

Abstract
In an era of precision medicine important treatment decisions are dictated by expression of clinically informative tumor protein biomarkers. These biomarkers can be detected by immunohistochemistry (IHC) performed in tumor tissue sections obtained from biopsies or resections. Like all experimental procedures, IHC needs optimization for several of its steps. However, the investigator must avoid optimizing the IHC procedure using valuable human biopsy samples which may be difficult to obtain. Ideally, valuable biopsy samples should only be subjected to IHC once the IHC protocol has been optimized. In this chapter, we describe a procedure for IHC optimization using tri-dimensional (3D) cellular spheroids created from cultured cells. In this approach, cultured cells are pelleted into 3D spheroids, which are then processed just like a tissue sample, namely, fixed, embedded, sectioned, mounted on slides, and stained with IHC just like a human tissue sample. These 3D cellular spheroids have a tissue-like architecture and cellularity resembling a tumor section, and both cellular and antigen structure are preserved. This method is therefore acceptable for IHC optimization before proceeding to the IHC staining of human tumor samples.
Keywords: spheroids block, immunohistochemistry optimization, p39, lung cancer, cell lines, formalin-fixed-paraffin-embedded tissues
1. Introduction
Clinically relevant protein biomarkers are needed to improve lung cancer management. Many important clinical decisions regarding disease management are guided by the detection by immunohistochemistry (IHC) staining of these biomarkers in tumor tissue obtained from biopsies or resections. For example, expression of several isoforms of cytokeratin, as detected by IHC, have diagnostic value for non-small cell lung carcinomas (NSCLC) [1]. As another example, a panel of IHC markers is routinely used in pathology laboratories for the sub-classification of NSCLC into squamous cell carcinomas or adenocarcinomas subtypes [2, 3].
In the context of basic and translational cancer research, IHC staining for any given protein should ideally be preceded by extensive optimization of the IHC workflow in order to ensure a strong staining signal that is specific, reproducible and of sufficient quality for publication. This involves optimizing parameters such as antibody concentration and incubation time, conditions for effective antigen retrieval, blocking to minimize background, number and duration of washes, signal development conditions, etc. Even when these parameters are optimized, they may still have to be re-checked when changing the batch of the antibody or of any other IHC reagent.
The optimization stage may involve a certain degree of trial and error. Therefore, it is strongly recommended that valuable human tumor tissue samples are not used during this stage. Pre-made tumor microarrays (TMAs) are commercially available for many cancer types, but these can be too expensive and also too valuable to be used for optimization purposes. Therefore, regardless of their origin, valuable human samples should ideally only be subjected to IHC analysis once the IHC protocol has been optimized.
In this chapter we describe a procedure for IHC optimization using tri-dimensional (3D) cellular spheroids created from cultured cells. This approach is relatively inexpensive since it starts with cultured cells. Briefly, cells are grown in culture to form confluent monolayers, then they are detached from culture plates, centrifuged and pelleted. The cell pellet is then processed like a tissue biopsy would normally be, namely, fixed, embedded, sectioned into tissue sections and mounted on glass slides that can then be used for IHC staining just like any other tissue biopsy. The resulting 3D cellular spheroids have a tissue-like architecture and cellularity resembling a tumor section, and the cellular structure and antigen availability are preserved during their preparation. They are therefore acceptable as an inexpensive biological material that can be used for IHC optimization.
We have successfully used the protocol described in this chapter to optimize an IHC staining procedure to detect p39 protein expression in lung cancer cell culture-derived tri-dimensional (3D) spheroids. We have previously reported elevated p39 expression as a biomarker for advanced stage lung squamous cell carcinomas (SCC) [4], and slides prepared from 3D spheroids have been instrumental in our laboratory to optimize the IHC protocol for p39 and for other antigens. After optimizing the p39 IHC staining with the 3D spheroids, we successfully used the optimized protocol to stain lung cancer TMAs for p39 expression by IHC [4]. It is important that the cell line chosen to prepare the 3D spheroids matches the tumor tissue type that will be eventually studied. In our case, we created the 3D spheroids using the NSCLC cell line H520. This cell line is of the SCC NSCLC subtype and is patient-derived from SCC tumors. We have identified p39 as part of a metastasis-associated proteomic biomarker signature in SCC [4] and therefore we optimized the IHC procedure using H520 3D spheroids before actually conducting the IHC p39 analyses using human lung TMAs. Therefore, choice of the appropriate cell line to prepare the 3D spheroids is of great importance.
