

Abstract
Angiotensin‐converting enzyme (ACE) inhibitors are first‐line therapy for the treatment of hypertension, congestive heart failure, and diabetic nephropathy. ACE inhibitors are associated with adverse side effects such as persistent dry cough (ACE‐cough) and, rarely, life‐threatening angioedema (ACE‐AE). The authors investigated the influence of ACE I/D polymorphism in combination with serum ACE activity, B 2 receptor −9/+9 polymorphism, and B2 receptor C‐58T single nucleotide polymorphism (SNP) on the development of ACE‐AE and ACE‐cough. The frequencies of ACE I/D as well as B2 receptor +9/−9 and C‐58T polymorphisms were compared in patients with ACE‐AE, ACE‐cough, and ACE inhibitor–exposed controls, and serum ACE activity was measured. There were 52 cases of ACE‐AE, 36 cases of ACE‐cough, and 77 controls. The genotyping revealed a significant association between the B2 −9 allele and ACE inhibitor–induced AE (62% vs 38%, P=.008), and ACE inhibitor–induced cough (61% vs 38%, P=.02) when compared with controls. There was no significant association between ACE I/D polymorphism as well as the B2 C‐58T SNP with both ACE‐induced AE and cough. ACE activity was significantly higher in controls compared with patients with ACE‐AE (34.5±1.14 mU/mL vs 17.8±0.86 mU/mL, P=.0001) and ACE‐cough (34.5±1.14 mU/mL vs 23.3±1.88 mU/mL, P=.0001). Thus, our data suggest that the B2 −9 allele and reduced ACE activity are associated with both ACE‐AE and ACE‐cough.
Angiotensin‐converting enzyme (ACE) inhibitors are used by more than 40 million people worldwide for the treatment of hypertension, congestive heart failure, myocardial infarction, and diabetic nephropathy.1 Although clinically effective, ACE inhibitors are commonly associated with adverse side effects such as persistent dry cough (ACE‐cough) and, more rarely, a potentially life‐threatening angioedema (ACE‐AE).2 Clinical risk factors of ACE‐AE include the black race, female sex, smoking, seasonal allergies, and immunosuppressant therapy,3, 4 while diabetes may be protective.4, 5 However, there is an urgent need to have more reliable predictors of ACE‐AE given its potential to cause death.
Clinically ACE‐AE presents as swelling of the face, tongue, and neck region that may obstruct the upper airways and cause death. The vasodilator peptide bradykinin (BK) plays a central role in the pathophysiology of both ACE‐AE and ACE‐cough.6, 7 BK is primarily degraded by ACE and secondarily by aminopeptidase P (APP).8, 9 During ACE inhibition, BK is primarily degraded by APP.10 BK exerts its pharmacologic activities through the constitutively expressed kinin B2 receptor, whereas its vasoactive metabolite des‐arg9‐BK binds to the B1 receptor.10 B1 and B2 activation results in several physiological and pathological responses, including vasodilation, increased vascular permeability, bronchospasm, edema, pain, and cellular proliferation.11
Identification of genes associated with ACE‐AE could help elucidate its mechanism and underlying predisposition. The ACE gene is located on chromosome 17 in humans and the polymorphism is detected in intron 16 based on the insertion [I]/deletion [D] of 287 bp.12, 13 The average serum levels of ACE‐DD genotype are higher than that of ACE‐II genotype,14 but these polymorphisms in Caucasians have not being associated with ACE‐AE.14 In contrast, in blacks, in whom ACE‐AE is more prevalent, there is a link with ACE II genotype.15
Further determinants that are potentially involved in genetic predisposition to ACE‐AE are BK receptors. A variant of the B2 receptor gene in which the presence or absence of a 9‐basepair repeat in exon 1 affects B2 transcription has been identified.16 The +9/+9 genotype is associated with decreased vasodilation and tissue plasmin activator production in response to BK during ACE inhibition compared with the −9/−9 allele.17 Furthermore, the −9/−9 allele has been associated with increased severity of AE in cases of type‐1 hereditary angioedema.18 The C‐58T B2 promoter single nucleotide polymorphism (SNP) has been associated with increased risk of essential hypertension in African Americans and also with ACE‐cough in Chinese patients.19, 20 However, both −9/+9 and C‐58T have never been investigated and associated with ACE‐AE.
