

Abstract
Native agarose gel electrophoresis-based particle gel assay has been commonly used for examination of hepatitis B virus (HBV) capsid assembly and pregenomic RNA encapsidation in HBV replicating cells. Interestingly, treatment of cells with several chemotypes of HBV core protein allosteric modulators (CpAMs) induced the assembly of both empty and DNA-containing capsids with faster electrophoresis mobility. In an effort to determine the physical basis of CpAM-induced capsid mobility shift, we found that the surface charge, but not the size, of capsids is the primary determinant of electrophoresis mobility. Specifically, through alanine scanning mutagenesis analysis of twenty-seven charged amino acids in core protein assembly domain and hinge region, we showed that except for K7 and E8, substitution of glutamine acid (E) or aspartic acid (D) on the surface of capsids reduced their mobility, but substitution of lysine (K) or arginine (R) on the surface of capsids increased their mobility in variable degrees. However, alanine substitution of the charged amino acids that are not exposed on the surface of capsid did not apparently alter capsid mobility. Hence, CpAM-induced electrophoresis mobility shift of capsids may reflect the global alteration of capsid structure that changes the exposure and/or ionization of charged amino acid side chains of core protein. Our findings imply that CpAM inhibition of pgRNA encapsidation is possibly due to the assembly of structurally altered nucleocapsids. Practically, capsid electrophoresis mobility shift is a diagnostic marker of compounds that target core protein assembly and predicts sensitivity of HBV strains to specific CpAMs.
1. Introduction
Hepatitis B virus (HBV) chronically infects 240 million people worldwide. Approximately 15-40% of these chronic HBV carriers ultimately develop severe liver diseases, including cirrhosis, hepatocellular carcinoma (HCC) and liver failure, and results in 680,000 deaths annually (Ott et al., 2012). The currently available antiviral therapeutics, including two formulations of alpha-interferon (standard and pegylated) and six nucleos(t)ide analogues (NUCs) that inhibit HBV DNA polymerase, fail to induce a durable off-drug control of HBV replication in the vast majority of patients (Dienstag, 2009; Perrillo, 2009). Discovery and development of novel antiviral therapeutics to stop HBV replication, eradicate or functionally inactivate the covalently closed circular (ccc) DNA as well as activate a functional host antiviral immune response against HBV are essential to achieve a functional cure of chronic hepatitis B (Chang et al., 2014; Tang et al., 2017).
HBV contains a 3.2 kb, partially double-stranded, relaxed circular (rc) DNA genome. However, unlike other DNA viruses, HBV replicates its genomic DNA via viral DNA polymerase self-primed reverse transcription of a RNA pregenome in cytoplasmic nucleocapsid (Block et al., 2007). Briefly, HBV genomic DNA synthesis begins by packaging viral pregenomic (pg) RNA and DNA polymerase complex with 120 copies of core protein dimers to form a nucleocapsid. Inside the nucleocapsid, viral DNA polymerase converts the pgRNA first to a single-stranded DNA and then to rcDNA. The rcDNA-containing mature nucleocapsids can either acquire an envelope and be secreted out of cells as infectious virions or deliver the rcDNA into the nucleus to amplify cccDNA pool, the sole transcription template supporting HBV replication (Guo and Guo, 2015; Seeger and Mason, 2015).
HBV core protein is a 183-aa polypeptide consisting of N-terminal assembly domain and C-terminal domain (CTD). While the assembly domain is sufficient to assemble empty capsids, the CTD is essential for packaging pgRNA and DNA polymerase into nucleocapsids (Zhao et al., 2018). Due to the unique structure and essential role in viral replication, disruption of nucleocapsid assembly with small molecular core protein allosteric modulators (CpAMs) represents a new frontier in development of novel antiviral agents against chronic HBV infection (Zlotnick et al., 2015). While heteroaryldihydropyrimidines (HAP), or type I CpAM, such as Bay 41-4109 and GLS4, misdirect capsid assembly to form non-capsid core proteins polymer (Bourne et al., 2006; Stray et al., 2005; Stray and Zlotnick, 2006; Wu et al., 2017), all the other reported CpAMs, including sulfamoylbenzamides (SBAs), benzamides (BAs) and phenylpropenamides (PPAs), induce the formation of variable sizes of morphologically normal capsids devoid of viral pgRNA and DNA polymerase and categorized as type II CpAM (Campagna et al., 2013; King et al., 1998; Wang et al., 2015; Wu et al., 2017; Yang et al., 2016). Capsid assembly is primarily driven by weak hydrophobic interactions at the interdimer interface of core proteins (Venkatakrishnan and Zlotnick, 2016). In spite of the difference in assembled products, both types of CpAMs disrupt core protein assembly by binding to the hydrophobic pocket, designated as the HAP pocket, at the dimer-dimer interface to induce large scale allosteric conformational changes in core protein subunits and accelerate the assembly kinetics (Katen et al., 2013; Klumpp et al., 2015; Venkatakrishnan et al., 2016; Zhou et al., 2017). In addition to CpAM treatment, substitution of selected amino acid residues at wall of the HAP pocket, such as V124W, can also enhance the thermodynamics of core protein assembly and alter capsid assembly and pgRNA packaging (Tan et al., 2015).
While the effects of CpAMs on the kinetics of core protein assembly and structure of capsids can be investigated in cell-free assembly system and examined by advanced biophysical technologies (Pierson et al., 2014; Stray and Zlotnick, 2006), native agarose gel electrophoresis-based particle gel assay has been commonly used to determine the effects of CpAMs on capsid assembly, pgRNA packaging and reverse transcriptional DNA synthesis in HBV infected cells (Guo et al., 2017; Xu et al., 2010). We reported recently that treatment of HBV replicating cells with CpAMs resulted in the formation of empty and DNA-containing capsids with faster electrophoresis mobility in the particle gel assay (Guo et al., 2017; Wu et al., 2017). The electrophoresis mobility of capsids can be attributed to changes of capsids in the sizes, shapes and/or surface charges, which all reflect the global alterations of capsid structure. It is thus conceivable that better understanding the biophysical basis of CpAM-induced capsid electrophoresis mobility shift will shed light on the mechanism underlying CpAM inhibition of pgRNA encapsidation. To this end, we determined the sizes and morphology of capsids purified from CpAMs-treated cells and investigated the effects of charged amino acids on capsid mobility by alanine scanning mutagenesis of 27 charged amino acid residues in the assembly domain and hinge region of core protein. We found that the surface charge, but not the size, of capsid is the major determinant of electrophoresis mobility. Hence, the observed electrophoresis mobility shift of HBV capsids in CpAM treated cells indicate that CpAMs induced the formation of structurally distinct empty and DNA-containing capsids with altered surface exposure and/or ionization of charged amino acid residues. Our results thus imply that CpAM inhibition of pgRNA encapsidation is possibly due to the accelerated assembly kinetics of empty capsids as well as formation of structurally altered nucleocapsids.
