

Abstract
RNA splicing and spliceosome assembly in eukaryotes occur mainly during transcription. However, co-transcriptional splicing has not yet been explored in plants. Here, we built transcriptomes of nascent chromatin RNAs in Arabidopsis thaliana and showed that nearly all introns undergo co-transcriptional splicing, which occurs with higher efficiency for introns in protein-coding genes than for those in noncoding RNAs. Total intron number and intron position are two predominant features that correlate with co-transcriptional splicing efficiency, and introns with alternative 5′ or 3′ splice sites are less efficiently spliced. Furthermore, we found that mutations in genes encoding trans-acting proteins lead to more introns with increased splicing defects in nascent RNAs than in mature RNAs, and that introns with increased splicing defects in mature RNAs are inefficiently spliced at the co-transcriptional level. Collectively, our results not only uncovered widespread co-transcriptional splicing in Arabidopsis but also identified features that may affect or be affected by co-transcriptional splicing efficiency.
Keywords: chromatin, co-transcriptional splicing, trans-acting proteins, Arabidopsis
INTRODUCTION
RNA splicing, the central processing step in eukaryotic RNA maturation that defines the identity and function of RNAs, plays a pivotal role in regulating development, cell differentiation, and responses to the environment. The recognition of cis-acting elements in pre-mRNA sequences by trans-acting splicing factors is crucial to spliceosome assembly, and this concept formed the foundation for the study of splicing regulation and the deciphering of the splicing code (Will and Luhrmann, 2011; Xiong et al., 2014; Jaganathan et al., 2019). However, the splicing code, which is based on RNA sequence features, accounts only partially for the observed patterns of alternative splicing (Reddy, 2007), indicating that additional splicing regulation factors exist.
Indeed, a growing body of studies have reported evidence for the functions of chromatin structures and histone modifications in alternative splicing, including the coupling of transcription with splicing (Beyer and Osheim, 1988; Bauren and Wieslander, 1994; Bentley, 2014), the exonic enrichment of several epigenetic modifications (Schwartz et al., 2009; Spies et al., 2009; Luco et al., 2011; Li et al., 2015), and regulation of H3K4me3 and H3K36me3 in alternative splicing (Luco et al., 2010, 2011; Guo et al., 2014). The epigenetic regulation of splicing in principle requires splicing to occur on chromatin-associated transcripts. Profiling of biochemically fractionated chromatin-associated nascent transcriptomes in model systems, such as yeast, insects, and mammalian cells, has led to a general agreement that splicing occurs predominantly at the co-transcriptional level (Carrillo Oesterreich et al., 2010; Khodor et al., 2011, 2012; Tilgner et al., 2012). The depletion of intron reads observed from global nuclear run-on sequencing indicates the existence of co-transcriptional splicing in Arabidopsis thaliana (Hetzel et al., 2016); nevertheless, no global studies on co-transcriptional splicing have been reported in plants. Although RNA splicing is a conserved pre-mRNA processing event in eukaryotes, there are many plant-specific features of this process, including a low proportion of genes undergoing alternative splicing, a high level of intron retention and low level of exon skipping (ES), a small number of introns per gene, genes with long exons and short introns, splice site recognition occurring predominantly by intron definition, and the presence of a large number of SR proteins (a family of conserved splicing factors) (Reddy, 2007).
Spliceosome assembly involves numerous trans-acting regulatory protein factors. The MOS4-associated complex (MAC) in Arabidopsis (containing MAC7, MAC3A, MAC3B, PRL1, PRL2, and other factors), also known as the Prp19 complex (Prp19C) in yeast or NineTeen Complex (NTC) in humans, is a conserved complex associated with the spliceosome and is involved in both steps of intron removal (Hogg et al., 2010; Jia et al., 2017). Global splicing defects are observed in pp4r3a-1 and pp4r3a-2, two mutants for a gene encoding the regulatory subunit of one form of the PROTEIN PHOSPHATASE 4 (PP4) complex, which is a conserved serine/threonine-specific phosphoprotein phosphatase (Wang et al., 2019). SNW/Ski-interacting protein (SKIP), a splicing factor and component of the spliceosome, links alternative splicing with both the circadian clock and salt tolerance (Wang et al., 2012; Feng et al., 2015). PRMT5 is a conserved type II protein arginine methyltransferase that methylates core spliceosome proteins to regulate pre-mRNA splicing (Deng et al., 2010; Bezzi et al., 2013; Hernando et al., 2015). The RNA recognition motif (RRM)-containing protein RRM25, which copurifies with intact spliceosomes, is a putative global splicing factor (Zhan et al., 2015; Carlson et al., 2017). Polypyrimidine tract binding protein homologs (PTBs) are RNA-binding proteins that regulate splicing through the nonsense-mediated-decay RNA surveillance pathway (Ruhl et al., 2012). Although global regulation of RNA splicing by these proteins has been revealed through mRNA-seq, which is used to examine the outcome of splicing at the mature mRNA level, knowledge of the connection between the splicing outcome at the mature RNA level and co-transcriptional splicing remains limited.
In this study, we performed directional RNA sequencing (RNA-seq) of chromatin RNAs to profile nascent transcriptomes and uncovered global co-transcriptional splicing in Arabidopsis. We also discovered inefficient co-transcriptional splicing of introns close to transcription end sites and in genes with fewer introns or alternative introns. Utilizing public mature transcriptomes and the nascent transcriptomes from this study, we discovered that co-transcriptional splicing efficiency for mature RNAs, but not nascent RNAs, negatively correlates with the degree of splicing defects caused by mutations in genes encoding trans-acting proteins.
RESULTS
Profiling of Nascent Transcriptomes in Arabidopsis
To capture nascent transcripts, we first isolated the chromatin fraction from Arabidopsis. Nuclei were separated from the cytoplasm using a well-established protocol (Wang et al., 2011). The nuclear pellets were treated with urea and nonionic detergents to remove loosely associated RNAs and proteins without disrupting ternary Pol II complexes (Pandya-Jones and Black, 2009). The chromatin fraction was precipitated, with the supernatant being the nucleoplasmic fraction. We evaluated the purity of the fractions by western blotting using antibodies against GAPDH, SERRATE, and histone H4 (Figure 1A). Histone H4, a chromatin protein, was present only in the chromatin fraction, and GAPDH, a cytoplasmic protein (Zaffagnini et al., 2013), was found only in the cytoplasmic fraction, whereas the nuclear speckle protein SERRATE (Reddy et al., 2012) was highly abundant in the nucleoplasm and was found in trace amounts in the chromatin and cytoplasmic fractions. These data confirmed that the fractionation process was effective. We further evaluated the distribution of unspliced RNAs in these cell fractions. We carried out RT–qPCR to amplify both unspliced and spliced RNAs using primer pairs spanning either exon–exon or exon–intron junctions for four introns and quantified the levels of unspliced RNAs normalized by the levels of spliced RNAs (Supplemental Table 1). We observed a higher level of unspliced RNAs in chromatin when compared with levels in the total cell extract, cytosol, and nucleoplasm (Figure 1B).
Figure 1. Construction of Nascent Transcriptomes in Arabidopsis.
