

SUMMARY
Here we report a phase 1b clinical trial testing the impact of oncolytic virotherapy with talimogene laherparepvec on cytotoxic T cell infiltration and therapeutic efficacy of the anti-PD-1 antibody pembrolizumab. Twenty-one patients with advanced melanoma were treated with talimogene laherparepvec followed by combination therapy with pembrolizumab. Therapy was generally well tolerated, with fatigue, fevers, and chills as the most common adverse events. No dose-limiting toxicities occurred. Confirmed objective response rate was 62%, with a complete response rate of 33% per immune-related response criteria. Patients who responded to combination therapy had increased CD8+ T cells, elevated PD-L1 protein expression, as well as IFN-γ gene expression on several cell subsets in tumors after talimogene laherparepvec treatment. Response to combination therapy did not appear to be associated with baseline CD8+ T cell infiltration or baseline IFN-γ signature. These findings suggest that oncolytic virotherapy may improve the efficacy of anti-PD-1 therapy by changing the tumor microenvironment.
In Brief
In combination with anti-PD-1 therapy, intratumoral injection of an oncolytic virus engineered to enhance immune recognition of cancer resulted in a high response rate in patients with advanced melanoma.
Graphical Abstract
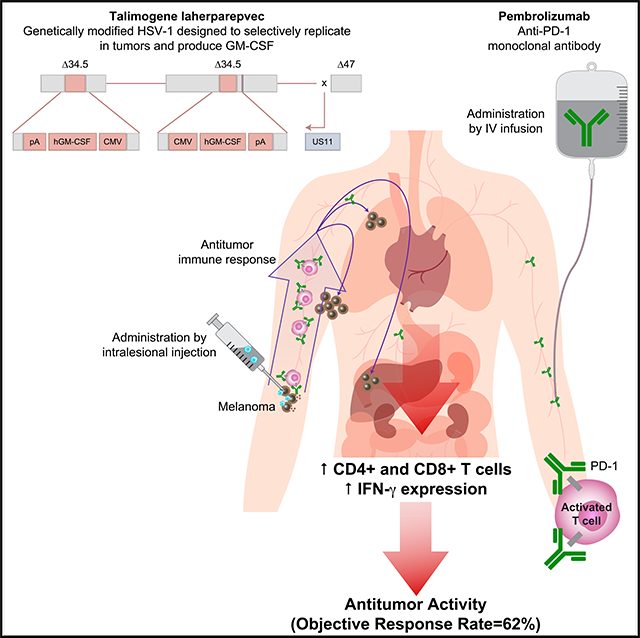
INTRODUCTION
Treatment with anti-programmed death protein 1 (PD-1) or anti-PD ligand 1 (PD-L1) antibodies results in long-lasting antitumor responses in patients with a variety of cancers, and it is becoming standard of care treatment for patients with metastatic melanoma, carcinomas of the head and neck, lung, kidney, and bladder, Merkel cell carcinoma, and Hodgkin disease (Sharma and Allison, 2015). However, in all of these indications, only a subset of patients respond to therapy, with the majority of patients being primarily resistant to PD-1 blockade. By analyzing baseline biopsies of patients treated with anti-PD-1 antibodies, it was previously observed that patients who did not respond were more likely to lack CD8+ T cells inside the tumor lesions (Herbst et al., 2014; Tumeh et al., 2014). If there are no CD8+ T cells within a tumor that are inhibited by the PD-1:PD-L1 interaction, then PD-1 blockade therapy is unlikely to work (Pardoll, 2012; Ribas, 2015; Spranger et al., 2013). In this setting, combination immunotherapy designed to attract CD8+ T cells into tumors by altering the immune-suppressive tumor microenvironment may improve the antitumor activity of PD-1 blockade therapy.
We hypothesized that the intratumoral administration of an oncolytic virus optimized to attract immune cells might favorably change the tumor microenvironment in the injected lesions and increase CD8+ T cell infiltration. Furthermore, reactive expression of PD-L1 in the tumor microenvironment could be a mechanism of resistance to oncolysis, which would be obviated by concurrent PD-1 blockade. After combined therapy, tumor antigen-specific CD8+ T cells that were fully stimulated in the injected lesion would be able to traffic to and infiltrate distant metastatic lesions to exert systemic antitumor activity, thereby reversing primary resistance to PD-1 blockade therapy.
Talimogene laherparepvec is a genetically modified herpes simplex virus type 1 designed to selectively replicate in tumors and produce granulocyte-macrophage colony-stimulating factor (GM-CSF) to enhance antigen release, presentation, and systemic antitumor immune response (Liu et al., 2003). In a prior phase 3 clinical trial, the intratumoral injection of talimogene laherparepvec into melanoma metastases improved the durable response rate compared with subcutaneous GM-CSF in patients with advanced melanoma (Andtbacka et al., 2015). Promising antitumor activity was demonstrated in a phase 1 study of talimogene laherparepvec combined with the checkpoint inhibitor ipilimumab, which blocks the cytotoxic T cell-associated antigen 4 (CTLA-4) (Chesney et al., 2016; Puzanov et al., 2016), and was confirmed in a phase 2 randomized trial comparing the same combination with ipilimumab alone (Chesney et al., 2017). There was a significant increase in the confirmed objective response rate by immune-related response criteria (irRC) with the combination compared with ipilimumab alone (39% versus 18%, respectively, p = 0.002).
We designed a phase 1b trial in patients with advanced melanoma combining the intratumoral injection of talimogene laherparepvec with the systemic administration of the anti-PD-1 antibody pembrolizumab, with baseline and repeated on-therapy biopsies; the primary objective was to test the safety of this combination and to explore its ability to boost inflammatory status of tumors. Specifically, we evaluated the ability of talimogene laherparepvec to reverse the low baseline presence of intratumoral CD8+ T cells in some of the metastatic lesions and then mediate increased objective tumor responses systemically.
RESULTS
A Phase 1b Clinical Trial Combining Talimogene Laherparepvec with Pembrolizumab
The phase 1b trial included a baseline biopsy before initiation of intratumoral talimogene laherparepvec injections, with a first injection of up to 4 mL × 106 plaque-forming units (pfu) per mL with the goal of inducing seroconversion and a protective immune response to the oncolytic viral vector, followed 3 weeks later with repeated injections of the full dose of up to 4 mL × 108 pfu/mL of talimogene laherparepvec every 2 weeks (Figure 1A). A second tumor biopsy was performed before administration of the second full dose of talimogene laherparepvec and before commencing treatment with pembrolizumab 200 mg intravenously every 2 weeks coinciding with subsequent doses of talimogene laherparepvec. The run-in period with single-agent talimogene laherparepvec administration was designed to analyze how intratumoral injection of this agent alters the tumor microenvironment before combination therapy began. A third tumor biopsy was planned, if feasible, during the combination therapy part of the study (Figures 1A and S1). The clinical trial enrolled 21 patients with advanced melanoma and dermal, subcutaneous, or nodal melanoma lesions amenable to intratumoral injection between December 2014 and March 2015 (see Table S1 for full patient characteristics); seven (33%) had received prior anticancer therapy (including adjuvant therapy) and four (19%) had received prior radiotherapy. Patients had a median (range) potential follow-up time of 18.6 (17.7–20.8) months at the time of reporting.