2. Materials and Methods
2.1. Cell culture and reagents
Human hepatoblastoma cell line HepG2 were maintained in MEM medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin. An AML12-derived stable cell line AML12HBV10 which can support replication of a stably-transfected envelope protein-deficient HBV genome in a tetracycline-inducible manner (39, 40) was maintained in DMEM/F12 medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml tetracycline and 400 μg/ml G-418. Entecavir (ETV) was provided by Dr. William S. Mason at Fox Chase Cancer Center, Philadelphia. Bay 41-4109 was purchased from MedChemExpress (MCE). ENAN-34017, BA-38017 and AT-61 were synthesized in house (Campagna et al., 2013; Wu et al., 2017). 17-(Dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG) was purchased from InvivoGen. Rabbit anti-HBc antibody was obtained from Dako (Cat. No. B0586).
2.2. Plasmids
pCMV-HBV expressing HBV pgRNA under the control of CMV immediate early promoter, wild-type HBV replicon pHBV1.3mer, pHBV-1.3-Y63F and pCMV-HBc have been described previously (Mao et al., 2011; Summers et al., 1990; Zoulim et al., 1994). Plasmids pHBV-genotype A-clone 4B or clone 8.22 and pHBV-genotype C-geno27.2 were gifts of Dr, Shuping Tong at Brown University (Ludgate et al., 2016). pHBV-genotype A- clone 4B-derived plasmid expressing mutant core protein Q77E was generated by GenScript. Information on the sequence and construction of the wild-type genotype B HBV plasmid pHY536207 has been published previously (Zhang et al., 2005).
The pCMV-HBc-derived plasmids expressing D2A, D4A, K7A, E8A and E14A mutant core proteins were generated by PCR and plasmids expressing D22A, R28A, D29A, D29E, D29W, D29L, D29G, D32A, R39A, E40A, E43A, E46A, R56A, E64A, E77A, E77Q, D78A, R82A, E83A, K96A, R98A, R112A, E113A, E117A, R127A, R127E, R127K, R127V, R133A, and E145A mutant core proteins were generated by overlapping PCR strategy. For instance, to generate the D2A mutant core protein, DNA was amplified with sense primer D2A-F (5’- GAGCTCGGATCCACTAGTCCAGTGTGGTGGAATTGCCCTTACCATGGCTATCGACCCTTATAA -3’) and HBc antisense primer R (5’- AACCGCGGGCCCTCTAGACTCGA -3’). The PCR products were then purified, digested with BamH I and Apa I and cloned into a pCMV-HBc vector. For generating the D22A mutant core protein, two DNA fragments were amplified with sense primer F (5’- GAGCTCGGATCCACTAGTCCAGTGTGGT -3’) and D22A antisense primer (5’- GTACTGAAGGAAAGAAGGCAGAAGGCAAAAACGA -3’), and with D22A sense primer (5’- TCGTTTTTGCCTTCTGCCTTCTTTCCTTCAGTAC -3’) and antisense primer R (5’- AACCGCGGGCCCTCTAGACTCGA-3’), respectively. The PCR productions were purified and mixed together in a molar ratio of 1:1. The mixture was amplified with F and R primers. Then the resultant DNA were purified and ligated into pCMV-HBc after digestion with BamH I and Apa I.
Using a similar strategy, pHBV-1.3- and pHBV-1.3-Y63F-derived plasmid expressing deficient core protein (Δcore) were generated by changing the 38th amino acid Y of core gene into a stop codon. Briefly, two DNA fragments were amplified with sense primer F172 (5’- ATATATGCATGCGTGGAACCTTTTCGGCTCCTCTG -3’) and Y38stop antisense primer (5’- CTAAGGCTTCCCGCTACAGAGCTGAGGC -3’), and with Y38stop sense primer (5’- GCCTCAGCTCTGTAGCGGGAAGCCTTAG -3’) and antisense primer R2116 (5’- ATATATGAATTCCACTGCATGGCCTGAGGATGAGTGTT -3’), respectively. After purification, the mixture of two DNA fragments was amplified with F172 and R2116 primers. The resultant DNA were digested with Sph I and EcoR I and ligated to pHBV-1.3 / Sph I + EcoR I and pHBV-1.3-Y63F/ Sph I + EcoR I, respectively. All plasmids were confirmed by DNA sequencing. Sequence of the primers used in the construction of those recombinant plasmids will be provided upon requests.
2.3. Transient transfection
HepG2 cells were seeded in 24-well plates and grown to approximately 80% confluence. The medium was replaced by MEM without antibiotics before transfection. Cells were then transfected with 0.25 μg desired plasmid(s) using 1.25 μl Lipofectamine 2000 (Invitrogen) per well. To introduce two plasmids into cells, 0.13 μg of each plasmid were mixed with 1.25 μl of Lipofectamine 2000 and applied to cells. Six hours post transfection, the culture media were replaced with fresh media or media containing desired concentration of compounds and cultured for additional 72 h. Intracellular HBV core protein, capsids, core DNA and pgRNA were examined with the assays specified below.
2.4. Analyses of HBV core DNA by hybridization and qPCR assays
Intracellular HBV core DNA from transfected HepG2 cells were extracted as described previously (Guo et al., 2007) and quantified by a real-time PCR assay using TransStart Tip Green qPCR SuperMix (TransGen Biotech) with forward primer 5’-GGCTTTCGGAAAATTCCTATG-3’ and reverse primer 5’-AGCCCTACGAACCACTGAAC-3’. The PCR was carried out using the ABI 7500 Fast Real-Time PCR system (Applied Biosystems) under the following conditions: denatured at 94°C for 30 seconds, followed by 40 cycles of amplification: 94°C, 5 seconds; 60°C, 30 seconds. The antiviral efficacy of a compound was expressed as the concentration that reduced the amount of HBV DNA by 50% (EC50) in comparison with the levels of the mock-treated controls. The HBV core DNA were also analyzed by Southern blot hybridization as described previously (Guo et al., 2007)
2.5. Electron microscopic analysis of capsids
AML12HBV10 cells were mock-treated or treated with 5 μM of ENAN-34017 for 2 days. HBV capsids in the cell lysates were purified by sucrose gradient centrifugation (Guo et al., 2010) and detected by EM after negatively stained with Uranyless (Electron Microscopy Sciences, Cat. No. 22409). Negatively stained sample were imaged on an FEI Tecnai 12 Spirit/Biotwin (LaB6 filament), operating at 100kV, with an AMT 2k x 2k CCD camera.
For EM analyses of D29 mutant capsids, HepG2 cells were seeded in 10-cm dishes and cultured for 18 - 24 h. The cells were transfected with 5 μg of plasmid using 25 μL of Lipofectamine 2000. After 3 days post-transfection, HBV capsids in the cell lysates were concentrated by 100KDa cut-off ultrafiltration (Millipore) and then purified by sucrose gradient centrifugation (Guo et al., 2010) and detected by EM after negatively stained with phosphotungstic acid on continuous carbon grids.
2.6. Western blot assay
Proteins were extracted from the transfected HepG2 cells and the expression of HBc (Dako) were determined by Western blot as described previously (Xu et al., 2010). β-actin was detected (Cell Signaling Technology) as loading control.