It is important to keep in mind that 3D spheroid models may not be suitable if preserving the heterogeneity of the tumor microenvironment if of the essence. The spheroids produced with the procedure herein described are relatively homogeneous in terms of cellular composition and lack the cellular variety that typically represents the complex interactions between the tumor and its microenvironment. Therefore, these spheroid structures may not be ideal to reliably assess protein expression that is strongly affected by intercellular signaling events. Neither they provide a suitable model to address the issue of tumor multi-clonal heterogeneity. However, it must be kept in mind that the purpose of this approach is not to study oncogenesis-related biological process, but it is rather aimed at the optimization of the immunological detection of an epitope within the context of a 3D tumor-like tissue architecture, and to mimic as much as possible the mechanical aspects of antigen accessibility in the context of 3D tissue structures. This chapter describes the complete procedure, from the formation of the 3D spheroids from cell cultures, preparation of spheroid sections, staining of the sections with hematoxylin and eosin to verify their cellular density, and finally, IHC staining of the spheroid sections for p39 protein expression.
2. Materials
All the solutions should be prepared using distilled water, unless otherwise specified. Reagents and solutions should be stored at room temperature, unless otherwise specified. Follow all waste disposal regulations for waste materials.
2.1. Cell lines and Cell Culture Reagents
Cell lines. We optimized this protocol using the H520 non-small cell lung carcinoma (NSCLC) cell line, which is of the squamous cell carcinoma subtype. Keep in mind that the purpose of the procedure described in this chapter is to use cell lines to optimize the IHC protocol that will then be applied to FFPE tissue sections. Therefore, the cell lines for the optimization of the IHC protocol should be as histologically equivalent as possible to the tumor tissue that will be subsequently tested. For example, H520 and H1666 cell lines originated from squamous cell lung carcinomas and from lung adenocarcinomas, respectively, therefore they can be used for IHC optimization for these lung tumor types. We culture cells in 100 mm culture plates or T75 flasks.
Cell culture medium. This depends of the cell type of your choice. We culture H520 cells in RPMI medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Phosphate-Buffered Saline (PBS), 1X. You can prepare 1X PBS as follows: dissolve 8.0 g of NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4.2 H2O and 0.24 g of KH2PO4, in of 800 mL of water. Adjust the pH to 7.2 with HCl (start with concentrated HCL and then use more diluted HCL as you approach the desired pH) and add distilled water to complete the volume to 1 L. Alternatively, ready-to-use commercially available PBS is also acceptable. Use cold.
0.25% Trypsin-EDTA solution. We buy in ready-to-use pre-mixed form. We use this solution to detach cells from the culture flasks or plates. Alternatively, you can scrape cells with a rubber scraper or spatula.
2.2. Histology Reagents and Supplies
Histology tissue embedding and processing cassettes.
10% neutral buffered formaldehyde solution. We purchase this as a ready-to-use pre-mixed solution, it is available from several vendors.
Ethanol. You need absolute (100%) ethanol, anhydrous, histological grade, and you also need 95%, 90%, 85%, 80% and 70% ethanol dilutions in distilled water (dH2O).
Xylene or xylene substitute.
Paraffin or tissue embedding medium, we use Paraplast X-TRA®, but other alternatives are acceptable. Check the manufacturer’s instruction for melting temperature.
Tissue embedding base metal molds, with dimensions of 32 x 25 x 12 mm.
Embedding rings, be sure they fit into the embedding base molds.
Microscope glass slides, 25 x 75 mm, including coverslips of the appropriate size.
Bluing reagent solution, ready-to-use solution.
Acid alcohol, 1%. Prepare a solution that is 1% hydrochloric acid in 70% ethanol. You can dilute 2 mL of concentrated 12.1 M HCl in 200 mL 70% ethanol.
Eosin staining solution, we use Eosin Y solution 1%, alcoholic, ready-to-use.
Hematoxylin stain, we use Harris Modified Hematoxylin.
Mounting medium, we use Cytoseal 60, but other alternatives can be used.
Standard slide racks and histology staining and washing jars.
