis or hepatocellular carcinoma. Provided that this activity translates from the preclinical DIO NASH model to clinical efficacy, counteracting established fibrosis in progressed NASH holds particular promise for future NASH treatment and is a likely superior effect to reversing steatosis with the cost of fibrosis progression and worsening of ballooning degeneration. These observations highlight the great potential of dual FXR/sEH modulation and present designed polypharmacology as appealing treatment approach for the multifactorial condition NASH.
Materials and Methods
Chemistry
General
All chemicals and solvents were of reagent grade and used without further purification unless otherwise specified. All reactions were conducted in oven-dried glassware under an argon atmosphere and in absolute solvents. NMR spectra were recorded on a Bruker AV 500, Bruker AV 300, or a Bruker am250xp spectrometer (Bruker Corporation, Billerica, MA). Chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS) as reference. Multiplicity is reported as follows: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; hept, heptet; m, multiplet. Approximate coupling constants (J) are given in hertz (Hz). High-resolution mass spectra were recorded on a MALDI LTQ ORBITRAP XL instrument (Thermo Fisher Scientific). Compound purity was analyzed on a Waters 600 Controller HPLC (Waters, Milford, MA) equipped with a Waters 2487 Dual Absorbance Detector and a Waters 717 plus Autosampler or on a VWR Chromaster (VWR, Radnor, PA) equipped with a 5160 pump system, a DAD 5430, a 5260 Autosampler, and a MultoHigh100 RP18–5 μ, 250 × 4 mm2 column (CS-Chromatographie Service GmbH, Langerwehe, Germany) using a gradient (H2O + 0.1% formic acid/MeOH 80:20 isocratic for 5 min to MeOH after an additional 45 min and MeOH for an additional 10 min) at a flow rate of 1 mL/min or a gradient (H2O + 0.1% formic acid/MeOH 60:40 isocratic for 5 min to MeOH after an additional 25 min and MeOH for an additional 10 min) at a flow rate of 1 mL/min with UV detection at 245 and 280 nm.
Synthesis of MH
1-(4-Amino-2-chlorophenyl)methanamine
LiAlH4 (1 M in THF, 16.4 mL, 16.4 mmol, 2.5 equiv) was cooled to 0 °C, and 4-amino-2-chlorobenzonitrile (1.0 g, 6.6 mmol, 1.0 equiv) in THF (absolute, 3.0 mL) was slowly added. After evolution of H2 had ceased, the mixture was allowed to warm to room temperature and then refluxed for 16 h. After cooling to room temperature, the mixture was diluted with 10 mL of THF and then cooled to 0 °C. Aqueous NaOH solution (10% w/w, 1 mL) and water (1.8 mL) were added dropwise. The colorless precipitate was filtered through Celite and washed with 15 mL of diethyl ether. Evaporation of the organic solvents from the filtrate in vacuo yielded 1-(4-amino-2-chlorophenyl)methanamine as a yellow oil (0.77 g, 75%). 1H NMR (500 MHz, DMSO-d6) δ = 7.11 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 2.2 Hz, 1H), 6.48 (dd, J = 8.2, 2.2 Hz, 1H), 5.19 (s, 2H), 3.59 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ = 148.56, 132.33, 129.67, 127.74, 113.63, 112.72, 42.88.
N-(4-Amino-2-chlorobenzyl)-5-(tert-butyl)thiophene-2-carboxamide
1-(4-Amino-1-chlorophenyl)methanamine (0.46 g, 3.0 mmol, 1.5 equiv), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (0.45 g, 2.4 mmol, 1.2 equiv), and 4-(dimethylamino)pyridine (0.02 mmol, 0.03 g, 0.1 equiv) were dissolved in a mixture of CHCl3 (absolute, 10 mL) and DMF (absolute, 3.0 mL). 5-tert-Butyl-thiophene-2-carboxylic acid (0.35 g, 2.0 mmol, 1.0 equiv) dissolved in DMF (absolute, 2.0 mL) was slowly added over 10 min, and the mixture was stirred overnight at 60 °C. After cooling to room temperature, aqueous hydrochloric acid (10%, 10 mL) was added, and phases were separated. The aqueous layer was brought to pH 10 with Na2CO3 solution and extracted three times with 20 mL of EtOAc each time. The combined organic layers were dried over Na2SO4, and the solvents were evaporated in vacuo. Further purification was carried out by column chromatography with hexane/EtOAC/acetic acid (65:32:2) as the mobile phase to obtain N-(4-amino-2-chlorobenzyl)-5-(tert-butyl)thiophene-2-carboxamide as a yellow solid (0.239 g, 35%). 1H NMR (300 MHz, methanol-d4) δ = 7.52 (d, J = 3.9 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 6.89 (d, J = 3.9, 1H), 6.75–6.72 (d, J = 2.3 Hz, 1H), 6.60 (dd, J = 8.3, 2.3 Hz, 1H), 4.48 (s, 2H), 1.41 (s, 9H). 13C NMR (75 MHz, MeOD) δ = 163.25, 162.90, 148.52, 135.17, 133.43, 129.85, 128.31, 123.72, 122.23, 115.00, 113.38, 40.56, 34.43, 31.25.
