

Abstract
Guanine nucleotide-binding proteins (G proteins) transduce extracellular signals received by G protein-coupled receptors (GPCRs) to intracellular signaling cascades. While GPCRs represent the largest class of drug targets, G protein inhibition has only recently been recognized as a novel strategy for treating complex diseases such as asthma, inflammation, and cancer. The structurally similar macrocyclic depsipeptides FR900359 (FR) and YM-254890 (YM) are potent selective inhibitors of the Gq subfamily of G proteins. FR and YM differ in two positions, FR being more lipophilic than YM. Both compounds are utilized as pharmacological tools to block Gq proteins in vitro and in vivo. However, no detailed characterization of FR and YM has been performed, which is a prerequisite for the compounds’ translation into clinical application. Here, we performed a thorough study of both compounds’ physicochemical, pharmacokinetic, and pharmacological properties. Chemical stability was high across a large range of pH values, with FR being somewhat more stable than YM. Oral bioavailability and brain penetration of both depsipeptides were low. FR showed lower plasma protein binding and was metabolized significantly faster than YM by human and mouse liver microsomes. FR accumulated in lung after chronic intratracheal or intraperitoneal application, while YM was more distributed to other organs. Most strikingly, the previously observed longer residence time of FR resulted in a significantly prolonged pharmacologic effect as compared to YM in a methacholine-induced bronchoconstriction mouse model. These results prove that changes within a molecule which seem marginal compared to its structural complexity can lead to crucial pharmacological differences.
Keywords: FR900359, Gq inhibitor, metabolic stability, physicochemical properties, pharmacokinetic behavior, YM-254890
1. Introduction
The macrocyclic depsipeptide FR900359 (FR, 1) isolated from the higher plant Ardisia crenata,1 in the leaves of which it is produced by the bacterial endophyte Candidatus Burkholderia crenata,2 acts as a selective Gq protein inhibitor.3 The structurally closely related natural product YM-254890 (YM, 2), which had been isolated from a culture broth of Chromobacterium sp. QS3666 during the search for novel platelet aggregation inhibitors,4 was reported to block Gq proteins with similar potency as 1. These Gq protein inhibitors have become indispensable tool compounds to study Gq protein-mediated signaling induced by G protein-coupled receptors (GPCRs).5−12 In contrast to the Gi protein inhibitor pertussis toxin and the Gs protein activator choleratoxin, proteins which have been essential tools for studying GPCR signaling for decades, FR and YM are small, macrocyclic, druglike molecules. Several studies provided strong evidence for the potential of Gq protein inhibitors as novel pharmacological agents to treat complex diseases, such as obesity,13 asthma,14 and cancer.17−20 Analogs have been isolated from bacteria21−23 or prepared by chemical synthesis,12,24−26 but none of them were significantly more potent than the natural products; in fact, most of them displayed much lower Gq-inhibitory potency or were even inactive.
FR and YM differ only in two residues (see Figure 1), and both inhibitors have been generally regarded as exchangeable due to their almost identical structures. However, in a recent study, we observed that tritiated FR binding to Gq proteins displayed a significantly longer residence time than radiolabeled YM suggesting pseudoirreversible binding of FR but not of YM.27 Thus, despite their striking structural similarity, these results indicated that both compounds may, in fact, behave differently. However, not much is known about FR’s and YM’s physicochemical and pharmacokinetic properties, and whether these might result in pharmacodynamic consequences. Such information would be important for guiding biological experiments including preclinical studies and for translation of this class of compounds into the clinics. In the present study, we present essential data providing a basis for future in vivo investigations utilizing FR and YM as tool compounds and promising therapeutic drugs.
Figure 1.
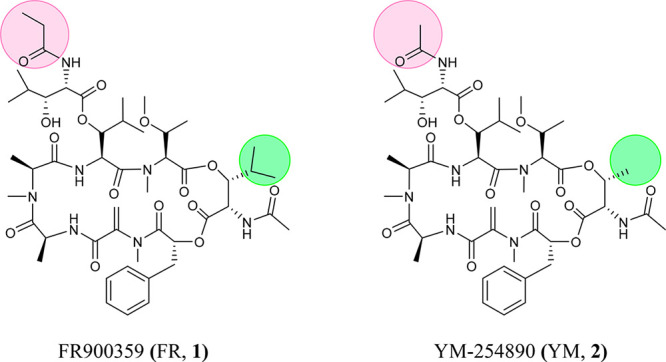
Chemical structures of the macrocyclic depsipeptidic Gq protein inhibitors FR (1) and YM (2).
2. Results and Discussion
In a first step, we collected, calculated, and/or determined the physicochemical properties of FR and YM, which are relevant for their application as drug molecules (see Table 1).
Table 1. Physicochemical and Druglike Properties of FR and YM.
| FR | YM | ||
|---|---|---|---|
| exact mass (Da) | 1001.53 | 959.49 | |
| heavy atoma count | 71 | 68 | |
| number of defined stereocenters | 11 | 11 | |
| specific rotationb ([α]D20) | –54.8 | –64.5 | |
| rotatable bond count | 15 | 13 | |
| polar surface area | 285 Å2 | 285 Å2 | |
| calculated logP valuec | 1.86 | 1.37 | |
| number of hydrogen bond donors | 5 | 5 | |
| number of hydrogen bond acceptors | 22 | 22 | |
| solubility in phosphate-buffered saline containing 1% dimethyl sulfoxide (DMSO)d | 189 ± 17 μM | 88 ± 12 μM | |
| plasma protein binding | 35% | 79% | |
| residence time of tritiated derivative at human platelet membranese | at 37 °C | 92.1 min | 3.8 min |
| at 21 °C | 343.3 min | 13.4 min | |
2.1. Physicochemical and Druglike Properties
Macrocyclic compounds have recently gained much attention in drug development due to their favorable pharmacokinetic properties despite their large molecular weight, which does not conform to Lipinsky’s rule of 5.28 The macrocyclic Gq protein inhibitors FR and YM, which are depsipeptides, differ only in two substituents: FR contains a propionyl instead of an acetyl group and an isopropyl instead of a methyl group (see Figure 1). FR is therefore larger (exact mass: 1001.53 Da) than YM (959.49 Da) and somewhat more lipophilic. Calculated logP values are 1.37 for YM-254890 compared to 1.86 for FR900359 (Table 1). FR and YM are highly complex molecules containing 11 stereocenters and 15 (FR) or 13 (YM) rotatable bonds, respectively. Both compounds were predicted not to be brain-permeable due to their high polar surface area (285 Å2) and their large number of hydrogen bond donors (5) and acceptors (22). N-Methylation of three of the peptide bonds increases their lipophilicity, which is, however, altogether moderate. The compounds are sufficiently and similarly water-soluble (FR, 189 μM; YM, 88 μM; kinetic solubility). Both display low to moderate plasma protein binding; interestingly, the bound proportion of the more lipophilic FR was significantly lower (35%) compared to that of the more polar analog YM (79%). This property of FR and its relatively high water solubility may be due to the bulky isopropyl substituent, which likely affects the molecule’s conformation, crystal packing, and interactions.