HistoCore Arcadia C-Cold Plate from Leica Biosystems, or any other histology cold plate.
HistoCore Arcadia H-Heated Paraffin Embedding Station, or any other heated histology embedding station.
Microtome with blade (follow all necessary precautions when handling sharp microtome blades).
2.3. Immunohistochemistry Reagents
Citrate antigen retrieval solution. Prepare by dissolving 1.92 g of trisodium citrate dehydrate and 0.74 g of EDTA in 800 ml of H2O, adjust pH to 6.2 with 1 N HCl, add 0.5 ml of Tween 20® and complete to a final volume of 1 L. This solution can be stored at 4°C for up to 6 months.
Hydrogen peroxide solution, 3%. Start with 30% hydrogen peroxide (H2O2) and dilute to 3% with distilled water.
PAP pen, or any other hydrophobic pen or marker, this is needed for drawing hydrophobic barriers around the 3D spheroid section in microscope slides.
Primary antibody of your choice, depending on the antigen you want to test. We optimized this protocol using a rabbit monoclonal antibody against p39, clone EPR5074 from Abcam. (Cat. No. 124896) (see Note 1). For a 1:50 dilution add 20 µl of antibody to 980 µl of 1X PBS.
Super sensitive Link-Label IHC kit from BioGenex (LP000-ULE). This immunohistochemistry kit includes a biotin-labeled anti-rabbit secondary antibody and a streptavidin conjugated horseradish peroxidase (HRP). Use a 1:20 dilution for the secondary antibody and a 1:10 dilution for the streptavidin. Other immunohistochemistry detection kits are acceptable but be sure to use according to manufacturer’s instructions, and that the secondary antibody should be compatible with your primary antibody (see Note 2).
Diaminobenzidine (DAB) reagent. This is the substrate for the HRP, it produces a dark brown precipitate when it is oxidized by the HRP. We use the BioGenex Two Components DAB-Pack (HK542-XAKE), following product instructions. To prepare 1 ml of DAB reagent solution, add 2 drops of DAB reagents to 1 ml of DAB buffer, vortex the solution and store at 4°C (see Note 3).
2.4. General Laboratory Equipment
Incubator set at 37°C, 5% CO2.
Humid chamber. This is to prevent the slides from drying up due to evaporation during antibody incubations. It can be commercially purchased or can be prepared in the laboratory using a tight sealing container. Humidity inside the chamber can be maintained by including absorbent paper soaked in distilled water.
Microscope with camera, and suitable image acquisition software.
Lint-free tissue paper.
Water bath, set to 80-85°C.
Tabletop centrifuge with a rotor capable of holding 50 ml tubes, preferably refrigerated. We use an Eppendorf 5810R, but equivalents are acceptable.
Conical tubes, 50 mL.
Filter paper, 180 µm.
Lens paper. Wax paper or weighting paper can also be used.
Oven, set to 37°C, then to 64°C.
3. Methods
3.1. Spheroid Preparation from Cell Cultures
For the preparation of 3D spheroid blocks with tissue-like organization from lung cancer H520 cell line (or the cell line of your choice), you should start with at least 5-6 confluent T75 flasks or 100 mm cell culture plates for each cell line in order to obtain a good-sized cell pellet from which the spheroid will be formed. Procedures related to spheroid fixation and preparation of the spheroid blocks (described in detail in Sections 3.1 and 3.2) are adaptations of previously published protocols [5, 6].
Collect cells by scraping them from the culture plate in 5 ml of 1X PBS. We recommend this volume for T75 flasks; you should adjust the volume of 1X PBS when using plates. It is important that the entire surface of the bottom of the flask or plate is covered with thin layer of 1X PBS. Alternatively, detach cells from the plate by incubating the cultures with a 0.25% Trypsin-EDTA solution at 37°C for 5-10 min.
Transfer the cell suspension to a 50 mL tube and pellet the cells by centrifugation (up to 300 xg) for 5 min, at room temperature (RT). If the cells were detached by trypsinization, dilute the trypsin solution 1:10 with fresh culture medium to ensure inactivation of the trypsin. Do this dilution before the centrifugation step. Keep in mind that at this point the size of the pellet will affect the cellularity of the spheroid. If you end up with spheroid tissue sections in which cells are sparse, you need to increase the number of culture plates in order to obtain a larger pellet.