5-(tert-Butyl)-N-(2-chloro-4-(methylsulfonamido)benzyl)thiophene-2-carboxamide (MH)
N-(4-Amino-2-chlorobenzyl)-5-(tert-butyl)thiophene-2-carboxamide (0.23 g, 0.71 mmol, 1.0 equiv) was dissolved in THF (absolute, 10 mL), and pyridine (1.0 mL) was added. Mesyl chloride (0.28 mL, 3.6 mmol, 5.0 equiv) was carefully added, and the mixture was stirred for 16 h at 60 °C. Aqueous hydrochloric acid (10%, 15 mL) was then added and the mixture was extracted three times with 30 mL of EtOAc each time. The combined organic layers were dried over Na2SO4, and the solvents were evaporated in vacuo. Further purification was carried out by column chromatography using hexane/EtOAc/acetic acid (59:39:2) as the mobile phase to obtain MH as a colorless solid (0.145 g, 41%). 1H NMR (500 MHz, methanol-d4) δ = 7.54 (d, J = 3.8 Hz, 1H), 7.36–7.29 (m, 2H), 7.15 (m, 1H), 6.90 (d, J = 3.8 Hz, 1H), 4.56 (s, 2H), 2.95 (s, 3H), 1.39 (s, 9H). 13C NMR (126 MHz, MeOD) δ = 164.81, 164.60, 139.91, 136.26, 134.81, 133.01, 130.95, 129.93, 123.73, 121.70, 119.60, 41.86, 39.35, 35.86, 32.65. HRMS (MALDI): m/z calculated for C17H22ClN3O3S2 401.07549. Found 401.07535.
The preparation of DM and its precursors has been described previously.26 The batches of MH and DM used in the DIO NASH model had a purity of >95% as determined by HPLC-UV analysis at λ = 245 and 280 nm. The batch of obeticholic acid used for the in vivo study was obtained commercially.
In Vitro Pharmacological Characterization
The in vitro pharmacological profile of DM has already been reported.26 In vitro profiling of MH for FXR modulation,35,36 sEH inhibition,36,37 selectivity,26,38,39 FXR LBD binding affinity,26,40 microsomal stability,26 and effects on FXR-regulated gene expression26 were carried out as described previously and in uniformity with the characterization of DM.
Hybrid Reporter Gene Assays for Selectivity Profiling
Plasmids
The Gal4-fusion receptor plasmids pFA-CMV-hPPARα-LBD,41 pFA-CMV-hPPARδ-LBD,41 pFA-CMV-hPPARγ-LBD,41 pFA-CMV-hLXRα-LBD,42 pFA-CMV-hLXRβ-LBD,42 pFA-CMV-hRXRα-LBD,43 pFA-CMV-hRXRβ-LBD,43 pFA-CMV-hRXRγ-LBD,43 pFA-CMV-hRARα-LBD,43 pFA-CMV-hRARβ-LBD,43 pFA-CMV-hRARγ-LBD,43 pFA-CMV-hVDR-LBD,43 pFA-CMV-hTHRα-LBD,39 and pFA-CMV-hTHRβ-LBD39 coding for the hinge region and LBD of the canonical isoform of the respective nuclear receptor have been reported previously. pFR-Luc (Stratagene) was used as reporter plasmid, and pRL-SV40 (Promega) was used for normalization of transfection efficiency and cell growth.
Assay Procedure
HEK293T cells were grown in high-glucose DMEM, supplemented with 10% fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2. The day before transfection, HEK293T cells were seeded in 96-well plates (3 × 104 cells/well). Before transfection, the medium was changed to Opti-MEM without supplements. Transient transfection was carried out using Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol with pFR-Luc (Stratagene), pRL-SV40 (Promega), and the corresponding Gal4-fusion nuclear receptor plasmid. At 5 h after transfection, medium was changed to Opti-MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and now additionally containing 0.1% DMSO and the respective test compound or 0.1% DMSO alone as untreated control. Each concentration was tested in duplicate, and each experiment was repeated independently five times. Following overnight (12–14 h) incubation with the test compounds, cells were assayed for luciferase activity using Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol. Luminescence was measured with a Spark 10 M luminometer (Tecan Deutschland GmbH). Normalization of transfection efficiency and cell growth was done by division of firefly luciferase data by renilla luciferase data and multiplying the value by 1000, resulting in relative light units (RLU). Fold activation was obtained by dividing the mean RLU of a test compound at a respective concentration by the mean RLU of the untreated control. Relative activation was obtained by dividing the fold activation of a test compound at a respective concentration by the fold activation of a respective reference agonist at 1 μM (PPARα: GW7647; PPARγ: pioglitazone; PPARδ: L165,041; LXRα/β: T0901317; RXRα/β/γ: bexarotene; RARα/β/γ: tretinoin; VDR: calcitriol; and THRα/β: triiodothyronine).