Figure 1. Melanoma Study Design and Clinical Response to Combination of Talimogene Laherparepvec and Pembrolizumab.

(A) Phase 1b study design schema. Stars indicate the time of scheduled tumor biopsies.
(B) Computed tomography scans of two patients with response to the combination therapy. Melanoma metastases are marked with a blue arrow at baseline.
(C) Waterfall plot of best response change in tumor burden from baseline. Patients were required to have baseline and ≥ 1 postbaseline tumor assessments to be included.
(D) Change in tumor burden over time.
(E) Kaplan-Meier analysis of progression-free survival.
(F) Kaplan-Meier analysis of overall survival.
See also Figures S1 and S2.
Combined Talimogene Laherparepvec and Pembrolizumab Did Not Increase the Toxicities from Single-Agent Therapy
With the combined therapy, there were no novel or dose-limiting toxicities in any of the 21 patients (see Table S2 for full details on toxicities). The most common treatment-related toxicities were fatigue (62%), chills (48%), and fever (43%), which are anticipated with the intratumoral injection of talimogene laherparepvec (Andtbacka et al., 2015). Frequently occurring and partially overlapping pembrolizumab-related adverse events were fatigue (62%), rash (33%), arthralgia (33%), fever (29%), and chills (29%), which are anticipated with this agent (Ribas et al., 2016). One event of grade 1 cytokine-release syndrome resulted in hospitalization and was described as possibly related to the combination. The only other serious adverse events were attributed solely to pembrolizumab and included grade 3 autoimmune hepatitis, grade 3 aseptic meningitis, and grade 4 pneumonitis (one patient each). In the patient with treatment-related aseptic meningitis, no herpes simplex virus was detected in the cerebrospinal fluid; the patient had stopped therapy with talimogene laherparepvec and pembrolizumab 1 month earlier and had already switched therapy to dabrafenib and trametinib at the time of first presentation of this adverse event.
Antitumor Activity with Combined Talimogene Laherparepvec and Pembrolizumab
The confirmed objective response rate as evaluated by investigators per irRC (Wolchok et al., 2009) was 61.9% (95% CI, 38.4%–81.9%), with a confirmed complete response rate of 33.3% (95% CI, 14.6%–57.0%) (Table 1). Responses occurred across all substages of melanoma (Figures 1B and 1C). Nine patients presented a transient increase in overall tumor size during the administration of talimogene laherparepvec, in particular after the first dose (106 pfu/mL) and before receiving the 108-pfu/mL dose in combination with pembrolizumab; however, these lesions later responded to combined therapy (Figure 1D). Median progression-free survival (PFS) and overall survival (OS) were not reached at the time of last follow up (Figures 1E and 1F). The combination treatment resulted in a >50% reduction in 82% of injected, 43% of noninjected nonvisceral, and 33% of noninjected visceral lesions (Figure S2). Interestingly, among the seven patients with stage IIIB/IIIC disease, four patients had noninjected nonvisceral lesions. In these patients, there were a total of 16 injected and 10 noninjected nonvisceral lesions (baseline and new) that were evaluable for assessment of percentage change in tumor area from baseline. Fifteen injected lesions (93.7%) showed any reduction; 6 noninjected lesions (60%) showed any reduction (Table S3).
Table 1.
Best Overall Responsea
| Talimogene Laherparepvec Plus Pembrolizumab (N = 21) | ||
|---|---|---|
| Totalb | Confirmedb | |
| Patients with a response | 15 | 13 |
| Response rate, % (95% CI) | 71 (48–89) | 62 (38–82) |
| Best overall response, n (%) | ||
| Complete response | 8 (38) | 7 (33) |
| Partial response | 7 (33) | 6 (29) |
| Stable diseasec | 1 (5) | 3 (14) |
| Progressive disease | 5 (24) | 5 (24) |
| Disease control rate, n (%) | 16 (76) | 16 (76) |
Response was evaluated per immune-related response criteria by investigators; data cutoff was August 31, 2016.
Responses were confirmed by a subsequent assessment at least 4 weeks later.
A best overall response of stable disease required an evaluation of stable disease no earlier than 77 days after enrollment.
Tumor Responses Independent of Baseline CD8+ Infiltration, PD-L1 Status, and Interferon-γ Signature
PD-L1 is induced by interferon gamma (IFN-γ) produced by tumor-infiltrating, antigen-specific T cells, in what is termed adaptive immune resistance allowing cancer cells to avoid the cytotoxic activity of T cells (Pardoll, 2012; Ribas, 2015). Because these T cells are then blocked by PD-1:PD-L1 interactions, it is not surprising that patients who respond to single-agent PD-1 blockade therapy have higher densities of baseline CD8+ infiltration, IFN-γ gene expression signatures, and PD-L1 expression (Herbst et al., 2014; Ribas et al., 2015; Tumeh et al., 2014). We analyzed baseline biopsies of patients in this study for CD8+ T cell density, PD-L1 positivity, and IFN-γ gene signature. As opposed to prior experience with single-agent pembrolizumab therapy (Ribas et al., 2015; Tumeh et al., 2014), responses in this clinical trial were evident in patients whose baseline biopsies had very low CD8+ T cell infiltrates or negative IFN-γ gene signature. Among 13 patients in whom biopsies revealed a CD8+ density < 1,000 cells/mm2, 9 patients went on to respond to therapy and 4 patients had disease progression (Figure 2A). Out of the five patients with baseline biopsies with a low IFN-γ signature, three patients went on to have a complete response and two had disease progression (Figure 2B). There was only one baseline biopsy that was scored as PD-L1 negative, but that patient went on to have a complete response to the combined therapy (Figure 2B).
Figure 2. Combination of Talimogene Laherparepvec and Pembrolizumab Is Effective in Patients with Low Tumor CD8+ Density.

(A) Baseline CD8+ density in tumor biopsies according to response rate. Magnitude of bars indicates baseline tumor CD8+ density in each patient’s baseline biopsy, and best overall response is indicated on x axis and by bar color. Gold, CR; pink, PR; blue, PD.
(B) Baseline PD-L1 by IHC status (1% cutoff) and IFN-γ signature score by NanoString analysis is shown under each patient’s CD8 result. Best overall response per investigator is shown as of cutoff date of August 2016. Abbreviations: CR, complete response; IFN-γ, interferon γ; IHC, immunohistochemistry; NA, result not available; PD, progressive disease; PR, partial response.