2.7. Particle gel assay
HBV capsids and associated viral DNA were analyzed by a native agarose gel electrophoresis-based particle assay as described previously (Xu et al., 2010).
2.8. Northern blot assay
Total and encapsidated viral pgRNA were extracted and determined by Northern blot asssay as described previously (Wu et al., 2017).
2.9. Computational analyses of structural simulation and surface charge
HBV core protein structure used in this study was downloaded from the protein data bank (PDB), identification number 5e0i (Berman et al., 2000). The protein structure was prepared using the Protein Preparation Wizard, a module in Schrödinger’s Small Molecule Drug Discovery Suite (2016). Location of the charged amino acids on capsid are showed.
2.10. Position Frequency of Amino acids of HBV core protein sequences deposited in GenBank.
HBV core sequences were downloaded from NCBI web site https://www.ncbi.nlm.nih.gov/ by keywords searching with “HBV core” and species “virus”. There are 38,430 sequences in total. Those sequences which cover less than 75% of query sequence (HBV core protein, GenBank accession number P03146) were filtered out. The remaining 15,871 sequences (HBVcore75.fasta) were used for multiple sequence alignment. Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was applied on the fasta data set to do multiple sequence alignment using default parameter setting. The percentage of an amino acid at each position (i) was calculated from the multiple sequence alignment by excluding gaps, given in the following equation.
Here N is the number of sequences in the multiple sequence alignment. #Gaps is the number of gaps (no amino acids aligned) at position i. #AAi is the number of an amino acid at position i.
3. Results
3.1. Type II CpAMs induce the assembly of capsids with faster electrophoresis mobility
We and others showed recently that capsids in HBV replicating cells can be resolved into two species with distinct migration mobility by increasing agarose concentration from 1.0% to 1.5 or 1.8% in particle gel assays and DNA-containing capsids, or nucleocapsids, co-migrate with the slower migrating capsids (Wu et al., 2017; Yang et al., 2016). As shown in Fig. 1A, while Bay 41-4109 treatment inhibited capsid assembly, treatment of AML12HBV10 cells with three chemotypes of type II CpAMs, i.e., SBA derivative ENAN-34017, BA derivative BA-38017 and PPA derivative AT-61, induced the formation of both “empty” and DNA-containing capsids with faster electrophoresis mobility. As expected, treatment of the cells with heat shock protein 90 (HSP90) ATPase inhibitor 17-DMAG that inhibits HBV DNA polymerase binding of pgRNA to prevent its encapsidation (Hu et al., 2004) or HBV DNA polymerase inhibitor entecavir (ETV) efficiently reduced the amounts of HBV DNA, but did not alter the electrophoresis mobility of capsids. These findings imply that binding of type II CpAMs at the HAP pocket between the dimer-dimer interfaces of core protein induces the assembly of not only structurally altered “empty” capsids, but also DNA-containing capsids. This later observation implies that type II CpAM inhibition of pgRNA encapsidation might be due to not only the accelerated capsid assembly kinetics as speculated in a previous study (Katen et al., 2010), but also the assembly of structurally altered nucleocapsids.
Fig. 1. Particle gel assay of capsids assembled in cells treated with representative type II CpAMs.

(A) AML12HBV10 cells were culture in the presence of tetracycline (tet +) or in the absence of tetracycline and mock-treated (tet −) or treated with the indicated concentrations of the compounds Bay 41-4109 (2 μM), ENAN-34017 (5 μM), BA-38017 (5 μM), AT-61 (25 μM), 17-DMAG (0.3 μM), or ETV (1 μM) for 2 days. The total amounts of capsids were separated by a particle gel assay in a 1.5% agarose gel electrophoresis. Capsid-associated HBV DNA was detected by hybridization with a 32P-labeled full-length HBV minus-strand specific riboprobe upon alkaline treatment of nucleocapsids on the membrane following the particle gel assay. The slow and fast migrating capsids are indicated by arrows. (B) Electronic microscopic graphs of capsids assembled in AML12HBV10 cells mock-treated or treated with 5 μM of ENAN-34017 for 2 days are presented. White scale bar indicates 100 nm.
Because the electrophoresis mobility of capsids can be affected, in theory, by their sizes, shapes, total mass and surface charges, we first determined the sizes of capsids purified from CpAMs-treated cells. The results presented in Fig. 1B demonstrated that ENAN-34017 treatment induced formation of morphologically normal capsids that are similar in size with the capsids prepared from mock-treated cells. Moreover, the previously published studies by others and us also suggested that BA-38017 and PPA derivative AT-130 treatment induced the assembly of capsids that are similar to or slightly larger than the capsids derived from mock treated samples, respectively (Katen et al., 2013; Wu et al., 2017). These results thus indicate that the increased electrophoresis mobility of capsids may not be due to their reduced sizes and altered morphology, but most likely the alteration of surface charges, resulted from CpAMs-induced capsid structural changes (Bourne et al., 2006; Katen et al., 2013; Venkatakrishnan et al., 2016).
3.2. A naturally occurring E77Q point mutation in genotype A HBV drastically reduces capsid mobility
In support of the speculated role of charged amino acids on the electrophoresis mobility of HBV capsids, we incidentally found that a naturally occurring E77Q point mutation in HBV core protein drastically reduces the electrophoresis mobility of capsids. Briefly, examination of HBV capsids formed in HepG2 cells transfected with HBV replicon plasmids derived from genotypes A, B, C and D by the particle gel assay revealed that the capsids from two strains of genotype A replicons, Clones 4B and 8.22, migrated significantly slower than the capsids of other genotypes of HBV (Fig. 2A, comparing lanes 11-14 with lanes 1 - 10). Subsequent sequence alignment analyses identified several amino acid substitutions that are unique in the core proteins of the two genotype A HBV strains (Fig. 2B). However, compared to the core protein of a representative genotype A HBV (GenBank Accession number AY233274), only the E77Q substitution was shared by clones 4B and 8.22 of genotype A HBV and might be responsible for the reduced electrophoresis mobility of capsids. Indeed, substitution of Q77 with E in the core protein of genotype A clone 8.22 resulted in the production of capsids with similar electrophoresis mobility of genotypes B, C and D (Fig. 2A, lanes 15 and 16). The results thus indicate that a single charged amino acid residue of HBV core protein has a strong impact on the electrophoresis mobility of capsids.
Fig. 2. Mapping of HBV genotype A mutations confer slow migration of capsids.

(A) Particle gel assay results. HepG2 cells were transiently transfected with the indicated plasmids and the cells were harvested at 72 h post transfection. The capsids were separated on a 1.5% agarose gel electrophoresis, transferred on to a Nylon membrane and detected by a rabbit polyclonal antibody against HBV core protein (Dako). Capsid-associated HBV DNA was detected by hybridization with a 32P-labeled full-length HBV minus-strand specific riboprobe upon alkaline treatment of the membrane following the particle gel separation. (B) Sequence alignment of genotype A HBV core proteins with genotype B, C and D HBV core proteins.