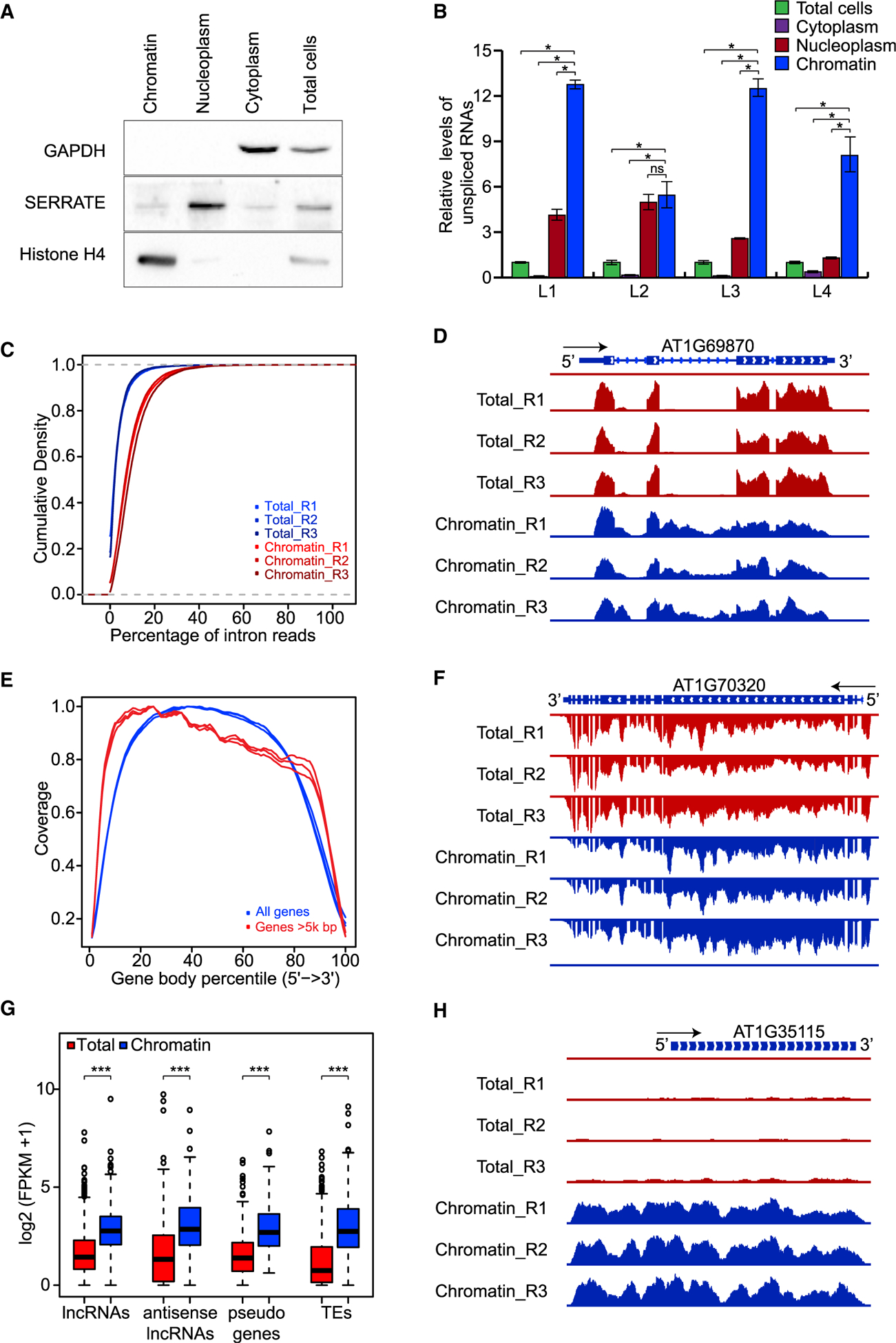
(A) Western blot analysis of GAPDH, SERRATE, and histone H4 in chromatin, nucleoplasm, cytoplasm, and total cells. Experiments were repeated three times independently with similar results.
(B) Relative levels of unspliced RNAs in chromatin, nucleoplasm, cytoplasm, and total cells were quantified using RT–qPCR. Error bars were calculated based on three technical replicates. Experiments were repeated two times independently with similar results.
(C) Cumulative density plot showing global enrichment of intron reads in chromatin RNAs. p = 0 (one-tailed Wilcoxon test).
(D) Genome browser view showing reads for retained introns in chromatin RNAs. The black arrow indicates the direction of transcription; reads from the positive strand of the reference genome were detected.
(E) Coverage of chromatin RNA-seq reads over the gene body for all genes (blue lines) and genes longer than 5000 bp (red lines).
(F) Genome browser view showing a decreasing gradient of reads from 5′ to 3′ for chromatin RNAs. The black arrow indicates the direction of transcription; reads from the negative strand of the reference genome were detected.
(G) Boxplot showing the enrichment of different types of ncRNAs in chromatin: lncRNAs, antisense lncRNAs, and transcripts from pseudogenes and transposable elements (TEs).
(H) Genome browser view showing enrichment of ncRNAs in chromatin RNA-seq data. The black arrow indicates the direction of transcription; reads from the positive strand of the reference genome were detected. Throughout, R1, R2, and R3 denote three biological replicates.
nsp > 0.1, *p < 0.1, ***p < 0.001 (one-tailed Wilcoxon test).
Next, we extracted RNAs from both the chromatin fraction and unfractionated samples and subjected them to rRNA depletion followed by RNA-seq library construction, using a method that preserves the strandedness of RNAs. Three separate experiments gave highly reproducible results (Supplemental Figure 1A). It is expected that, as compared with the total RNAs, nascent RNAs might contain higher levels of unspliced introns and might display decreasing read intensity from 5′ to 3′ for long transcripts (Carrillo Oesterreich et al., 2010). We calculated the percentage of intron reads for every non-overlapping gene and found that the percentage was significantly higher for chromatin RNAs than for total RNAs (Figure 1C and 1D). In addition, we observed a pattern of decreasing read density from 5′ to 3′ for long transcripts, but not for all transcripts (Figure 1E and 1F).
Noncoding RNAs (ncRNAs) act as pivotal regulators of genome structure and gene expression through their interaction with chromatin-modifying enzymes and nucleosome-remodeling factors (Saxena and Carninci, 2011), so it is expected that ncRNAs should be depleted from cytosolic RNAs and enriched in chromatin RNAs. The Araport 11 annotation defines several types of ncRNAs, including long noncoding RNAs (lncRNAs), antisense lncRNAs, pseudogene transcripts, and RNAs from transposable elements. We examined the abundance of these categories of RNAs separately and found that their enrichment in the chromatin fraction was significantly higher than that in total RNAs, consistent with their roles in chromatin regulation (Figure 1G and 1H).
Global Co-transcriptional Splicing in Arabidopsis
The level of retained introns is an indicator of intron splicing efficiency, so we calculated the level of retained introns as the percentage of introns (PI) (the higher the PI value, the lower the intron splicing efficiency) using a web-based pipeline called SQUID. SQUID provides two methods for calculating PI: PI_Junction based on intron-inclusion and intron-skipping reads, and PI_Density based on reads mapping to intronic regions and reads mapping to exonic regions (https://github.com/Xinglab/SQUID) (Figure 2A and Methods). Because calculation of intron-inclusion and intron-skipping reads is skewed for alternative introns and PI_Junction performed better than PI_Density for constitutive introns (Supplemental Figure 2A and 2B and Methods), we calculated PI using PI_Junction for constitutive introns and PI_Density for alternative introns.
Figure 2. Global Co-transcriptional Splicing in Arabidopsis.
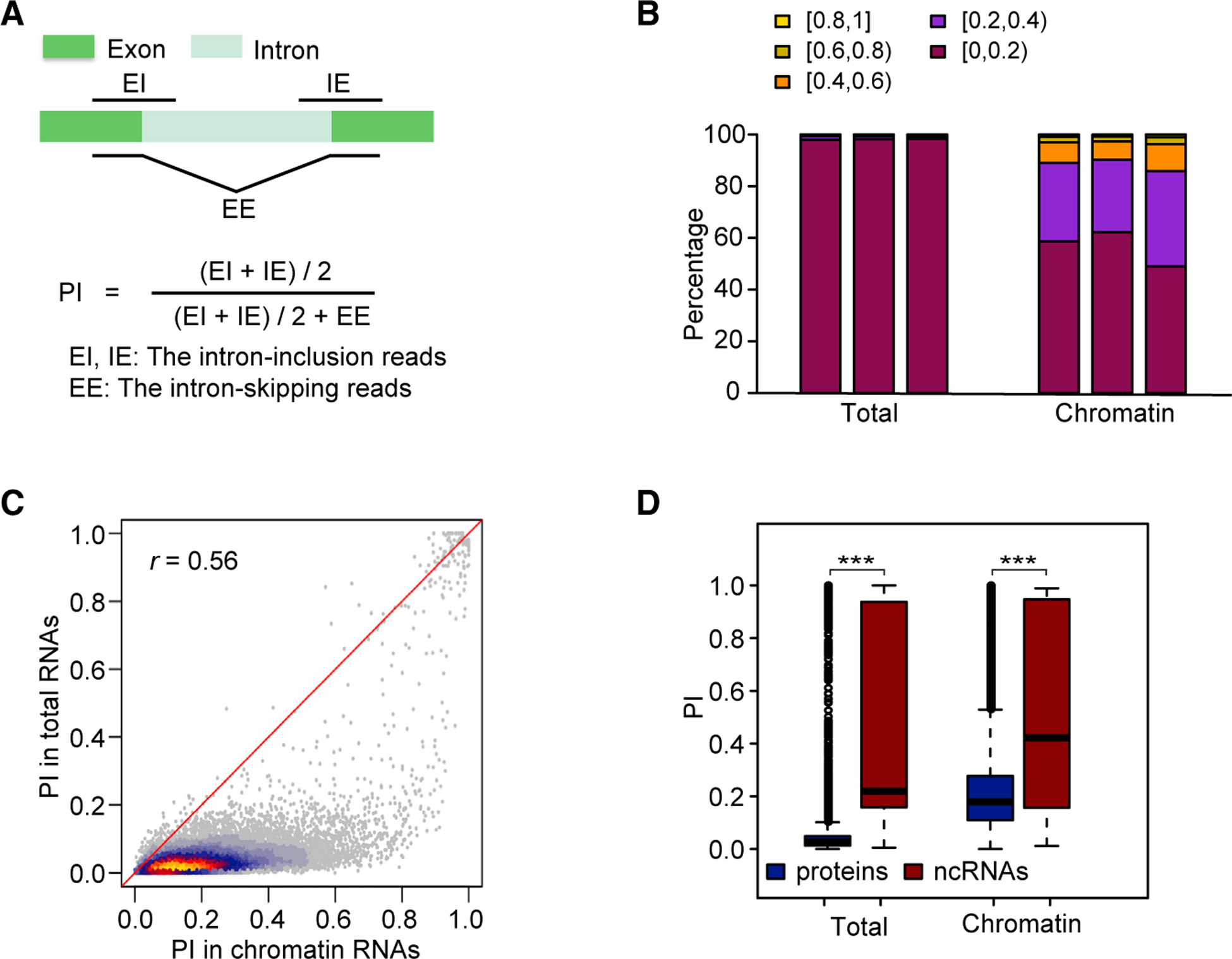
(A) Diagram showing the PI_Junction calculation using intron-inclusion and intron-skipping reads.