Talimogene Laherparepvec Intratumoral Injections Increase CD8+ T Cell Infiltration in Patients Who Respond to Combined Therapy
Because some patients whose baseline biopsies had relatively low CD8+ cell density and were not positive for an IFN-γ gene signature went on to have an objective response, we analyzed whether the run-in period with single-agent talimogene laherparepvec had changed the tumor microenvironment by bringing T cells into metastatic melanoma lesions in patients who responded to therapy. Indeed, immunohistochemical (IHC) analysis comparing baseline biopsies with biopsies performed after talimogene laherparepvec alone showed an increase in the density of infiltrating CD8+ T cells in 8 out of 12 injected lesions available for analysis, which further increased in several of the biopsies obtained at the time of combined therapy (Figures 3A and 3B). In three patients with a response to therapy, the CD8+ density decreased in the on-therapy biopsy, and one additional patient had no change in CD8+ density. The three patients without a response all had a decrease in CD8+ density in the on-therapy biopsies. Overall, the increase in CD8+ density was most evident in the injected lesions of the patients who went on to respond to therapy (Figure 3B), a relationship supported by logistic regression analysis (p = 0.0048; Figure S3A; logistic regression described in STAR Methods). The change in CD8+ infiltration density was variable in the noninjected lesions at week 6 even in patients who later responded to therapy, with the caveat that there are only three such biopsies available for interpretation (Figures 3B and S3B). Some posttreatment tumor-depleted samples were not initially analyzed because of histologic absence of tumor in the sample, but upon reevaluation were found to have evidence of prior tumor content (indicated by open symbols in Figures 3B and 3C). In the five patients with tumor-depleted samples at week 6, the CD8+ cell density was much higher in the injected lesions from the four responding patients as compared with the single nonresponder. We also performed IHC for the cytotoxic granule component granzyme B (associated with the cytotoxic subset of CD8+ T cells and natural killer cells), which has been shown to increase in tumors after PD-1 blockade (Tumeh et al., 2014). A trend suggesting increased granzyme B in tumors after talimogene laherparepvec and combination treatment was also observed, in particular for the biopsies with low residual tumor content (Figure 3C). Furthermore, on analysis of tumor gene expression data, we found that CD8α and IFN-γ mRNAs were elevated after treatment, providing additional supporting evidence for treatment-related change in the tumor microenvironment increasing the number of IFN-γ-producing cytotoxic T cells (Figures 3D and 3E). CD8α increased 1.7-fold (p = 0.01) in injected lesions at week 6 compared with baseline and 1.44-fold (p = 0.0012) in noninjected lesions. Similarly, the IFN-γ fold increases for injected and noninjected lesions were 1.63 (p = 0.0004) and 1.41 (p = 0.17), respectively.
Figure 3. Talimogene Laherparepvec Increases Tumor CD8+ Density in Patients Responding to Combination of Talimogene Laherparepvec and Pembrolizumab.

(A) Examples of pre (week 1) and post (week 6) talimogene laherparepvec and talimogene laherparepvec plus pembrolizumab (week 30) CD8+ density in tumor biopsies: visualization of cells stained with CD8 antibody with red chromogen. Staining was quantified for tissue regions of interest including CD8+ density in the tumor as shown for talimogene laherparepvec-injected tumors.
(B and C) CD8+ density (B) and granzyme B H-score (C) is shown for baseline and postbaseline biopsies. The left side in each panel shows postbaseline results from injected lesions, and the right side in each panel shows results from noninjected lesions. Open circles indicate results from tumor biopsies that were depleted of melanoma cells but had pathologic features of having previously been infiltrated by melanoma cells such as melanin deposits. Response is color coded for best overall response per investigator (complete or partial response in red and nonresponse in blue).
(D and E) CD8α (D) and IFN-γ normalized (E) mRNA transcript count were measured in the NanoString Pan Cancer Immune Profiling Panel. IFN-γ = interferon γ.
See also Figure S3.
Characterization of Changes in Immune Cell Infiltrates in Talimogene Laherparepvec Injected and Noninjected Lesions
To further characterize changes in tumors, we performed multiplexed immunofluorescence staining of paired biopsies at different time points from 13 patients. We observed broad changes in tumor inflammation after talimogene laherparepvec at week 6, including increased infiltration by immune cells and a clear increase in cells expressing PD-L1 in eight out of ten injected tumors and in two out of four noninjected tumors (Figure 4A). Changes in immune infiltrates in the on-treatment biopsies from some patients included an influx of a large proportion of CD4+ and CD8+ T cells, many coexpressing PD-1, as well as CD56+-expressing cells and CD20+ B cells (the full set of immunofluorescence analyses in biopsies is reported in Table S4). Increases were also observed in the density of cells expressing the memory T cell marker CD45RO and in cells expressing the regulatory T cell (Treg) marker Foxp3 (Figure 4A). The magnitude of effector T cell (Teff) increases, however, was much larger relative to Treg cells, resulting in an overall decrease in the Treg to Teff ratio in tumors after talimogene laherparepvec (Figure S4) consistent with previous reports (Kaufman et al., 2010). Table S4 additionally shows that there was no apparent change in the density of macrophages based on CD68 staining. An example of increased CD8+ and PD-L1 density by immunofluorescence at weeks 6 and 30 relative to baseline is shown in Figure 4B. At weeks 6 and 30, tumor cells costaining for S100 (blue) and PD-L1 (red) are evident along with CD8+ T cells (green), showing coexpression of PD-L1. The biopsy taken during combined therapy in a responding patient was nearly completely infiltrated by CD8+ T cells. Additional representative images are shown in Figure S5. Finally, to address potential changes in dendritic cell subsets, we assessed CD141 (marker of Batf3 cross-priming dendritic cells) and CD123 (a marker of plasmacytoid dendritic cells) mRNA levels. We did not observe significant changes in either marker in biopsies from talimogene laherparepvec-injected and -noninjected lesions from week 1 to 6 (Figure S6).
Figure 4. Talimogene Laherparepvec Increases Tumor-Infiltrating Lymphocyte Density and PD-L1 Expression in Tumors.

Twelve-color immunofluorescence staining was performed on a single slide from paired pre- and post-talimogene laherparepvec tumor biopsies from each of 13 patients. Markers evaluated included S100 (as melanoma segmentation marker), CD3, CD4, CD8, PD-1, PD-L1, CTLA-4, CD45RO, Foxp3, CD56, CD68, and CD20.
(A) A subset of changes at week 6 from baseline in marker cell positive cell density for results with statistical significance (PD-L1, PD-1, CD8, CD4, CD56, CD20, CD45RO, and Foxp3) are graphed for noninjected (left) and injected (right) samples. Median change for each subset is shown with a horizontal line. Response is color coded for best overall response per investigator: complete or partial response in red and nonresponse in blue.