3.3. Identification of charged amino acid residues that affect the electrophoresis mobility of capsids
We have already demonstrated that the C-terminal domain of core protein as well as its phosphorylation status do not affect CpAM-induced capsid mobility shift (Wu et al., 2017). As showed in Table 1, the assembly domain and hinge region of HBV core protein has 27 highly conserved charged amino acid residues. It is conceivable that due to their distinct locations in capsids and ionization potentials, each of the charged amino acids should not equally affect the capsid electrophoresis mobility. To determine the effects of each charged amino acid residue on capsid assembly, DNA replication and capsid mobility, we performed an alanine scanning mutagenesis analysis of the charged amino acids. Specifically, capsid assembly and viral DNA replication in HepG2 cells co-transfected with pHBV-1.3-derived plasmid deficient in core protein expression and pCMV-HBc-derived plasmids expressing wild-type or an indicated alanine substituted mutant core protein were examined by particle gel assay. As shown in Fig. 3, alanine (A) substitution of glutamine acid (E) or aspartic acid (D) either did not alter or reduced capsid mobility in variable degrees while only E8A substituted core protein assembled capsids with increased mobility. On the contrary, alanine substitution of lysinse (K) or arginine (R) either did not alter or increased capsid mobility in variable degrees, except for K7A substituted core protein that assembled capsids with decreased mobility. Specifically, while K7A, D22A, D29A, D32A, E64A, E77A, D78A and D83A mutant core proteins assembled slower migrating capsids, E8A, R28A, R82A, K96A, R98A, R127A and R133A mutant core proteins assembled faster migrating capsids (Fig. 3A, C, E, G, and I). On the contrary, alanine substitution of charged amino acids at N-terminal region (D2A and D4A) and regions spanning R39 to R56 (R39A, E40A. E43A. E46A and R56A) and R112 to E117 (R112A, E113A and E117A) did not apparently alter capsid mobility. In another words, except for K7 and E8, each of the charged amino acid residues located in three sequence-continuing stretches of charged amino acid residues of core protein, including D22 to D32, E64 to R98 and R127 to R133, affects capsid mobility in a variable degree. Obviously, the charged amino acids at the tip of spike of core protein dimer, such as E77, D78, R82 and D83, have the strongest impacts on capsid mobility.
Table 1.
Sequence alignment analysis of 27 charged amino acids in assembly domain from 16,432 HBV core protein sequences deposited in GenBank.
| AA position | 17 negatively charged AAs |
AA position | 10 positively charged AAs |
||
|---|---|---|---|---|---|
| D | E | K | R | ||
| 2 | 99.597# | ||||
| 4 | 99.603 | 0.019 | |||
| 7 | 99.691 | 0.019 | |||
| 8 | 99.653 | ||||
| 14 | 99.118 | 0.132 | |||
| 22 | 99.754 | 0.044 | |||
| 28 | 0.277 | 98.815 | |||
| 29 | 99.206 | 0.271 | |||
| 32 | 99.590 | 0.063 | |||
| 39 | 0.013 | 99.483 | |||
| 40 | 13.055 | 86.434 | |||
| 43 | 0.069 | 99.055 | |||
| 46 | 0.056 | 98.866 | |||
| 56 | 0.057 | 99.691 | |||
| 64 | 5.211 | 94.058 | |||
| 77 | 3.176 | 92.338 | |||
| (Q=3.232) | |||||
| 78 | 98.834 | 0.063 | |||
| 82 | 0.151 | 98.563 | |||
| 83 | 44.566 | 53.903 | |||
| 96 | 98.135 | 0.088 | |||
| 98 | 0.315 | 97.889 | |||
| 112 | 0.989 | 97.580 | |||
| 113 | 3.944 | 92.395 | |||
| 117 | 0.151 | 98.910 | |||
| 127 | 99.168 | ||||
| 133 | 0.101 | 99.395 | |||
| 145 | 0.025 | 98.847 | |||
Frequency expressed as percentage of the indicated amino acid at the position in the 16,432 HBV core protein sequences deposited in GenBank
Fig. 3. Systematic mutagenesis of charged amino acids in HBV core assembly domain.


HepG2 cells were transfected with pHBV1.3 or co-transfected with pHBV1.3-Δcore and pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with indicated charged amino acid mutation. Cells were also co-transfected with pHBV1.3-Δcore and the vector plasmid as a negative control. The cells were harvested at 72 h post transfection. (A, C, E, G, I) Cytoplasmic capsids were detected by a particle gel assay. (B, D, F, H, J) The HBV core DNA accumulation was determined by Southern blotting assay. A 32P-labeled full-length minus-strand specific riboprobe was used for Southern blot analysis. (K) Core protein (Cp) expression was detected by Western blotting with a rabbit polyclonal antibody (Dako) and the β-actin served as loading controls. Lane * in panels B and D, loaded with DNA side markers, 3.2 and 2.0 kb.
In addition to capsid mobility, the studies also showed that alanine substitution of majority of the charged amino acids did not dramatically reduced the amounts of capsids. However, D2A, R56A, D78A, R127A and R133A mutant core proteins assembled significantly less amounts of capsids (Fig. 3A, C, E, and I) and support significantly reduced levels of HBV DNA synthesis in transfected HepG2 cells (Fig. 3B, D, F and I). Western blot analysis indicated that D2A, R56A, D78A mutant core proteins were not stable and accumulated to significantly reduced levels (Fig. 3K), which is in agreement with previously published results (Ponsel and Bruss, 2003).
3.4. Structural basis of differential impacts of different charged amino acid residues on capsid assembly and electrophoresis mobility
As illustrated in Fig. 4, except for K7/E8, all other charged amino acids that affect capsid electrophoresis mobility are located on the surface of capsid. More specifically, while the charged amino acids between residues 64–98 locate on the spike of core protein dimer, the charged amino acids between residues 22-32 as well as R127 and R133 are at the bottom of valley below spike near the dimer-dimer interface. On the contrary, all the 12 charged amino acids that do not affect capsid electrophoresis mobility are located in the lumen side of capsid or at the interface of core protein subunits.
Fig. 4. Location of charged amino acid residues in capsids.

45° view of five-fold symmetry of HBV capsid from structure 2G33. (A) Charged residues that cause slower migration of capsids upon alanine substitution - D22, D29, D32, E64, E77, D78 and D83 are shown in pink and the residue K7 represented in light pink that faces the three-fold axis result in faster migration. (B) Charged residues resulting in faster migration of capsids upon alanine substit - R28, R82, K96, R98, R127 and R133 are shown in orange and residue E8 shown in light orange resides in the three-fold axis result in slower migration, (C) Charged residues that does not alter the migration of capsids upon alanine substit - D2, D4, E14, E39, E40, E43, E46, R56, R112, E113, E117, E145 are shown in grey and they reside either in the dimer interface or in the lumen of capsids.