One striking difference between FR and YM is their residence time at Gq proteins (Table 1). FR binds pseudoirreversibly to Gq proteins and displays a residence time of 92.1 min at 37 °C compared to 3.8 min for YM.27 This finding was the first indication that FR and YM are not exchangeable with respect to therapeutic applications despite their high degree of structural similarity.
Thus, FR and YM are macrocyclic molecules with very similar physicochemical properties. The main differences are their lipophilicity, percentage of plasma protein binding, and target residence time.
2.2. Caco-2 Cell Permeation
To experimentally assess the potential of the two Gq protein inhibitors for oral bioavailability, Caco-2 cell permeability assays were performed.29,30 Caco-2 cells, a human colon adenocarcinoma cell line, are cultured on transwell cell culture plates. After differentiation, they resemble intestinal epithelial cells characterized by the formation of a polarized monolayer with a well-defined brush border on the apical surface expressing various transporters and enzymes as well as intercellular junctions. Results of Caco-2 permeability tests are used to predict intestinal absorption and consequently the bioavailability of a drug when orally administered. Both FR and YM possessed a low apparent permeability coefficient (Papp) of 0.4 × 10–6 cm/s and 0.1 × 10–6 cm/s, respectively (see Table 2). Apical to basolateral transport rates for both compounds were found to be well below the transport rate of the well-penetrating reference drug testosterone and in a similar range as those for the control drugs atenolol and erythromycin, both of which display low peroral absorption. Perhaps due to its higher lipophilicity, Caco-2 cell permeation was somewhat higher for FR as compared to YM. Interestingly, YM showed a Papp of 20.9 × 10–6 cm/s for basolateral to apical transport resulting in a ratio of 182, indicating very high efflux. This ratio was even higher than that determined for erythromycin included as a reference compound for drugs that are substrates of P-glycoprotein 1 (Pgp, multidrug resistance protein MDR1). In general, a ratio of >2 indicates Pgp transport.31 The determined ratio shows that YM is indeed a substrate of an efflux transporter such as Pgp. The more lipophilic FR displayed an efflux/influx ratio of 29 indicating that it possesses Pgp substrate properties too, although not as strong as observed for YM. Lacking or low permeation could be advantageous to avoid intoxication upon local, e.g., inhalative treatment. Thus, low absorption combined with high efflux will result in very low oral bioavailability.
Table 2. Apparent Transport Rates (Papp) of YM-254890, FR900359, and Control Compounds in Caco-2 Cells.
| compound | direction | Papp·10-6 (cm/s)a | ratio (b-a/a-b)b | permeability |
|---|---|---|---|---|
| YM-254890 | a-b | 0.1 ± 0.0 | 182 | low |
| b-a | 20.9 ± 2.8 | |||
| FR900359 | a-b | 0.4 ± 0.0 | 29 | low |
| b-a | 11.5 ± 0.4 | |||
| testosterone | a-b | 19.2 ± 1.0 | 2 | high |
| b-a | 39.7 ± 5.2 | |||
| erythromycin | a-b | 0.1 ± 0.1 | 106 | low |
| b-a | 11.2 ± 0.4 | |||
| atenolol | a-b | 0.4 ± 0.0 | 7 | low |
| b-a | 2.8 ± 0.4 |
Apparent permeability (values represent means ± SD, n = 3).
High ratio indicates efflux by transporter proteins.
2.3. Chemical Stability
Next, we studied the Gq protein inhibitors’ chemical stability at different pH values including simulated gastric fluid32 (pH 1), weakly basic (pH 9), and more strongly basic conditions (pH 11, see Figure 2). The chemical stability of both compounds, FR and YM, was assessed in aqueous solution at 37 °C. A straightforward high-performance liquid chromatography–mass spectrometry (HPLC-MS) method was developed to detect and quantify both compounds and their potential degradation products. In short, HPLC on a reversed-phase column coupled to a single-quadrupole mass spectrometer equipped with an electrospray ionization source was applied for chromatographic separation, and the extract ion chromatograms (EICs) of both compounds were used for identification and quantification (for details see Methods).
Figure 2.

Chemical stability of FR and YM A. in simulated gastric fluid, pH 1, B. at pH 9, and C. at pH 11 (100 μM starting concentration). Solutions were prepared by adding 50 μL of a 1 mM stock solution in DMSO of FR or YM to 450 μL of an aqueous solution A, B, or C. Values represent means ± SEM from three independent experiments. For details, see Methods.
We did not observe significant degradation of FR and YM in simulated gastric fluid containing hydrochloric acid (pH 1) and the peptidase pepsin indicating high stability in acid and toward proteolytic cleavage. Both compounds were also stable in an aqueous solution of pH 9. While FR appeared to be completely stable, a slight degradation of YM could be observed (see Figure 2). These results show that both Gq inhibitors would likely survive peroral application. In contrast, at a strongly basic pH value of 11, both compounds were degraded, the more lipophilic FR being significantly more stable than YM. YM decomposed rapidly and completely within 20 min, whereas more than 75% of FR was still present after 4 h of incubation. Interestingly, under all conditions, both depsipeptides formed a small amount of an isomer with equal mass as the parent compound. The isomer could be separated by analytical HPLC from the parent compound, but its structure remains unknown. We propose that the formed isomer constitutes a rearrangement product caused by intramolecular transesterification involving the secondary alcoholic function of the compounds (see Figure 3), a hypothesis that needs to be tested in future studies.
Figure 3.