(B) Example of the combination of S100 (blue), CD8 (green), and PD-L1 (red) staining is shown at low (top) and high (bottom) magnification for a baseline biopsy from a patient who went on to have a partial response (week 1), week 6 after injection of talimogene laherparepvec, and at week 30 after long-term treatment with the combination of talimogene laherparepvec and pembrolizumab. Abbreviations: CTLA-4, cytotoxic T cell-associated antigen 4; I, biopsy of an injected metastasis; PD-1, programmed death protein 1; PD-L1, programmed death ligand 1; NI, biopsy of a noninjected metastasis.
See also Figures S4–S6.
Changes in the Functional Phenotype of Circulating T Cells with Combined Therapy
We also analyzed changes in immune cells in peripheral blood as a potential pharmacodynamic effect of single-agent and combined therapy. After talimogene laherparepvec single-agent therapy, the majority of patients had an increase in the number of circulating CD8+ and CD4+ T cells in peripheral blood, which did not increase further when pembrolizumab was added (Figures 5A and 5B). However, the addition of pembrolizumab tended to increase the number of dividing CD8+ T cells in circulation as indicated by increases in Ki67+CD3+CD8+ T cells (Figure 5C). Analysis of the expression of different immune checkpoint receptors in circulating CD3+CD8+ T cells revealed an increase in PD-1 and TIM-3 (a molecule expressed on IFN-γ-producing CD8+ and CD4+ T cells) with single-agent talimogene laherparepvec therapy (Figures 5D and 5E), whereas there was no change in B- and T-lymphocyte attenuator protein (BTLA; Figure 5F). No associations of response with baseline cell levels or changes over time passed our false discovery controls.
Figure 5. Circulating T Cell Subsets and Expression of Activation Markers.
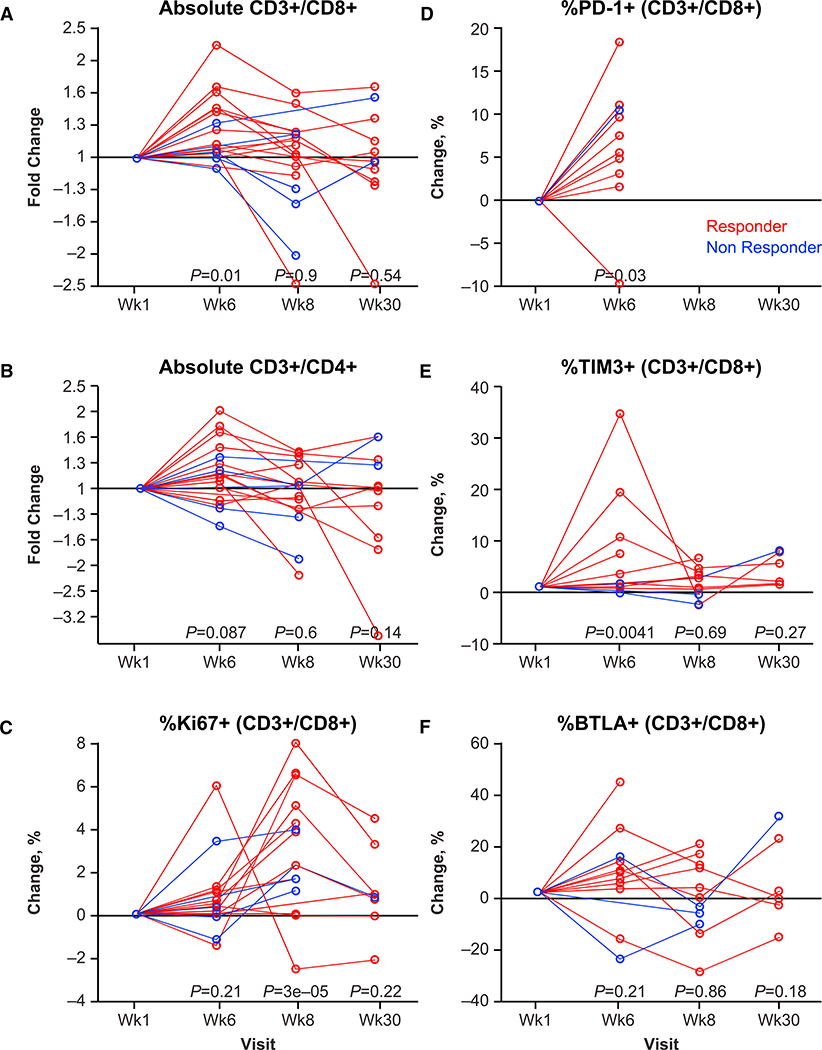
Peripheral blood cells obtained from baseline, week 1, week 6, week 8, and week 30 were analyzed by flow cytometry.
(A) Fold change in absolute CD3+CD8+ cells.
(B) Fold change in absolute CD3+CD4+ cells.
(C) Percentage change in Ki67+ (CD3+CD8+) cells.
(D) Percentage change in PD-1+ (CD3+CD8+) cells at week 1 and week 6 only; after starting on pembrolizumab, the staining antibody competed for the same epitope.
(E) Percentage change in TIM3+ (CD3+CD8+) cells.
(F) Percentage change in BTLA+ (CD3+CD8+) cells.
p values for comparison with baseline are shown below data for each postbaseline visit, based on contrasts from linear mixed-effects modeling. Response is color-coded for best overall response per investigator (complete or partial response in red and nonresponse in blue). Abbreviations: BTLA, B- and T-lymphocyte attenuator; PD-1, programmed death protein 1.
DISCUSSION
This first-in-human combination immunotherapy clinical trial demonstrates a high overall and complete response rate in patients with advanced melanoma, which was associated with changes in tumor biopsies that were mechanistically correlated with the hypothesis that the injection of the oncolytic virus talimogene laherparepvec would change the tumor microenvironment by attracting T cells that may induce a systemic response in distant metastases after subsequent blockade of PD-1 with pembrolizumab. Indeed, during the run-in period of the study with single-agent talimogene laherparepvec intratumoral administration, there was evidence of a systemic increase in circulating CD4+ and CD8+ T cells and increased CD8+ T cell infiltration into tumors. These T cells expressed PD-1 and the tumor cells expressed PD-L1, likely limiting the antitumor activity of single-agent talimogene laherparepvec, which benefitted from PD-1 blockade, thereby resulting in clinical activity beyond what would be expected with either therapy alone. The benefit of increased responses was achieved with a low rate of toxicities, most of which were expected with the single-agent use of talimogene laherparepvec or pembrolizumab (Andtbacka et al., 2015; Ribas et al., 2016).