The K7-E8 dipeptide is located on a loop near the monomer-monomer interface at the base of the four-helical bundle close to the lumen side of the capsid. K7 faces the capsid pore at the symmetry axes. Solvent accessible surface area analysis shows that K7 is 10-20% more solvent exposure than K96, the only other lysine residue in the core protein. In contrast, E8 is buried in the monomer-monomer interface and has about the same solvent exposure when compared to the other glutamic acid residues in the core protein. E8 forms a salt bridge with R56 on the other core monomer. The K7A/E8A mutations having opposite effects on capsid mobility could be due to their unique location around the pore at symmetry axes or dynamic molecular motion of capsid that transiently exposes the dipeptide on the surface of capsids (Hadden et al., 2018).
D29 and D32 reside on the 2a helix composed of the non-canonical dimer-dimer interface. R127 is on the 5 helix and R133 is on the loop downstream of the 5 helix. R127, in many of the crystal structures, appears to have bifurcated hydrogen bonds to both D29 and D32. This strong salt bridge interaction likely enhances the stability of non-canonical dimer-dimer interaction. It is thus conceivable that mutation of R127 disrupts the salt bridge interaction (Fig. 5A and B) and renders the capsid instability, as revealed by particle gel assay (Fig. 3I and J).
Fig. 5. Interaction of R127 with D29 and D32 at core protein inter-dimer interface.

(A) The bifurcated hydrogen bond of Arg127 at the dimer-dimer interface (subunits B and C from PDB ID 2G33). (B) The bifurcated hydrogen bond at this interface is broken when a CpAM, AT-130, is bound (subunits B and C from PDB ID 4g93). In panels B and C, subunit B is colored green and subunit C is colored coral pink.
3.5. Not a single charged amino acid is fully responsible for CpAM-induced capsid mobility shift
CpAMs disrupt capsid assembly by binding to the hydrophobic HAP pocket between the core protein dimer-dimer interfaces. To investigate the role of the individual charged amino acids on CpAM-induced capsid mobility shift, HepG2 cells were transfected with pCMV-HBc or pCMV-HBc-derived plasmids expressing mutant core proteins with an indicated charged amino acid residues substituted with alanine. The transfected cells were mock-treated or treated with the representative compounds of three chemotypes of CpAMs. Particle gel assay revealed that Bay 41-4109 efficiently abolished the accumulation of capsids in the cells expressing all alanine substituted core proteins (Fig. 6), except R127A (as shown in Fig. 8B). However, BA-38017 and ENAN-34017 treatment induced the assembly of fast migrating capsids in the cells expressing each of the alanine-substituted core proteins examined, except that the capsid mobility shifts in cells expressing D22A and D29A were less profound (Fig. 6 and Fig. 7E). These results clearly indicate that no single charged amino acid residue is fully responsible for CpAM-induced capsid mobility shifts.
Fig. 6. Effects of alanine-substitution of individual charged amino acid on CpAM-induced capsid mobility shift.

HepG2 cells were transfected with pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with indicated charged amino acid mutation. Six hours later, the cells were left untreated or treated with 5 μM of BA-38017, 5 μM of ENAN-34017 or 2 μM of Bay 41-4109 for 72 h. The cytoplasmic capsids were analyzed by a particle gel assay.
Fig. 8. R127 plays a critical role in CpAM interaction with core protein.

HepG2 cells were transfected with pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with R127K, R127E, R127A, or R127V mutation. The cells were harvested at 72 h post transfection. (A) Core protein (Cp) expression was detected by a Western blotting assay. (B) Capsids were detected by particle gel assay. (C) HepG2 cells were transfected with pHBV1.3-Y63F or pHBV1.3-Y63F-Δcore or co-transfected with pHBV1.3-Y63F-Δcore and plasmids expressing the indicated core proteins. The total and encapsidated pgRNA were determined by Northern blot assay, 28S and 18S rRNA served as loading controls (upper panel). HepG2 cells were transfected with pHBV1.3 or pHBV1.3-Δcore or co-transfected with pHBV1.3-Δcore and plasmids expressing the indicated core proteins. The core DNA were determined by Southern blot assay (lower panel). (D) HepG2 cells were transfected with the indicated core protein-expressing plasmid. Six hours post transfection, the cells were left untreated or treated with 5 μM of BA-38017, 5 μM of ENAN-34017 or 2 μM of Bay 41-4109 for 72 h. The cytoplasmic capsids were analyzed by a particle gel assay.
Fig. 7. D29 plays a critical role in CpAM interaction with core protein.
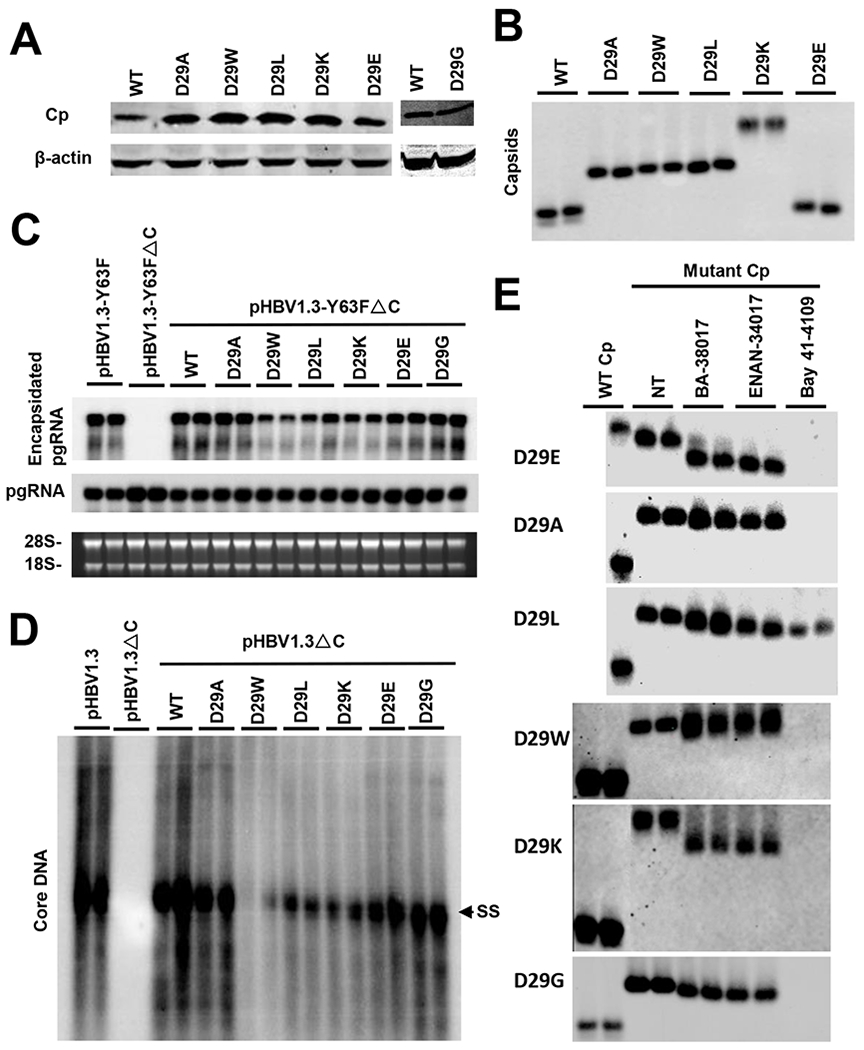
HepG2 cells were transfected with pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with D29A, D29W, D29L, D29K, D29G, or D29E mutation. The cells were harvested at 72 h post transfection. (A) Core protein (Cp) expression was detected by a Western blotting assay. (B) Capsids were detected by a particle gel assay. (C) HepG2 cells were transfected with pHBV1.3-Y63F or pHBV1.3-Y63F-Δcore or co-transfected with pHBV1.3-Y63F-Δcore and pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with and D29A, D29W, D29L, D29K, D29E or D29G. The total and encapsidated pgRNA were determined by Northern blot assay, 28S and 18S rRNA served as loading controls. (D) HepG2 cells were transfected with pHBV1.3 or pHBV1.3-Δcore or co-transfected with pHBV1.3-Δcore and pCMV-HBc or pCMV-HBc-derived plasmids expressing core protein with D29A, D29W, D29L, D29K, D29G or D29E mutation. The HBV core DNA were determined by Southern blot assay. (E) HepG2 cells were transfected with the indicated core protein-expressing plasmid. Six hours post transfection, the cells were left untreated or treated with 5 μM of BA-38017, 5 μM of ENAN-34017 or 2 μM of Bay 41-4109 for 72 h. The total amounts of capsids were determined by a particle gel assay in a 1.5% agarose gel electrophoresis.