Reporter Gene Assay for Human Full-Length FXR
Plasmids
pcDNA3-hFXR contains the sequence of human FXR and was already published elsewhere.44 pGL3basic (Promega Corporation, Fitchburg, WI) was used as a reporter plasmid, with a shortened construct of the promotor of the bile salt export protein (BSEP) cloned into the SacI/NheI cleavage site in front of the luciferase gene.45 pRL-SV40 (Promega) was transfected as a control for normalization of transfection efficiency and cell growth. pSG5-hRXR was also already published elsewhere.46
Assay Procedure
HeLa cells were grown in high-glucose DMEM supplemented with 10% FCS, sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2. At 24 h before transfection, HeLa cells were seeded in 96-well plates with a density of 8000 cells/well. At 3.5 h before transfection, the medium was changed to high-glucose DMEM, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.5% charcoal-stripped FCS. Transient transfection of HeLa cells with BSEP-pGL3, pRL-SV40, and the expression plasmids pcDNA3-hFXR and pSG5-hRXR was carried out using the calcium phosphate transfection method. At 16 h after transfection, the medium was changed to high-glucose DMEM supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.5% charcoal-stripped FCS. At 24 h after transfection, the medium was changed to DMEM without phenol red, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), l-glutamine (2 mM), and 0.5% charcoal-stripped FCS, now additionally containing 0.1% DMSO and the respective test compound or 0.1% DMSO alone as untreated control. Each concentration was tested in triplicate, and each experiment was repeated independently five times. Following 24 h of incubation with the test compounds, cells were assayed for luciferase activity using Dual-Glo luciferase assay system (Promega) according to the manufacturer’s protocol. Luminescence was measured with a Spark 10 M luminometer (Tecan Deutschland GmbH). Normalization of transfection efficiency and cell growth was done by division of firefly luciferase data by renilla luciferase data multiplied by 1000 resulting in RLU. Fold activation was obtained by dividing the mean RLU of the tested compound at a respective concentration by the mean RLU of untreated control. Relative activation was obtained by dividing the fold activation of the tested compound at a respective concentration by the fold activation of FXR full agonist GW4064 at 1 μM. EC50 and standard error of the mean values were calculated from the mean relative activation values by SigmaPlot 2001 (Systat Software GmbH, Erkrath, Germany) using a four-parameter logistic regression). The assay was validated with FXR agonists OCA (EC50 = 0.16 ± 0.02 μM, 87 ± 3% max relative activity), CDCA (EC50 = 18 ± 1 μM, 88 ± 3% max relative activity), and GW4064 (EC50 = 0.51 ± 0.16 μM, 1 μM defined as 100%).47
sEH Activity Assay
sEH inhibitory potency was determined in a fluorescence-based 96-well sEH activity assay using purified recombinant human enzyme.37 Nonfluorescent (3-phenyloxiranyl)acetic acid cyano-(6-methoxynapthalen-2-yl)methyl ester48,49 (PHOME) was used as substrate which is hydrolyzed by sEH to fluorescent 6-methoxynaphthaldehyde. Purified recombinant human sEH (in BisTris buffer, pH 7, with 0.1 mg/mL BSA containing a final concentration of 0.01% Triton-X 100) was preincubated with test compounds (in DMSO, final DMSO concentration: 1%) for 30 min at room temperature. Then, substrate was added (final concentration 50 μM), and hydrolysis of the substrate was determined by measuring fluorescent product formation on a Tecan Infinite F200 Pro (λem = 330 nm, λex = 465 nm) for 30 min (one point per minute). A blank control (no protein and no compound) as well as a positive control (no compound) were executed. All experiments were conducted in triplicate and repeated in five independent experiments. For IC50 calculation, the dose–response curves of increasing compound concentrations were recorded.
Quantification of FXR- and PPAR-Regulated Gene Expression in HepG2 Cells
FXR target gene quantification was carried out as described previously.47 HepG2 cells were grown in high-glucose DMEM, supplemented with 10% FCS, 1 mM SP, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2. Cells were seeded in 6-well plates (2 × 106 per well). at 24 h after seeding, the medium was changed to MEM supplemented with 1% charcoal stripped FCS, penicillin (100 U/mL), streptomycin (100 μg/mL), and 2 mM l-glutamine. After an additional 24 h, cells were incubated with test compound 41 (0.1, 0.3, and 1 μM), obeticholic acid (1 μM), or elafibranor (1 μM) dissolved in the same medium with 0.1% DMSO or medium with 0.1% DMSO alone as untreated control for 12 h. The cells were then harvested, washed with cold phosphate-buffered saline (PBS), and directly used for RNA extraction. Total RNA was extracted using the total RNA Mini Kit (R6834–02, Omega Bio-Tek, Inc., Norcross, GA). A total of 2 μg of RNA was then reverse-transcribed into cDNA using a high-capacity cDNA reverse transcription kit (4368814, Thermo Fischer Scientific, Inc.) according to the manufacturer’s protocol. FXR-regulated gene expression was studied by quantitative real-time PCR analysis with a StepOnePlus System (Life Technologies, Carlsbad, CA) using PowerSYBRGreen (Life Technologies; 12.5 μL/well). Each sample was set up in duplicate and repeated in five independent experiments. Data was analyzed by the comparative ΔΔCT method with glycerinaldehyde 3-phosphate dehydrogenase (GAPDH) as the reference gene. The following primers (for the human genes) were used: hGAPDH: 5′-ATA TGA TTC CAC CCA TGG CA (forward), 5′-GAT GAT GAC CCT TTT GGC TC (reverse); hSHP: 5′-GCT GTC TGG AGT CCT TCT GG (forward), 5′-CCA ATG ATA GGG CGA AAG AAG AG (reverse); hCYP7A1:5′-CAC CTT GAG GAC GGT TCC TA (forward), 5′-CGA TCC AAA GGG CAT GTA GT (reverse); hBSEP: 5′-CAT GGT GCA AGA AGT GCT GAG T (forward), 5′-AAG CGA TGA GCA ACT GAA ATG AT (reverse); hSREBP1c: 5′-GGA GGG GTA GGG CCA ACG GCC T (forward), 5′-CAT GTC TTC GAA AGT GCA ATC C (reverse).