In this study we investigated the role the ACE I/D polymorphism in combination with serum ACE activity as well BK B2 receptor −9/+9 and C‐58T polymorphisms in the development of ACE‐AE and ACE‐cough.
Methods
Participants
The study protocol was approved by the Human Research Ethics Committee at the University of Cape Town. Three different patient cohorts were recruited for the study from the hypertension clinic at Groote Schuur Hospital and Victoria Hospital, Wynberg Cape Town: cohort 1 (control group) included hypertensive patients currently taking treatment with the ACE inhibitor enalapril for more than 2 years with no side effects; cohort 2 included patients with ACE‐AE); and cohort 3 included patients ACE‐cough who had discontinued treatment. Participants self‐identified themselves as of African ancestry (black), Cape mixed ancestry, or Caucasian (white) and demographic details were obtained regarding smoking, seasonal allergies, and comorbidities such as diabetes, congestive heart failure, and kidney failure. The date of the episode, frequency, localization, and severity of AE were recorded.
DNA and serum samples were collected from 77 control patients and 52 ACE‐AE and 36 ACE‐cough patients. Serum samples for ACE activity were taken from controls taking enalapril and 48 hours after discontinuation.
ACE I/D Genotyping
Genomic DNA was extracted from peripheral blood leucocytes using QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according the manufacturer's instructions.
The ACE I/D polymorphism was amplified by polymerase chain reaction (PCR) using the following primers: forward (5′gcttgcagcatgtggcccca3′) and reverse (5′gccatcctccagttttacccgaa3′). Amplification was performed with the initial DNA denaturation step at 98°C for 2 minutes, then underwent 30 cycles of 96°C for 30 seconds, annealing at 65°C for 30 and 45 seconds at 72°C for DNA extension. After the last cycle, the reaction was completed at 72°C for 8 minutes. PCR products were separated using 4% agarose gel electrophoresis. Amplification of this primer pair resulted in 607 and 320 bp corresponding to the ACE I and ACE D alleles, respectively.
B2 −9/+9 Genotyping
The bradykinin B2 receptor exon 1 −9/+9 polymorphism was amplified by PCR using the following primers: forward (5′tctggcttctgggctccgag3′) and reverse (5′actgaagtgcccatgccgct3′). Cycling conditions for PCR were initial DNA denaturation at 95°C for 10 minutes, followed by 30 seconds of denaturation at 95°C for 30 cycles, annealing at 60°C for 30 and 30 seconds at 72°C for DNA extension, followed by final extension of 1 cycle for 8 minutes. PCR products were separated using 7.5% polyacrylamide gel electrophoresis. The presence of the −9/+9 is confirmed by the presence of 102 and 93 bp bands.
The B2 C‐58T SNP in the promoter region of the B2 gene located on chromosome14q32.1–q32.2 was detected using PCR. Because the C‐58T substitution does not alter a recognition sequence for a restriction enzyme, a partial recognition site for TSP45I was introduced by introduction of a single mismatch base (bold) in the forward primer. The primer used was a follows: forward (5′cccaggaggctgatgacgtcac3′ and reverse (5′tctggcttctgggctccgag3′). Thermocycling included an initial denaturation step of 95°C for 10 minutes, followed by 30 seconds of denaturation at 95°C for 30 cycles, annealing at 60°C for 30 seconds and 30 seconds at 72°C for DNA extension, followed by final extension of 1 cycle for 8 minutes. PCR products were digested with restriction enzyme TSP45I for 2 hours at 37°C. The digested PCR product was separated using 4% agarose gel electrophoresis. The presence of the C‐58T allele is confirmed by the presence of 86 bp band, whereas the C‐58C allele is confirmed by PCR product of 66 and 20 bp bands, respectively.
Serum ACE Activity
Serum ACE activity was determined using a spectroflourimetric assay described by Danilov adapted from the method by Friedland and Silverstein.21, 22
Because ACE is inhibited by unidentified low molecular weight endogenous inhibitors in serum, urine and other tissues,21 serum was diluted in 1× phosphate buffered saline (PBS) in a dilution series of 1:2, 1:4, and 1:8 simultaneously with undiluted serum so that the effect of these endogenous inhibitors was minimized.
