

Abstract
Motivation
One avenue to address the paucity of clinically testable targets is to reinvestigate the druggable genome by tackling complicated types of targets such as Protein-Protein Interactions (PPIs). Given the challenge to target those interfaces with small chemical compounds, it has become clear that learning from successful examples of PPI modulation is a powerful strategy. Freely accessible databases of PPI modulators that provide the community with tractable chemical and pharmacological data, as well as powerful tools to query them, are therefore essential to stimulate new drug discovery projects on PPI targets.
Results
Here, we present the new version iPPI-DB, our manually curated database of PPI modulators. In this completely redesigned version of the database, we introduce a new web interface relying on crowdsourcing for the maintenance of the database. This interface was created to enable community contributions, whereby external experts can suggest new database entries. Moreover, the data model, the graphical interface, and the tools to query the database have been completely modernized and improved. We added new PPI modulators, new PPI targets and extended our focus to stabilizers of PPIs as well.
Availability and implementation
The iPPI-DB server is available at https://ippidb.pasteur.fr The source code for this server is available at https://gitlab.pasteur.fr/ippidb/ippidb-web/ and is distributed under GPL licence (http://www.gnu.org/licences/gpl). Queries can be shared through persistent links according to the FAIR data standards. Data can be downloaded from the website as csv files.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
As the drug discovery sector is facing tremendous challenges (DiMasi et al., 2016) in terms of pharmaceutical innovation (Teague, 2011), attrition rates (Swinney and Anthony, 2011) and stronger regulations, new routes must be taken to expand the pharmacological options at our disposal. To this end, designing drugs to target Protein-Protein Interactions is a powerful strategy (Kuenemann et al., 2015), as these PPIs are the key regulators of biological processes. Yet, given the difficulty to identify chemical probes (Azzarito et al., 2013) that modulate these biological interfaces, several studies have adopted the strategy of deriving chemical guidance from publicly available successful examples of PPI modulation by small chemical compounds (Kim et al., 2016; Laraia et al., 2015; Sperandio et al., 2010). Over the last decade, the emergence of the ’big data’ era has promoted a great interest in exploiting publicly available chemical information for drug discovery (Kim, 2016) through major initiatives including PubChem (Kim et al., 2019) and ChEMBL (Gaulton et al., 2017). Both databases offer an impressive number of online tools and web services to query and analyze both chemical and pharmacological data on all types of therapeutic targets. In the PPI community, two databases paved the way in the late 2000’s. The first one is the Timbal database (Higueruelo et al., 2013) which automatically extracts relevant data from ChEMBL following the identification of PPI targets. The second is the 2P2I database (Basse et al., 2016) which is manually curated and exclusively focused on experimental structures of PPI modulators derived from the Protein DataBank (PDB) (Berman, 2000). We first reported iPPI-DB in 2013 (Labbé et al., 2013), and improvements were made on a subsequent version by adding more PPI modulators and more PPI targets, as well as a chemical similarity mode for querying the database (Labbé et al., 2016). Here, we present a new generation of this database. It now includes a graphical interface that facilitates the entry of new data. This improvement enables the provision of a community contribution mode, whereby experts external to our group can enter new data based on any published relevant work. After a review by the core curators of the database, these contributions are integrated into the database and become available in the web interface (see Fig. 1).Moreover, the architecture of this application has been entirely redesigned. The database and web application now support complex queries over thousands of compounds, easily accessible from an interactive exploration interface.
Fig. 1.
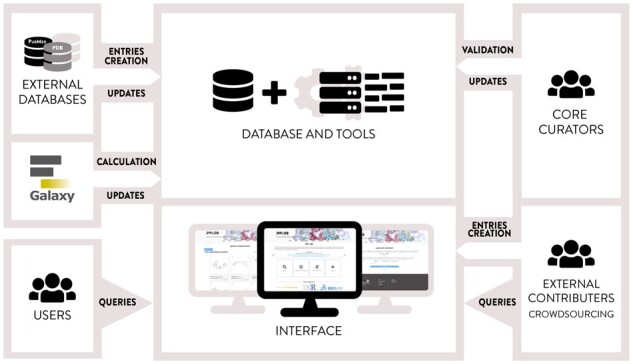
The iPPI-DB database. The new integrated interface allows users to build tailored queries and contributors to make new entries on iPPI-DB using intuitive and assisted tools
By providing an intuitive interface to add new data and robust querying tools, this reinvented iPPI-DB represents a unique portal for chemists and biologists willing to initiate drug discovery projects against PPI targets or to contribute to the sharing of pharmacological data by a community of experts.