(B) Distribution of PI for three replicates (shown as three bars) of RNA-seq data from total RNAs and chromatin RNAs.
(C) Heat scatter plot showing pairwise comparisons of PI for chromatin and total RNAs. Pearson correlation coefficient (r) is at top left.
(D) Boxplot showing PI distribution for introns in protein-coding genes and in ncRNAs.
***p < 0.001 (one-tailed Wilcoxon test).
Since 85% of introns from Araport 11 are constitutive introns, we calculated PI_Junction using data from chromatin RNA-seq and total RNA-seq. A total of 19 737 constitutive introns were interrogated (Supplemental Table 2). As expected, the levels of retained introns in chromatin RNAs were much higher than those in total RNAs (Figure 2B). The facts that the median PI for total RNAs was 0.02 and that PI was lower than 0.2 for most introns in total RNAs are indicative of relatively complete intron removal in total RNAs. By contrast, the facts that the median PI for chromatin RNAs was 0.18, and that PI was lower than 0.8 for most introns in chromatin RNAs, indicate that there is genome-wide co-transcriptional splicing in Arabidopsis, which matches the patterns previously reported in yeast (Carrillo Oesterreich et al., 2010), fly (Khodor et al., 2011), mouse (Bhatt et al., 2012), and human cells (Tilgner et al., 2012). The positive correlation between PI values for chromatin and total RNAs suggests that the efficiency of co-transcriptional splicing may positively impact the final splicing outcome (Figure 2C). Inefficient and delayed intron splicing of lncRNAs has been reported in human cells (Tilgner et al., 2012). In our study, higher PI values for both chromatin and total RNAs was also observed for introns from ncRNAs, which is indicative of similar inefficient splicing of ncRNAs in Arabidopsis (Figure 2D).
Inefficient Splicing of Alternative Introns with Alternative 5′ or 3′ Splice Sites
The splicing of constitutive introns is more likely to be co-transcriptional than that of alternative introns (Brugiolo et al., 2013). To examine the effects of alternative splicing in Arabidopsis, we used PI_Density to calculate the splicing efficiency for alternative and constitutive introns. To eliminate the bias caused by overlapping exons, we excluded alternative introns that overlapped with exons from the analysis (Figure 3A and Supplemental Table 3). In Arabidopsis, there are three major types of alternative introns: introns with alternative 5′ splicing sites (A5SS), introns with alternative 3′ splicing sites (A3SS), and introns with ES (Figure 3A and 3B). The proportion of introns with ES is much lower than that of introns with A5SS or A3SS (Keren et al., 2010). Consistent with previous reports (Brugiolo et al., 2013), the level of retained introns was higher for alternative introns than for constitutive introns in both chromatin RNAs and total RNAs (Figure 3C). We then calculated PI for each type of alternative intron and observed higher PI for introns with A5SS and A3SS than for constitutive introns in both chromatin and total RNAs, while for introns with ES, we observed significantly higher PI only for those in total RNAs (Figure 3C). To eliminate potential effects caused by differences between sets of genes, we analyzed constitutive introns and alternative introns residing in the same set of genes and confirmed that the pattern was similar to that seen when all introns were analyzed together (Supplemental Figure 3). These results indicate that introns with alternative 5′ or 3′ splice sites are less efficiently spliced than constitutive introns.
Figure 3. Inefficient Splicing of Alternative Introns.
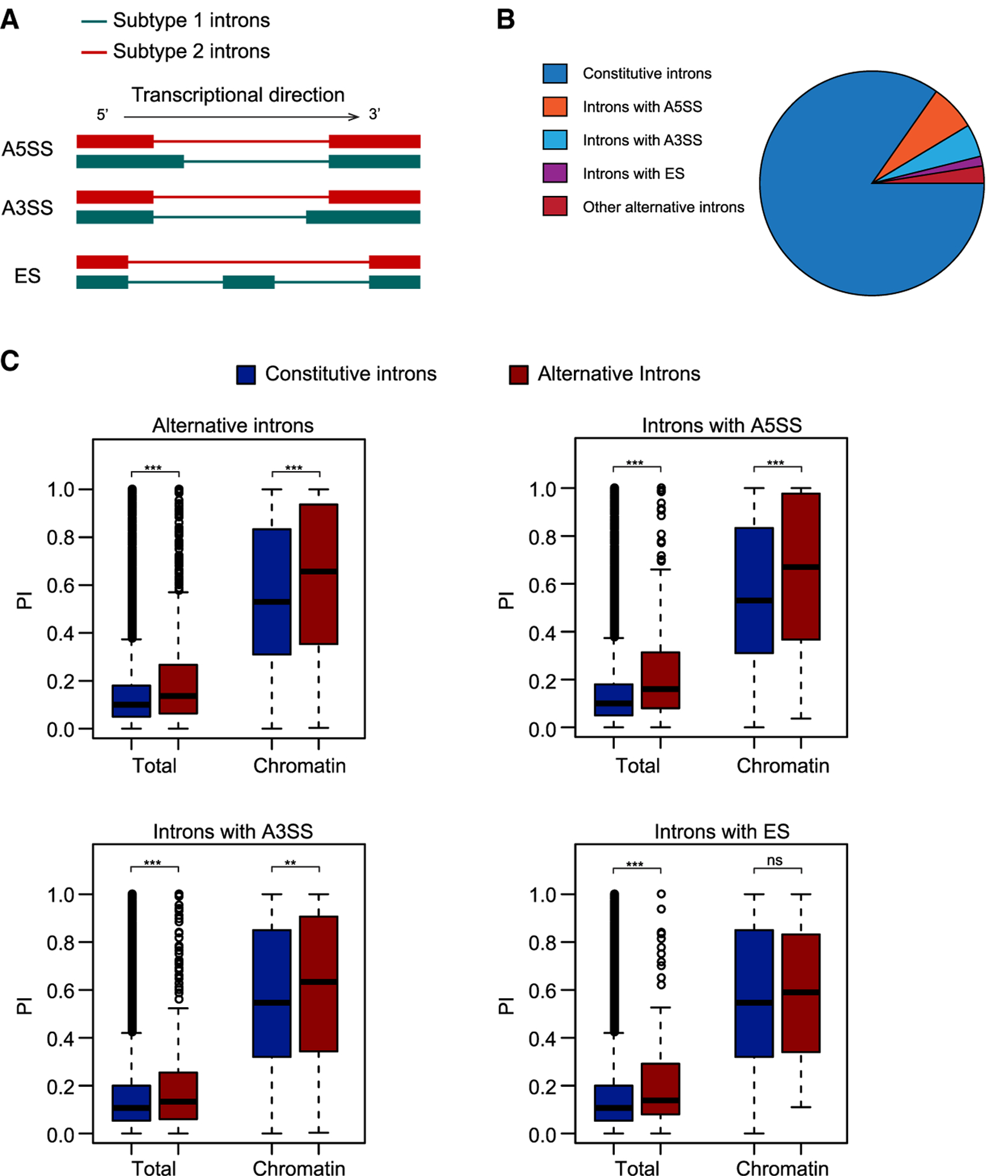
(A) Diagram showing introns undergoing three major types of alternative splicing: A5SS, A3SS, and ES. Subtype 1 introns are those that do not overlap with exons and were therefore used for later analysis. Subtype 2 introns overlap with exons and were discarded from later analysis to eliminate bias.