Serum ACE Inhibition
Lisinopril (10 mg) was dissolved in distilled water to make a 1‐mm stock. Different concentrations of lisinopril (0, 0.1, 0.3, 1, 3, 10, and 30 nm) were then made up in 1× PBS. Serum was diluted 1:5 in 1× PBS and 180 μL was added to the Eppendorf tube containing 20 μL of each inhibitor concentration in triplicates. The mixture was mixed and preincubated for 2 hours at room temperature so that the enzyme and inhibitor reached equilibrium. The residual ACE activity was determined with 5 mm HHL and 2 mm Z‐FHL, as previously described. The Z‐FHL/HHL ratio was then calculated and plotted against the concentration of lisinopril to determine the levels of ACE bound to inhibitor.
Statistical Analysis
Statistical analysis was carried out using SPSS 18.0 (SPSS Inc, Chicago, IL) for categorical variables or Statistica 10 (StatSoft, Tulsa, OK) for numerical variables. The controls were tested for Hardy‐Weinberg equilibrium (HWE) for all genotypes in this study. For categorical variables, chi‐square test was used to test for significance between cases and controls. For numeric variables, Student t test and one‐way analysis of variance was used to test for the difference. Data are presented as mean±standard deviation.
Results
A total of 165 participants were enrolled in the study to investigate the association of genetic polymorphisms with ACE‐cough and ACE‐AE in a South African population (Table 1). The majority of patients with ACE‐AE were of Cape mixed ancestry (61%), and this cohort mirrored the demographic profile of the patients with ACE‐cough (72%). In both the ACE‐AE (79%) and ACE‐cough (92%) groups, the majorities were women and in the ACE‐AE and ACE‐cough cohorts, 25% and 46% were diabetic, respectively. All participants in the control group received 10 mg of enalapril twice daily.
Table 1.
Characteristics of All Patients Enrolled in the Study
| Characteristics | Controls (n=77) | ACE‐AE (n=52) | ACE‐Cough (n=36) |
|---|---|---|---|
| Race | |||
| Cape mixed ancestry | 56 (71) | 32 (61) | 27 (72) |
| Black | 19 (26) | 17 (33) | 4 (14) |
| White | 2 (3) | 3 (6) | 5 (14) |
| Sex | |||
| Male | 33 (43) | 11 (21) | 3 (8) |
| Female | 44 (57) | 41 (79) | 33 (92) |
| Mean age (IQR) | 59 (50–70) | 49 (49–70) | 59 (50–70) |
| Comorbidities | |||
| Diabetic | 23 (30) | 13 (25) | 12 (46) |
| Nondiabetic | 54 (70) | 39 (75) | 24 (54) |
Abbreviations: ACE‐AE, angiotensin‐converting enzyme inhibitor–associated angioedema; ACE‐cough, angiotensin‐converting enzyme inhibitor–associated cough; IQR, interquartile range. Values are expressed as number (percentage).
ACE Insertion/Deletion Polymorphism
The distribution of ACE I/D polymorphism genotypes for 77 control patients was: ACE‐II 17 (22%), ID 34 (44%), and DD 26 (34%) (Table 2). In a group of 36 patients with ACE‐cough, the distribution of the 3 genotypes was: ACE‐II 7 (19%), ID 20 (56%), and DD 9 (25%). The frequencies of the ACE I/D genotypes in 52 patients with ACE‐AE were: ACE‐II 11 (21%), ID 20 (39%), and DD 21 (40%). There was no significant difference in the distribution of ACE I/D polymorphism between men and women (P=.910) or ethnic groups (P=.087) (data not shown). No significant difference was observed for the ACE‐II genotype when comparing controls with patients with ACE‐cough (19% vs 22%, P=.942) and patients with ACE‐AE (21% vs 22%, P=.926). There was also no significant association between ACE‐ID genotype and patients with ACE‐cough (56% vs 44%, P=.353) and patients with ACE‐AE (39% vs 34%, P=.644) when compared with controls. Similarly, there was no significant difference in the distribution of the ACE‐DD genotype in patients with ACE‐cough (25% vs 34%, P=.471) and patients with ACE‐AE (40% vs 34%, P=.562) when compared with controls. The ACE I/D genotypes were in HWE for all controls.
Table 2.