Carefully remove the supernatant after the centrifugation, being careful not to disrupt the cell pellet.
Resuspend the cell pellet in 20–25 ml cold 1X PBS and centrifuge at 300 xg for 10 min at RT.
Remove the supernatant, then carefully add 20–25 ml of the 10% neutral buffered formalin (NBF). Pour the NBF down the tube’s inner wall to avoid disrupting the cell pellet which will constitute the spheroid.
To produce the fixed spheroid, fix the cell pellet by incubating in NBF overnight at 4°C.
After overnight fixation carefully remove the NBF (see Note 4).
Resuspend the spheroid in 10–15 ml cold 1X PBS and centrifuge at 300 xg for 10 min at RT. Repeat this step for a total of three 1X cold PBS washes.
3.2. Preparation of Spheroid Paraffin Blocks
Carefully transfer the spheroids from 50 mL tube to a piece of filter paper. You can use a spatula or a pipette, as long as you do not break the spheroid. Allow the spheroids to air-dry on the filter paper. During this step the filter paper will remove the excess moisture from the spheroid. If there are any remnants of cells in the tube you can wash the tube with cold 1X PBS using a pipette and deposit the wash in the filter paper.
Collect spheroids from the filter paper by gently scraping the paper with a flat spatula. Be careful not to break the filter paper to avoid losing sample, or to scrape too hard that pieces of the filter paper could detach together with the spheroid.
Prepare a small envelope or pocket using lens paper (Fig. 1), prepare one of these for each spheroid. You can also use wax paper or weighing paper. Carefully transfer the dry spheroid to the lens paper envelope and place it into a tissue processing cassette.
Dehydrate and clear the spheroids by following the schedule shown in Table 1. Use glass jars for the washes indicated in the table. Carefully label each glass jars with each solution. Place the tissue cassette into the corresponding glass jar for the indicated time. Proceed to the following paraffin embedding steps below in this section.
Heat the HistoCore Paraffin Embedding station. To determine the appropriate temperature, look for the melting temperature recommended for the specific paraffin or embedding medium of your choice, as indicated in the product instructions. Melt the paraffin or embedding medium and add to a heated a metal mold, ensuring to cover the entire bottom of the mold. Place and keep the mold in the hot surface.
Using tweezers, carefully remove the spheroid from the cassette (leave it in the paper envelope) and place it in the molten paraffin within the metal mold. Allow it to cool down.
Once the paraffin solidifies (Fig. 1, left panel), remove the block from the metal mold and cut around the spheroid using a scalpel, in such way that you obtain a piece of paraffin containing inside the embedded cell spheroid.
Using the trimmed embedded spheroid, repeat step 6 (immersion in molten paraffin) and let the paraffin to slightly cool down, but not to become entirely solid (Fig 1. center and right panels).
Before the paraffin entirely solidifies, place a tissue embedding cassette without the lid (or use an O ring) over the metal mold and fill with molten paraffin. This will create a paraffin block in which the cassette encases and holds the embedded spheroid together. This set-up is illustrated in Fig. 2.
Allow the paraffin block to solidify by placing it in the cold plate for 1-2 min.
Leave the block to continue solidification at room temperature overnight (see Note 5).
Fig. 1.

Paraffin embedding of the cell spheroid. The spheroid cell pellet is placed inside an envelope made of lens paper (the envelope can be appreciated inside the paraffin in the mold in the left panel), which in turn is submerged in the molten paraffin in the metal mold. After a few minutes at room temperature the paraffin solidifies in the mold. The spheroid can be seen in the paraffin block (arrow in the right panel).
Table 1.
Steps for the dehydration and clearing of cell 3D spheroids formed from cell pellets. RT, room temperature.
| Procedure | Steps | Solution | Time/Temperature |
|---|---|---|---|
| Dehydration | 1 | 70% alcohol | 15 min/RT |
| 2 | 80% alcohol | 15 min/RT | |
| 3 | 95% alcohol | 15 min/RT | |
| 4 | 100% alcohol | 15 min/RT | |
| Clearing | 1 | Xylene | 10min/RT |
| 2 | Xylene | 10min/RT | |
| 3 | Xylene | 10min/RT | |
| 4 | Xylene | 10min/RT |
Fig. 2.