(B) Pie chart showing the composition of constitutive and alternative introns in Arabidopsis.
(C) Boxplots showing PI distributions for constitutive and alternative introns. The plots show all alternative introns as well as introns with A5SS, A3SS, and ES separately.
nsp > 0.1, **p < 0.01, ***p < 0.001 (one-tailed Wilcoxon test).
Predominant Roles of Total Intron Number and Intron Position in Co-transcriptional Splicing
Features of introns, flanking exons, and intron-harboring genes may affect splicing, so we calculated Pearson correlation coefficients between PI and these features. Among all of the features tested, total intron number and gene length of intron-harboring genes showed the highest correlations with chromatin RNA PI values (Figure 4A and Supplemental Figure 4A–4C). Since total intron number correlates tightly with gene length (Supplemental Figure 4D), we attempted to differentiate the influence of gene length and total intron number on co-transcriptional splicing. We divided all tested introns into 10 deciles according to increasing chromatin RNA PI value (grouping 1), increasing total intron number (grouping 2), and increasing gene length (grouping 3) (Supplemental Figure 5). The correlations between total intron number and gene length and chromatin RNA PI, and total intron number and chromatin RNA PI were calculated for each decile in groupings 1, 2, and 3, respectively. The correlations between total intron number and gene length were not affected by chromatin RNA PI value (grouping 1), and the correlations between total intron number and chromatin RNA PI were not affected by gene length (grouping 3), while the correlations between gene length and chromatin RNA PI were diminished when total intron number was within a narrow range (grouping 2) (Figure 4B). This suggested that total intron number, but not gene length, plays a major role in determining co-transcriptional splicing efficiency.
Figure 4. Predominant Roles of Total Intron Number and Intron Position in Co-transcriptional Splicing.
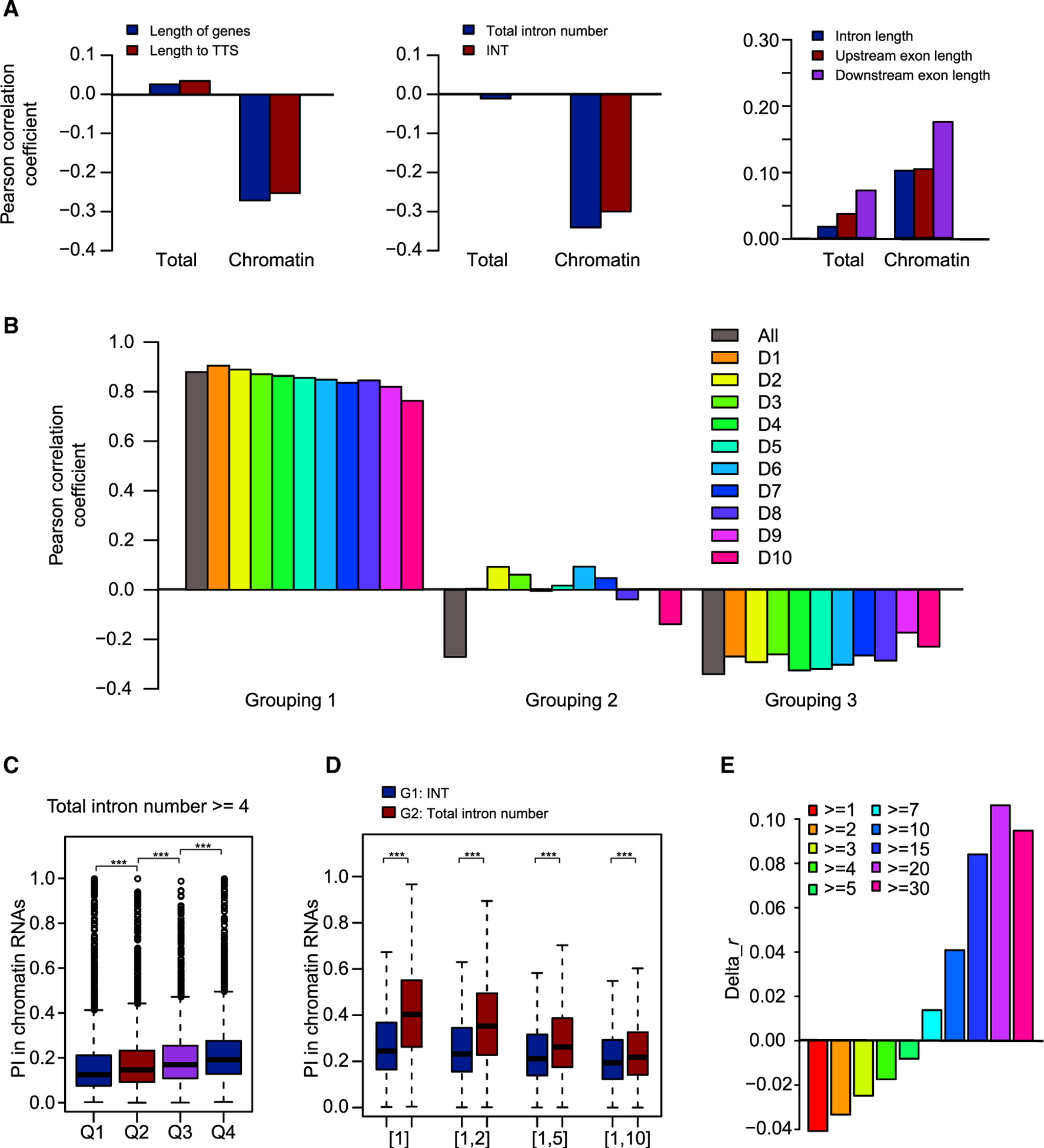
(A) Correlations between PI values for total and chromatin RNAs with features of introns, flanking exons, and intron-harboring genes.
(B) Pearson correlation coefficients for three groupings. In grouping 1, introns were divided into 10 deciles according to increasing chromatin RNA PI value. Correlations between total intron number and gene length were calculated for all introns and for each of the 10 deciles. In grouping 2, introns were divided into 10 deciles according to increasing total intron number. Correlations between chromatin RNA PI value and gene length were calculated for all introns and for each of the 10 deciles. In grouping 3, introns were divided into 10 deciles according to increasing gene length. Correlations between chromatin RNA PI value and total intron number were calculated for all introns and for each of the 10 deciles. All, all introns; D1–D10, the 10 deciles.
(C) All introns in genes with total intron number ≥4 were divided into four quartiles, Q1–Q4, with decreasing INT. The distribution of PI values for chromatin RNAs is shown in the boxplot.
(D) Chromatin RNA PI value/123 is shown for introns with INT (G1) or total intron number (TIN) (G2) values within certain ranges: [1], TIN/INT = 1; [1,2], 1 ≤ TIN/INT ≤2; [1,5], 1 ≤ TIN/INT ≤5; [1,10], 1 ≤ TIN/INT ≤10.
(E) Bar plot showing the difference in Pearson correlation coefficients (Pearson’s r). Delta_r was calculated as the Pearson’s r between INT and chromatin RNA PI value minus Pearson’s r between total intron number and chromatin RNA PI value.
***p < 0.001 (one-tailed Wilcoxon test).
Co-transcriptional splicing efficiency is reported to decrease from 5′ to 3′ along genes: i.e., introns close to transcription start sites (TSSs) are more efficiently spliced than introns close to TTSs (Bentley, 2014; Tilgner et al., 2012). We also found that PI for chromatin RNAs negatively correlates the distance to TTSs measured either in intron number or length of nucleotide (Figure 4A). Since INT also correlates with total intron number, we further differentiated the roles of total intron number and INT. First, we eliminated the effects of total intron number to examine the role of INT. When we examined the chromatin RNA PI value for introns in genes with two or three introns, we found that PI increased from the first intron to the last intron (Supplemental Figure 4E and 4F). We then divided introns in genes with more than three introns into four quartiles, Q1–Q4, with decreasing INT, and we found that chromatin RNA PI increased from Q1 to Q4 (Figure 4C). The decreasing co-transcriptional splicing efficiency along genes validated the role of INT, which is consistent with the previously proposed “first come, first served model” of splicing (Bentley, 2014). Next, we eliminated the effects of INT to examine the role of total intron number. We selected two groups of introns by restricting INT (in G1) or total intron number (in G2) to a certain range. Since INT is also limited by total intron number, G2 is a subset of G1 with lower total intron number. If total intron number plays a role in co-transcriptional splicing efficiency, chromatin RNA PI values should be higher for G2 than for G1. As expected, chromatin RNA values were higher for G2 than G1 (Figure 4D), which supports a role of total intron number. Since both total intron number and INT correlate with chromatin RNA PI value, we further compared their effects by calculating the Pearson correlation coefficients for groups of introns with different minimal total intron numbers. The difference between the Pearson correlation coefficients between INT and chromatin RNA PI and between total intron number and chromatin RNA PI increased with increasing total intron number (Figure 4E). The results suggest that total intron number plays a dominant role for introns in genes with a small total intron number, whereas intron position relative to TTSs plays a dominant role for introns in genes with a large total intron number.