Genotype Frequencies of ACE I/D and Bradykinin B2 Receptor −9/+9 Polymorphisms and B2 C‐58T SNP
| Gene Polymorphism | Controls (n=77) | ACE‐Cough (n=36) | ACE‐AE (n=52) |
|---|---|---|---|
| ACE I/D | |||
| II | 17 (22) | 7 (19) | 11 (21) |
| ID | 34 (44) | 20 (56) | 20 (39) |
| DD | 26 (34) | 9 (25) | 21 (40) |
| B2 −9/+9 | |||
| −9/−9 | 8 (10) | 4 (11) | 8 (15) |
| −9/+9 | 21 (28) | 18 (50) | 24 (46) |
| +9/+9 | 48 (62) | 14 (39) | 20 (39) |
| B2 C‐58T SNP | |||
| TT | 11 (14) | 6 (16) | 5 (10) |
| CT | 19 (25) | 15 (42) | 24 (46) |
| CC | 47 (61) | 15 (42) | 23 (44) |
Abbreviations: ACE, angiotensin‐converting enzyme; ACE‐AE, angiotensin‐converting enzyme inhibitor–associated angioedema; ACE‐cough, angiotensin‐converting enzyme inhibitor–associated cough; SNP, single nucleotide polymorphism. Values are expressed as number (percentage).
B2 −9/+9 Insertion/Deletion Polymorphism
Genotyping results for the B2 −9/+9 polymorphism results are presented in Table 2. Genotypes were not in HWE among controls (P=.03). The B2 −9/+9 polymorphism genotype frequencies were similar between men and women (P=.37) and ethnic groups (P=.09) in all patients (data not shown). The −9/−9 genotype was not significantly associated with ACE‐AE (15% vs 10%, P=.11) or ACE‐cough (11% vs 10%, P=.87) when comparing these groups with controls (Figure 1A). However, for the −9/+9 genotype there was a significant association with both ACE‐AE (46% vs 28%, P=.01) and ACE‐cough (50% vs 28%, P=.01). The frequency of the +9/+9 genotype was significantly lower in ACE‐AE patients compared with controls (39% vs 62%, P=.008), as well as between ACE‐cough patients and controls (39% vs 62%, P=.02). When the B2–9 allele was treated as dominant (−9/−9+−9/+9) (Figure 1B) in ACE‐AE patients compared with controls, the B2–9 allele was significantly associated with ACE inhibitor–induced AE (62% vs 38%, P=.008). Similarly, if the B2–9 was treated as dominant in ACE‐cough patients vs controls, the results also displayed a significant association (61% vs 38%, P=.02).
Figure 1.
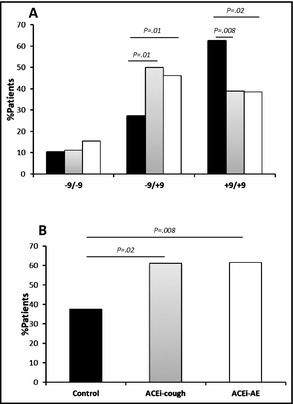
Analysis of the B2 −9/+9 insertion/deletion polymorphism. (A) Comparison of the B2 −9/+9 polymorphism in controls (black bars) with angiotensin‐converting enzyme inhibitor–associated cough (ACE‐cough) (grey bars) and angiotensin‐converting enzyme inhibitor–associated angioedema (ACE‐AE) (white bars). (B) Analysis of the dominant −9 variant in controls, ACE‐AE, and ACE‐cough.
B2 C‐58T Genotype in ACE‐AE and ACE‐Cough
B2 C‐58T genotype distributions were in HWE. Patient characteristics are shown in Table 2. The C‐58T SNP genotype frequencies displayed no significant association with men and women (P=.388) and ethnic groups (P=.422) in the study (data not shown). The TT genotype frequency was similar in ACE‐AE patients compared with controls (10% vs 14%, P=.6) as well as ACE‐cough patients compared with controls (16% vs 14%, P=.96) (Table 2 and Figure 2A). The CT heterozygote was significantly associated with ACE‐AE (46% vs 25%, P=.01), but there was no significant association with ACE‐cough (42% vs 25%, P=.07). The CC genotype frequency was similar in both ACE‐AE (44% vs 61, P=.089) and ACE‐cough (42% vs 61%, P=.08) compared with controls. When the T allele was treated as a dominant (ie, TT+CT), no significant association with the −58T allele was observed in both ACE‐AE (56% vs 39%, P=.06) and ACE‐cough (58% vs 39%, P=.06) patients vs controls (Figure 2B).
Figure 2.