PD-1 blockade therapy with pembrolizumab or nivolumab leads to an objective response of approximately 35% to 40% for treatment-naive patients with metastatic melanoma (Ribas et al., 2016; Robert et al., 2015a, 2015b). Although the need to select patients who had injectable lesions may have skewed the population toward those with a good prognosis, an overall response rate of 62% and a CR rate of 33% is unlikely to be a result of anti-PD-1 therapy alone. In a study of 655 patients treated with pembrolizumab, there were 34 patients who had only skin and nodal metastases (stage M1a), and the overall response rate in this group of patients was 38% (Ribas et al., 2016). When evaluating the efficacy outcomes of the current study, it is important to note that the primary objective of this study was to evaluate the safety of the combination of talimogene laherparepvec and pembrolizumab in patients with advanced melanoma. Therefore, we acknowledge that the interpretation of the efficacy outcomes is limited by the small size of the study population (n = 21) and the limited number of enrolled patients with stage IV M1c disease. Only a randomized trial would be able to definitively demonstrate that the combination is better than either single-agent pembrolizumab or talimogene laherparepvec. An ongoing phase 3 clinical trial is currently comparing systemic administration of pembrolizumab with intralesional injection of talimogene laherparepvec or placebo in patients with stage IIIB-IV melanoma (ClinicalTrials.gov: NCT02263508).
To significantly increase the response rate to single-agent anti-PD-1, a new combination therapy should address the major mechanism for primary resistance. Patients whose baseline biopsies had low densities of CD8+ T cells, lack of significant IFN-γ expression, and resulting low PD-L1 expression would be unlikely to respond (Postow et al., 2015; Ribas et al., 2015; Topalian et al., 2012; Tumeh et al., 2014). Therefore, the combination therapy should increase the intratumoral infiltration by CD8+ T cells, which may attract enough T cells with tumor specificity to reverse the primary resistance to PD-1 blockade therapy (Chen et al., 2016; Ribas, 2015). Our data suggest that talimogene laherparepvec may provide this combinatorial effect. In this study, the number of patients with tumors with low baseline CD8+ density and a low IFN-γ signature who had an objective response to combined therapy was high compared with prior trials of single-agent pembrolizumab (Ribas et al., 2015; Tumeh et al., 2014).
Evidence that local administration of talimogene laherparepvec contributed to a systemic antitumor effect was provided by the increase in circulating CD8+ and CD4+ T cells and the increase in inflammation observed in tumors not injected with talimogene laherparepvec before the introduction of pembrolizumab. In the pivotal single-agent study of talimogene laherparepvec, a decrease in tumor size was observed in 15% of evaluable, noninjected, measurable visceral lesions (Andtbacka et al., 2015, 2016). We also observed reductions in dimensions of noninjected lesions, including both visceral and nonvisceral lesions (including in patients with stage IIIB/IIIC disease). Approximately two out of the four week 6 noninjected lesions showed increased CD8+ density and PD-L1 (by immunofluorescence), and three out of five for IFN-γ mRNA. Alternatively, talimogene laherparepvec’s unique properties (incorporating local GM-CSF for dendritic cell recruitment together with its own innate immune stimulation via toll-like receptors and cytoplasmic sensing pathways to promote adaptive immune responses) may provide a unique set of signals, making it ideal for immunotherapy combinations, including checkpoint inhibitors. Although we did not detect any differences in dendritic cell subset markers from week 1 to week 6 in the either injected or noninjected lesions (Figure S6), it is possible that the late timing of the biopsies (with week 6 occurring 2 weeks after the previous talimogene laherparepvec injection) was not optimal to address this question. Another possibility is that the selected marker, CD141 (mRNA), was not specific enough to accurately represent Batf3 DC abundance. Therefore, we also evaluated additional Batf3 DC markers, IRF8 and XCR1, but significant changes were not observed for these markers either. Future studies evaluating biopsies soon after talimogene laherparepvec injection will be needed to determine the timing of dendritic cell recruitment and to address the role of local GM-CSF. Further information on events leading to CD8+ infiltration is provided by preclinical studies. Administration of OncoVEXmGM-CSF (talimogene laherparepvec with the mouse GM-CSF transgene) alone or in combination with checkpoint blockade in an A20 contralateral murine tumor model resulted in increased tumor-specific CD8+ T cells and also anti-AH1 T cells and systemic efficacy (Moesta et al., 2017).
We will seek confirmation of the conclusions from this 21-patient phase 1b study (e.g., lack of requirement for baseline tumor infiltration) in the ongoing phase 3 study of the combination of talimogene laherparepvec plus pembrolizumab, which is currently accruing 660 patients, half receiving combination therapy and half receiving pembrolizumab with intratumoral placebo in the control arm (ClinicalTrials.gov: NCT02263508). Also, to further evaluate systemic effects of talimogene laherparepvec, a separate biomarker study is ongoing to evaluate baseline and post-talimogene laherparepvec noninjected tumors from more than 100 patients (ClinicalTrials.gov: NCT02366195). This will help provide follow-up data on findings from the small set of tumor biopsies not injected with talimogene laherparepvec in this series, many of which showed increased tumor inflammation.
In conclusion, the high response rate in this phase 1 clinical trial and the mechanistic changes documented in patient biopsies suggest that the combination of talimogene laherparepvec and pembrolizumab may be able to overcome some limitations of either single-agent therapy and provide responses beyond what would be expected with either talimogene laherparepvec or pembrolizumab administered alone.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and/or data may be directed to the Lead Contact, Toni Ribas (aribas@mednet.ucla.edu). Any sharing of materials or data may be subject to material transfer agreements and/or data-sharing agreements per the requirements of the study sponsors and applicable legislation.
EXPERIMENTAL MODEL AND PATIENT DETAILS
Eligible patients (≥18 years) had histologically confirmed, surgically unresectable, stage IIIB to IV cutaneous melanoma; measurable disease (≥1 melanoma lesion with longest diameter ≥ 10 mm); and ≥ 1 injectable cutaneous, subcutaneous, or nodal melanoma lesion(s) ≥ 10 mm in longest diameter, either alone or in aggregate, for which surgery was not recommended. Patients were required to have adequate performance status and hematologic, hepatic, renal, and coagulation function. Patients were excluded if they had uveal/mucosal melanoma; had previously received talimogene laherparepvec or any prior systemic anticancer treatment (ie, chemotherapy, immunotherapy, targeted therapy) given in a nonadjuvant setting for unresectable, stage IIIB to IV melanoma; Eastern Cooperative Oncology Group performance status ≥ 2; active brain metastases; active herpetic skin lesions; prior complications from herpetic infection; or required systemic antiherpetic treatment other than intermittent topical use. Of the 21 patients included in this study, 13 (62%) were female and 8 (38%) were male. The median (range) age of patients was 58 (37–89) years. All patients provided written informed consent. Study procedures were approved by an institutional ethics committee at each site.