In addition, we observed positive correlations between PI for chromatin RNAs and the lengths of introns and flanking exons. Since the length of introns and flanking exons is negatively correlated with total intron number (Supplemental Figure 4G), we carried out a test similar to the one done for gene length to differentiate their roles: all introns were divided on the basis of chromatin RNA PI values (grouping1), total intron number (grouping 2), and length of introns or flanking exons (grouping 3). Similar to what we found for gene length, we did not observe correlations between chromatin RNA PI value and length of introns and flanking exons (grouping 2) (Supplemental Figure 6), suggesting that it is total intron number, and not the length of introns and flanking exons, that plays a major role in determining co-transcriptional splicing efficiency. In addition, since ncRNAs tend to have fewer introns (Supplemental Figure 7A), we tested whether the higher PI for ncRNAs is due to a low total intron number. We compared PI for ncRNAs and genes with equal ranges of total intron number, and we still observed higher PI for ncRNAs in both total and chromatin RNAs (Supplemental Figure 7B and 7C), which suggests that other factors lead to inefficiency of ncRNA intron splicing.
Moreover, we observed a weak negative correlation between splice junction strength and PI values for total and chromatin RNAs, which is consistent with the fact that splice junction strength, which is based on maximum entropy, is commonly used for predicting 5′ and 3′ splice sites (Yeo and Burge, 2004) (Supplemental Figure 4A). The weak positive correlations between PI values for total and chromatin RNAs and intron GC content, and weak negative correlations between PI values for total and chromatin RNAs and exon GC content, may be explained by the influence of GC content on pre-mRNA secondary structure (Supplemental Figure 4A) (Zhang et al., 2011). We also observed weak negative correlations both between gene expression level and PI values for chromatin RNAs and between gene expression level and PI values for total RNAs (Supplemental Figure 4A).
Chromatin RNA PI Correlates with the Degree of Splicing Defects Caused by Mutations in Genes Encoding trans-Acting Proteins
Spliceosome assembly on pre-mRNAs depends on the recognition of cis-elements in the pre-mRNAs by trans-acting proteins. Here, we sought to explore the connection between co-transcriptional splicing and the action of these trans-acting proteins. As a first step, we identified introns that are regulated by trans-acting proteins by analyzing splicing defects in the corresponding mutants using public mRNA-seq data. We selected four mutants for which public mRNA-seq datasets were available (Supplemental Table 4): mac3a mac3b (MAC3A and MAC3B are two homologous MAC proteins with E3 ligase activity in vitro) (Jia et al., 2017), prl1 prl2 (PRL1 and PRL2 are two homologous nuclear WD-40 proteins in the MAC) (Jia et al., 2017), pp4r3a (PP4R3A is a conserved serine/threonine-specific phosphoprotein phosphatase) (Wang et al., 2019), and skip (SKIP is a splicing factor and component of the spliceosome) (Wang et al., 2012). Global splicing defects were found in these mutants, and differentially spliced introns (DSIs) were identified using SQUID (Supplemental Figure 8). Since these four trans-acting proteins with transcriptome data are splicing activators, we only included DSIs with increased PI in this study. To quantify the degree of splicing defects at these DSIs, we calculated delta_PI (PI in mutants minus PI in wild type), and used it as an indicator of splicing defects: the higher the delta_PI, the higher the degree of splicing defects.
Having identified the DSIs regulated by trans-acting proteins, the next step was to examine the PI for these DSIs in the nascent transcriptomes we generated in this study. We found that these DSIs had a significantly higher chromatin RNA PI values compared with all introns (Figure 5A), suggesting that the introns regulated by these trans-acting proteins tend to have lower co-transcriptional splicing efficiencies. Consistent with this, we found that these DSIs had both lower total intron number and lower INT compared with all introns (Figure 5B and 5C). Having shown that the DSIs tend to be poorly spliced at the co-transcriptional level, we next sought to determine whether the splicing defects in DSIs in the mutants correlated with the efficiency of co-transcriptional splicing. We performed Pearson correlation analysis between delta_PI and chromatin RNA PI value, total intron number, and INT. We discovered that chromatin RNA PI value positively correlates with delta_PI (Figure 5D), whereas such correlations were not found for total intron number or INT. This suggests that chromatin RNA PI is the predominant factor that correlates with the splicing regulation by trans-acting proteins. We also extended our analysis to DSIs regulated by more trans-acting proteins and DSIs responsive to environmental stress using other public datasets (Supplemental Table 4), and found that these DSIs always had higher chromatin RNA PI values, but that they were not necessarily from genes with a lower total intron number and INTs (Supplemental Figure 9), which is consistent with the dominant role of chromatin RNA PI correlated with splicing defects caused by mutations in genes encoding trans-acting proteins.
Figure 5. Introns Regulated by trans-acting Proteins in Mature RNAs Have Higher Chromatin RNA PI values.
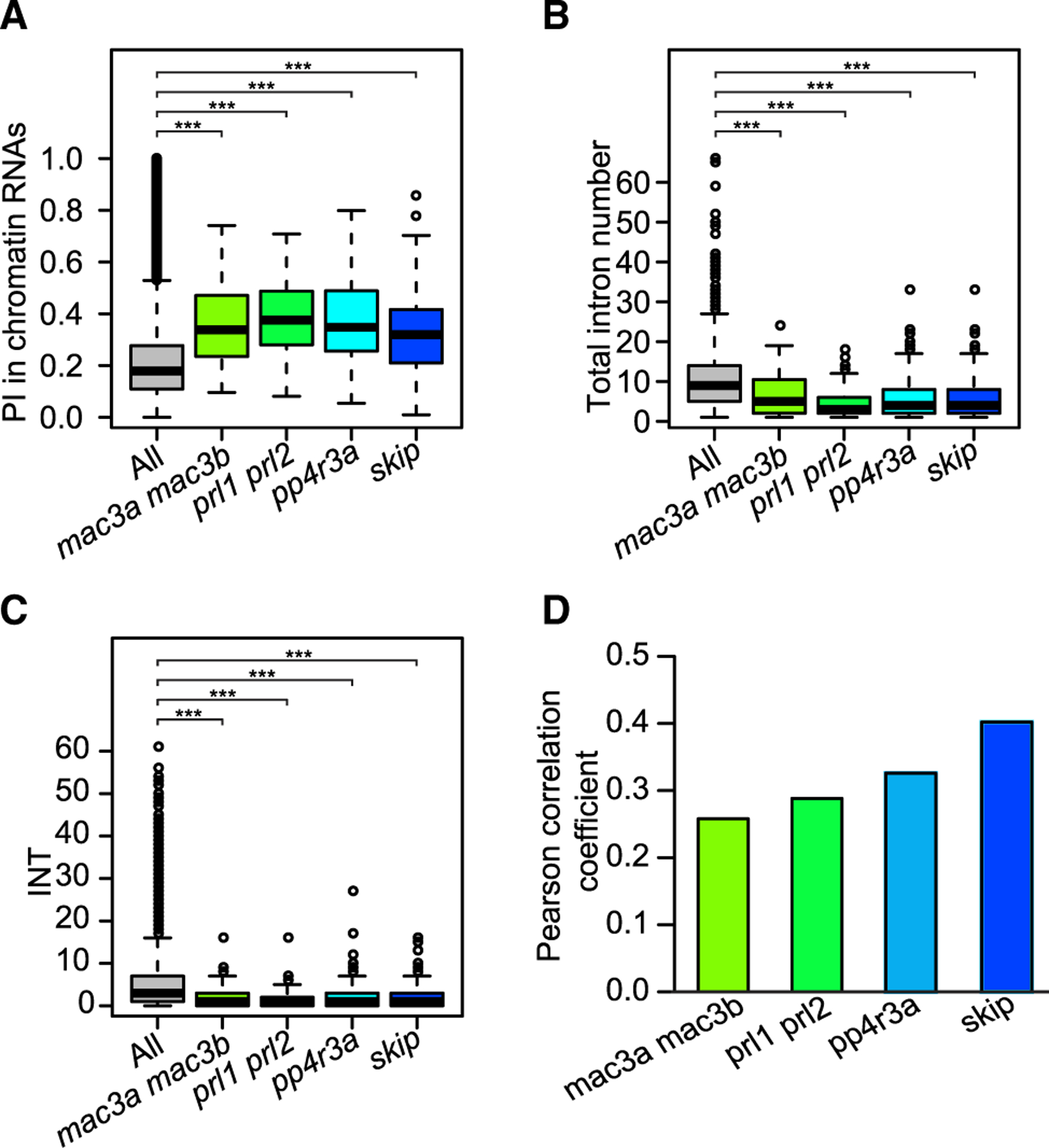
(A–C) Boxplot showing chromatin RNA PI, total intron number, and INT values for DSIs in mac3a mac3b, prl1 prl2, pp4r3a, and skip.