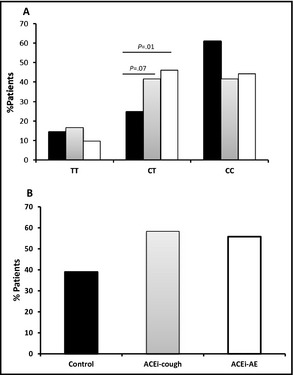
B2 C‐58T genotype in angiotensin‐converting enzyme inhibitor–associated angioedema (ACE‐AE) and angiotensin‐converting enzyme inhibitor–associated cough (ACE‐cough). (A) Evaluation of B2 C‐58T SNP in controls (black bars) with ACE‐cough (grey bars) and ACE‐AE patients (white bars). (B) Analysis of the dominant B2 C‐58T allele in ACE‐AE and ACE‐cough patients vs controls.
Analysis of Serum ACE Activity in Patients With ACE‐AE and ACE‐Cough
Endogenous ACE inhibitor in serum adversely affects the enzymatic activity of ACE.21, 23 Furthermore, the determination of ACE activity using two substrates (Z‐FHL and HHL) and the calculation of the ratio of ZPHL/HHL activity provides a sensitive method for the detection of ACE inhibitors in biological samples.21 21 ACE activity and Z‐FHL/HHL ratios indicated that there was minimal inhibition of ACE at serum dilutions of 1:4 and 1:8 (data not shown). Thus, a 1:5 dilution of serum was found to be most convenient for the measurement of maximal ACE activity. ACE activity was determined in 29 controls, 52 ACE‐AE, and 35 ACE‐cough patients. For controls, ACE activity was determined during and 48 hours after discontinuation of enalapril. The mean ACE activity of patients off and on enalapril was approximately 3‐fold higher (34.5±1.14 mU/mL vs 9.0 mU/mL, P=.0001) (Figure 3A). The Z‐FHL/HHL ratio was significantly higher in controls taking or not taking enalapril (3.25 vs 0.91, P=.0001), confirming adherence with enalapril (Figure 3B).
Figure 3.
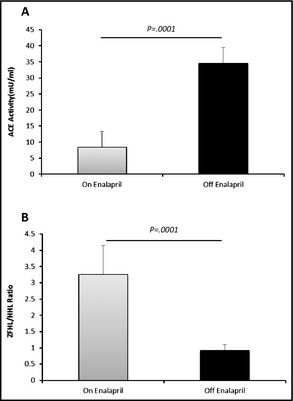
Serum angiotensin‐converting enzyme (ACE) activity in controls on and off treatment. (A) Paired test comparing serum mean ACE activity of patients on and off treatment. Values are mean±standard deviation. (B) ZFHL/HHL ratio to check for patient compliance.
The mean ACE activity of patients with ACE‐AE and ACE‐cough was 50% and 30% less than that of controls, respectively (Figure 4). There was a smaller, but significant, difference in the mean ACE activity of cough vs AE patients (P=.0003). The ACE activity in serum was correlated with ACE genotype in controls and ACE‐AE and ACE‐cough patients (Figure 5). ACE activities of controls, ACE‐AE, and ACE‐cough were similar between, ACE‐II, ID, and DD. However, ACE‐AE and ACE‐cough II homozygotes had significantly lower mean ACE activities compared with controls (P=.0001 and P=.01, respectively). In contrast, the lower mean activity ACE‐AE compared with ACE‐cough ACE‐II homozygotes was not significant (17.05±5.16 mU/mL vs 23.05±10.19 mU/mL, P=.11). This profile of ACE activity was similar in ACE ID and DD genotypes, but the difference between the mean ACE activity of ACE‐AE and ACE‐cough was less pronounced in the ACE‐DD genotype for cough‐AE, P=.188 for ACE‐DD genotype; P=.0016 for ACE‐ID; and P=.115 for ACE‐II).
Figure 4.
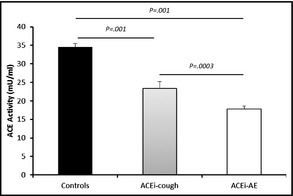
Analysis of serum angiotensin‐converting enzyme (ACE) activity in patients with ACE‐associated angioedema (ACE‐AE) and ACE‐associated cough (ACE‐cough). One‐way analysis of variance comparing ACE enzymatic activity of controls with patients with ACE‐AE and ACE‐cough. Values are mean±standard deviation.