METHOD DETAILS
Study Design
The phase 1b portion of the MASTERKEY-265 study was an open-label, multicenter, single-arm study that primarily evaluated the safety of intralesional talimogene laherparepvec in combination with intravenous pembrolizumab (Figure S1). Briefly, to seroconvert herpes simplex virus–negative patients, intralesional talimogene laherparepvec 106 pfu/mL was administered on day 1 of study week 1. Subsequent doses of talimogene laherparepvec 108 pfu/mL were administered on day 1 of weeks 4 and 6 and every 2 weeks thereafter. Up to 4 mL (total volume) of talimogene laherparepvec could be administered by intralesional injection at each treatment visit; the volume delivered to each injected lesion was contingent on the diameter of the lesion (Hoffner et al., 2016). The injected volume per lesion ranged from 0.1 mL for lesions ≤ 0.5 cm to 4.0 mL for lesions > 5 cm in longest diameter. Talimogene laherparepvec administration continued until disappearance of injectable lesions, CR, confirmed disease progression (PD) per modified irRC (Wolchok et al., 2009), treatment intolerance, 24 months from the first dose of pembrolizumab, or end of study, whichever occurred first. If toxicity occurred, talimogene laherparepvec doses could be delayed for up to 4 weeks; delays > 4 weeks resulted in permanent discontinuation.
Pembrolizumab (200 mg) was administered intravenously every 2 weeks beginning on day 1 of week 6 (ie, at the time of the third dose of talimogene laherparepvec). Pembrolizumab treatment was to be continued until confirmed PD by irRC, treatment intolerance, 24 months from the first dose of pembrolizumab, or end of study, whichever occurred first. Pembrolizumab could be withheld or discontinued per protocol-specified rules consistent with the US prescribing information. If pembrolizumab was withheld > 12 weeks, pembrolizumab treatment was permanently discontinued.
The primary endpoint was incidence of dose-limiting toxicities (DLTs) starting from when both agents were given in combination. Incidence of DLTs in the first 6 DLT-evaluable patients and additional safety data from all patients were evaluated by a dose-level review team comprising investigators and representatives of the Amgen/Merck study teams. The combination would be declared tolerable if the incidence of DLTs was < 33% during the DLT evaluation period. Secondary endpoints included confirmed objective response rate (ORR; the rate of CR plus partial response [PR]) as evaluated by investigators per irRC, (Wolchok et al., 2009) best overall response, and incidence of adverse events.
Dose-limiting toxicities were defined as any of the following treatment-related toxicities occurring during the 6-week period from the beginning of pembrolizumab treatment: grade 4 nonhematologic toxicity; grade 3/4 pneumonitis; grade 3 nonhematologic toxicity lasting > 3 days despite optimal supportive care (except grade 3 fatigue); grade 3/4 nonhematologic laboratory value requiring medical intervention/hospitalization or persisting > 1 week; grade 3/4 febrile neutropenia; thrombocytopenia < 25 × 109/L if associated with a life-threatening bleeding event or bleeding event requiring platelet infusion; any grade 5 toxicity; or any toxicity requiring permanent discontinuation of talimogene laherparepvec or pembrolizumab.
Study Clinical Assessments
Adverse events occurring from week 1 to 30 days after the last dose of study treatment were recorded and graded using Common Terminology Criteria for Adverse Events version 4.0.
Tumor response was evaluated per modified irRC (Wolchok et al., 2009) by investigators. CR was defined as the disappearance of all lesions; PR was defined as a decrease in tumor area ≥ 50% relative to baseline; PD was defined as an increase in tumor area ≥ 25% relative to nadir; and SD was defined as any outcome not meeting the criteria for response or PD with ≥ 77 days elapsed after enrollment. Responses were confirmed within 4 weeks from the date of first documentation of response. Tumor assessments were performed at screening, week 6 (prior to initiation of pembrolizumab), week 18, and every 12 weeks thereafter. Radiographic imaging for assessment of lesions was performed using computed tomography, positron emission tomography, magnetic resonance imaging, or ultrasound. Clinical measurement of cutaneous, subcutaneous, and palpable nodal tumor lesions was conducted with calipers. Initial measurement of PD was confirmed by assessment of measureable/nonmeasureable new lesions as well as index lesions ≥ 4 weeks later. If clinically stable, patients continued treatment while awaiting confirmation of PD.
Biomarker Analysis
Flow Cytometry
T cell subsets were analyzed in three immunophenotyping assays using fresh blood samples evaluated in regional flow cytometry labs (LabCorp, Cranford, NJ, USA; Mechelin, Belgium; Singapore). First T cell counts were derived from a BD TruCOUNT assay including the CD45, CD3, CD4, and CD8 markers. Additionally, checkpoint markers on T cell subsets were assessed in a second assay including PD-1, Tim3, and BTLA. Finally, a third assay evaluated T cell subsets for intracellular markers including Ki67.
RNA Profiling and Interferon- γ Gene Signature
Total RNA was isolated from 5-μm thick formalin-fixed paraffin-embedded sections fixed on positively charged slides. Percentage tumor area was first assessed and either all tissue was scraped for isolation or if < 50% tumor area was present, tumor tissue was macrodissected for isolation. RNA isolation was performed using the High Pure FFPET RNA isolation kit from Roche Diagnostics (Indianapolis, IN, USA). NanoString gene expression profiling was conducted using 50 ng of RNA run on the nCounter PanCancer Immune Profiling Panel (NanoString Technologies, Seattle, WA, USA) per manufacturer’s instructions. An IFN-γ gene signature score was obtained using a calculation that compared a calculated normalized value to a predefined weighted score for each gene within the signature. To determine these normalized gene expression values from the NanoString assay, the log10 transformed raw counts for each gene were subtracted from the log10 calculated mean of the housekeeping genes. Of note, the normalized reference value determined here was used solely to derive the IFN-γ gene signature score and not for any statistical analyses.
Immunohistochemistry
PD-L1 expression in tumors was assessed using IHC as described previously (Daud et al., 2016) using an investigational version of the Dako PD-L1 22C3 assay (Carpinteria, CA, USA).
CD8 and granzyme B IHC analysis was performed at Mosaic Laboratories (Lake Forest, CA, USA). A hematoxylin and eosin stain was performed and reviewed by a pathologist to verify the presence of melanoma and to define tumor areas as regions of interest for analysis. The anti-CD8 mouse monoclonal antibody clone C8/144B was used for CD8 IHC. The anti-granzyme B mouse monoclonal antibody clone GrB-7 (Dako) was used for granzyme B IHC. Immunohistochemical detection was performed with a polymer-based detection method and a red chromogen. Slides were scanned using a ScanScope CS or AT Turbo system (Aperio, Vista, CA, USA), the region of tumor was circled, and the density of positive cells (eg, CD8+ cells per mm2) was evaluated by automated image analysis. To address the potential for tumor heterogeneity and ensure that it did not unduly influence the assessment of tumor biopsies, the IHC analysis plan required the pathologist to identify tumor areas by hematoxylin and eosin stain and for CD8 IHC analysis, for example, entire slides were scanned and automated cell counting applied to all tumor areas. Thus, the numerical data are a reflection of the entire specimen.