Final embedding set-up of the cell spheroid to obtain the paraffin block that will be subsequently cut. The block is put again in molten paraffin (left), and before the paraffin solidifies the O-ring or an embedding cassette without the lid is placed (center), and the whole set-up is allowed to solidify to create the final block that will be cut in the microtome (right).
3.3. Microtome Sectioning of Spheroid Paraffin Blocks
Carefully remove the spheroid block from the metal mold. Keep the paraffin-embedded spheroid block on ice at all times before sectioning (see Note 6).
Using a microtome, cut the spheroid blocks to get ribbon spheroid sections (Fig. 3). The sections should have a thickness of 5 μm (see Note 7).
Mount the spheroid sections on glass slides.
Fig. 3.

The final block shown in Fig. 2 is cut into paraffin ribbons 5 µm thick.
3.4. Hematoxylin and Eosin (H&E) Staining
The spheroid sections generated in Section 3.3 will be used for immunohistochemistry (IHC) staining as described in Section 3.5 below. However, before proceeding with the IHC some of the sections must first be stained with Hematoxylin & Eosin (H&E). The purpose of the H&E staining is to allow you to observe under the microscope if the spheroid sections have the appropriate cellular density and architecture to resemble a tumor tissue section. Do not proceed to the IHC staining steps described in Section 3.5 until you have achieved spheroid sections with sufficient cellularity.
Place slides in a slide rack and incubate at 37°C overnight in an oven or incubator.
Next day, and right before staining, incubate the slides at 64°C for 1 h.
Inside a fume hood, submerge the slide rack in xylene substitute for 2 min. Do two additional xylene substitute submersions, also 2 min each, for a total of 3. Use fresh xylene for each submersion. This and the remaining steps in this section must be performed inside the fume hood.
Hydrate the slides by submerging the slide rack into the jars that contain a series of ethanol solution as follows: two consecutive washes in 100% alcohol, 1 min each; one 95% ethanol, 1 min; and a final rinse in tap water for 30 seconds. Do this rinse carefully to avoid the spheroid sections to detach from the slide.
Submerge the slide rack in the jar that contain Hematoxylin for 2–4 min.
Rinse the slides with tap water for 30 seconds. Do this rinse carefully to avoid the spheroid sections to detach from the slide.
Submerge the slide rack into a jar containing 1% acid alcohol for 15–20 seconds.
Rinse slides with tap water for 30 seconds. Do this rinse carefully to avoid the spheroid sections to detach from the slide.
Submerge the slide rack in a jar containing Bluing reagent for 30 seconds.
Carefully rinse the slides with tap water for 30 seconds, always avoiding direct contact with the tissue section.
Dehydrate the spheroid sections by submerging the slide rack into jars with ethanol washes as follows: 70% alcohol for 1 min, then 95% alcohol for 1 min.
Submerge the slide rack into a jar that contains the Eosin solution for 2 min.
Submerge the slide rack into a jar with 95% alcohol for 10 seconds, followed by a 100% alcohol wash for 1 min.
Submerge the slide rack into a jar containing xylene substitute for 2 min. Repeat this step a second time.
Drain the xylene from the slides and allow the slides to dry inside the hood.
Add 2–3 drops of mounting media on top of each spheroid section and carefully place a cover glass on top of the section, avoiding the formation of air bubbles (see Note 8).
Analyze the slides under a light microscope. Capture images at various magnifications. Fig. 4 illustrates a spheroid section with good cellular density. The cellular density should be as similar as possible to the cellularity of a tumor tissue and should have similar architecture in terms of cell-to-cell contacts.
Fig. 4.