2 A paradigm shift in our model of data curation
The pharmacological results that are valuable for specialized databases like iPPI-DB are retrieved from peer-reviewed publications including scientific articles and patents. This type of information has been recently defined as DARCLP relationships in a very interesting review (Southan, 2020). This definition illustrates the relationships that bioscientists reading papers or patents have to establish in order for the data to become tractable: a document (D) in which bioactivities (A) have to be associated with quantitative results (R), chemical compounds (C), explicit location references (L) for the compounds within the document (e.g. ‘compound 10b’) and a protein target (P), here a PPI target. A rapid survey of iPPI-DB-related published studies available on PubMed illustrates that there are now more than 2000 publications a year that are of potential interest for iPPI-DB (Fig. 2). This represents a large amount of data published annually for which DARCLP relationships need to become tractable.
Fig. 2.

Number of annual publications available in PubMed related to the topics of inhibitors of protein-protein interactions. To carry out this estimation, a combination of terms present either in the titles or the abstracts was used to look for potential iPPI-DB-related research articles. Terms such as inhibitors, protein-protein interactions and the names of existing PPI targets were used
In this context, our previous model of database curation that relied essentially on manual intervention is no longer sustainable. Two complementary solutions can be adopted to ensure the sustainable maintenance of databases such as iPPI-DB: a fully automated process relying on automated chemical and biomedical entity recognition from text (e.g. via Natural Language Processing and artificial intelligence) (Southan, 2020); or a community curation based on a cohort of experts.
Artificial intelligence efforts have clearly made great progresses toward the fully automated extraction of explicit DARCLP relationships from documents. Nonetheless, those procedures are not yet exhaustive, and still leave unaddressed data. Among the difficulties which contribute to this persistent gap, one can identify the access to paywalled literature, or the complexity of the automation when interpreting affinity tables that rely on R-groups to describe chemical series and structure-activity-relationships.
Moreover, as opposed to the users of generic public chemistry databases like PubChem or ChEMBL, users of specialized databases like iPPI-DB, are not only interested in associating a protein target with compound activity, they need tractable pharmacological data about compounds that are capable of modulating an interaction between several protein partners. Therefore, this implies more complex DARCLP relationships that are hardly reachable by fully automated processes.
These persistent limitations stand in sharp contrast to community curation approaches whereby biocurators have the ability to discern such relationships from a paper in minutes (Southan, 2020). The fact a significant portion of the iPPI-DB compounds are missing from major public generic databases (13% are missing from PubChem and up to 51% are missing from ChemBL) is also a good illustration of the importance of combining different data sources and that community curation of specialized database like iPPI-DB is a strong strategy. A growing number of life science data resources adopt community contribution to support data collection and curation, and to ensure their long term sustainability (Jassal et al., 2020; Lee et al., 2018; Lock et al., 2020).
Yet, fully automated processes and community curation both required substantial developments in order to become efficient. Although it is obvious for automated curation procedures especially when they rely on artificial intelligence, community curation also mandates the development of adapted technologies that enable the automation of every process that does not require human supervision and the management of numerous entry sources from different groups of experts.
3 A web application for maintenance relying on community curation
In order for iPPI-DB to rely on community curation, major developments were necessary to design a web application for expert curators. It was built to assist them in their extraction of DARCLP relationships from bibliographic sources (see Fig. 3). It follows the pillars of DARCLP: bibliographic source, biological assays, bioactivities, compound and compound location and PPI target. This constitutes a significant improvement over the previous version of the database, which was a complex and largely manual process relying on numerous scripts and external data sheets. This interface has been designed with the aim of facilitating the contributions as much as possible, even from experts who are not familiar with the technical aspects of this database.
Fig. 3.