Microsomal Stability Assay
The solubilized test compound (5 μL, final concentration 10 μM in phosphate buffer (0.1 M, pH 7.4)) was preincubated at 37 °C in 432 μL of phosphate buffer (0.1 M, pH 7.4) together with a 50 μL of NADPH regenerating system (30 mM glucose-6-phosphate, 4 U/mL glucose-6-phosphate dehydrogenase, 10 mM NADP, 30 mM MgCl2). After 5 min, the reaction was started by the addition of 13 μL of microsome mix from the liver of Sprague–Dawley rats (Invitrogen; 20 mg protein/mL in 0.1 M phosphate buffer) in a shaking water bath at 37 °C. The reaction was stopped by addition of 250 μL of ice-cold methanol at 0, 15, 30, and 60 min. The samples were diluted with 250 μL of DMSO and centrifuged at 4 °C and 10 000g for 5 min. The supernatants were analyzed, and the test compound was quantified by HPLC: mobile phase: MeOH 83%/H2O 17%/formic acid 0.1%; flow-rate: 1 mL/min; stationary phase: MultoHigh Phenyl phase, 5 μm, 250 × 4, precolumn, phenyl, 5 μm, 20 × 4; detection wavelength: 330 and 254 nm; injection volume: 50 μL. Control samples were also analyzed to check the test compound’s stability in the reaction mixture. The first control was without NADPH, which is needed for the enzymatic activity of the microsomes; the second control was with inactivated microsomes (incubated for 20 min at 90 °C). The third control was without test compound (to determine the baseline). The amounts of the test compound were quantified by an external calibration curve, where data are expressed as the mean ± SEM of single determinations obtained in five independent experiments.
DIO NASH Model in Mice
The DIO NASH model was carried out by the contract research organization Gubra (Hørsholm, Denmark) on a fee-for-service basis. All animal experiments were conducted in accordance with Gubra’s bioethical guidelines, which are fully compliant to internationally accepted principles for the care and use of laboratory animals. The animals were checked at minimum once daily for signs of abnormal behavior, abnormal locomotor activity, ataxia, or clinical signs of disease (lack of grooming, raised fur, signs of pain upon handling, loss of excessive body weight).
Animals and Treatment
Male C57BL/6JRj mice (purchased from Janvier Laboratories, France at 5 weeks of age) were used for the study. The animals were housed in a temperature- and humidity-controlled room (19–23 °C and 40–60%) and maintained in a 12 h light/12 h dark cycle. Mice received the Gubra AMLN NASH (GAN; D09100310, Research Diet, US; 40% fat (primarily palm oil), 40% carbohydrate (20% fructose), and 2% cholesterol) diet for 41 weeks before treatment start. Prior to treatment, all animals underwent liver biopsy for histological conformation of liver disease (steatosis score ≥ 2 and fibrosis stage ≥ 1) using the nonalcoholic fatty liver disease activity scoring (NAS) and fibrosis staging system. Randomization and stratification to treatment was carried out according to quantitative collagen staining (Picro-Sirius Red). GAN DIO-NASH mice (n = 16 per group) received treatment (p.o.) with vehicle, MH (10 mg/kg, b.i.d.), DM (10 mg/kg, b.i.d.), or obeticholic acid (30 mg/kg, q.d., plus vehicle) for 12 weeks. Water containing 1% hydroxypropyl methylcellulose (HPMC)/Tween 80 (99:1) served as vehicle. Mice were then sacrificed by cardiac puncture under isoflurane anesthesia for histopathological and biochemical analysis.
Evaluation
For prebiopsies, mice were anaesthetized by inhalation anesthesia using isoflurane (2–3%). A small abdominal incision was made in the midline, and the left lateral lobe of the liver was exposed. A cone-shaped wedge of liver tissue (approximately 50 mg) was excised from the distal portion of the lobe and fixated in 10% neutral buffered formalin (10% NBF) for histology. The cut surface of the liver was instantly electrocoagulated using bipolar coagulation (ERBE VIO 100 electrosurgical unit). The liver was returned to the abdominal cavity, and the abdominal wall was sutured. The skin was closed with staples. For postoperative recovery, mice received carprofen (5 mg/kg) administered subcutaneously on operation day and post-operation days 1 and 2. Triglycerides (TG), total cholesterol (TC), alanine transaminase (ALT), and aspartate transaminase (AST) levels were determined from blood samples collected at week 4 of treatment in heparinized tubes. Plasma glucose and insulin were determined from blood samples collected at week 11 of treatment. Plasma was separated and stored at −80°C until analysis. TG, TC, ALT, AST, glucose, and insulin were measured using commercial kits (Roche Diagnostics) on the cobas c 501 autoanalyzer according to the manufacturer’s instructions. After termination by heart puncture, livers were collected and weighed. Specific liver samples and biopsies were dissected and processed. The liver was divided into left lateral lobe, medial lobe, right lateral lobe, and caudal lobe. The left lateral lobe was used for the prebiopsy (not applicable at termination). The liver post biopsy (∼200 mg, less than 0.7 × 0.5 cm2) was cut 4 mm from the prebiopsy site and with an edge. The tissue was collected in paraformaldehyde. The medial lobe was sectioned and snap-frozen in liquid nitrogen for later analysis. One piece (25 ± 5 mg) was dissected and used for TG/TC analysis. Biopsy tissues were cut at 10 μm on a Cryostat, and the sections were mounted on precooled PEN membrane frame slides (ThermoFisher), quickly transferred to precooled 75% EtOH, and stored at −20 °C. Liver samples were fixed in formalin, paraffin-embedded, and sections were stained. For H&E staining, the slides were incubated in Mayer’s Hematoxylin (Dako), washed in tap water, stained in Eosin Y solution (Sigma-Aldrich), hydrated, and cover-slipped. For PSR staining, the slides were incubated in Weigert’s iron hematoxylin (Sigma-Aldrich), washed in tap water, stained in PSR (Sigma-Aldrich), and washed twice in acidified water. Excess water was removed by shaking the slides, and the slides were then dehydrated in three changes of 100% ethanol, cleared in xylene, and cover-slipped. For type I collagen (Southern Biotech, catalog no. 1310–01), alpha-smooth muscle actin (α-SMA; Abcam, catalog no. Ab124964) and galectin-3 analysis (Biolegend, catalog no. 125402), immune-histochemistry (IHC) was carried out using standard procedures. Briefly, after antigen retrieval and blocking of endogenous peroxidase activity, slides were incubated with primary antibody. The primary antibody was detected using a polymeric HRP-linker antibody conjugate. Next, the primary antibody was visualized with DAB as chromogen. Finally, sections were counterstained in hematoxylin and coverslipped. Samples were scored for NAS by a specialist using the clinical criteria outlined by Kleiner et al.500 The total NAS score represents the sum of scores for steatosis, inflammation, and ballooning, and ranges from 0 to 8.