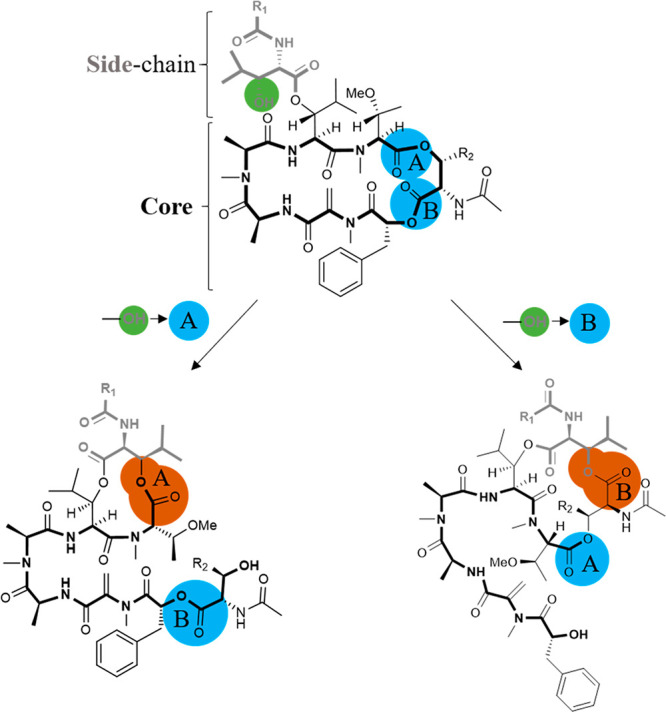
Proposed isomerization reactions (FR: R1 = CH2CH3, R2 = CH(CH3)2; YM: R1 = CH3, R2 = CH3). The secondary alcohol is displayed in green. Esters which possibly take part in the isomerization reaction are highlighted in blue and labeled A or B. Ester bonds newly formed after the reaction are highlighted in dark orange.
Apart from the mass peaks of the parent compounds and the rearrangement products, a rising number and amount of degradation products were observed with an increased incubation time. Under all conditions, but predominantly at pH 11, hydrated products (FR/YM+H2O; 1020.19 g/mol; 978.11 g/mol) and the FR/YM “core” structure, lacking the 3-hydroxy-4-methylpentanoate side chain, were detected (816.95 g/mol and 788.9 g/mol, respectively; see Figure 3). The hydrated compounds (FR/YM+H2O) are likely the result of ester hydrolysis; alternatively, Michael addition reaction to the α,β-unsaturated ketone might occur. These results show that the ester functions of YM and FR are reactive toward nucleophiles, and they support the proposed mechanism for the formation of the observed isomers by intramolecular transesterification. Our results indicate that the chemical stability of both FR and YM under physiological conditions is high. Degradation was mainly observed at strongly basic pH values, and the more lipophilic FR was found to be more stable than YM.
2.4. Metabolic Stability
2.4.1. Stability in Liver Microsomes and Cytochrome P450 Enzyme Inhibition
Next, we studied the metabolic stability of the compounds in liver microsomal preparations; results are summarized in Table 3. Incubation of FR or YM with human (Figure 4A) and mouse (Figure 4B) liver microsomes resulted in degradation of both compounds, which was more pronounced in mouse as compared to human liver. FR revealed a higher apparent intrinsic clearance (CLint, app) as compared to YM. Because of its low half-life of only 5.8 min in mouse and 8.1 min in human liver microsomes, FR was completely metabolized within 60 min in both preparations. YM was more stable with CLint, app values of 82.2 μL/min/mg protein in mouse and 50.8 μL/min/mg protein in human liver microsomes translating into half-lives of 16.9 min (mouse) and 27.3 min (human). There was approximately one-quarter of YM left after 60 min of incubation in human liver microsomes. The apparent intrinsic clearance of verapamil, which is known to be rapidly metabolized33 and was therefore included as a positive control, was in a similar range as that of FR. Based on the measured CLint, app value, an in vivo CLint can be estimated using suitable scaling factors, i.e., microsomal protein content per gram of liver to be 45 mg of microsomal protein per gram of liver tissue and 26 g (human) or 87 g (mouse) of liver tissue per kilogram of body weight.34,35 CLint values of FR and YM in humans were determined to be 200.1 mL/min/kg and 59.4 mL/min/kg, respectively. Typically, a CLint (human) below 15 mL/min/kg, between 15–45 mL/min/kg, and above 45 mL/min/kg can be classified as low, intermediate, and high metabolic degradation, respectively.34,36 Thus, YM and FR were found to be high clearance compounds and do not appear to be suitable for systemic application based on their short half-lives. Due to its somewhat slower metabolic clearance, YM would be preferred for systemic treatment; however, a very short duration of action has to be expected.
Table 3. Metabolic Stability of FR and YM in Human and Mouse Liver Microsomes.
| CLint,app (μL/min/mg protein) |
t1/2 (min) |
|||
|---|---|---|---|---|
| human | mouse | human | mouse | |
| FR900359 | 171.0 | 237.4 | 8.1 | 5.8 |
| YM-254890 | 50.8 | 82.2 | 27.3 | 16.9 |
| verapamil (control) | 134.6 | 335.7 | 10.3 | 4.1 |
Figure 4.

Stability of G protein inhibitors under various conditions: A. human liver microsomes (FR, YM); B. mouse liver microsomes (FR, YM); C. stability in mouse plasma (FR); and D. mouse lung tissue (FR). Data represent means ± SEM (n = 3).
Subsequent metabolite identification studies revealed that N/O-dealkylation at various positions of the depsipetides, as well as hydroxylation (observed only for FR), constituted the main metabolic pathways. Structures of specific metabolites have not been identified due to the structural complexity of the compounds which feature many functional groups that may be susceptible to metabolism.
Subsequently, we investigated potential inhibition of cytochrome P450 (CYP enzymes) that is important for drug metabolism by FR and YM. At a concentration of 1 μM, both compounds exhibited only negligible effects on the investigated CYP enzymes. At a very high concentration of 10 μM, inhibition was still negligible to low (Figure S1 of the Supporting Information), except for CYP3A4, which was inhibited by about 50% (FR: 50%, YM: 56%). FR at 10 μM also displayed moderate inhibition of CYP2C8 (30%) and CYP2C19 (38%), whereas inhibition by 10 μM of YM was below 25%. These results indicate that both Gq inhibitors are not expected to interfere with hepatic metabolism of other molecules.
2.4.2. Stability in Lung Tissue and Blood Plasma
FR, which had been found to be less metabolically stable in liver microsomes than YM, was selected for stability testing in lung tissue and blood plasma of mice. The compound was found to be highly stable in both matrices, with more than 90% of the unaltered compound still present after 4 h of incubation in mouse plasma or lung tissue, respectively. A very small amount was converted to its isomer with equal mass (for the presumed structure see Figure 3). This clearly shows that FR is well suited for local, bronchial application.