(D) Bar plot showing Pearson correlation coefficients between chromatin RNA PI values and delta_PI for constitutive introns. Delta_PI was calculated as PI_Junction in mutants minus PI_Junction in wild type.
*p < 0.1, ***p < 0.001 (one-tailed Wilcoxon test).
One potential problem in the analyses above is that the public RNA-seq datasets were from different tissues or plants grown under different conditions as compared with the samples used in our chromatin RNA-seq. To evaluate whether RNA-seq data from different samples gives different intron splicing efficiencies, we determined the splicing efficiencies using wild-type transcriptomes from different datasets. We found that the level of splicing of constitutive introns was comparable between wild-type transcriptomes despite differences in growth conditions or tissue types or stages (Supplemental Figure 10 and Supplemental Table 4). Thus, we focused only on constitutive introns in the identification of DSIs in the analyses above.
Mutations in MAC Lead to Different Splicing Defects in Nascent Versus Mature RNAs
We further explored the regulation of splicing by trans-acting proteins by constructing libraries with two biological replicates of rRNA-depleted nascent chromatin RNAs and poly-A mature RNAs from two MAC double mutants: mac3a mac3b and prl1 prl2 (Supplemental Figure 1B). We found that levels of unspliced introns in the mutants were higher in both nascent and mature RNAs, and that the splicing defects were more severe in nascent RNAs than in mature RNAs (Figure 6A and Supplemental Table 5). To further examine the different splicing defects in nascent and mature RNAs, we identified DSIs regulated by MAC3A and MAC3B or PRL1 and PRL2 from both nascent and mature RNAs. When we quantified the splicing defects for these DSIs, we found that the degree of defects was similar in nascent and mature RNAs for DSIs identified from mature RNAs, but was significantly lower in mature RNAs than in nascent RNAs for DSIs identified from nascent RNAs (Figure 6B and 6C). When we examined co-transcriptional splicing efficiencies in the wild type, we observed higher chromatin RNA PI values for DSIs identified from mature RNAs but not for DSIs identified from nascent RNAs (Figure 6D). Our results indicate that defects in splicing of nascent RNAs in mac3a mac3b and prl1 prl2 can be rescued in mature RNAs when introns have regular chromatin RNA PI values, but not when introns have higher chromatin RNA PI values.
Figure 6. Different Splicing Defects in Nascent and Mature RNAs Caused by MAC Mutation.
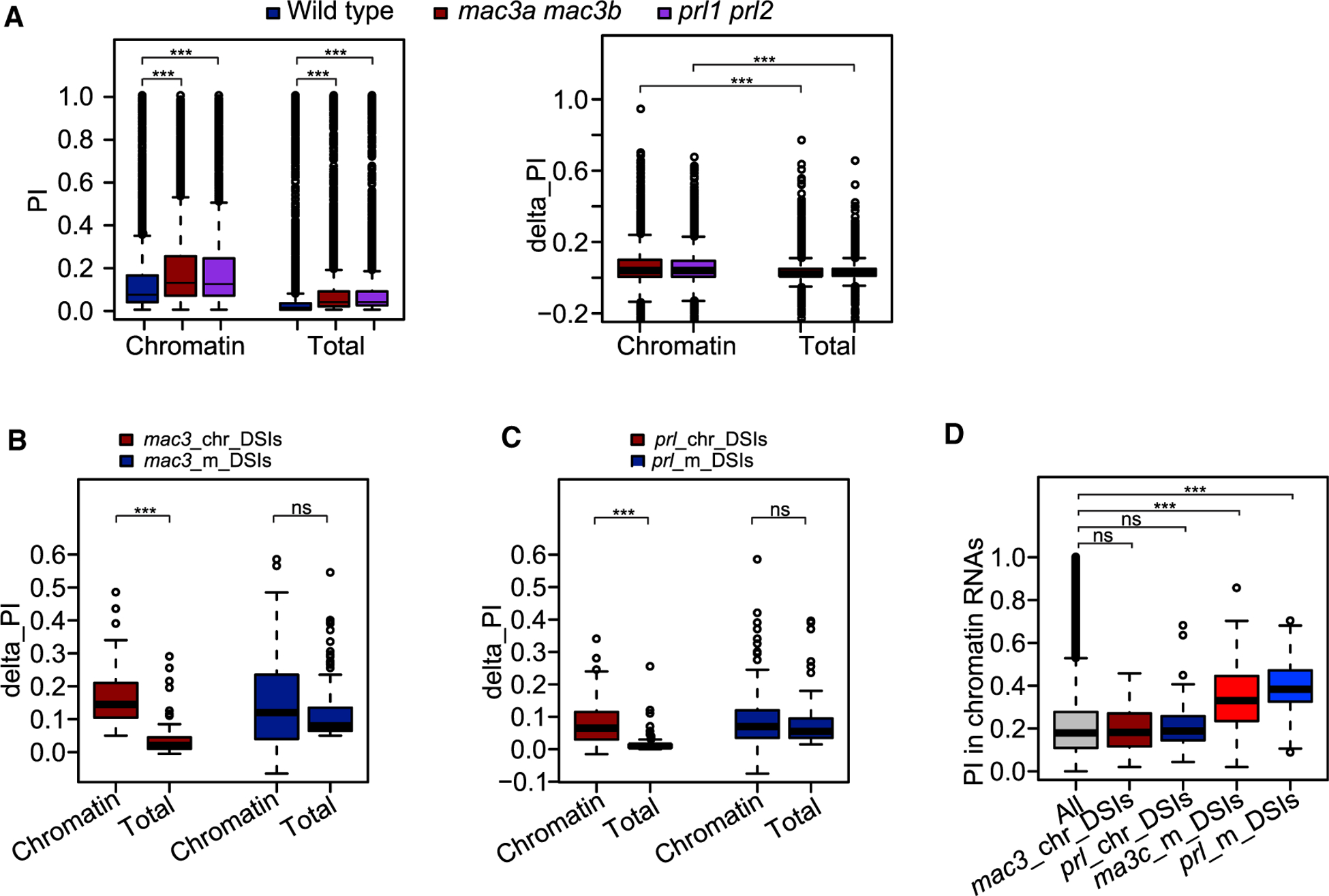
(A) PI distribution in wild type, mac3a mac3b, and prl1 prl2.
(B–D) mac3_chr_DSIs denotes DSIs identified in mac3a mac3b nascent RNAs, mac3_m_DSIs denotes DSIs identified in mac3a mac3b mature RNAs, prl_chr_DSIs denotes DSIs identified in prl1 prl2 nascent RNAs, and prl_m_DSIs denotes DSIs identified in prl1 prl2 mature RNAs. Delta_PI was calculated as PI_Junction in each mutant minus PI_Junction in wild type. (B and C) Boxplot showing delta_PI for these DSIs. (D) Boxplot showing chromatin RNA PI values for these DSIs.
nsp > 0.1, ***p < 0.001 (one-tailed Wilcoxon test).