iPPI-DB contribution mode page (https://ippidb.pasteur.fr/contribute)—during the process the contributor: (1) provides the bibliographic identifier of the publication he will describe (Pubmed ID, DOI, Patent ID); (2) confirms the fetched publication metadata and specifies which type of test data are included in this publication; (3) provides a PDB code that contains the structure of the full PPI complex, including all protein partners; (4) selects its architecture from a list of proposed schematics; (5) assigns the proteins and domains included in the PDB entry to the bound and partner complexes forming the PPI; (6) specifies the PPI target family as well as an associated disease (7) specifies the chemical structure and name of the compounds; (8) and describes the tests performed and their results
Each contribution is based on a description of the content of a publication or a patent. A wizard-based web interface assists the expert curators in retrieving the DARCLP relationships disseminated in the document. Indeed, contributors indicate (step-by-step), the architecture of the PPI complex(es), the chemical compounds tested for modulation and the various assays in which these compounds were tested. An important step consists for the contributor in indicating a PDB ID for the entry containing the investigated PPIs and select the associated class of PPI architecture from a list of provided schematics as described in (Zarzycka et al., 2016). This input from the expert curator is a key element to accurately define the PPI target and the mode of action of the active compounds. The combination of the expert’s insight and the automated processes allows to efficiently translate the DARCLP relationships into tractable database entries.
The contribution interface only requires minimal information from contributors. Whenever they indicate references to some entities such as bibliographic references (DOI, PubMed ID or Patent), PDB codes, proteins or compounds, the server automatically retrieves additional details from other reference databases using web services. This contributes to reducing the risk of errors and facilitates contributions. When the process is over, the contribution is deposited into the administration portal of the core curators for an ultimate validation that will eventually launch final calculations such as compound properties and push the contributions into production. This validation process makes the contribution data publicly visible, and updates all plots within the query interface including chemical space, efficiency plots, etc.
Scientists willing to contribute to iPPI-DB are invited to do so directly from the iPPI-DB website. We recommend that users log into the system using their ORCID ID (Haak et al., 2012) on https://ippidb.pasteur.fr/accounts/login/, to get an immediate access to the contribution mode. Contributors will be credited at each release of the database.
4 Versatile and interactive query capabilities
While the new web application for contribution was designed to assist expert curators in feeding iPPI-DB with pharmacological data and DARCLP, the querying interface was entirely redesigned to access these data in the most tractable manner. Thus, the design of new querying interface was also articulated around the key elements of DARCLP relationships: bibliographic source, biological assays, bioactivities, compound and compound location and PPI target. The compounds available in iPPI-DB can be queried through an interface (see Fig. 4) that allows for the exploration of the data through the combination of multiple chemical and pharmacological filters. These filters include the properties of the compounds themselves (e.g. the chemical similarity to a query compound), or the compliance with chemistry rules such as the Lipinski’s RO5 (Lipinski et al., 2001), as well as the properties of their targets, and the data available to assess their mode of action (e.g. X-ray crystallographic structure, cellular assay, pharmacokinetic data). Depending on the nature of the data, the filters will be displayed with different widgets used to pick values from a list, to specify ranges for numeric values or to draw chemical compound structures, using MarvinJS (www.chemaxon.com, version 20.5.0). Users can easily combine multiple filters to constitute complex queries, and the modification of the filter criteria is immediately reflected in the results page. This interactive approach enables users to iteratively refine their queries by adding new filters.
Fig. 4.

iPPI-DB query mode page (https://ippidb.pasteur.fr/compounds/)—the query page provides filters (upper left part of the figure) which cover the properties of the compounds and the properties of their targets. Filters allow to select values from lists, specify ranges for numeric values or to draw chemical compound structures. Users can combine multiple filters (here two are defined for a range of AlogP values and a given PPI family) to constitute complex queries. Query results can be sorted (top right of the main page) on multiple criteria and displayed as thumbnails, a list of cards or as a table. Results can be downloaded in the CSV file format. Clicking on a specific compound leads to a detailed page (bottom part of the figure) with compound pharmacology, physico-chemistry properties and drug similarities
Query results can be sorted on multiple criteria, and displayed as thumbnails, as a list of cards or as a table. Each of these successive views corresponds to various levels of detail for the properties of the compounds and their targets:
the thumbnail view provides a quick overview of the results, focused on essential properties of the compounds (chemical structure, PPI target family and molecular weight),
the list view offers more details for each entry, including the multiple identifiers, cross links to other databases, bibliography references, compliance with chemistry rules and a summary of the pharmacological profile of the compound.
the table view displays a number of properties which may be further customized in different columns and exported as a CSV file.