2.5. In Vivo Bioavailability and Organ Distribution
Next, we studied the concentrations of intact FR and YM in vivo after different application schemes in mice utilizing a recently developed sensitive LC-MS/MS method combined with an optimized extraction procedure.37
2.5.1. Intratracheal Application
FR or YM (5 μg) was intratracheally (i.t.) applied to mice on 7 consecutive days. The organs were harvested approximately 45 min after the last FR/YM application, subsequently extracted, and analyzed for FR or YM concentration. High levels of both Gq protein inhibitors were detected in lung and kidney (Figure 5A,B). Only low concentrations of YM and FR were found in brain and blood plasma. Liver, intestine, and lung showed significantly different drug levels of FR as compared to YM. While YM concentrations in liver and kidney were significantly higher than those of FR, the opposite was true for lung, in which higher FR levels than YM levels were detected. These differences are likely due to the different metabolic stabilities of the compounds in liver (compare Figure 4B), FR being less metabolically stable than YM. Moreover, different lipophilicities and different residence times may contribute. FR, which displays a very slow dissociation kinetic,27 shows higher accumulation in lung as compared to YM likely because it displays pseudoirreversible binding to the Gq proteins and therefore sticks to its targets at the point of entry. Both compounds are preferentially eliminated via the kidneys and, to a smaller but still measurable extent, through feces.
Figure 5.

A. Concentration of FR ± SEM and B. concentration of YM ± SEM in mouse tissues after intratracheal application of 5 μg of drug on 7 consecutive days. FR and YM levels in organs from three mice were determined.
We subsequently studied FR, the preferred Gq inhibitor for intratracheal application, for 21 days. Even after a 3-week treatment, FR levels in lung remained high and were at about the same level as those measured after 1 week (see Figure 6A). These results show that long-term treatment in lung, e.g., for antiasthmatic therapy, is feasible resulting in constantly high drug levels.
Figure 6.

A. Concentration of FR ± SEM in various mouse tissues after intratracheal application of 2.5 μg of FR twice a day for 3 weeks. B. Concentration of FR ± SEM in various mouse tissues after intraperitoneal application of 10 μg of FR for 3 weeks (administration from Monday to Friday). FR levels in organs from three mice were determined.
2.5.2. Intraperitoneal Application
In further in vivo studies, FR was administered intraperitoneally for 3 weeks. Subsequent extraction and analysis showed that the highest concentrations of FR were again found in the lung (Figure 6B). Direct comparison of i.p. versus i.t. application of FR is shown in Figure 6. In general, FR displayed a similar distribution in the body after both routes of application, but FR levels were overall slightly higher after intratracheal as compared to i.p. application despite slightly lower applied doses. Interestingly, systemic i.p. application resulted in FR levels in the lung comparable to those after local i.t. application. This may be due to high Gq expression in the lung combined with pseudoirreversible binding of FR. FR concentrations determined in the kidneys were significantly higher after intratracheal application, whereas moderate amounts of FR in the gut could only be measured after i.p. application. Independent of the way of application, FR could also be found in the eyes, liver, and intestine. Again, only low to marginal concentrations were found in the brain.
Both in vivo studies showed that FR and YM are not or only marginally able to cross the blood-brain barrier but can be detected in other vital organs, partly in relatively high concentrations in organs which most likely take part in their metabolism or excretion (liver, kidney). Because FR and YM possess low nanomolar IC50/Ki values at their target protein,22,27 the determined drug concentrations can be expected to be sufficient for pharmacological activity. Besides lung, eye diseases, e.g., uveal melanoma,16−18 would be the primary targets for local or systemic FR treatment.
2.6. Pharmacological Effects of FR and YM in Mouse Lung Determined by Plethysmography
The results obtained so far in in vitro and in vivo studies indicated that FR and YM may be particularly suited for treating lung disease, since both drugs accumulate in this organ (see Figures 5 and 6). To find out whether the structurally similar Gq inhibitors FR and YM display the same pharmacological properties in vivo, we studied and compared their effect on airways in mice by plethysmography.
Enhanced pause (Penh) displays an index which indicates changes of the airflow waveform in a whole-body plethysmograph that can be correlated with pulmonary reactivity.38 This noninvasive method enables long-term measurements in vivo in specific individuals and allows assessment of kinetic profiles over a long time course. Previous studies had demonstrated that FR is able to potently induce airway relaxation in mice.14 Since radiolabeled FR was recently shown to have a much longer residence time than radiolabeled YM27 (also see Table 1), we wanted to compare the effects of both Gq protein inhibitors on bronchial function. Figure 7 shows the results of repeated Penh measurements with increasing concentrations of methacholine (MCh), a muscarinic acetylcholine receptor agonist inducing bronchoconstriction, after a one-time intratracheal application (i.t.) of FR or YM, respectively. Both Gq protein inhibitors were able to reduce a methacholine-induced Penh increase 4 h after application. However, 48 h after application, mice that had been treated with YM showed a similar reaction as control mice treated with dimethyl sulfoxide (DMSO), in which the Gq protein inhibitors had been dissolved. This indicates, that after 48 h, the antiasthmatic effects of YM were terminated. Interestingly, the effect of FR persisted much longer and could be observed even 96 h after FR application.
Figure 7.

Repeated enhanced pause (Penh) measurements after intratracheal application of FR and YM. Bronchoconstriction was induced by application of methacholine (MCh). FR/YM (2.5 μg) or DMSO was administered intratracheally on day 0. Whole-body plethysmography was carried out after 4 h, 24 h, 48 h, 72 h, and 96 h in the presence of increasing concentrations of MCh which was applied as an aerosol by a nebulizer in the plethysmograph. Data represent means ± SEM (FR, DMSO n = 5; YM n = 4). Statistical significance was assessed by two-way repeated measures ANOVA with Bonferroni’s post hoc test. ****, ***, and ** represent a p value < 0.0001, 0.001, and 0.01, respectively.
These data demonstrate that the significantly longer residence time of the Gq inhibitor FR as compared to YM translates into much longer persistence of the antiasthmatic action.
3. Conclusion
Until now, great efforts have been made to evaluate the biochemical and pharmacological profiles of the Gq protein inhibitors FR and YM in order to fully understand their effects on a molecular level.5,39−41,25,42 The current project focused on a comparison between both compounds and provides first insights into their pharmacokinetic behavior which is a prerequisite for their translational development as therapeutic drugs. Both compounds are Pgp substrates. Interestingly, we found that FR was metabolized significantly faster than YM in human and mouse liver microsomal preparations, which correlated with higher YM levels in vital organs after intratracheal application (Figure 5A,B). Another striking difference was the effect of FR on methacholine-induced bronchoconstriction upon Penh measurements which clearly lasted significantly longer than the effect of YM. This can be explained by the slower dissociation kinetic of FR. These data, along with the determined high FR levels in the lung after local and systemic applications, imply that FR is a prime compound for targeting Gq-based signaling in the respiratory system. However, local administration of the Gq protein inhibitor will be preferred since systemic application can be expected to block Gq signaling throughout the body and is therefore not suitable for use in the clinic. In addition, our results prove that changes within a molecule which seem marginal compared to its structural complexity can lead to crucial pharmacological differences. Additionally, our work demonstrates that differences which were identified by utilizing an artificial and simplified test system like measurement of target residence time of drugs can be transferred to and confirmed in more complex models.