Statistical Evaluation
Data from in vitro experiments are expressed as mean ± SEM; data from in vivo experiments are expressed as mean ± SD. To determine statistical significance between groups, data was analyzed using GraphPad Prism 7 (GraphPad Software Inc., USA) as follows: Normality of data distribution was analyzed by Shapiro–Wilk’s test (p < 0.05 for normal distribution). For normally distributed data, one-way ANOVA with Dunnett’s test was carried out. For not normally distributed data, Kruskal–Wallis one-way analysis of variance was applied followed by Dunn’s test to calculate significance. Pre- to post-treatment comparisons were analyzed by one-sided Fisher’s exact test with Bonferroni correction (scores) or paired t-test: p values < 0.05 were considered as statistically significant, and p values ≤ 0.1 were assumed a trend or tendency. Significance levels are denoted as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Acknowledgments
This research was financially supported by the Else-Kroener-Fresenius-Foundation. The authors thank Jason Wang, Martin Raditsch, and Matthias Götz for support.
Glossary
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BSEP
bile salt export protein
- CCR
C–C chemokine receptor
- CDCA
chenodeoxycholic acid
- CYP7A1
cholesterol 7α hydroxylase
- DIO
diet-induced obesity
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- Gal3
Galectin-3
- GAPDH
glycerinaldehyde 3-phosphate dehydrogenase
- GLP-1
glucagon-like peptide
- HP
hydroxyproline
- LBD
ligand binding domain
- LXR
liver X receptor
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFL activity score
- NASH
nonalcoholic steatohepatitis
- OCA
obeticholic acid
- PHOME
(3-phenyloxiranyl)acetic acid cyano-(6-methoxynaphthalen-2-yl)methyl ester
- PPAR
peroxisome proliferator-activated receptor
- PSR
Picrosirius Red
- PK
pharmacokinetics
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- sEH
soluble epoxide hydrolase
- SGLT-2
sodium-dependent glucose transporter 2
- SHP
small heterodimer partner
- SREBP1c
sterol regulatory elements binding protein 1c
- TC
total cholesterol
- TG
triglycerides
- THR
thyroid hormone receptor
- VDR
vitamin D receptor
The authors declare the following competing financial interest(s): M.H., J.S., E.P., and D.M. are inventors of WO2018215610A1 which claims compounds described in this study.
References
- Buzzetti E.; Pinzani M.; Tsochatzis E. A. (2016) The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metab., Clin. Exp. 65 (8), 1038–1048. 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- MacHado M. V.; Diehl A. M. (2016) Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 150 (8), 1769–1777. 10.1053/j.gastro.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl A. M.; Day C. (2017) Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 377 (21), 2063–2072. 10.1056/NEJMra1503519. [DOI] [PubMed] [Google Scholar]
- Tilg H.; Adolph T. E.; Moschen A. R. (2021) Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 73 (2), 833–842. 10.1002/hep.31518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida Y.; Okanoue T.; Nakajima A. (2019) Phase 3 Drug Pipelines in the Treatment of Non-Alcoholic Steatohepatitis. Hepatol. Res. 49 (11), 1256–1262. 10.1111/hepr.13425. [DOI] [PubMed] [Google Scholar]
- Sumida Y.; Yoneda M.; Toyoda H.; Yasuda S.; Tada T.; Hayashi H.; Nishigaki Y.; Suzuki Y.; Naiki T.; Morishita A.; Tobita H.; Sato S.; Kawabe N.; Fukunishi S.; Ikegami T.; Kessoku T.; Ogawa Y.; Honda Y.; Nakahara T.; et al. (2020) Common Drug Pipelines for the Treatment of Diabetic Nephropathy and Hepatopathy: Can We Kill Two Birds with One Stone?. Int. J. Mol. Sci. 21 (14), 4939. 10.3390/ijms21144939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. T.; Mao Y. M. (2020) Development of New Drugs for the Treatment of Nonalcoholic Steatohepatitis. J. Dig. Dis. 21 (1), 3–11. 10.1111/1751-2980.12830. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri B. A.; Loomba R.; Sanyal A. J.; Lavine J. E.; Van Natta M. L.; Abdelmalek M. F.; Chalasani N.; Dasarathy S.; Diehl A. M.; Hameed B.; Kowdley K. V.; McCullough A.; Terrault N.; Clark J. M.; Tonascia J.; Brunt E. M.; Kleiner D. E.; Doo E. (2015) Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 385 (9972), 956–965. 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V.; Harrison S. A.; Francque S.; Bedossa P.; Lehert P.; Serfaty L.; Romero-Gomez M.; Boursier J.; Abdelmalek M.; Caldwell S.; Drenth J.; Anstee Q. M.; Hum D.; Hanf R.; Roudot A.; Megnien S.; Staels B.; Sanyal A.; Mathurin P.; et al. (2016) Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 150 (5), 1147–1159e5. 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- Mullard A. (2020) FDA Rejects NASH Drug. Nat. Rev. Drug Discovery 19 (8), 501. 10.1038/d41573-020-00126-9. [DOI] [PubMed] [Google Scholar]
- Genfit. (2020) GENFIT: Announces Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis, https://ml-eu.globenewswire.com/Resource/Download/38e085e1-66f5-4251-8abe-648d0e7b9ed1.