Figure 5.
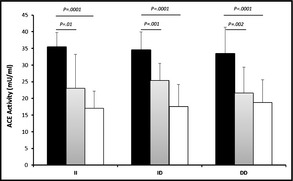
Analysis of serum angiotensin‐converting enzyme (ACE) activity in ACE insertion/deletion genotypes. Correlation between serum ACE activity and ACE ID genotype in controls (black bars) with patients with ACE‐associated cough (ACE‐cough) (grey bars) and ACE‐associated angioedema (ACE‐AE) (white bars).
Discussion
Much research has been done on the clinical and molecular bases of the ACE inhibitor side effects. However, the mechanisms that cause ACE‐AE and ACE‐cough are still not fully understood. In the present study we investigated whether ACE I/D and bradykinin B2 receptor −9/+9 polymorphism and the B2 C‐58T SNP are associated with the occurrence of ACE‐AE and ACE‐cough. Previous studies have shown that the ACE I/D polymorphism is associated with diseases such asthma, myocardial infarction, and idiopathic nephritic syndrome.24, 25, 26 In this study, there was no significant association between the ACE I/D polymorphism and ACE inhibitor–associated AE or cough. These findings are in agreement with recent work investigating the impact of the ACE I/D polymorphism in Caucasian patients with documented episodes of ACE‐AE,27 but in contrast to previous work suggesting that the ACE‐II genotype was dominant in black patients with ACE‐AE.15 However, serum ACE activity was significantly decreased in both ACE‐AE and ACE‐cough patients, and not related to ACE I/D polymorphisms. This confirms a previous report that ACE genotype has limited impact on ACE activity in a black male South African cohort.28 We found that ACE activity is higher in controls than in ACE‐AE and ACE‐cough patients across all genotypes and this suggests that it may be a clinically useful risk marker for ACE‐AE and ACE‐cough. The reason for the altered ACE activity is unclear; however, it is possible that this might be due to aberrant processing of the membrane‐bound form of ACE as opposed to changes in activity or protein expression.29, 30 A soluble isoform of ACE produced by alternative splicing was identified in human umbilical vein endothelial cells.31 However, this form has not been identified in patients during or after ACE inhibitor treatment.
BK increases vascular permeability and stimulates cough reflex in the lung by stimulating the B2 receptor, and is probably the underlying mechanism for cough and angioedema. Genetic mutations in the BK metabolic pathway are attractive candidate genes for ACE‐cough and ACE‐AE. The B2 −9/+9 polymorphisms have been linked to increased vasodilation during ACE inhibition,17 and hereditary AE in cases of C‐1 esterase inhibitor deficiency. To our knowledge, this is the first study to associate both ACE‐AE and ACE‐cough with the −9/+9 B2 receptor polymorphism. There was a significant association with B2–9 variant with both ACE‐AE and ACE‐cough. The C‐58T receptor promoter SNP has been associated with ACE‐cough in Asian patients.20 In this study, however, no correlation was shown between C‐58T SNP and ACE‐AE and ACE‐cough.
Study Limitations
There are a number of limitations in this study that are worth highlighting. By chance, white patients and men with angioedema were underrepresented, making it difficult to carry out meaningful ethnic and sex comparisons. Van Guilder and colleagues17 have reported that the −9/+9 genotype is associated with increased systolic blood pressure in normotensive white Americans and with increased forearm vascular resistance in normotensive black Americans. Thus, the findings of this study are not applicable to other ethnic groups. While the association of the ACE B2 −9/+9 genotype with ACE‐AE and ACE‐cough is of interest, greater numbers are required to further validate these findings. Finally, previous studies have reported an increase in ACE activity following prolonged ACE inhibitor treatment in hypertensive patients.32 However, there was no change in ACE expression in rats following lisinopril administration.33 Consequently, while it is possible that the increased ACE activity in the controls could be partly due to an ACE inhibitor–induced increase in ACE expression, the ACE activity of the patients with ACE‐AE was significantly lower than that of patients with ACE‐cough.
Conclusions
In the present study we showed a significant correlation between B2 −9/+9 and low ACE activity and the development of ACE‐AE and ACE‐cough. Furthermore, no significant association was shown between ACE I/D or B2 C‐58T gene polymorphisms and ACE‐AE and ACE‐cough. The difficulty in elucidating the genetic basis of complex diseases is entrenched in multiple factors that have an effect in the development of a disease. Therefore, further studies should be carried out to determine whether the combination of genetic polymorphisms might be risk factors for the development of both ACE‐AE and ACE‐cough.