4. Methods
4.1. Caco-2 Permeability
Caco-2 permeability assays were performed by Pharmacelsus GmbH (Saarbruecken, Germany) in a differentiated Caco-2 cell monolayer-based test system.15 Transport rates of FR and YM were determined at 10 μM and at a pH of 6.5 (apical, A) and 7.4 (basolateral, B). As reference compounds, testosterone (high transport rates, high bioavailability), atenolol (low transport rates, low bioavailability) and erythromycin (Pgp substrate) were included. Measurements were made at the following time points: 0, 15, 45, and 90 min. Caco-2 cells were differentiated for 21 days in Transwell plates. Bidirectional permeation experiments were performed according to Pharmacelsus in-house protocols. Data represent means ± SEM from three experiments.
4.2. Stability in Simulated Gastric Fluid
Artificial gastric fluid (450 μL, prepared according to the European Pharmacopoeia (Ph. Eur. 10) consisting of 0.32 g of pig pepsin, 0.2 g of NaCl, 8 mL of 1 M HCl, and 100 mL of H2O) was spiked with 50 μL of FR or YM (1 mM stock solution prepared in DMSO). The mixture was subsequently incubated at 37 °C. Samples (50 μL each) were drawn after 0, 20, 40, 60, 120, 180, and 240 min, treated with ice-cold acetonitrile (1:1), vortexed, and centrifuged for 3 min at 15,000 g. An aliquot (50 μL) of the supernatant was transferred to a vial and subjected to LC-MS/MS analysis (see Section Quantitative Analysis of FR and YM by LC-MS/MS for in Vitro Stability Studies). Each experiment was repeated 3 times for each compound.
4.3. Stability in Alkaline Solutions
Alkaline aqueous solutions were prepared by adjusting the pH value of deionized water with NaOH until a pH of 9.0 or 11.0, respectively, was reached. To the alkaline solutions (450 μL) was added 50 μL of FR or YM (1 mM stock solution in DMSO), and the mixtures were incubated at 37 °C. Samples (50 μL each) were drawn after 0, 20, 40, 60, 80, 120, 180, and 240 min. Samples were treated with ice-cold acetonitrile (1:1), vortexed, and centrifuged for 3 min at 15,000 g. Aliquots (50 μL) of the supernatant were transferred to a vial and subjected to LC-MS/MS analysis (see Section Quantitative Analysis of FR and YM by LC-MS/MS for in Vitro Stability Studies). Each experiment was repeated 3 times for each compound.
4.4. Quantitative Analysis of FR and YM by LC-MS/MS for in Vitro Stability Studies
Measurements were performed on an Agilent 1260 Infinity HPLC coupled to an Agilent Infinity Lab LC/MSD Single Quadrupole mass spectrometer with an electrospray ion source. Chromatographic separation was performed on an EC 50/3 Nucleodur C18 Gravity, 3 μm (Macherey-Nagel, Dueren, Germany). Mobile phase A consisted of methanol containing 2 mM ammonium acetate and 0.1% formic acid, and mobile phase B consisted of water with 2 mM ammonium acetate and 0.1% formic acid. The run started with 60% A and 40% B for 1 min, followed by a gradient that reached 100% of eluent A after 9 min. Then, the column was flushed for 5 min with 100% of mobile phase A. The flow rate was adjusted to 0.4 mL/min. Positive full scan MS was observed from 200 to 1500 m/z. The peak appeared at 5.7 min for YM-254890 and at 7.4 min for FR900359. For identification and quantification using the Data Analysis program on OpenLab CDS software 2.4, the extracted ion chromatogram (EIC) of 960.5 ± 0.7 m/z was used for YM, and the EIC of 1002.5 ± 0.7 m/z was used for FR. This method was used for the stability studies. Peak areas were evaluated from the EICs and normalized to the FR and YM areas at zero time, and the percentage of remaining compound was calculated. Three independent experiments were performed.
4.5. Metabolic Stability in Human and Mouse Liver Microsomes
Metabolic stability of FR and YM in human and mouse liver microsomes was assessed by Pharmacelsus GmbH (Saarbruecken, Germany). FR or YM (1 μM) was incubated with pooled human or mouse liver microsomes (0.5 mg/mL) in phosphate buffer pH 7.4 in the presence of NADPH and MgCl2. Samples were drawn after 0, 10, 30, and 60 min. The percentage loss of the parent compound was determined by LC-MS analysis. Subsequent metabolite identification in mouse liver microsomes was carried out using HPLC-HRMS by determining accurate masses and fragmentation patterns.
4.6. Metabolic Stability in Mouse Lung Tissue
For stability testing in lung tissue, lungs from four CD1 wild-type mice were pooled, weighed, and subsequently homogenized in a TissueLyzer (Qiagen, Venlo, Netherlands) for 8 min at 50 strokes/min in a precooled tube holder. HEPES buffer, 50 mM, pH 7.4, was added to the homogenate (1 mL of buffer was added to 300 mg of tissue), and the mixture was transferred to a reagent tube. Then, 50 μL of FR solution (1 mM dissolved in DMSO) was added to 450 μL of the lung homogenate and incubated at 37 °C. Samples (6 samples, 80 μL each) were drawn after 0, 15, 30, 60, 120, and 240 min, mixed with ice-cold acetonitrile (1:1), vortexed, and centrifuged for 3 min at 15,000 g. An aliquot of 50 μL of the supernatant was transferred to a suitable vial and subjected to LC-MS/MS analysis.
4.7. Stability in Mouse Blood Plasma
Blood plasma of three wild-type CD1 mice was pooled, and 90 μL of the plasma was mixed with 10 μL of FR solution (1 mM in DMSO) and incubated at 37 °C. Samples (6 samples, 10 μL each) were drawn at 0, 15, 30, 60, 120, and 240 min, mixed with ice-cold acetonitrile (1:1), vortexed, and centrifuged for 3 min at 15,000 g. Aliquots of 10 μL of the supernatant were transferred to a suitable vial and subjected to LC-MS/MS analysis.