- Harrison S. A.; Alkhouri N.; Davison B. A.; Sanyal A.; Edwards C.; Colca J. R.; Lee B. H.; Loomba R.; Cusi K.; Kolterman O.; Cotter G.; Dittrich H. C. (2020) Insulin Sensitizer MSDC-0602K in Non-Alcoholic Steatohepatitis: A Randomized, Double-Blind, Placebo-Controlled Phase IIb Study. J. Hepatol. 72 (4), 613–626. 10.1016/j.jhep.2019.10.023. [DOI] [PubMed] [Google Scholar]
- Lefere S.; Devisscher L.; Tacke F. (2020) Targeting CCR2/5 in the Treatment of Nonalcoholic Steatohepatitis (NASH) and Fibrosis: Opportunities and Challenges. Expert Opin. Invest. Drugs 29 (2), 89–92. 10.1080/13543784.2020.1718106. [DOI] [PubMed] [Google Scholar]
- Safadi R.; Konikoff F. M.; Mahamid M.; Zelber-Sagi S.; Halpern M.; Gilat T.; Oren R.; Safadi R.; Konikoff F. M.; Hershkovitz A.; et al. (2014) The Fatty Acid-Bile Acid Conjugate Aramchol Reduces Liver Fat Content in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 12 (12), 2085–2091.e1. 10.1016/j.cgh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- Mudaliar S.; Henry R. R.; Sanyal A. J.; Morrow L.; Marschall H. U.; Kipnes M.; Adorini L.; Sciacca C. I.; Clopton P.; Castelloe E.; Dillon P.; Pruzanski M.; Shapiro D. (2013) Efficacy and Safety of the Farnesoid x Receptor Agonist Obeticholic Acid in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 145 (3), 574–582.e1. 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Dang H.; Li D.; Pang W.; Hammock B. D.; Zhu Y. (2012) Inhibition of Soluble Epoxide Hydrolase Attenuates High-Fat-Diet-Induced Hepatic Steatosis by Reduced Systemic Inflammatory Status in Mice. PLoS One 7 (6), e39165. 10.1371/journal.pone.0039165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T. R.; Bettaieb A.; Kodani S.; Dong H.; Myers R.; Chiamvimonvat N.; Haj F. G.; Hammock B. D. (2015) Inhibition of Soluble Epoxide Hydrolase Attenuates Hepatic Fibrosis and Endoplasmic Reticulum Stress Induced by Carbon Tetrachloride in Mice. Toxicol. Appl. Pharmacol. 286 (2), 102–111. 10.1016/j.taap.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Vicario C.; Alcaraz-Quiles J.; García-Alonso V.; Rius B.; Hwang S. H.; Titos E.; Lopategi A.; Hammock B. D.; Arroyo V.; Clària J. (2015) Inhibition of Soluble Epoxide Hydrolase Modulates Inflammation and Autophagy in Obese Adipose Tissue and Liver: Role for Omega-3 Epoxides. Proc. Natl. Acad. Sci. U. S. A. 112 (2), 536–541. 10.1073/pnas.1422590112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D.; Kalish B. T.; Huang S.; Bielenberg D. R.; Le H. D.; Yang J.; Edin M. L.; Lee C. R.; Benny O.; Mudge D. K.; Butterfield C. E.; Mammoto A.; Mammoto T.; Inceoglu B.; Jenkins R. L.; Simpson M. A.; Akino T.; Lih F. B.; Tomer K. B.; et al. (2013) Epoxyeicosanoids Promote Organ and Tissue Regeneration. Proc. Natl. Acad. Sci. U. S. A. 110 (33), 13528–13533. 10.1073/pnas.1311565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C. C.; Zhang C. Y.; Duan J. X.; Guan X. X.; Yang H. H.; Jiang H. L.; Hammock B. D.; Hwang S. H.; Zhou Y.; Guan C. X.; Liu S. K.; Zhang J. (2020) PTUPB Ameliorates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease via Inhibiting NLRP3 Inflammasome Activation in Mice. Biochem. Biophys. Res. Commun. 523 (4), 1020–1026. 10.1016/j.bbrc.2019.12.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J.; Hardesty J.; Zirnheld K.; McClain C.; Warner D.; Kirpich I. (2020) Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology (Basel) 9 (6), 124. 10.3390/biology9060124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. (2014) Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 57 (19), 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Proschak E.; Stark H.; Merk D. (2019) Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 62 (2), 420–444. 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Drenth J. P. H., and Schattenberg J. M. (2020) The Nonalcoholic Steatohepatitis (NASH) Drug Development Graveyard: Established Hurdles and Planning for Future Success, in Expert Opinion on Investigational Drugs, Taylor and Francis Ltd. [DOI] [PubMed] [Google Scholar]