Immunofluorescence
Available paired, pre- and post-treatment biopsies were evaluated using MultiOmyx technology to stain 12 biomarkers using a single slide. Repeated cycles of staining using a pair of antibodies directly conjugated to either Cy3 or Cy5, followed by imaging and dye inactivation were performed according to published methods (Q. Au et al., 2016, Amer. Assoc. Cancer Res., conf., Abstract 4146; Gerdes et al., 2013). The staining was performed by NeoGenomics (Aliso Viejo, CA) to interrogate modulation of immune cells and checkpoint markers in tumors after treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Using a 6+3 trial design, 6 to 9 DLT-evaluable patients were required to assess the DLT profile of talimogene laherparepvec in combination with pembrolizumab, assuming a true DLT incidence rate between 11% and 33%. Additional patients were enrolled to evaluate the association between biomarkers and response.
The DLT analysis set included all DLT-evaluable patients enrolled in phase 1b who had the opportunity to be on treatment ≥ 6 weeks from the initial dose of pembrolizumab and who received ≥ 2 doses of talimogene laherparepvec and 2 doses of pembrolizumab in combination, or who experienced a DLT within 6 weeks of starting combination therapy. The safety analysis sets included all patients who received ≥ 1 dose of talimogene laherparepvec or pembrolizumab. Predictive biomarker analyses included all patients with a baseline biomarker result; analyses of biomarker changes included all patients with a baseline biomarker result and ≥ 1 subsequent biomarker result.
Corresponding exact 95% CIs were calculated for ORR and disease control rate. PFS (time from enrollment to disease progression per modified irRC or death) and OS (time from enrollment to death) were estimated using the Kaplan-Meier method.
For cell density or H-score results from IHC, change from baseline was assessed with the sign-test of the log2 ratio of postbaseline over baseline (week 1) in injected and noninjected lesions separately. For flow cytometry results, change from baseline was assessed with a linear mixed-effects model with baseline as a covariate for log10 ratio (absolute counts or molecules of equivalent soluble fluorochrome [MESF]) or percentage difference to/from baseline. For immunofluorescence-based multiparameter imaging, effects on cell density were assessed with linear mixed-effects models for cube root of density with visit and injection status as factors. For NanoString, gene expression changes from baseline were assessed with linear mixed effects models for normalized transcript count log2 ratio to baseline with covariates of total pre-normalization transcript count, and baseline. Injected and noninjected lesions were evaluated separately. NanoString total transcript counts were normalized to 1 million prior to statistical analysis. Only a single visit after baseline was fit with the mixed effects model, and only a single lesion per patient was fit by the model. Some patients had two (but not more than two) Nanostring results for a given visit and lesion. In these instances, the patient was included in the model each time to account for the correlation in results of these potential duplicate tests. Two rounds of testing were conducted: week 0 change from baseline for injected lesions, and week 0 change from baseline for noninjected lesions.
Association with unconfirmed best response per investigator as of August 2016 was evaluated with logistic regression of response (CR or PR) versus continuous biomarker results at either baseline or change from baseline at a given visit. Transformed results were used for analyses. Injected and noninjected lesions were analyzed separately. The Kruskal-Wallis test was also evaluated in cases of small sample size.
The false discovery rate (FDR) was controlled at 5% with the Benjamini-Hochberg procedure, and flow cytometry analysis was stratified by a priority set of endpoints and reporting metrics (Abs, MESF, %).
DATA AND SOFTWARE AVAILABILITY
The lead contact should be contacted for dataset requests. As noted previously, sharing may be subject to data-sharing agreement.
ADDITIONAL RESOURCES
The ClinicalTrials.gov Identifier for this study is NCT02263508; the study protocol is provided as supplemental material.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PD-L1 | Dako | 22C3 |
| CD8 | Dako | C8/144B |
| Granzyme B | Dako | GrB-7 |
| Bacterial and Virus Strains | ||
| Talimogene laherparepvec (modified Herpes simplex virus type 1) | Amgen Inc. | http://www.imlygic.com/ |
| Chemicals, Peptides, and Recombinant Protein | ||
| BD Multitest CD3/CD8/CD45/CD4 | BD | 340499 |
| TruCount Tubes | BD | 340334 |
| CD45 V500 | BD Horizon | HI30 |
| CD3 AF700 | BioLegend | UCHT1 |
| CD4 PerCP | BD | SK3 |
| CD8 Biotin | BioLegend | HIT8a |
| PD-1 APC | BioLegend | EH12.2H7 |
| TIM3 APC | Miltenyi Biotec | F38-2E2 |
| BTLA (CD272) APC | BioLegend | MIH26 |
| CD8 BV510 | BioLegend | RPA-T8 |
| CD4 Pacific Blue | BD PharMingen | RPA-T4 |
| Ki67 AF647 | BioLegend | Ki-67 |
| MsIgG1 PE | BD | MOPC-21 |
| MsIgG1 APC | BD PharMingen | MOPC-21 |
| MsIgG1 PerCP | BD | X40 |
| MsIgG1 AF647 | BioLegend | MOPC-21 |
| MsIgG1 V450 | BD Horizon | MOPC-21 |
| MsIgG1 Biotin | BioLegend | MOPC-21 |
| Streptavidin-BV605 | BD Horizon | 563260 |
| Per-Fix nc kit (no centrifuge assay) | Beckman Coulter | B31167 |
| Critical Commercial Assays | ||
| High Pure FFPET RNA isolation kit | Roche Diagnostics | 06650775001 |
| nCounter PanCancer Immune Profiling Panel | NanoString Technologies | XT-CSO-HIP1-12 |
| MultiOmyx platform, TIL panel | NeoGenomics | https://neogenomics.com/portals/0/pdf/pharma/MultiOmyx-Tumor-Infiltrating-Lymphocyte-Panel.pdf |
| Software and Algorithms | ||
| SAS version 9.4 | SAS Institute | https://www.sas.com/en_us/software/sas9.html |
| MATLAB R2015a | The MathWorks, Inc. | https://www.mathworks.com/products/new_products/release2015a.html |
Highlights.
Oncolytic virus plus anti-PD-1 therapy favorably changed the tumor microenvironment
A high overall response rate of 62% to the combination in metastatic melanoma
A high complete response rate of 33% to the combination in metastatic melanoma
Responses to this combination appeared independent of baseline CD8+ infiltration
ACKNOWLEDGMENTS
The authors thank the following individuals: Richard Scolyer, Robyn Saw, Andrew Spillane, Kenneth Lee, and Omgo Neiwig of the Melanoma Institute Australia for tumor excision biopsies to maximize tissue acquisition; Hajime Hiraragi (Amgen Inc.) for peer review of tumor biopsy analyses, Jessica Stern (Amgen Inc.) for flow cytometry peer review and organization of IHC image galleries for preparation of figures, David Kaufman and Christine Gause from Merck & Co for many hours of designing the study and the statistical plan for both the phase 1b and 3 portions, and with special acknowledgment to Jeffrey Chou, MD, PhD, for his medical and scientific contributions to the design and execution of this study during his tenure as the Amgen medical monitor. A.R. is supported by the NIH grant R35 CA197633. I.P. is supported by NIH grant P20CA016056 to Roswell Park Cancer Institute. G.V.L. is supported by an Australian NHMRC fellowship (APP1119059) and the Medical Foundation K8625, University of Sydney. Lastly, we wish to thank Meghan Johnson, PhD (Complete Healthcare Communications, LLC, West Chester, PA), whose work was funded by Amgen Inc., Emily Plummer, PhD (Amgen Inc.), and Mee Rhan Kim, PhD (Amgen Inc.) for assistance in the preparation of this manuscript. The study was funded by Amgen Inc.