4.8. In Vivo Experiments
Animal experiments were approved by the local ethics committee and carried out in accordance to the guidelines of the German law of protection of animal life with approval by the local government authorities (Landesamt für Natur, Umwelt and Verbraucherschutz Nordrhein-Westfalen, NRW, Germany).
4.8.1. Quantification of FR and YM in Mouse Tissues
In an initial in vivo study, FR or YM (5 μg) was administered intratracheally on 7 consecutive days. In further studies, 10 μg and 2.5 μg of FR, respectively, were administered intraperitoneally (Monday to Friday) and intratracheally (twice a day, Monday to Sunday), respectively, for 3 weeks. In each study, organs from three different mice were harvested and snap-frozen approximately 45 min after the last FR or YM application. FR and YM were extracted by a three-step liquid–liquid extraction method and quantified as previously described by HPLC-ESI-MS/MS.37 Chromatographic separation was performed using a Dionex Ultimate 3000 (Thermo Fisher Scientific, MA, USA) equipped with an integrated variable wavelength detector coupled to a micrOTOF-Q mass spectrometer (Bruker, MA, USA) with an electrospray ion source. An EC50/2 Nucleodur C18 Gravity 3 μm column (Macherey-Nagel, Dueren, Germany) was used for chromatographic separation. The two mobile phases were A (40% aq. methanol containing 2 mM ammonium acetate and 0.1% formic acid) and B (methanol, 2 mM ammonium acetate, 0.1% formic acid). The run started with 100% A. After 1 min, a gradient was started reaching 100% eluent B within 9 min. Then, the column was flushed for 5 min with solvent B. Positive full scan MS was recorded from 200 to 1500 m/z. The extract ion chromatogram (EIC) of 1002.54 ± 0.01 m/z was used for the identification and quantification of FR by the QuantAnalysis program (Bruker, MA, USA). An EIC of 960.49 ± 0.01 m/z was employed for the identification and quantification of YM.
4.8.2. Whole Body Plethysmography
For i.t. drug application, mice were anaesthetized with isoflurane (5%) and orotracheally intubated using an i.v. cannula (22 G, Vasofix Safely, B. Braun, Melsungen, Germany). The correct positioning of the endotracheal tube was checked under mechanical ventilation with 1.5% isoflurane via a small animal ventilator (MiniVent, Hugo Sachs, Germany). Then, the tube was disconnected from the ventilator, and Gq inhibitors FR or YM (2.5 μg, 1% DMSO in 0.9% NaCl, 50 μL) or the solvent (1% DMSO in 0.9% NaCl, 50 μL) was applied into the tube by a pipet. Thereafter, mechanical ventilation was continued for about 30 s to allow uptake of the liquid. Whole body plethysmography was performed at 4, 24, 48, 72, and 96 h after extubation. Therefore, the awake and unrestrained mice were placed into cylindrical Plexiglas chambers of the whole body plethysmograph (emka Technologies, France). Penh was recorded for 40 s under resting conditions (baseline) and after nebulization of increasing concentrations of methacholine (0, 12.5, 25, and 50 mg/mL).
4.9. Calculation of Compound Properties
LogP, PSA, and peroral and CNS bioavailability were calculated using StarDrop (Optibrium, Cambridge, UK, 2013).
4.10. Data Analysis
4.10.1. Statistical Analysis
Statistical significance was determined using a two-way repeated measures ANOVA with a subsequent Bonferroni’s post hoc test. Data analysis and plotting were performed using GraphPad PRISM, Version 7.0 (GraphPad, San Diego, CA, USA).
4.10.2. Metabolic Stability
The metabolic degradation process was defined as a first-order decay (eq 1). To obtain a straight line, the natural log of the remaining compound (%) was plotted against time (min). The slope was used to calculate half-lives (eq 2), and CLint,app was determined by using eq 3.35 Subsequently, eq 4 was applied to obtain CLint,app based on 45 mg of microsomal protein per g of liver tissue and 87 g (mouse) or 26 g (human) of liver tissue per kg body mass.
| 1 |
| 2 |
| 3 |
| 4 |
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (FOR2372, MU-1665/7-2, WE 4461/2-1 and -2, and FL 276/8-1 and -2).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00021.
Inhibition of CYP450 enzymes by FR and YM (PDF)
Author Contributions
⊥ J.G.S. and M.T. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Fujioka M.; Koda S.; Morimoto Y.; Biemann K. (1988) Structure of FR900359, a cyclic depsipeptide from Ardisia crenata Sims. J. Org. Chem. 53, 2820–2825. 10.1021/jo00247a030. [DOI] [Google Scholar]
- Carlier A.; Fehr L.; Pinto-Carbó M.; Schäberle T.; Reher R.; Dessein S.; König G.; Eberl L. (2016) The genome analysis of Candidatus Burkholderia crenata reveals that secondary metabolism may be a key function of the Ardisia crenata leaf nodule symbiosis. Environ. Microbiol. 18, 2507–2522. 10.1111/1462-2920.13184. [DOI] [PubMed] [Google Scholar]
- Takasaki J.; Saito T.; Taniguchi M.; Kawasaki T.; Moritani Y.; Hayashi K.; Kobori M. (2004) A novel Gαq/11-selective inhibitor. J. Biol. Chem. 279, 47438–47445. 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- Taniguchi M.; Nagai K.; Arao N.; Kawasaki T.; Saito T.; Moritani Y.; Takasaki J.; Hayashi K.; Fujita S.; Suzuki K.-i.; Tsukamoto S.-i. (2003) YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J. Antibiot. 56, 358–363. 10.7164/antibiotics.56.358. [DOI] [PubMed] [Google Scholar]