DISCUSSION
In this study, we found that, for nearly all introns in Arabidopsis thaliana, splicing begins during transcription, and this occurs with higher efficiency for introns in protein-coding genes than for those in ncRNAs. Many intron-surrounding features are reported to correlate with co-transcriptional splicing efficiency, including intron position, intron length, gene length, and alternative splicing (Khodor et al., 2011, 2012; Tilgner et al., 2012). Here, we conducted a thorough investigation of the impact of these features on co-transcriptional splicing in Arabidopsis. Intron position is a common and striking feature observed in Drosophila melanogaster, mice, and human cells, as well as in Arabidopsis: co-transcriptional splicing efficiency decreases with decreasing distance from the TTS. Intron length negatively correlates with co-transcriptional splicing efficiency in Drosophila and human cells, and gene length negatively correlates with co-transcriptional splicing efficiency in Drosophila and mice. In Arabidopsis, we initially observed a negative correlation between gene length and PI values for chromatin RNAs and a positive correlation between PI values for chromatin RNAs and length of introns and flanking exons, but we found that the correlation disappeared once the effects of total intron number were eliminated. Single-intron genes are inefficiently spliced in Drosophila and mice. In Arabidopsis, we found that total intron number is another striking feature positively correlated with co-transcriptional splicing efficiency beside intron position. Total intron number is more important for introns in genes with a small total intron number, whereas intron position is more important for introns in genes with a large total intron number. Given that the cap-binding complex functions in the recognition of the first exon, that there is reciprocal interaction between 3′-end processing and last-intron splicing, and that spliceosome assembly involves stages of snRNP assembly and release (Herzel et al., 2017), it is expected that spliceosome assembly on introns in transcripts with a small total intron number will take longer. Since there are on average four introns per gene in Arabidopsis versus eight introns per gene in humans (Reddy, 2007), the role of total intron number in Arabidopsis is more striking. Inefficiently spliced ncRNAs and alternative introns are two conserved features in both plants and mammals (Tilgner et al., 2012).
Features of pre-mRNA sequences lead to different efficiencies of co-transcriptional splicing; however, most introns that are not spliced during transcription are spliced later, after the completion of transcription, either while transcripts are still attached to chromatin or after they are released to nuclear speckles (Bhatt et al., 2012; Girard et al., 2012). An important question thus arises: does co-transcriptional splicing efficiency matter? To address this point, we examined the role of PI values for chromatin RNAs in the context of the regulation of splicing by trans-acting proteins. Transcriptome profiling of mature RNAs is an approach that is widely used to identify the regulatory role of trans-acting proteins. We observed higher chromatin RNA PI values for DSIs identified from the transcriptomes of eight mutants with trans-acting protein mutations as well as from four abiotic stress transcriptomes, suggesting that introns with poor co-transcriptional splicing tend to be targets of splicing regulation. We further profiled the transcriptomes of nascent and mature RNAs from plants with MAC protein mutations. For introns with regular co-transcriptional splicing efficiency, the splicing defects in nascent RNAs are rescued in mature RNAs, probably through post-transcriptional splicing or post-transcriptional RNA removal through the RNA surveillance pathway (Girard et al., 2012; Drechsel et al., 2013). Interestingly, for introns with low co-transcriptional splicing efficiency, the splicing defects remain in mature RNAs in the MAC mutants. These results lead us to propose the following model: RNAs recruit trans-acting proteins to regulate splicing co-transcriptionally; but only the RNAs with low co-transcriptional splicing efficiency (i.e., with high chromatin RNA PI values) require the trans-acting proteins for the final outcome of mRNA splicing. Thus, the regulatory role of trans-acting proteins cannot be detected in RNA-seq data for a portion of transcripts with binding sites for those proteins (Meyer et al., 2017). Our findings thus provide novel insights into the substrate repertoire of trans-acting splicing regulators.
METHODS
Plant Materials and Growth Conditions
All tissues used in the study were from unopened flower buds, and all Arabidopsis thaliana strains used were in the Columbia (Col-0) ecotype. Plants were cultivated at 22°C with a 16-h light/8-h dark cycle. The mac3a mac3b and prl1 prl2 mutants in the Col-0 ecotype were described previously (Monaghan et al., 2009; Ji et al., 2015).
Chromatin–Nucleoplasmic–Cytoplasmic Fractionation
The cytosolic and nuclear fractions were isolated following a published protocol with β-mercaptoethanol replaced by DTT (Wang et al., 2011). The chromatin fraction was further precipitated with urea and nonionic detergents as described by Pandya-Jones and Black (2009). The nucleus pellets were suspended in 500 μl of glycerol buffer (20 mM Tris–Cl [pH 7.9], 75 mM NaCl, 0.5 mM EDTA, 0.85 mM DTT, 50% glycerol, 0.125 mM PMSF, 10 mM β-mercaptoethanol, and 160 unit/ml RNase inhibitor) and carefully overlaid on top of 500 μl of urea buffer (10 mM HEPES [pH 7.6], 1 mM DTT, 7.5 mM MgCl2, 0.2 mM EDTA, 0.3 M NaCl, 1 M urea, 1% NP-40, 0.5 mM PMSF, proteinase inhibitor cocktail, 10 mM β-mercaptoethanol, and 160 unit/ml RNase inhibitor). The tube was gently vortexed two times for 2 s, incubated on ice for 5 min, and then centrifuged for 5 min at 4°C at 13 000 rpm. The nucleoplasm supernatant and the chromatin pellets were retained for later experiments.
Antibodies Used in the Study
SERRATE: Agrisera AS09 532A; histone H4: Millipore, 04–858; GAPDH: Santa Cruz, SC365062.
RT–qPCR
RNAs were extracted from total cells as well as cell fractions (cytosol, nucleoplasm, and chromatin) using the TRI Reagent (MRC, TR118) and treated with DNase I (Roche, 04716728001). cDNAs were synthesized using a random hexamer (Invitrogen, N8080127) with SuperScript III Reverse Transcriptase (Invitrogen, 18080085). RT–qPCR was performed with three technical replicates using iTaq Supermix (Bio-Rad, 1725120) on the Bio-Rad CFX96 Real-Time PCR System. The information for primer sequences and internal controls is listed in Supplemental Table 1.
Construction and Processing of RNA-Seq Libraries
RNAs were extracted from total tissues and chromatin pellets using TRIzol. DNA-free RNAs (10 μg) were either subjected to rRNA removal using a Ribominus Kit (Thermo Fisher Scientific, A1083808) for RNA-seq libraries or subjected to mRNA purification using the mRNA magnetic isolation module (NEB, E7490) for mRNA-seq libraries. These RNAs were used to construct NGS libraries using the NEBNext Ultra Directional RNA Library Prep Kit (NEB, E7420) and sequenced in 150-nt paired-end mode using Illumina HiSeq X Ten. The sequence data were deposited in the NCBI database under accession number GEO: GSE126064.
RNA-seq reads were collapsed into nonredundant reads and mapped back to the TAIR10 reference genome with Araport 11 annotation using STAR aligner with a maximum of eight mismatches per paired-end read (Dobin et al., 2013). The gene expression level for chromatin and total RNAs was quantified with Cuffdiff (Trapnell et al., 2013). The intronic and exonic reads were calculated as described previously (Gaidatzis et al., 2015), and the percentage of intron reads was calculated as intronic reads divided by the sum of intronic and exonic reads for all non-overlapping genes.
Definition of Constitutive Introns and Alternative Introns
In this study, constitutive introns were defined as introns that do not overlap with other splicing events, and all other introns were defined as alternative introns (Supplemental Figure 2A). Note that retained introns were not defined as alternative introns in this study, since most of the chromatin RNAs have retained intron reads and the PI_Junction calculation is not confounded by retained intron reads.
Quantification of Intron Retention
Araport 11-annotated introns with a minimum length of 50 nt were obtained using an in-house python script. The levels of retained introns were calculated as the PI using a web-based algorithm, SQUID (https://github.com/Xinglab/SQUID). In brief, there are two ways of calculating PI: PI_Junction is calculated as intron-inclusion reads divided by the sum of intron-inclusion reads and intron-skipping reads (Figure 2A), and PI_Density is calculated as observed intron reads (normalized intronic reads) divided by expected intron reads (normalized exonic reads). For alternative introns, both intron-inclusion reads and intron-skipping reads can be skewed by reads from alternative splicing transcripts, while for alternative introns that do not overlap with other exons, both the intronic and exonic reads are not skewed (Supplemental Figure 2A). Theoretically, both PI_Junction and PI_Density should perform well on constitutive introns and PI_Density should perform better on alternative introns that do not overlap with other exons. To evaluate their performance at constitutive introns, both PI_Junction and PI_Density were calculated for RNA-seq data simulated using Flux Simulator with a known true PI (Griebel et al., 2012). Only introns with enough reads were kept, that is the sum of intron-inclusion reads and intron-skipping reads was no less than 20 for PI_Junction calculation and introns in genes had a minimum of 10 fragments per kilobase of transcript per million mapped reads for PI_Density calculation. The results suggested that PI_Junction performed better than PI_Density on constitutive introns (Supplemental Figure 2B), so in this study we used PI_Junction for analysis of constitutive introns and PI_Density for analysis of alternative introns.