3.6. D29 and R127 play critical roles in CpAM modulation of capsid assembly
Because D29 and R127 are located in the vicinity of HAP pocket and the results presented above suggest that those two residues may involve in the interaction of selected CpAMs with core proteins, we thus performed additional mutagenesis analyses to further investigate the role of those two charged amino acid residues in CpAM disruption of capsid assembly. Western blot analysis indicated that all of these D29 mutant core proteins were stable (Fig. 7A). As shown in Fig. 7B to D, substitution of D29 with another negatively charged amino acid glutamic acid (E) did not apparently alter the amounts and electrophoresis mobility of capsids, pgRNA packaging and viral DNA replication. However, as shown in Table 2, D29E core protein conferred 5 and 6 fold resistance to Bay 41-4109 and BA-38017 inhibition of HBV DNA replication, respectively, but had no impact on ENAN-34017 inhibition of viral DNA replication. On the contrary, substitution of D29 with a positively charged amino acid lysine (K) resulted in formation of capsids with slowest mobility, reduced amounts of packaged pgRNA and viral DNA replication. D29K mutant core protein conferred more than 5 fold resistance on HBV DNA replication to all three chemotypes of CpAMs tested. Interestingly, while substitution of D29 with all the four non-charged amino acids resulted in the formation of capsids with similarly slower migrating capsids, substitution with amino acids with larger hydrophobic side chain, such as L and W, significantly reduced pgRNA packaging and viral DNA replication. Consistent with the particle gel assay results that D29L capsids were resistant to Bay 41-4109-induced decay as well as BA-38017 and ENAN-34017 treatment induced less dramatic mobility shifts of capsids with D29 substitutions by any of the four non-charged amino acids (Fig. 7E), those mutations also conferred significant resistance to the CpAMs inhibition of HBV DNA replication (Table 2).
Table 2.
Sensitivity of mutant core proteins to three chemotypes of CpAMs.
| AA | CpAMs |
|||||
|---|---|---|---|---|---|---|
| Substitution | Bay 41-4109 |
BA-38017 |
ENAN-34017 |
|||
| EC50* | Folds of R# | EC50* | Folds of R# | EC50* | Folds of R# | |
| WT | 0.05±0.01 | 1 | 0.46±0.06 | 1 | 0.09±0.01 | 1 |
| R127K | 0.32±0.08 | 6 | 0.59±0.09 | 1 | 0.48±0.02 | 5 |
| R127A | 0.47±0.18 | 9 | >10 | >20 | >10 | >100 |
| R127V | >2 | >40 | 5.34±0.94 | 11 | 0.37±0.05 | 4 |
| D29E | 0.28±0.06 | 5 | 2.79±0.43 | 6 | 0.11±0.02 | 1 |
| D29G | 0.12±0.01 | 2 | 0.74±0.11 | 2 | 0.09±0.01 | 1 |
| D29A | 0.16±0.04 | 3 | 1.89±0.34 | 4 | 0.43±0.24 | 5 |
| D29L | >2 | >40 | 5.30±0.94 | 13 | 0.33±0.03 | 4 |
| D29W | 0.61±0.02 | 12 | 4.92±0.98 | 11 | 0.28±0.05 | 3 |
| D29K | 0.44±0.04 | 9 | 1.85±0.43 | 4 | 0.49±0.14 | 5 |
| D22A | 0.05±0.01 | 1 | 0.69±0.13 | 1 | 0.46±0.11 | 5 |
| R28A | 0.11±0.01 | 2 | 1.67±0.32 | 4 | 0.21±0.06 | 2 |
| D32A | 0.20±0.02 | 4 | 0.61±0.11 | 1 | 0.40±0.12 | 4 |
| E77A | 0.12±0.01 | 2 | 1.24±0.21 | 3 | 0.10±0.02 | 1 |
| E77Q | 0.04±0.01 | 1 | 0.25±0.04 | 0.5 | 0.03±0.01 | 0.3 |
| R82A | 0.04±0.01 | 1 | 0.53±0.06 | 1 | 0.03±0.01 | 0.3 |
| K96A | 0.02±0.01 | 0.5 | 0.27±0.08 | 0.5 | 0.08±0.01 | 1 |
| R98A | 0.12±0.01 | 1 | 0.70±0.14 | 1 | 0.22±0.03 | 2 |
| R112A | 0.02±0.01 | 0.5 | 0.57±0.10 | 1 | 0.09±0.03 | 1 |
| E113A | 0.02±0.01 | 0.5 | 0.21±0.04 | 2 | 0.02±0.01 | 0.4 |
| E117A | 0.02±0.01 | 0.5 | 0.26±0.03 | 0.5 | 0.06±0.01 | 1 |
EC50 (μM), Mean value ± standard deviation (n = 4) is presented.
Fold of drug resistance
Similarly, as shown in Fig. 8B to C, while substitution of R127 with a negatively charged glutamic acid (E) results in drastically reduced capsid accumulation in HepG2 cells, substitution of R127 with another positively charged amino acid lysine (K) did not apparently affect capsid assembly, pgRNA packaging and DNA replication. However, R127K core protein conferred 6 and 5 folds of resistance to Bay 41-4109 and ENAN-34017 inhibition of HBV DNA replication, respectively, but not BA-38017 (Table 2). Furthermore, substitution of R127 with non-charged amino acids, such as alanine and valine, resulted in slightly faster migrating capsids that are resistant to Bay 41-4109-induced decay. Both R127A and R127V mutant core protein demonstrated reduced ability to support pgRNA packaging and HBV DNA replication as well as conferred significant resistance to the inhibition of HBV DNA replication by all the three CpAMs examined (Table 2).