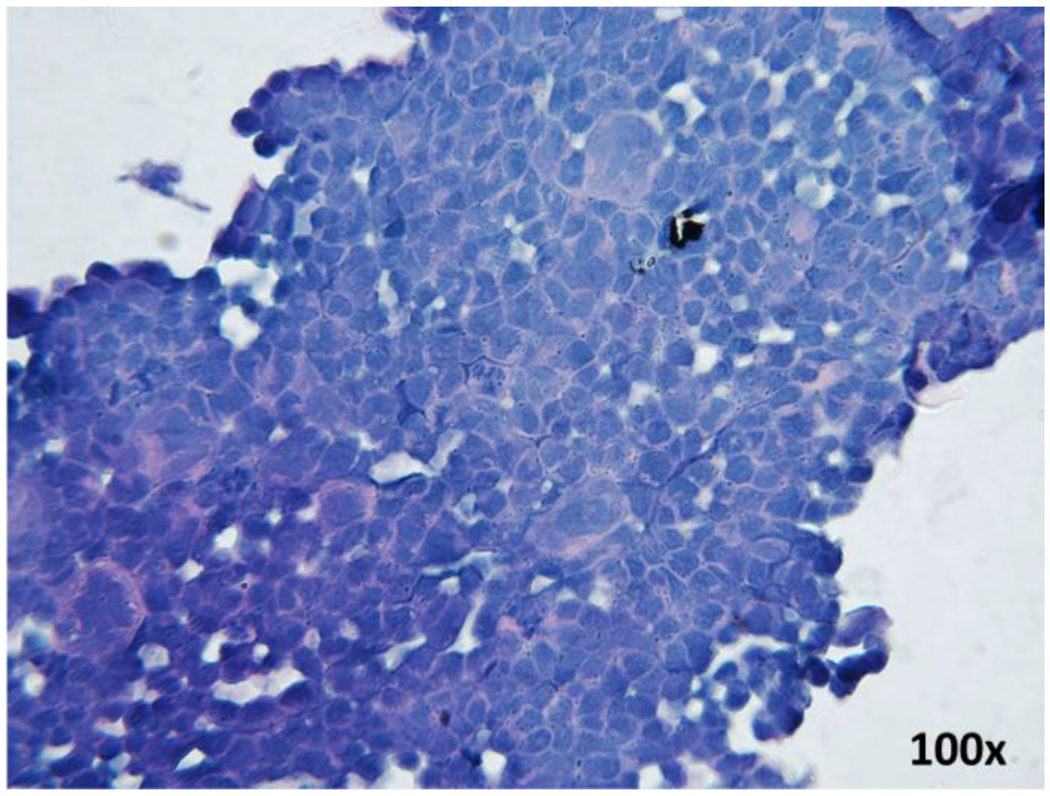
H&E staining of a spheroid slide prepared from H520 cells, observed under light microscope at magnification of 100x. The figure shows the preservation of cellular structure with intact nuclei and cell membranes, and a monolayer that resembles a tissue-like organization.
3.5 Immunohistochemistry (IHC) of Spheroid Sections
Once the H&E staining has allowed you to determine that the spheroid sections are of sufficient cellularity, you can proceed to stain with IHC some of the slides generated in Section 3.3.
Place the slides in a slide rack and incubate at 37°C overnight. Omit this step if you have performed this previously as described in step 1 of Section 3.4.
Incubate slides at 64°C for 1 h. Omit this step if you have performed this previously as described in step 2 of Section 3.4.
Remove the paraffin from the spheroid sections by submerging slides in a slide rack in xylene for 5 min. Remember that this step involving xylene and all volatile solvents should be performed inside the fume hood.
Hydrate slides by performing the following ethanol washes: 100% alcohol for 3 min, 95% ethanol for 3 min, and 70% ethanol for 2 min.
Perform a 1-hour antigen retrieval step with the antigen retrieval citrate buffer heated to 80–85°C. The slides should be completely submerged in the buffer, and the jar should in turn be tightly covered and submerged into a water bath set at 80–85°C (see Note 9).
Remove the whole jar from the water bath and allow it to cool down to room temperature for 20–30 min (see Note 10).
Transfer the slides to 1X PBS, incubate for 10 min.
Place slides flat in the humid chamber. From this point forward, prevent the slides form drying out. In the subsequent steps, ensure that the tissue sections are completely soaked in indicated solutions.
Cover the spheroid sections in the slides with a layer of 3% hydrogen peroxide block to inactivate endogenous peroxide activity. Incubate for 30 min at room temperature.
Rinse sections with 1X PBS for 10 min.
Carefully dry the slides with a lint-free absorbent paper. Dry only in the areas surrounding the spheroid sections, do not touch directly the sections with the absorbent paper.
After drying the areas surrounding the sections, use the PAP pen or any hydrophobic pen to make a hydrophobic barrier surrounding the sections. Again, be careful not to damage the spheroid sections.