Calculation of DSIs
DSIs were obtained using a very stringent method in SQUID using the cutoff: combined_FDR < 0.1, Diff_PI_Junction > 0.05, Diff_PI_Density > 0.05.
Calculation of Splice Junction Strength
The splice junction strength was assessed with MaxEntScan based on the 9-mer sequences at 5′ splice sites and 23-mer sequences at 3′ splice sites (Yeo and Burge, 2004).
Parameters for Boxplots Used in this Study
The horizontal line in the box represents the median value, and the bottom and top of the box represent the lower (Q1) and upper quartiles (Q3), respectively. The upper whisker is min(max(x), Q3 + 1.5 3 IQR), and the lower whisker is max(min(x), Q1 − 1.5 3 IQR). IQR (interquartile range) = Q3 – Q1. Black dots located outsides the whiskers are outliers.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr Yi Xing for comments on PI calculation and Dr. Wu Zhe for communication and coordination and the authors declare no conflict of interest.
FUNDING
This work was supported by the Fundamental Research Funds for the Central Universities, China and National Science Foundation of China (91740202). Y.W. was supported by a fellowship from Shenzhen University.
Footnotes
ACCESSION NUMBERS
The sequencing data and processed files are available at the Gene Expression Omnibus under accession number GEO: GSE126064. Reviewer links to deposited GEO data with token ezipukgmldkzhqf https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126064.
SUPPLEMENTAL INFORMATION
Supplemental Information is available at Molecular Plant Online.
REFERENCES
- Bauren G, and Wieslander L (1994). Splicing of Balbiani ring 1 gene pre-mRNA occurs simultaneously with transcription. Cell 76:183–192. [DOI] [PubMed] [Google Scholar]
- Bentley DL (2014). Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet 15:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer AL, and Osheim YN (1988). Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev 2:754–765. [DOI] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, and Guccione E (2013). Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27:1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, and Smale ST (2012). Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiolo M, Herzel L, and Neugebauer KM (2013). Counting on co-transcriptional splicing. F1000Prime Rep. 5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SM, Soulette CM, Yang Z, Elias JE, Brooks AN, and Gozani O (2017). RBM25 is a global splicing factor promoting inclusion of alternatively spliced exons and is itself regulated by lysine mono-methylation. J. Biol. Chem 292:13381–13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo Oesterreich F, Preibisch S, and Neugebauer KM (2010). Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell 40:571–581. [DOI] [PubMed] [Google Scholar]
- Deng X, Gu L, Liu C, Lu T, Lu F, Lu Z, Cui P, Pei Y, Wang B, Hu S, et al. (2010). Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc. Natl. Acad. Sci. U S A 107:19114–19119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel G, Kahles A, Kesarwani AK, Stauffer E, Behr J, Drewe P, Ratsch G, and Wachter A (2013). Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell 25:3726–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Li J, Gao Z, Lu Y, Yu J, Zheng Q, Yan S, Zhang W, He H, Ma L, et al. (2015). SKIP confers osmotic tolerance during salt stress by controlling alternative gene splicing in Arabidopsis. Mol. Plant 8:1038–1052. [DOI] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Florescu M, and Stadler MB (2015). Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol 33:722–729. [DOI] [PubMed] [Google Scholar]
- Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, and Luhrmann R (2012). Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun 3:994. [DOI] [PubMed] [Google Scholar]
- Griebel T, Zacher B, Ribeca P, Raineri E, Lacroix V, Guigo R, and Sammeth M (2012). Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 40:10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al. (2014). BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell 56:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando CE, Sanchez SE, Mancini E, and Yanovsky MJ (2015). Genome wide comparative analysis of the effects of PRMT5 and PRMT4/CARM1 arginine methyltransferases on the Arabidopsis thaliana transcriptome. BMC Genomics 16:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol 18:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzel J, Duttke SH, Benner C, and Chory J (2016). Nascent RNA sequencing reveals distinct features in plant transcription. Proc. Natl. Acad. Sci. U S A 113:12316–12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg R, McGrail JC, and O’Keefe RT (2010). The function of the NineTeen Complex (NTC) in regulating spliceosome conformations and fidelity during pre-mRNA splicing. Biochem. Soc. Trans 38:1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176:535–548.e24. [DOI] [PubMed] [Google Scholar]
- Ji H, Wang S, Li K, Szakonyi D, Koncz C, and Li X (2015). PRL1 modulates root stem cell niche activity and meristem size through WOX5 and PLTs in Arabidopsis. Plant J 81:399–412. [DOI] [PubMed] [Google Scholar]
- Jia T, Zhang B, You C, Zhang Y, Zeng L, Li S, Johnson KCM, Yu B, Li X, and Chen X (2017). The Arabidopsis MOS4-associated complex promotes MicroRNA biogenesis and precursor messenger RNA splicing. Plant Cell 29:2626–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, and Ast G (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet 11:345–355. [DOI] [PubMed] [Google Scholar]
- Khodor YL, Menet JS, Tolan M, and Rosbash M (2012). Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA 18:2174–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodor YL, Rodriguez J, Abruzzi KC, Tang CH, Marr MT 2nd, and Rosbash M, (2011). Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 25:2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Vandivier LE, Tu B, Gao L, Won SY, Li S, Zheng B, Gregory BD, and Chen X (2015). Detection of Pol IV/RDR2-dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res. 25:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, and Misteli T (2011). Epigenetics in alternative pre-mRNA splicing. Cell 144:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, and Misteli T (2010). Regulation of alternative splicing by histone modifications. Science 327:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Koster T, Nolte C, Weinholdt C, Lewinski M, Grosse I, and Staiger D (2017). Adaptation of iCLIP to plants determines the binding landscape of the clock-regulated RNA-binding protein AtGRP7. Genome Biol. 18:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan J, Xu F, Gao M, Zhao Q, Palma K, Long C, Chen S, Zhang Y, and Li X (2009). Two Prp19-like U-box proteins in the MOS4-associated complex play redundant roles in plant innate immunity. PLoS Pathog. 5:e1000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, and Black DL (2009). Co-transcriptional splicing of constitutive and alternative exons. RNA 15:1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AS (2007). Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol 58:267–294. [DOI] [PubMed] [Google Scholar]
- Reddy AS, Day IS, Gohring J, and Barta A (2012). Localization and dynamics of nuclear speckles in plants. Plant Physiol. 158:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl C, Stauffer E, Kahles A, Wagner G, Drechsel G, Ratsch G, and Wachter A (2012). Polypyrimidine tract binding protein homologs from Arabidopsis are key regulators of alternative splicing with implications in fundamental developmental processes. Plant Cell 24:4360–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, and Carninci P (2011). Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays 33:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Meshorer E, and Ast G (2009). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol 16:990–995. [DOI] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, and Burge CB (2009). Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 36:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, and Guigo R (2012). Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ye R, Xin Y, Fang X, Li C, Shi H, Zhou X, and Qi Y (2011). An importin beta protein negatively regulates MicroRNA activity in Arabidopsis. Plant Cell 23:3565–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wu F, Xie Q, Wang H, Wang Y, Yue Y, Gahura O, Ma S, Liu L, Cao Y, et al. (2012). SKIP is a component of the spliceosome linking alternative splicing and the circadian clock in Arabidopsis. Plant Cell 24:3278–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Quan L, Li S, You C, Zhang Y, Gao L, Zeng L, Liu L, Qi Y, Mo B, et al. (2019). The protein Phosphatase4 complex promotes transcription and processing of primary microRNAs in Arabidopsis. Plant Cell 31:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, and Luhrmann R (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 3. 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. (2014). The human splicing code reveals new insights into the genetic determinants of disease. Science 347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol 11:377–394. [DOI] [PubMed] [Google Scholar]
- Zaffagnini M, Fermani S, Costa A, Lemaire SD, and Trost P (2013). Plant cytoplasmic GAPDH: redox post-translational modifications and moonlighting properties. Front. Plant Sci 4:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Qian B, Cao F, Wu W, Yang L, Guan Q, Gu X, Wang P, Okusolubo TA, Dunn SL, et al. (2015). An Arabidopsis PWI and RRM motif-containing protein is critical for pre-mRNA splicing and ABA responses. Nat. Commun 6:8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kuo CC, and Chen L (2011). GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics 12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.