Taken together, the results presented in Figs. 6 to 8 and Table 2 collectively imply that although the selected charged amino acid residues affect the capsid mobility in variable degrees, not a charged amino acid residue is single-handedly responsible for CpAM-induced capsid mobility shifts. However, as anticipated, while the charged amino acid residues located near the HAP pocket, particularly D22, R28, D29, D32 and R127, play more direct roles in CpAM disruption of capsid assembly, other charged amino acid residues located at far distance of the HAP pocket might not have a significant impact on CpAM modulation of pgRNA encapsidation/DNA synthesis and thus did not confer resistance to any of the tested CpAMs.
3.7. Additional evidence supporting that the size of capsids is not the major determinant of its electrophoresis mobility
To further determine the biophysical parameters determining capsid mobility in the particle gel assay, cytoplasmic capsids were purified by ultracentrifugation from HepG2 cells transfected with pCMV-HBc (wild-type) and pCMV-HBc-derived plasmids expressing D29 substituted core proteins that form capsids with drastically different electrophoresis mobility (Fig. 7B) and subjected for electronic microscopic analyses. As shown in Fig. 9, the capsids assembled from D29E, D29A, D29L, D29W, and D29G mutant core protein are similar in size and morphology to those from wide-type core protein. These results further indicate that the size of capsids is not correlated with their electrophoresis mobility.
Fig. 9. Electronic micrographs studies of selected capsids.

HepG2 cells were transfected with plasmid expressing WT HBV core protein or core protein with D29E, D29A, D29L, D29W and D29G mutation for 3 days. (A) HBV capsids in the cell lysates were purified by sucrose gradient centrifugation and detected by EM after negatively stained with phosphotungstic acid. Scale bar indicates 100 nm. (B) The diameters of the capsids are plotted. Average diameters (and standard deviation) of capsids are indicated.
4. Discussion
The genome of every viral species is wrapped with capsid protein(s) to form nucleocapsids. Viral capsid proteins are structurally distinct from all the cellular proteins and are thus highly selective antiviral targets (Zlotnick and Mukhopadhyay, 2011). Despite no capsid-targeting antiviral drug has been approved for treatment of viral diseases in clinic, disruption of nucleocapsid assembly and/or disassembly with small molecular compounds is a new frontier in development of novel antiviral agents against several medically important viruses including HBV (Zlotnick et al., 2015), human immunodeficiency virus (Klumpp and Crepin, 2014) and dengue virus (Byrd et al., 2013; Scaturro et al., 2014). Particularly, several chemotypes of CpAMs are currently under preclinical development or in clinical trials for treatment of chronic hepatitis B (Liang et al., 2015), a disease that curative therapeutics are still not available.
In an effort toward understanding the mechanism underlying CpAM disruption of capsid assembly and inhibition of pgRNA packaging in HBV replicating cells, we found that treatment of HBV replicating cells with the three major chemotypes of type II CpAMs induced the assembly of capsids with faster electrophoresis mobility. To our surprise, compared to the capsids derived from mock-treated cells, not only the “empty” capsids, but also the DNA-containing capsids, i.e., nucleocapsids, migrated faster (Fig. 1A). In principle, capsid electrophoresis mobility shift in the native agarose gel can be attributed to the changes of capsids in size, mass, shape and/or net charge. Interestingly, a recent study of HBV capsid structure and dynamics by all-atom molecular dynamics simulations revealed that capsid structure is flexible and has a propensity for asymmetric distortion (Hadden et al., 2018). It is thus possible that changes in the dynamic molecular motion of drug-treated or mutation-containing capsids could contribute to altered electrophoretic mobility. However, no matter whether the charge, mass, size, shape and/or dynamic molecular motion affect electrophoresis mobility of capsids, all those physical parameters are related to capsid structures. Therefore, the capsid electrophoresis mobility shift observed in this study should indicate structural alterations of capsids.
The finding that CpAM treatment alters the structure of not only empty capsids, but also DNA-containing capsids strongly indicates that in addition to accelerating empty capsid assembly kinetics and indirectly reducing pgRNA encapsidation (Katen et al., 2010), the possibility that CpAMs induce the assembly of structurally altered nucleocapsids that do not favor the encapsidation of viral pgRNA and DNA polymerase complex cannot be ruled out. Moreover, our recent studies also indicate that in addition to disrupting capsid/nucleocapsid assembly, CpAMs also specifically interact with nucleocapsids containing mature forms of HBV genomic rcDNA and induce their disassembly (Guo et al., 2017). Hence, better understanding the biophysical basis of CpAM-induced capsid mobility shift should help revealing the mechanism of CpAM-induced capsid/nucleocapsid structural changes and provide structural basis for discovery of more potent core protein-targeting antiviral therapeutics.
In this study, we provide two independent lines of evidence suggesting that the size of capsids is not the key determinant of capsid electrophoresis mobility in the particle gel assay. First, we showed that the capsids from CpAM-treated cells are at least similar or slightly larger in size, compared to the capsids from mock-treated cells (Fig. 1) (Wu et al., 2017; Yang et al., 2016). Second, the sizes of capsids assembled from different D29 mutant core proteins are not correlated with their electrophoresis mobility (Fig. 9). Hence, the increased mobility of CpAM-treated capsids is not due to their smaller sizes, but likely because of the increase of net “positive” surface charge.
Taking the alanine scanning mutagenesis approach, we further demonstrated that only the charged amino acid residues located within one of the four continuous charged amino acid stretches affect capsid mobility in variable degrees, as illustrated in Fig. 4. Except for K7 and E8, all other charged amino acid residues are located on the surface of capsids. However, alanine substitution of each of those amino acids altered capsid electrophoresis in a different degree. The results suggest that the factors affecting the impact of the individual amino acids on capsid mobility should depend on not only its surface exposure, but also its ionization potential as determined by its local structural environment.
Concerning the biophysical basis of CpAM-induced increase of capsid mobility, we demonstrated that not a single charged AA is fully responsible. Moreover, considering that usually the very dramatic mobility shift is induced by alanine substitution of a single charged AA, we believe that the most likely scenario of CpAM-induced modest capsid mobility shift is the results of the compound-induced global capsid structure changes as revealed by cryo-EM studies (Bourne et al., 2006; Katen et al., 2013; Venkatakrishnan et al., 2016), which may slightly alter the exposure or ionization potential of a few charged AAs at specific position. Of course, this hypothesis remains to be tested with structure biology studies of capsids.
Similar to CpAM treatment, mutations of AAs on the wall, or in the vicinity, of HAP pocket also resulted in formation of capsids with slightly faster electrophoresis mobility, such as V124L and V124W (Wu et al., 2017). Interestingly, similar to CpAM treatment, such mutant core proteins also favor the empty capsid assembly and thus have reduced ability to support HBV DNA replication (Wu et al., 2017). Hence, the subtle disruption of dimer-dimer interactions by CpAMs or mutations of core protein at the “HAP” pocket may induce relatively conserved structural responses.