- Schrage R.; Schmitz A.-L.; Gaffal E.; Annala S.; Kehraus S.; Wenzel D.; Büllesbach K. M.; Bald T.; Inoue A.; Shinjo Y.; Galandrin S.; Shridhar N.; Hesse M.; Grundmann M.; Merten N.; Charpentier T. H.; Martz M.; Butcher A. J.; Slodczyk T.; Armando S.; Effern M.; Namkung Y.; Jenkins L.; Horn V.; Stößel A.; Dargatz H.; Tietze D.; Imhof D.; Galés C.; Drewke C.; Müller C. E.; Hölzel M.; Milligan G.; Tobin A. B.; Gomeza J.; Dohlman H. G.; Sondek J.; Harden T. K.; Bouvier M.; Laporte S. A.; Aoki J.; Fleischmann B. K.; Mohr K.; König G. M.; Tüting T.; Kostenis E. (2015) The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 6, 10156. 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognini D.; Moss C. E.; Nilsson K.; Petersson A. U.; Donnelly I.; Sergeev E.; König G. M.; Kostenis E.; Kurowska-Stolarska M.; Miller A.; Dekker N.; Tobin A. B.; Milligan G. (2016) A novel allosteric activator of free fatty acid 2 receptor displays unique Gi-functional bias. J. Biol. Chem. 291, 18915–18931. 10.1074/jbc.M116.736157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr R.; Koziol-White C.; Zhang J.; Lam H.; An S. S.; Tall G. G.; Panettieri R. A.; Benovic J. L. (2016) Interdicting Gq activation in airway disease by receptor-dependent and receptor-independent mechanisms. Mol. Pharmacol. 89, 94–104. 10.1124/mol.115.100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H.; MacIntyre D. A.; Hanyaloglu A. C.; Blanks A. M.; Thornton S.; Bennett P. R.; Terzidou V. (2016) The oxytocin receptor antagonist, Atosiban, activates pro-inflammatory pathways in human amnion via Gαi signalling. Mol. Cell. Endocrinol. 420, 11–23. 10.1016/j.mce.2015.11.012. [DOI] [PubMed] [Google Scholar]
- Liao Y.; Lu B.; Ma Q.; Wu G.; Lai X.; Zang J.; Shi Y.; Liu D.; Han F.; Zhou N. (2016) Human neuropeptide S receptor is activated via a Gαq protein-biased signaling cascade by a human neuropeptide S analog lacking the C-terminal 10 residues. J. Biol. Chem. 291, 7505–7516. 10.1074/jbc.M115.704122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badolia R.; Inamdar V.; Manne B. K.; Dangelmaier C.; Eble J. A.; Kunapuli S. P. (2017) Gq pathway regulates proximal C-type lectin-like receptor-2 (CLEC-2) signaling in platelets. J. Biol. Chem. 292, 14516–14531. 10.1074/jbc.M117.791012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeil E. M.; Brands J.; Merten N.; Vögtle T.; Vescovo M.; Rick U.; Albrecht I.-M.; Heycke N.; Kawakami K.; Ono Y.; Ngako Kadji F. M.; Hiratsuka S.; Aoki J.; Häberlein F.; Matthey M.; Garg J.; Hennen S.; Jobin M.-L.; Seier K.; Calebiro D.; Pfeifer A.; Heinemann A.; Wenzel D.; König G. M.; Nieswandt B.; Fleischmann B. K.; Inoue A.; Simon K.; Kostenis E. (2020) Heterotrimeric G protein subunit Gαq is a master switch for Gβγ-mediated calcium mobilization by Gi-coupled GPCRs. Mol. Cell 80, 940. 10.1016/j.molcel.2020.10.027. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Nielsen A. L.; Strømgaard K. (2020) Recent achievements in developing selective Gq inhibitors. Med. Res. Rev. 40, 135–157. 10.1002/med.21598. [DOI] [PubMed] [Google Scholar]
- Klepac K.; Kilić A.; Gnad T.; Brown L. M.; Herrmann B.; Wilderman A.; Balkow A.; Glöde A.; Simon K.; Lidell M. E.; Betz M. J.; Enerbäck S.; Wess J.; Freichel M.; Blüher M.; König G.; Kostenis E.; Insel P. A.; Pfeifer A. (2016) The Gq signalling pathway inhibits brown and beige adipose tissue. Nat. Commun. 7, 10895. 10.1038/ncomms10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthey M.; Roberts R.; Seidinger A.; Simon A.; Schröder R.; Kuschak M.; Annala S.; König G. M.; Müller C. E.; Hall I. P.; Kostenis E.; Fleischmann B. K.; Wenzel D. (2017) Targeted inhibition of Gq signaling induces airway relaxation in mouse models of asthma. Sci. Transl. Med. 9, eaag2288. 10.1126/scitranslmed.aag2288. [DOI] [PubMed] [Google Scholar]
- van Breemen R. B.; Li Y. (2005) Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 1, 175–185. 10.1517/17425255.1.2.175. [DOI] [PubMed] [Google Scholar]
- Sun H.; Chow E. C.; Liu S.; Du Y.; Pang K. S. (2008) The Caco-2 cell monolayer: usefulness and limitations. Expert Opin. Drug Metab. Toxicol. 4, 395–411. 10.1517/17425255.4.4.395. [DOI] [PubMed] [Google Scholar]
- Onken M. D.; Makepeace C. M.; Kaltenbronn K. M.; Kanai S. M.; Todd T. D.; Wang S.; Broekelmann T. J.; Rao P. K.; Cooper J. A.; Blumer K. J. (2018) Targeting nucleotide exchange to inhibit constitutively active G protein α subunits in cancer cells. Sci. Signaling 11, eaao6852. 10.1126/scisignal.aao6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annala S.; Feng X.; Shridhar N.; Eryilmaz F.; Patt J.; Yang J.; Pfeil E. M.; Cervantes-Villagrana R. D.; Inoue A.; Häberlein F.; Slodczyk T.; Reher R.; Kehraus S.; Monteleone S.; Schrage R.; Heycke N.; Rick U.; Engel S.; Pfeifer A.; Kolb P.; König G.; Bünemann M.; Tüting T.; Vázquez-Prado J.; Gutkind J. S.; Gaffal E.; Kostenis E. (2019) Direct targeting of Gαq and Gα11 oncoproteins in cancer cells. Sci. Signal. 12, eaau5948. 10.1126/scisignal.aau5948. [DOI] [PubMed] [Google Scholar]
- Lapadula D.; Farias E.; Randolph C. E.; Purwin T. J.; McGrath D.; Charpentier T. H.; Zhang L.; Wu S.; Terai M.; Sato T.; Tall G. G.; Zhou N.; Wedegaertner P. B.; Aplin A. E.; Aguirre-Ghiso J.; Benovic J. L. (2019) Effects of oncogenic Gαq and Gα11 inhibition by FR900359 in uveal melanoma. Mol. Cancer Res. 17, 963–973. 10.1158/1541-7786.MCR-18-0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua V.; Lapadula D.; Randolph C.; Benovic J. L.; Wedegaertner P. B.; Aplin A. E. (2017) Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol. Cancer Res. 15, 501–506. 10.1158/1541-7786.MCR-17-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M.; Suzumura K.-i.; Nagai K.; Kawasaki T.; Takasaki J.; Sekiguchi M.; Moritani Y.; Saito T.; Hayashi K.; Fujita S.; Tsukamoto S.-i.; Suzuki K.-i. (2004) YM-254890 analogues, novel cyclic depsipeptides with Gαq/11 inhibitory activity from Chromobacterium sp. QS3666. Bioorg. Med. Chem. 12, 3125–3133. 10.1016/j.bmc.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Reher R.; Kuschak M.; Heycke N.; Annala S.; Kehraus S.; Dai H.-F.; Müller C. E.; Kostenis E.; König G. M.; Crüsemann M. (2018) Applying molecular networking for the detection of natural sources and analogues of the selective Gq protein inhibitor FR900359. J. Nat. Prod. 81, 1628–1635. 10.1021/acs.jnatprod.8b00222. [DOI] [PubMed] [Google Scholar]