The filters, sorting and display options corresponding to these queries are directly reflected in the URL of the results page, which can be easily bookmarked for later reuse, or shared between collaborators.
Each compound in iPPI-DB possesses its own ID card through a persistent URL, for example https://ippidb.pasteur.fr/compounds/1602 for JQ1, a pan-Bromodomain inhibitor. This page provides all available data on the compound, including its structure, its physicochemical/pharmacological profiles, but also the structures of its most chemically similar marketed or investigated drugs. Each of these pages includes links to the external sources of information that have either been used to collect the data or that might be of interest to the user, such as Uniprot (Bateman, 2019), PubChem, ChEMBL, ChemSpider (http://www.chemspider.com) or DrugBank (Wishart et al., 2018). Each of these cards, as well as the other main pages of the site, includes BioSchemas-based (Gray et al., 2017) schema.org metadata to facilitate search engine indexation. An overview of the contents of the database is provided in the about tab of the user interface, through a number of tables and plots. These elements provide general information about iPPI-DB data, pharmacological and physicochemical summaries, interactive plots to navigate through compound efficiency and chemical spaces.
5 Evolution of the iPPI-DB database contents and features
This new version of iPPI-DB represents a significant improvement over the previous versions, both in terms of data contents and features. The web application has been completely reinvented and designed as a community-centered database. The development of the new interface for community curation, which was released in July2020, has allowed us to rapidly add new data without having yet to rely on external expert curators (Fig. 5). Indeed, the number of compounds (2374) has increased of 48% from the first version and of 35% since July2020, while the number of bioactivities (3895) has increased of 84% since the first version and of 58% since July2020. Moreover, the number of PPI families of homologous PPIs (108), the number of PPI targets (115) and the number of publications (231), have increased of 246%, 342% and 114% respectively. Therefore, we now hope that, with the help the PPI community, we will rapidly cover the vast majority of published iPPI compounds and of their bioactivities. Among the new data, we added stabilizers of PPI targets (https://ippidb.pasteur.fr/compounds/?stabilisation_role=true), and epigenetic target modulators such as compounds active on Kme/Rme readers which are gaining attention recently for various therapeutic applications (https://ippidb.pasteur.fr/compounds/?family=523&family=525&page=3).
Fig. 5.

Evolution of the iPPI-DB database contents. The release of the new web application for community-curation in July2020 has dramatically changed the rate of data entry in the database, for compounds and pharmacological data (top panel) and for the diversity of PPI targets, families of homologous targets or number of bibliographic sources (bottom panel)
The querying interface has also gained a significant number of new features and has improved in ergonomics over the previous versions (Fig. 6). This last version now proposes new filter criteria, visualization tools and links to external resources. To further guarantee the openness and sustainability of this resource, the code is made publicly available on GitHub and the data are shareable, interoperable and downloadable, with the ultimate aim of reaching FAIR standards. Finally, the new contribution mode based on community curation is a major feature improvement and has clearly impacted the rate of data entries and will allow us to initiate new projects on special pathologies or disease-associated pathways mediated by specific PPIs.
Fig. 6.

Evolution of the iPPI-DB database features. Illustration of the evolution of iPPI-DB features since the first version in 2013. The number of compounds is indicated as well for each realease. * indicates the release of the community curation application to highlight the impact this had on the data content. Features are described by categories queries, Visualization of results, links, open science and contribution. These are then subcategorized in block sharing similar feature profiles along the years
6 User-Centered Design (UCD) of the new iPPI-DB
The iPPI-DB initiative aims to be centered on the community. Thus, this major update of the database had to place the database users and contributors at the center of the development strategy. We therefore adopted a User-Centered Design (UCD) approach (de Matos et al., 2013). We based this process on a constant dialogue with the project stakeholders, and the support of various participatory design techniques. It allowed us first to identify three different user roles: (i) data explorers, who browse the contents of the database; (ii) external contributors, who suggest new entries based on publications and (iii) core curators, who validate new suggestions.