Disclosure
This study was supported by the South African National Research Foundation (EDS), National Health Laboratory Service (BRR and EPO), and the University of Cape Town (EDS and BRR). The authors have no conflicts of interest to disclose.
J Clin Hypertens (Greenwich). 2013;15:413–419. ©2013 Wiley Periodicals, Inc.23730990
References
- 1. Blais C Jr, Rouleau JL, Brown NJ, et al. Serum metabolism of bradykinin and des‐Arg9‐bradykinin in patients with angiotensin‐converting enzyme inhibitor‐associated angioedema. Immunopharmacology. 1999;43:293–302. [DOI] [PubMed] [Google Scholar]
- 2. Israili ZH, Hall WD. Cough and angioneurotic edema associated with angiotensin‐converting enzyme inhibitor therapy. A review of the literature and pathophysiology. Ann Intern Med. 1992;117:234–242. [DOI] [PubMed] [Google Scholar]
- 3. Brown NJ, Ray WA, Snowden M, Griffin MR. Black Americans have an increased rate of angiotensin converting enzyme inhibitor‐associated angioedema. Clin Pharmacol Ther. 1996;60:8–13. [DOI] [PubMed] [Google Scholar]
- 4. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637–1642. [DOI] [PubMed] [Google Scholar]
- 5. Byrd JB, Touzin K, Sile S, et al. Dipeptidyl peptidase IV in angiotensin‐converting enzyme inhibitor associated angioedema. Hypertension. 2008;51:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cugno M, Nussberger J, Cicardi M, Agostoni A. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol. 2003;3:311–317. [DOI] [PubMed] [Google Scholar]
- 7. Nussberger J, Cugno M, Amstutz C, et al. Plasma bradykinin in angio‐oedema. Lancet. 1998;351:1693–1697. [DOI] [PubMed] [Google Scholar]
- 8. Molinaro G, Cugno M, Perez M, et al. Angiotensin‐converting enzyme inhibitor‐associated angioedema is characterized by a slower degradation of des‐arginine(9)‐bradykinin. J Pharmacol Exp Ther. 2002;303:232–237. [DOI] [PubMed] [Google Scholar]
- 9. Duan QL, Nikpoor B, Dube MP, et al. A variant in XPNPEP2 is associated with angioedema induced by angiotensin I‐converting enzyme inhibitors. Am J Hum Genet. 2005;77:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moreau ME, Garbacki N, Molinaro G, et al. The kallikrein‐kinin system: current and future pharmacological targets. J Pharmacol Sci. 2005;99:6–38. [DOI] [PubMed] [Google Scholar]
- 11. Braun A, Maier E, Kammerer S, et al. A novel sequence polymorphism in the promoter region of the human B2‐bradykinin receptor gene. Hum Genet. 1996;97:688–689. [DOI] [PubMed] [Google Scholar]
- 12. Soubrier F, Cambien F. The angiotensin I‐converting enzyme gene polymorphism: implication in hypertension and myocardial infarction. Curr Opin Nephrol Hypertens. 1994;3:25–29. [DOI] [PubMed] [Google Scholar]
- 13. Rigat B, Hubert C, Alhenc‐Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I‐converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gulec M, Caliskaner Z, Tunca Y, et al. The role of ace gene polymorphism in the development of angioedema secondary to angiotensin converting enzyme inhibitors and angiotensin II receptor blockers. Allergol Immunopathol (Madr). 2008;36:134–140. [PubMed] [Google Scholar]
- 15. Ducroix JP, Outurquin S, Benabes‐Jezraoui B, et al. [Angioedema and angiotensin converting enzyme inhibitors: a report of 19 cases]. Rev Med Interne. 2004;25:501–506. [DOI] [PubMed] [Google Scholar]
- 16. Braun A, Kammerer S, Maier E, et al. Polymorphisms in the gene for the human B2‐bradykinin receptor. New tools in assessing a genetic risk for bradykinin‐associated diseases. Immunopharmacology. 1996;33:32–35. [DOI] [PubMed] [Google Scholar]