- Hermes C.; Richarz R.; Wirtz D. A.; Patt J.; Hanke W.; Kehraus S.; Voß J. H.; Küppers J.; Ohbayashi T.; Namasivayam V.; Alenfelder J.; Inoue A.; Mergaert P.; Gütschow M.; Müller C. E.; Kostenis E.; König G. M.; Crüsemann M. (2021) Thioesterase-mediated side chain transesterification generates potent Gq signaling inhibitor FR900359. Nat. Commun. 12, 144. 10.1038/s41467-020-20418-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X.-F.; Zhang H.; Underwood C. R.; Harpsøe K.; Gardella T. J.; Wöldike M. F.; Mannstadt M.; Gloriam D. E.; Bräuner-Osborne H.; Strømgaard K. (2016) Total synthesis and structure-activity relationship studies of a series of selective G protein inhibitors. Nat. Chem. 8, 1035–1041. 10.1038/nchem.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X.-F.; Zhang H.; Boesgaard M. W.; Underwood C. R.; Bräuner-Osborne H.; Strømgaard K. (2019) Structure-activity relationship studies of the natural product Gq/11 protein inhibitor YM-254890. ChemMedChem 14, 865–870. 10.1002/cmdc.201900018. [DOI] [PubMed] [Google Scholar]
- Rensing D. T.; Uppal S.; Blumer K. J.; Moeller K. D. (2015) Toward the selective inhibition of G proteins: Total synthesis of a simplified YM-254890 analog. Org. Lett. 17, 2270–2273. 10.1021/acs.orglett.5b00944. [DOI] [PubMed] [Google Scholar]
- Kuschak M.; Namasivayam V.; Rafehi M.; Voss J. H.; Garg J.; Schlegel J. G.; Abdelrahman A.; Kehraus S.; Reher R.; Küppers J.; Sylvester K.; Hinz S.; Matthey M.; Wenzel D.; Fleischmann B. K.; Pfeifer A.; Inoue A.; Gütschow M.; König G. M.; Müller C. E. (2020) Cell-permeable high-affinity tracers for Gq proteins provide structural insights, reveal distinct binding kinetics and identify small molecule inhibitors. Br. J. Pharmacol. 177, 1898–1916. 10.1111/bph.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Lennernäs H. (1997) Human jejunal effective permeability and its correlation with preclinical drug absorption models. J. Pharm. Pharmacol. 49, 627–638. 10.1111/j.2042-7158.1997.tb06084.x. [DOI] [PubMed] [Google Scholar]
- Yee S. (1997) In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man--fact or myth. Pharm. Res. 14, 763–766. 10.1023/A:1012102522787. [DOI] [PubMed] [Google Scholar]
- Faassen F. (2003) Caco-2 permeability, P-glycoprotein transport ratios and brain penetration of heterocyclic drugs. Int. J. Pharm. 263, 113–122. 10.1016/S0378-5173(03)00372-7. [DOI] [PubMed] [Google Scholar]
- Federal Institute of Drugs and Medical Devices . (2020). European Pharmacopoeia, 10th ed., Bonn, Germany. [Google Scholar]
- Hamann S. R.; Blouin R. A.; McAllister R. G. (1984) Clinical pharmacokinetics of verapamil. Clin. Pharmacokinet. 9, 26–41. 10.2165/00003088-198409010-00002. [DOI] [PubMed] [Google Scholar]
- Słoczyńska K.; Gunia-Krzyżak A.; Koczurkiewicz P.; Wójcik-Pszczoła K.; Żelaszczyk D.; Popiół J.; Pękala E. (2019) Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 69, 345–361. 10.2478/acph-2019-0024. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. (2019) Clearance in drug design. J. Med. Chem. 62, 2245–2255. 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- McNaney C. A.; Drexler D. M.; Hnatyshyn S. Y.; Zvyaga T. A.; Knipe J. O.; Belcastro J. V.; Sanders M. (2008) An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Technol. 6, 121–129. 10.1089/adt.2007.103. [DOI] [PubMed] [Google Scholar]
- Kuschak M.; Schlegel J. G.; Schneider M.; Kehraus S.; Voss J. H.; Seidinger A.; Matthey M.; Wenzel D.; Fleischmann B. K.; König G. M.; Müller C. E. (2020) Sensitive LC-MS/MS method for the quantification of macrocyclic Gαq protein inhibitors in biological samples. Front. Chem. 8, 833. 10.3389/fchem.2020.00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelmann E.; Schwarze J.; Takeda K.; Oshiba A.; Larsen G. L.; Irvin C. G.; Gelfand E. W. (1997) Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 156, 766–775. 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- Inamdar V.; Patel A.; Manne B. K.; Dangelmaier C.; Kunapuli S. P. (2015) Characterization of UBO-QIC as a Gαq inhibitor in platelets. Platelets 26, 771. 10.3109/09537104.2014.998993. [DOI] [PubMed] [Google Scholar]
- Roszko K. L.; Bi R.; Gorvin C. M.; Bräuner-Osborne H.; Xiong X.-F.; Inoue A.; Thakker R. V.; Strømgaard K.; Gardella T.; Mannstadt M. (2017) Knockin mouse with mutant Gα11 mimics human inherited hypocalcemia and is rescued by pharmacologic inhibitors. JCI Insight 2, e91079. 10.1172/jci.insight.91079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietze D.; Kaufmann D.; Tietze A. A.; Voll A.; Reher R.; König G.; Hausch F. (2019) Structural and dynamical basis of G protein inhibition by YM-254890 and FR900359: An inhibitor in action. J. Chem. Inf. Model. 59, 4361–4373. 10.1021/acs.jcim.9b00433. [DOI] [PubMed] [Google Scholar]
- Boesgaard M. W.; Harpsøe K.; Malmberg M.; Underwood C. R.; Inoue A.; Mathiesen J. M.; König G. M.; Kostenis E.; Gloriam D. E.; Bräuner-Osborne H. (2020) Delineation of molecular determinants for FR900359 inhibition of Gq/11 unlocks inhibition of Gαs. J. Biol. Chem. 295, 13850. 10.1074/jbc.RA120.013002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.