Apply the primary antibody solution by adding a drop sufficiently large cover the entire area of the spheroid section. At this step the slides should be in the humid chamber.
Incubate with the primary antibody overnight or between 12–18 hours at 4–8°C. Remember to perform negative control sections in which the primary antibody is omitted. The negative control sections are incubated in 1X PBS instead of the antibody solution.
Remove the primary antibody and rinse the slides in 1X PBS for 5 min. Remove excess PBS with a lint-free absorbent paper. Remember that sections must never be allowed to get dry.
Incubate slides in the secondary antibody solution, ensuring you cover the whole section area with antibody solution. Incubation should be in the humid chamber at room temperature for 30 min.
Remove the secondary antibody solution and wash the slides in 1X PBS for 5 min. Remove the excess liquid with an absorbent paper.
Add the streptavidin-HRP mix to each section and incubate in the humid chamber for 30 min at room temperature.
Remove the streptavidin-HRP mix and wash the slides with 1X PBS for 5 min. Remove excess PBS with an absorbent paper.
Prepare the DAB solution and add a drop of solution to each section. Incubate for a maximum of 2 min (see Note 11).
Stop the DAB reaction by submerging the slides in two consecutive washes with distilled water.
Place the slides under running tap water for 5 min, being careful that the water does not directly touch the sections.
Dehydrate the slides with ethanol washes in the following order: 85%, 90%, 95%, and absolute 100% ethanol for 2 min each wash.
Incubate the slides in xylene for 2 min, inside the fume hood.
Drain the xylene from the slides and let them to air dry inside the fume hood.
Add 3 drops of mounting media on top of each section and carefully place a cover glass on top of the section, avoiding the formation of air bubbles inside the sample (see Note 8).
Analyze the slides under a light microscope. Take images at 10, 20, 40 up to 100x magnification. A positive p39 staining signal is appreciated as a brown precipitate (Fig. 5). The localization of the staining (nuclear, cytoplasmic, membrane, etc.) depends on the cellular localization of the antigen you are studying.
Fig. 5.
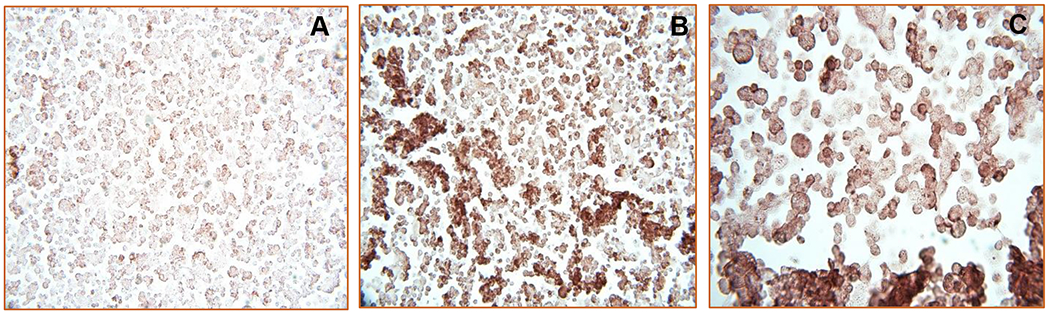
Spheroid section from H520 cells immunohistochemically stained using an anti-p39 antibody. (a) Negative control section in which the primary antibody was omitted. (b) Primary antibody at a 1:50 dilution and at 40x magnification. (c) Primary antibody at a 1:50 dilution and at 100x magnification. Staining is observed as a strong brown signal (b and c), which is absent from the negative control (a).
Acknowledgements
Work in our laboratory is supported by the U54 Moffitt Cancer Center-Ponce Health Sciences University Partnership NIH-NCI (#2U54CA163071-06), the NIMHD- NIAID funded Puerto Rico Clinical & Translational Research Consortium (#U54MD007587), the Molecular Genomics (MAGIC) Core (MBCL-RCMI Grant RR003050 MD007579) and its staff, the PHSU RCMI Program (Award Number #5G12MD007579-33 from The National Institute on Minority Health and Health Disparities), the PHSU-Moffitt Cancer Center Summer Research Program 2U54CA163071.07, and the PHSU RISE program under NIH Grant 2R25GM096955.