- Bechtold B., and Clarke J. (2021) Multi-Factorial Pharmacokinetic Interactions: Unraveling Complexities in Precision Drug Therapy, in Expert Opinion on Drug Metabolism and Toxicology, Taylor and Francis Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Rotter M.; Weiser T.; Wittmann S.; Weizel L.; Kaiser A.; Heering J.; Goebel T.; Angioni C.; Wurglics M.; Paulke A.; Geisslinger G.; Kahnt A.; Steinhilber D.; Proschak E.; Merk D. (2017) A Dual Modulator of Farnesoid X Receptor and Soluble Epoxide Hydrolase to Counter Nonalcoholic Steatohepatitis. J. Med. Chem. 60 (18), 7703–7724. 10.1021/acs.jmedchem.7b00398. [DOI] [PubMed] [Google Scholar]
- Hye Khan M. A.; Schmidt J.; Stavniichuk A.; Imig J. D.; Merk D. (2019) A Dual Farnesoid X Receptor/Soluble Epoxide Hydrolase Modulator Treats Non-Alcoholic Steatohepatitis in Mice. Biochem. Pharmacol. 166, 212–221. 10.1016/j.bcp.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen H. H.; Ægidius H. M.; Oró D.; Evers S. S.; Heebøll S.; Eriksen P. L.; Thomsen K. L.; Bengtsson A.; Veidal S. S.; Feigh M.; Suppli M. P.; Knop F. K.; Grønbæk H.; Miranda D.; Trevaskis J. L.; Vrang N.; Jelsing J.; Rigbolt K. T. G. (2020) Human Translatability of the GAN Diet-Induced Obese Mouse Model of Non-Alcoholic Steatohepatitis. BMC Gastroenterol. 20, 210. 10.1186/s12876-020-01356-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ægidius H. M.; Veidal S. S.; Feigh M.; Hallenborg P.; Puglia M.; Pers T. H.; Vrang N.; Jelsing J.; Kornum B. R.; Blagoev B.; Rigbolt K. T. G. (2020) Multi-Omics Characterization of a Diet-Induced Obese Model of Non-Alcoholic Steatohepatitis. Sci. Rep. 10, 1148. 10.1038/s41598-020-58059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J. D.; Veidal S. S.; Fensholdt L. K. D.; Rigbolt K. T. G.; Papazyan R.; Nielsen J. C.; Feigh M.; Vrang N.; Young M.; Jelsing J.; Adorini L.; Hansen H. H. (2019) Combined Obeticholic Acid and Elafibranor Treatment Promotes Additive Liver Histological Improvements in a Diet-Induced Ob/Ob Mouse Model of Biopsy-Confirmed NASH. Sci. Rep. 9, 9046. 10.1038/s41598-019-45178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tølbøl K. S.; Kristiansen M. N. B.; Hansen H. H.; Veidal S. S.; Rigbolt K. T. G.; Gillum M. P.; Jelsing J.; Vrang N.; Feigh M. (2018) Metabolic and Hepatic Effects of Liraglutide, Obeticholic Acid and Elafibranor in Diet-Induced Obese Mouse Models of Biopsy-Confirmed Nonalcoholic Steatohepatitis. World J. Gastroenterol. 24 (2), 179–194. 10.3748/wjg.v24.i2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouihan H.; Will S.; Guionaud S.; Boland M. L.; Oldham S.; Ravn P.; Celeste A.; Trevaskis J. L. (2017) Superior Reductions in Hepatic Steatosis and Fibrosis with Co-Administration of a Glucagon-like Peptide-1 Receptor Agonist and Obeticholic Acid in Mice. Mol. Metab. 6 (11), 1360–1370. 10.1016/j.molmet.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S.; Katsuno K.; Isaji M.; Nagasawa T.; Buehrer B.; Walker S.; Wilkison W. O.; Cheatham B. (2015) Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 5 (3), 190–198. 10.1016/j.jceh.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tølbøl K. S.; Kristiansen M. N. B.; Hansen H. H.; Veidal S. S.; Rigbolt K. T. G.; Gillum M. P.; Jelsing J.; Vrang N.; Feigh M. (2018) Metabolic and Hepatic Effects of Liraglutide, Obeticholic Acid and Elafibranor in Diet-Induced Obese Mouse Models of Biopsy-Confirmed Nonalcoholic Steatohepatitis. World J. Gastroenterol. 24 (2), 179–194. 10.3748/wjg.v24.i2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Klingler F.-M.; Proschak E.; Steinhilber D.; Schubert-Zsilavecz M.; Merk D. (2015) NSAIDs Ibuprofen, Indometacin, and Diclofenac Do Not Interact with Farnesoid X Receptor. Sci. Rep. 5, 14782. 10.1038/srep14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierle S.; Helmstädter M.; Schmidt J.; Hartmann M.; Horz M.; Kaiser A.; Weizel L.; Heitel P.; Proschak A.; Hernandez-Olmos V.; Proschak E.; Merk D. (2020) Dual Farnesoid X Receptor/Soluble Epoxide Hydrolase Modulators Derived from Zafirlukast. ChemMedChem 15 (1), 50–67. 10.1002/cmdc.201900576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blöcher R.; Lamers C.; Wittmann S. K.; Merk D.; Hartmann M.; Weizel L.; Diehl O.; Brüggerhoff A.; Boß M.; Kaiser A.; Schader T.; Göbel T.; Grundmann M.; Angioni C.; Heering J.; Geisslinger G.; Wurglics M.; Kostenis E.; Brüne B.; et al. (2016) N-Benzylbenzamides: A Novel Merged Scaffold for Orally Available Dual Soluble Epoxide Hydrolase/Peroxisome Proliferator-Activated Receptor γ Modulators. J. Med. Chem. 59 (1), 61–81. 10.1021/acs.jmedchem.5b01239. [DOI] [PubMed] [Google Scholar]