The redesign of the query interface was initiated using techniques such as ‘Six Up and One Up’ . During these workshops, each participant suggested multiple options in the form of sketches, later discussed until consensus was reached. The outcomes suggested a series of guidelines: (i) the need to combine multiple filters, as freely and as interactively as possible; (ii) the possibility to access results with multiple levels of detail, needed as users refine their search; (iii) a compound ‘card’ which provides all its details in a single page identified by a unique and persistent URL.
The design of the contribution interface, on the other hand, was guided by prototyping meetings and focus groups, in order to facilitate the dialogue between developers, designers and chemoinformatics experts. The key point of the resulting design is the minimization of the amount of information to be provided manually, in order to reduce both the time spent entering data, and the risk of data entry mistakes.
All initial designs of all interfaces were improved iteratively, starting from sketches, later refined as wireframes and finally interactive prototypes, before their implementation.
7 System architecture
The central component of iPPI-DB is a web application (described in Section 7.1), coupled to a relational database system. In addition, the retrieval of information from external databases and the computation of compound properties rely respectively on external web services (Section 7.2) and workflows available on our Galaxy server (see Section 7.3) (Fig. 7).
Fig. 7.
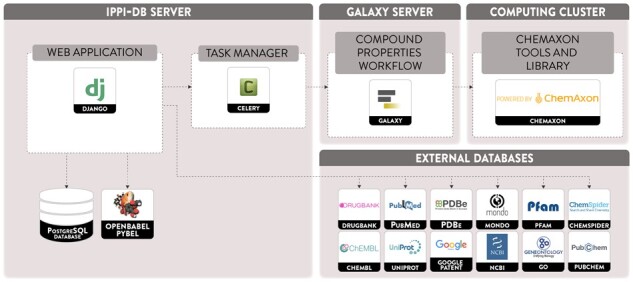
The iPPI-DB architecture. This view represents the various components of the system, including (1) the web server hosting the web application and the database, (2) a Galaxy server that enables the execution of a workflow computing the compound properties or an HPC server and (3) the various external databases accessed via web services
7.1 Web application
The iPPI-DB web application is based on Django, a framework we chose for its industry-grade robustness and the size of its community. Another factor for the choice of this framework is its use of the Python programming language, therefore easily interoperable with many existing Bioinformatics, Chemoinformatics and data processing libraries, such as Open Babel (O’Boyle et al., 2011) and pandas (McKinney, 2011). The data are stored in a PostgreSQL database.
Besides the HTML code generated by the Django templates, the interface of the application relies on a number of Javascript components for visualisation. Marvin JS (20.5.0, 2020, http://www.chemaxon.com) and SmilesDrawer (Probst and Reymond, 2018) are used to draw and display the molecular structure of the compounds. The dynamic charts that display the various statistics on iPPI-DB (see https://ippidb.pasteur.fr/about-pharmacology/ and https://ippidb.pasteur.fr/about-physicochemistry/), or let users explore the compounds in the context of their chemical space (see https://ippidb.pasteur.fr/about-pca/) or their efficiencies (see https://ippidb.pasteur.fr/about-le-lle/) use the ChartsJs javascript library (https://www.chartsjs.org).
7.2 Web services
The data structure stored in iPPI-DB is heavily linked to multiple existing reference databases, and data themselves reflect a formal description of the knowledge represented in the source publications. These data are provided through the contribution interface, which relies on external web services. These services query multiple databases (Agarwala et al., 2017; Ashburner et al., 2000; Carbon et al., 2019; Finn et al., 2016; Jupp et al., 2015; Mir et al., 2018; Shefchek et al., 2020) to retrieve various information during the contribution process, using HTTP/REST web services. These web services are either directly accessed from custom python code, or through the BioServices python package (Cokelaer et al., 2013).
7.3 Galaxy
The physicochemical properties of the compounds are computed using the Chemaxon library JChem (17.3.1, https://www.chemaxon.com). The java executables are available as Galaxy (Afgan et al., 2018) tools, from the Galaxy instance of the Institut Pasteur (Mareuil et al., 2017). A workflow coordinates their execution to compute all properties, from an input SDF file, and formats them as a machine-readable JSON file. Because the computation of these properties can be resource-intensive, it is performed asynchronously, as part of the contribution validation process, using the Celery task queue.