Footnotes
The appropriate dilution or concentration should be empirically determined for each antibody. We recommend that you start using the dilution or concentration recommended by the manufacturer, and depending on the results, you can use the antibody more concentrated if the staining is too weak, or more diluted, if background staining is too strong. When using antibodies against phosphorylated proteins, it is recommended that you use freshly cut tissue sections.
Many immunohistochemistry kits are commercially available. You have to ensure that the secondary antibody that is included with the kit matches the primary antibody that you are using (e. g. a goat anti-rabbit secondary antibody if you are using a rabbit polyclonal primary antibody, or a goat anti-mouse when using a mouse monoclonal primary antibody).
The DAB reagent and solution are photosensitive, prepare the solution in amber colored tubes or bottles, or wrap the tube with aluminum foil to avoid its exposure to light. Solution should be prepared right before using it and used fresh for the procedure to work optimally. Using a DAB solution that has been stored for days or weeks can result in poor staining intensity. When troubleshooting for weak staining signal, preparing a fresh DAB solution may be a good place to start.
Make sure you carefully remove the NBF, preferably by decanting or by slowly suctioning with a pipette. Try not to disrupt the spheroid. If the spheroid breaks apart during the removal of the NBF, spin again at 300 xg for 10 min at RT.
If the block is placed at −20°C for storage or to speed up solidification, cracks might form in the paraffin. This could complicate the sectioning process. Therefore, complete solidification is best conducted at room temperature overnight.
Try not to cover the blocks with the ice. Instead, place them with the paraffin facing down just over the ice. This will keep the surface cool and humid.
If the sections are to be placed in a water bath, make sure the temperature is not too high. If the water bath is too hot, the cell spheroid could detach from the paraffin resulting in loss of the sample. We used a room temperature water bath.
Gently slide the tip of a pencil into the cover glass to remove any air bubbles that might have formed over the sample section.
Citrate buffer should be pre-heated in a water bath (or microwave). Pre-heat the buffer before initiating the antigen retrieval step, do not submerge the slides in the buffer until it has reached the desired temperature.
At this point the jar can be slightly opened to allow the vapors to be released and ensure the buffer is cooled down to room temperature. Do this step inside a fume hood.
When we use the anti-p39 primary antibody diluted to 1:50, we noticed that the optimal DAB incubation time is around 1 minute. You must determine the optimal DAB incubation time for your cell lines and antibody of choice but bear in mind that prolonged reaction times can lead to high background.
5. References
- 1.Chen Y, Cui T, Yang L et al. (2011) The diagnostic value of cytokeratin 5/6, 14, 17, and 18 expression in human non-small cell lung cancer. Oncology 80 (5–6): 333–340 [DOI] [PubMed] [Google Scholar]
- 2.Rekhtman N, Ang DC, Sima CS et al. (2011) Immunohistochemical algorithm for differentiation of lung adenocarcinoma and squamous cell carcinoma based on large series of whole-tissue sections with validation in small specimens. Modern Pathology 24(10): 1348–1359 [DOI] [PubMed] [Google Scholar]
- 3.Righi L, Vavalà T, Rapa I et al. (2014) Impact of Non–Small-Cell Lung Cancer-Not Otherwise Specified Immunophenotyping on Treatment Outcome. Journal of Thoracic Oncology 9(10): 1540–1546 [DOI] [PubMed] [Google Scholar]
- 4.Pérez-Morales J, Mejías-Morales D, Rivera-Rivera S et al. (2018) Hyper-phosphorylation of Rb S249 together with CDK5R2/p39 overexpression are associated with impaired cell adhesion and epithelial-to-mesenchymal transition: Implications as a potential lung cancer grading and staging biomarker. PLoS ONE 13(11): e0207483. doi: 10.1371/journal.pone.0207483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathew EP, Nair V (2017) Role of cell block in cytopathologic evaluation of image-guided fine needle aspiration cytology. J Cytol 34 (3): 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poojan S, Han-Seong K, Ji-Woon Y, et al. (2018) Determination of Protein Expression Level in Cultured Cells by Immunocytochemistry on Paraffin-embedded Cell Blocks. Journal of Visualized Experiments 135: E57369. doi: 10.3791/57369 [DOI] [PMC free article] [PubMed] [Google Scholar]