- 17. Van Guilder GP, Pretorius M, Luther JM, et al. Bradykinin type 2 receptor BE1 genotype influences bradykinin‐dependent vasodilation during angiotensin‐converting enzyme inhibition. Hypertension. 2008;51:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lung CC, Chan EK, Zuraw BL. Analysis of an exon 1 polymorphism of the B2 bradykinin receptor gene and its transcript in normal subjects and patients with C1 inhibitor deficiency. J Allergy Clin Immunol. 1997;99:134–146. [DOI] [PubMed] [Google Scholar]
- 19. Gainer JV, Brown NJ, Bachvarova M, et al. Altered frequency of a promoter polymorphism of the kinin B2 receptor gene in hypertensive African‐Americans. Am J Hypertens. 2000;13:1268–1273. [DOI] [PubMed] [Google Scholar]
- 20. Mukae S, Itoh S, Aoki S, et al. Association of polymorphisms of the renin‐angiotensin system and bradykinin B2 receptor with ACE‐inhibitor‐related cough. J Hum Hypertens. 2002;16:857–863. [DOI] [PubMed] [Google Scholar]
- 21. Danilov SM, Balyasnikova IV, Albrecht RF, Kost OA. Simultaneous determination of ACE activity with 2 substrates provides information on the status of somatic ACE and allows detection of inhibitors in human blood. J Cardiovasc Pharmacol. 2008;52:90–103. [DOI] [PubMed] [Google Scholar]
- 22. Friedland J, Silverstein E. A sensitive fluorimetric assay for serum angiotensin‐converting enzyme. Am J Clin Pathol. 1976;66:416–424. [DOI] [PubMed] [Google Scholar]
- 23. Lieberman J, Sastre A. An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875. [DOI] [PubMed] [Google Scholar]
- 24. Benessiano J, Crestani B, Mestari F, et al. High frequency of a deletion polymorphism of the angiotensin‐converting enzyme gene in asthma. J Allergy Clin Immunol. 1997;99:53–57. [DOI] [PubMed] [Google Scholar]
- 25. Mehri S, Baudin B, Mahjoub S, et al. Angiotensin‐converting enzyme insertion/deletion gene polymorphism in a Tunisian healthy and acute myocardial infarction population. Genet Test Mol Biomarkers. 2010;14:85–91. [DOI] [PubMed] [Google Scholar]
- 26. Tsai IJ, Yang YH, Lin YH, et al. Angiotensin‐converting enzyme gene polymorphism in children with idiopathic nephrotic syndrome. Am J Nephrol. 2006;26:157–162. [DOI] [PubMed] [Google Scholar]
- 27. Bas M, Hoffmann TK, Tiemann B, et al. Potential genetic risk factors in angiotensin‐converting enzyme‐inhibitor‐induced angio‐oedema. Br J Clin Pharmacol. 2010;69:179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dhamrait SS, Payne JR, Li P, et al. Variation in bradykinin receptor genes increases the cardiovascular risk associated with hypertension. Eur Heart J. 2003;24:1672–1680. [DOI] [PubMed] [Google Scholar]
- 29. Eyries M, Michaud A, Deinum J, et al. Increased shedding of angiotensin‐converting enzyme by a mutation identified in the stalk region. J Biol Chem. 2001;276:5525–5532. [DOI] [PubMed] [Google Scholar]
- 30. Nesterovitch AB, Hogarth KD, Adarichev VA, et al. Angiotensin I‐converting enzyme mutation (Trp1197Stop) causes a dramatic increase in blood ACE. PLoS ONE. 2009;4:e8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sugimura K, Tian XL, Hoffmann S, et al. Alternative splicing of the mRNA coding for the human endothelial angiotensin‐converting enzyme: a new mechanism for solubilization. Biochem Biophys Res Commun. 1998;247:466–472. [DOI] [PubMed] [Google Scholar]
- 32. Stephan D, Grima M, Welsch M, et al. Interruption of prolonged ramipril treatment in hypertensive patients: effects on the renin‐angiotensin system. Fundam Clin Pharmacol. 1996;10:474–483. [DOI] [PubMed] [Google Scholar]
- 33. Ferrario CM, Jessup J, Gallagher PE, et al. Effects of renin‐angiotensin system blockade on renal angiotensin‐(1‐7) forming enzymes and receptors. Kidney Int. 2005;68:2189–2196. [DOI] [PubMed] [Google Scholar]