8 Conclusion and perspectives
In a context of increasing pressure to fast-track pharmaceutical innovation, it is essential to rely on powerful tools to apprehend the full extent of available data. This comes with the collection of properly annotated pharmacological information, cross-references with complementary databases and intuitive tools to navigate them. The pharmacological modulation of PPI targets offers endless applications and benefits for human health. To this end, we have completely redesigned iPPI-DB, our database of PPI modulators. First, we completely revised the layout and implementation of the query interface, in order to make it more intuitive, interactive and powerful. Second, in the big data era, it has become clear that both automated processes and community curation represent complementary solutions to guarantee the growth and sustainability of reference databases. In the case of a specialized database such as iPPI-DB, community curation represents the strongest strategy when assisted by appropriate tooling and automation, to facilitate the extraction of highly intricate DARCLP relationships.
In the near future, we plan to expand our links and connections with complementary initiatives and strengthen our network of partnerships. We aim to develop tailored projects around the iPPI-DB initiative by implicating communities of experts in specific fields such as antimicrobial resistance, or specific pathologies, and by focusing on specific families of PPI targets or disease-associated pathways. With regards to the mode of contribution, we plan to combine our current expert contribution interface with automated approaches such as text-mining, in order to further facilitate and accelerate the process, while preserving the high quality of expert-curated database. Finally, we also will explore various mechanisms to disseminate the information stored in this database to other resources, through linking from other databases. Along this line, we are currently finalizing the implementation of external links from Europe PMC to iPPI-DB, in order for each compound of our database to be associated with corresponding article accessible from Europe PMC.
By providing a community-driven interface to add new data and robust querying tools, the new iPPI-DB version represents a unique portal for chemists and biologists willing to initiate drug discovery projects against PPI targets.
Supplementary Material
Acknowledgements
This work used the computational and storage services (TARS cluster, VM Hosting) provided by the IT department at Institut Pasteur, Paris. The authors wish to acknowledge in particular the help and technical advice of Eric Deveaud, Emmanuel Guichard, Thomas Ménard and Youssef Ghorbal (IT Department, Institut Pasteur). They also want to acknowledge the technical help of Tru Huynh (Structural Bioinformatics Unit, Institut Pasteur). They thank Jon Ison, Benjamin Bardiaux and Pascal Campagne for their proofreading of the paper.
Marvin JS (20.5.0, 2020, http://www.chemaxon.com) is used for drawing and displaying chemical structures in both Query mode and Contribution mode of iPPI-DB. Pipeline Pilot (server 19.1) is used to prepare the DrugBank database from a SDF file prior to chemical similarity search 202 (2020).
Financial Support: none declared.
Conflict of Interest: none declared.
Contributor Information
Rachel Torchet, Hub de Bioinformatique et Biostatistique Département Biologie Computationnelle, Institut Pasteur, USR 3756 CNRS, Paris 75015, France.
Karen Druart, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
Luis Checa Ruano, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
Alexandra Moine-Franel, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
Hélène Borges, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
Olivia Doppelt-Azeroual, Hub de Bioinformatique et Biostatistique Département Biologie Computationnelle, Institut Pasteur, USR 3756 CNRS, Paris 75015, France.
Bryan Brancotte, Hub de Bioinformatique et Biostatistique Département Biologie Computationnelle, Institut Pasteur, USR 3756 CNRS, Paris 75015, France.
Fabien Mareuil, Hub de Bioinformatique et Biostatistique Département Biologie Computationnelle, Institut Pasteur, USR 3756 CNRS, Paris 75015, France.
Michael Nilges, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
Hervé Ménager, Hub de Bioinformatique et Biostatistique Département Biologie Computationnelle, Institut Pasteur, USR 3756 CNRS, Paris 75015, France.
Olivier Sperandio, Department of Structural Biology and Chemistry, Institut Pasteur, Paris 75015, France.