Practically, the simple particle gel assay measures not only the extent of CpAMs on capsid assembly and pgRNA packaging, but also the compound-induced global capsid structural alteration and hence serves as a convenient and reliable diagnostic maker for compounds that target core proteins assembly. Moreover, mutant core proteins that are resistant to type I CpAM-induced abolishment of capsids or type II CpAM-induced capsid mobility shifts also support viral DNA synthesis resistant to the respective CpAMs. However, as shown in this study, some of mutant core proteins that are sensitive to type II CpAM induced capsid mobility shifts support viral DNA synthesis modestly resistant to selected CpAMs. In spite of the imperfect correlation, the particle gel assay reveals distinct molecular features of CpAM interaction with viral capsids, it could thus be used as an alternative assay to evaluate the sensitivity of clinical strains of HBV to different CpAMs and investigate the mode of drug-resistance.
ACKNOWLEDGEMENTS
We thank Dr. Shuping Tong at Brown University for providing HBV genotypes A and C plasmids. This work was supported by a grant from the National Institutes of Health, USA (AI113267). Shuo Wu is supported by Peking Union Medical College Youth Fund (2017350008).
References
- 2016. Small-Molecule Drug Discovery Suite 2016-3. Schrödinger, LLC, New York, NY. [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, 2000. The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block TM, Guo H, Guo JT, 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11, 685–706, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne CR, Finn MG, Zlotnick A, 2006. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J Virol 80, 11055–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd CM, Dai D, Grosenbach DW, Berhanu A, Jones KF, Cardwell KB, Schneider C, Wineinger KA, Page JM, Harver C, Stavale E, Tyavanagimatt S, Stone MA, Bartenschlager R, Scaturro P, Hruby DE, Jordan R, 2013. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob Agents Chemother 57, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT, 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87, 6931–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Guo F, Zhao X, Guo JT, 2014. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta pharmaceutica Sinica. B 4, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstag JL, 2009. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 49, S112–121. [DOI] [PubMed] [Google Scholar]
- Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, Cuconati A, Wei L, Du Y, Li W, Chang J, Guo JT, 2017. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog 13, e1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT, 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81, 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Mao R, Block TM, Guo JT, 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Guo H, 2015. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res 122, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadden JA, Perilla JR, Schlicksup CJ, Venkatakrishnan B, Zlotnick A, Schulten K, 2018. All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Flores D, Toft D, Wang X, Nguyen D, 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78, 13122–13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katen SP, Chirapu SR, Finn MG, Zlotnick A, 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol 5, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katen SP, Tan Z, Chirapu SR, Finn MG, Zlotnick A, 2013. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 21, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ, 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-L- 2’,3’-dideoxy-3’-thiacytidine [published erratum appears in Antimicrob Agents Chemother 1999 Mar;43(3):726]. Antimicrob Agents Chemother 42, 3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K, Crepin T, 2014. Capsid proteins of enveloped viruses as antiviral drug targets. Curr Opin Virol 5, 63–71. [DOI] [PubMed] [Google Scholar]
- Klumpp K, Lam AM, Lukacs C, Vogel R, Ren S, Espiritu C, Baydo R, Atkins K, Abendroth J, Liao G, Efimov A, Hartman G, Flores OA, 2015. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A 112, 15196–15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS, 2015. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 62, 1893–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, Delaney W.E.t., Hu J, 2016. Cell-Free Hepatitis B Virus Capsid Assembly Dependent on the Core Protein C-Terminal Domain and Regulated by Phosphorylation. J Virol 90, 5830–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, Block TM, Guo JT, Guo H, 2011. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J Virol 85, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott JJ, Stevens GA, Groeger J, Wiersma ST, 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30, 2212–2219. [DOI] [PubMed] [Google Scholar]
- Perrillo R, 2009. Benefits and risks of interferon therapy for hepatitis B. Hepatology 49, S103–111. [DOI] [PubMed] [Google Scholar]
- Pierson EE, Keifer DZ, Selzer L, Lee LS, Contino NC, Wang JC, Zlotnick A, Jarrold MF, 2014. Detection of late intermediates in virus capsid assembly by charge detection mass spectrometry. J Am Chem Soc 136, 3536–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsel D, Bruss V, 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J Virol 77, 416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaturro P, Trist IM, Paul D, Kumar A, Acosta EG, Byrd CM, Jordan R, Brancale A, Bartenschlager R, 2014. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J Virol 88, 11540–11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Mason WS, 2015. Molecular biology of hepatitis B virus infection. Virology 479–480, 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A, 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 102, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray SJ, Zlotnick A, 2006. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit 19, 542–548. [DOI] [PubMed] [Google Scholar]
- Summers J, Smith PM, Horwich AL, 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. Journal of Virology 64, 2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A, 2015. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol 89, 3275–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Zhao Q, Wu S, Cheng J, Chang J, Guo JT, 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert opinion on drug discovery 12, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan B, Katen SP, Francis S, Chirapu S, Finn MG, Zlotnick A, 2016. Hepatitis B Virus Capsids Have Diverse Structural Responses to Small-Molecule Ligands Bound to the Heteroaryldihydropyrimidine Pocket. J Virol 90, 3994–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan B, Zlotnick A, 2016. The Structural Biology of Hepatitis B Virus: Form and Function. Annual review of virology 3, 429–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Lu D, Xu YB, Xing WQ, Tong XK, Wang GF, Feng CL, He PL, Yang L, Tang W, Hu YH, Zuo JP, 2015. A novel pyridazinone derivative inhibits hepatitis B virus replication by inducing genome-free capsid formation. Antimicrob Agents Chemother 59, 7061–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhao Q, Zhang P, Kulp J, Hu L, Hwang N, Zhang J, Block TM, Xu X, Du Y, Chang J, Guo JT, 2017. Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J Virol 91, e0051917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT, 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84, 9332–9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang YJ, Chen HJ, Shi LP, Tong XK, Zhang YM, Wang GF, Wang WL, Feng CL, He PL, Xu YB, Lu MJ, Tang W, Nan FJ, Zuo JP, 2016. Effect of a hepatitis B virus inhibitor, NZ-4, on capsid formation. Antiviral Res 125, 25–33. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Yao X, Wang YX, Liu F, Ma ZM, Weng XH, Wen YM, 2005. High replicative full-length lamivudine-resistant hepatitis B virus isolated during acute exacerbations. J Med Virol 77, 203–208. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Hu Z, Cheng J, Wu S, Luo Y, Chang J, Hu J, Guo JT, 2018. Hepatitis B virus core protein dephosphorylation occurs during pregenomic RNA encapsidation. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JA, Qin N, 2017. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci Rep 7, 42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A, Mukhopadhyay S, 2011. Virus assembly, allostery and antivirals. Trends Microbiol 19, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S, 2015. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res 121, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F, Saputelli J, Seeger C, 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol 68, 2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]