- Pollinger J.; Gellrich L.; Schierle S.; Kilu W.; Schmidt J.; Kalinowsky L.; Ohrndorf J.; Kaiser A.; Heering J.; Proschak E.; Merk D. (2019) Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 62 (4), 2112–2126. 10.1021/acs.jmedchem.8b01848. [DOI] [PubMed] [Google Scholar]
- Gellrich L.; Heitel P.; Heering J.; Kilu W.; Pollinger J.; Goebel T.; Kahnt A.; Arifi S.; Pogoda W.; Paulke A.; Steinhilber D.; Proschak E.; Wurglics M.; Schubert-Zsilavecz M.; Chaikuad A.; Knapp S.; Bischoff I.; Fürst R.; Merk D. (2020) L-Thyroxin and the Nonclassical Thyroid Hormone TETRAC Are Potent Activators of PPARγ. J. Med. Chem. 63 (13), 6727–6740. 10.1021/acs.jmedchem.9b02150. [DOI] [PubMed] [Google Scholar]
- Merk D.; Sreeramulu S.; Kudlinzki D.; Saxena K.; Linhard V.; Gande S. L.; Hiller F.; Lamers C.; Nilsson E.; Aagaard A.; Wissler L.; Dekker N.; Bamberg K.; Schubert-Zsilavecz M.; Schwalbe H. (2019) Molecular Tuning of Farnesoid X Receptor Partial Agonism. Nat. Commun. 10 (1), 2915. 10.1038/s41467-019-10853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau O.; Wurglics M.; Paulke A.; Zitzkowski J.; Meindl N.; Bock A.; Dingermann T.; Abdel-Tawab M.; Schubert-Zsilavecz M. (2006) Carnosic Acid and Carnosol, Phenolic Diterpene Compounds of the Labiate Herbs Rosemary and Sage, Are Activators of the Human Peroxisome Proliferator-Activated Receptor Gamma. Planta Med. 72 (10), 881–887. 10.1055/s-2006-946680. [DOI] [PubMed] [Google Scholar]
- Heitel P.; Achenbach J.; Moser D.; Proschak E.; Merk D. (2017) DrugBank Screening Revealed Alitretinoin and Bexarotene as Liver X Receptor Modulators. Bioorg. Med. Chem. Lett. 27 (5), 1193–1198. 10.1016/j.bmcl.2017.01.066. [DOI] [PubMed] [Google Scholar]
- Flesch D.; Cheung S.-Y.; Schmidt J.; Gabler M.; Heitel P.; Kramer J. S.; Kaiser A.; Hartmann M.; Lindner M.; Lüddens-Dämgen K.; Heering J.; Lamers C.; Lüddens H.; Wurglics M.; Proschak E.; Schubert-Zsilavecz M.; Merk D. (2017) Non-Acidic Farnesoid X Receptor Modulators. J. Med. Chem. 60 (16), 7199–7205. 10.1021/acs.jmedchem.7b00903. [DOI] [PubMed] [Google Scholar]
- Steri R.; Achenbach J.; Steinhilber D.; Schubert-Zsilavecz M.; Proschak E. (2012) Investigation of Imatinib and Other Approved Drugs as Starting Points for Antidiabetic Drug Discovery with FXR Modulating Activity. Biochem. Pharmacol. 83 (12), 1674–1681. 10.1016/j.bcp.2012.02.027. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan M.; Balasubramanian N.; Makishima M.; Mangelsdorf D. J.; Suchy F. J. (2001) Human Bile Salt Export Pump Promoter Is Transactivated by the Farnesoid X Receptor/Bile Acid Receptor. J. Biol. Chem. 276 (31), 28857–28865. 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- Seuter S.; Väisänen S.; Rådmark O.; Carlberg C.; Steinhilber D. (2007) Functional Characterization of Vitamin D Responding Regions in the Human 5-Lipoxygenase Gene. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1771 (7), 864–872. 10.1016/j.bbalip.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Merk D.; Lamers C.; Ahmad K.; Carrasco Gomez R.; Schneider G.; Steinhilber D.; Schubert-Zsilavecz M. (2014) Extending the Structure-Activity Relationship of Anthranilic Acid Derivatives as Farnesoid X Receptor Modulators: Development of a Highly Potent Partial Farnesoid X Receptor Agonist. J. Med. Chem. 57 (19), 8035–8055. 10.1021/jm500937v. [DOI] [PubMed] [Google Scholar]
- Jones P. D.; Wolf N. M.; Morisseau C.; Whetstone P.; Hock B.; Hammock B. D. (2005) Fluorescent Substrates for Soluble Epoxide Hydrolase and Application to Inhibition Studies. Anal. Biochem. 343 (1), 66–75. 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf N. M.; Morisseau C.; Jones P. D.; Hock B.; Hammock B. D. (2006) Development of a High-Throughput Screen for Soluble Epoxide Hydrolase Inhibition. Anal. Biochem. 355, 71–80. 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner D. E.; Brunt E. M.; Van Natta M.; Behling C.; Contos M. J.; Cummings O. W.; Ferrell L. D.; Liu Y.-C.; Torbenson M. S.; Unalp-Arida A. (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]