References
- (2020) BIOVIA, Dassault Systèmes , Pipeline Pilot,9.5. San Diego: Dassault Systèmes. [Google Scholar]
- Afgan E. et al. (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res., 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwala R. et al. (2017) Database resources of the national center for biotechnology information. Nucleic Acids Res., 46, D8–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. Nature Genetics,25, 25–29. [DOI] [PMC free article] [PubMed]
- Azzarito V. et al. (2013) Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem., 5, 161–173. [DOI] [PubMed] [Google Scholar]
- Basse M.J. et al. (2016) 2P2Idb v2: update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database, 2016, baw007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res., 47, D506–S515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S. et al. (2019) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res., 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokelaer T. et al. (2013) BioServices: a common Python package to access biological Web Services programmatically. Bioinformatics, 29, 3241–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Matos P. et al. (2013) The Enzyme Portal: a case study in applying user-centred design methods in bioinformatics. BMC Bioinformatics, 14, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi J.A. et al. (2016) Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ., 47, 20–33. [DOI] [PubMed] [Google Scholar]
- Finn R.D. et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res., 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A. et al. (2017) The ChEMBL database in 2017. Nucleic Acids Res., 45, D945–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray A.J. et al. (2017) Bioschemas: from potato salad to protein annotation. In ISWC 2017 Poster Proceedings. Vienna, https://iswc2017.semanticweb.org/paper-579/, ISSN 16130073.
- Haak L.L. et al. (2012) ORCID: a system to uniquely identify researchers. Learned Publish., 25, 259–264. [Google Scholar]
- Higueruelo A.P. et al. (2013) TIMBAL v2: update of a database holding small molecules modulating protein–protein interactions. Database, 2013, bat039–bat039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal B. et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res., 48, 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupp S. et al. (2015) A new ontology lookup service at EMBL-EBI. In CEUR Workshop Proceedings, vol. 1546, pp. 118–119.
- Kim J. et al. (2016) Diversity-oriented synthetic strategy for developing a chemical modulator of protein–protein interaction. Nat. Commun., 7, 13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. (2016) Getting the most out of PubChem for virtual screening.,Nat. Commun., 7,13196. [DOI] [PMC free article] [PubMed]
- Kim S. et al. (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res., 47, D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenemann M.A. et al. (2015) In silico design of low molecular weight protein–protein interaction inhibitors: Overall concept and recent advances. Prog. Biophys. Mol. Bio., 119, 20–32. [DOI] [PubMed] [Google Scholar]
- Labbé C.M. et al. (2013) IPPI-DB: a manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov. Today, 18, 958–968. [DOI] [PubMed] [Google Scholar]
- Labbé C.M. et al. (2016) IPPI-DB: an online database of modulators of protein–protein interactions. Nucleic Acids Res., 44, D542–D547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laraia L. et al. (2015) Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein–protein interactions. Chem. Biol., 22, 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.Y. et al. (2018) WormBase 2017: molting into a new stage. Nucleic Acids Res., 46, D869–D874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C.A. et al. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev., 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Lock A. et al. (2020) Community curation in PomBase: enabling fission yeast experts to provide detailed, standardized, sharable annotation from research publications. , Database, Volume 2020, 2020, baaa028, 10.1093/database/baaa028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareuil F. et al. (2017) A public Galaxy platform at Pasteur used as an execution engine for web services. F1000Research, 6, 157022. [Google Scholar]
- McKinney W. (2011) pandas: a foundational python library for data analysis and statistics. In: Python for High Performance and Scientific Computing, pp. 1–9.
- Mir S. et al. (2018) PDBe: towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res., 46, D486–D492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N.M. et al. (2011) Open Babel: an Open chemical toolbox. J. Cheminf., 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst D., Reymond J.L. (2018) SmilesDrawer: parsing and drawing SMILES-encoded molecular structures using client-side JavaScript. J. Chem. Inf. Model., 58, 1–7. [DOI] [PubMed] [Google Scholar]
- Shefchek K.A. et al. (2020) The Monarch Initiative in 2019: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res., 48, D704–D715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C. (2020) Opening up connectivity between documents, structures and bioactivity. Beilstein J. Org. Chem.,16, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio O. et al. (2010) Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov. Today, 15, 220–229. [DOI] [PubMed] [Google Scholar]
- Swinney D.C., Anthony J. (2011) How were new medicines discovered? Nat. Rev. Drug Discov., 10, 507–519. [DOI] [PubMed] [Google Scholar]
- Teague S.J. (2011) Learning lessons from drugs that have recently entered the market. Drug Discov. Today, 16, 398–411. [DOI] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res., 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzycka B. et al. (2016) Stabilization of protein-protein interaction complexes through small molecules. Drug Discov. Today, 21, 48–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.