A.R. has received consulting fees from Amgen Inc. and Merck. R.D. has intermittent, project-focused consulting and/or advisory relationships with Novartis, Merck Sharp & Dohme, Bristol-Myers Squibb, Roche, Amgen Inc., Takeda, and Pierre Fabre outside the submitted work. I.P. has received consulting fees and honoraria from Amgen Inc. A.VW. has received research grants from Amgen Inc. and consulting fees from Bristol-Myers Squibb, AstraZeneca, and Amgen Inc. R.H.I.A. has received consulting fees from Amgen Inc., Merck, Takara, and Provectus. O.M. has previously served on advisory boards for Bristol-Myers Squibb, Merck Sharp & Dohme, Roche, Novartis, and Amgen Inc. and has received travel assistance from Bristol-Myers Squibb, Merck Sharp & Dohme, and Amgen Inc. A.J.O. has received research grants from Takeda, Bristol-Myers Squibb, Kyowa, Immunocore, EMD Serono, Amgen Inc., Incyte, Eli Lilly, Advaxis, Mirati, Ignyta, Novartis, Pfizer, and Kura and has received consulting fees from Merck, Takeda, Bristol-Myers Squibb, G1 therapeutics, and Kyowa Hakko Kirin. J.M. has received research grants from Almirall, Amgen Inc., Roche, GlaxoSmithKline, and Bristol-Myers Squibb; has received consulting fees from Almirall, Amgen Inc., Pierre Fabre, and Leo Pharma; and has served on speakers’ bureaus for Amgen Inc., Almirall, Novartis, and Isdin. J.C. has received honoraria from GlaxoSmithKline, Bristol-Myers Squibb, Novartis, Merck Sharp & Dohme, and Amgen Inc.; has served in a nonremunerative position of influence for Austin Health Oncology Information System project Control Group, Olivia Newton-John Cancer & Wellness Centre Executive Committee, and Olivia Newton-John Cancer and Wellness Centre MRI Advisory Committee; has served as a consultant/advisor for Amgen Inc., Bionomics, Bristol-Meyers Squibb, Merck Sharp & Dohme, and Novartis; has received research funding to his institution from GlaxoSmithKline and CSL; has patents/intellectual property at GlaxoSmithKline, Bristol-Meyers Squibb, and Novartis; and is an employee of the Olivia Newton-John Cancer Research Institute. E.F. has received consulting fees from Amgen, Inc. J.M.K. has received research grants from Merck and Prometheus and has received consulting fees from Bristol-Myers Squibb, Green Peptide, Roche, Solaran RX, Checkmate Pharmaceuticals, and Novartis. T.F.G. has received research funding from Amgen Inc., Bristol-Myers Squibb, Merck, Roche/Genentech, Ono, Incyte, and Seattle Genetics; has served on advisory board or as a consultant for Merck, Roche/Genentech, Bayer, Abbvie, Aduro, and Jounce; and owns stock in Jounce. L.C., K.S.G., A.A.A., and J.G. are employees of, and own stock in, Amgen Inc. S.J.D. and M.L. are employees of, and own stock in, Merck. F.S.H. has received research funding from Bristol-Myers Squibb; has received consulting fees from Amgen Inc., Merck, Bristol-Myers Squibb, Novartis, EMD Serono, Celldex, and Genentech; and has received royalties to his institution (per institutional policy) from MICA; related disorders patent pending. G.V.L. has received consulting fees from Amgen Inc., Array, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Roche, and Pierre Fabre.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.08.027.
A video abstract is available at http://dx.doi.org/10.1016/j.cell.2017.08.027#mmc2.
REFERENCES
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. (2015). Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788. [DOI] [PubMed] [Google Scholar]
- Andtbacka RH, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, Amatruda T, Zager JS, Cranmer L, Hsueh E, et al. (2016). Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann. Surg. Oncol 23, 4169–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney J, Collichio F, Andtbacka RH, Puzanov I, Glaspy JA, Milhem M, Hamid O, Cranmer L, Saenger Y, Ross M, et al. (2016). Interim safety and efficacy of a randomized (1:1), open-label phase 2 study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected, stage IIIB- IV melanoma. Ann. Oncol 27, 10.1093/annonc/mdw379.04. [DOI] [Google Scholar]
- Chesney J, Puzanov I, Ross M, Collichio F, Milhem M, Chen L, Kim JH, Garbe C, Hauschild A, and Andtbacka RHI (2017). Primary results from a randomized (1:1), open-label phase II study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected stage IIIB- IV melanoma. J. Clin. Oncol 35, 9509–9509. [Google Scholar]
- Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, Hodi FS, Joshua AM, Kefford R, Hersey P, et al. (2016). Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J. Clin. Oncol 34, 4102–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. USA 110, 11982–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffner B, Iodice GM, and Gasal E (2016). Administration and handling of talimogene laherparepvec: an intralesional oncolytic immunotherapy for melanoma. Oncol. Nurs. Forum 43, 219–226. [DOI] [PubMed] [Google Scholar]
- Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, and Kim-Schulze S (2010). Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol 17, 718–730. [DOI] [PubMed] [Google Scholar]
- Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, et al. (2003). ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 10, 292–303. [DOI] [PubMed] [Google Scholar]
- Moesta AK, Cooke K, Piasecki J, Mitchell P, Rottman JB, Fitzgerald K, Zhan J, Yang B, Le T, Belmontes B, et al. (2017). Local delivery of OncoVEX(mGM-CSF) generates systemic anti-tumor immune responses enhanced by cytotoxic T-lymphocyte-associated protein blockade. Clin. Cancer Res. Published online July 13, 2017. 10.1158/1078-0432.CCR-17-0681. [DOI] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, and Wolchok JD (2015). Immune checkpoint blockade in cancer therapy. J. Clin. Oncol 33, 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J, et al. (2016). Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 34, 2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A (2015). Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 5, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Robert C, Hodi FS, Wolchok JD, Joshua AM, Hwu WJ, Weber JS, Zarour HM, Kefford R, Loboda A, et al. (2015). Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J. Clin. Oncol 33, 3001. [Google Scholar]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, et al. (2016). Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. (2015a). Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med 372, 320–330. [DOI] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. ; KEYNOTE-006 investigators (2015b). Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells . Sci. Transl. Med 5, 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, et al. (2009). Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin. Cancer Res. 15, 7412–7420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The lead contact should be contacted for dataset requests. As noted previously, sharing may be subject to data-sharing agreement.