

Abstract
Background
Stress during the adolescent period influences postnatal maturation and behavioral patterns in adulthood. Adolescent stress-induced molecular and functional changes in neurons are the key clinical features of psychiatric disorders including schizophrenia.
Objective
In the present study, we exposed genetically vulnerable mice to isolation stress to examine the molecular changes in the glutamatergic system involving N-methyl-d-aspartate (NMDA) receptors via dopaminergic disturbance in the prefrontal cortex (PFc).
Results
We report that late adolescent stress in combination with Disrupted-in-Schizophrenia 1 (DISC1) genetic risk elicited alterations in glutamatergic neurons in the PFc, such as increased expression of glutamate transporters, decreased extracellular levels of glutamate, decreased concentration of d-serine, and impaired activation of NMDA-Ca2+/calmodulin kinase II signaling. These changes resulted in behavioral deficits in locomotor activity, forced swim, social interaction, and novelty preference tests. The glutamatergic alterations in the PFc were prevented if the animals were treated with an atypical antipsychotic drug clozapine and a dopamine D1 agonist SKF81297, which suggests that the activation of dopaminergic neurons is involved in the regulation of the glutamatergic system.
Conclusion
Our results suggest that adolescent stress combined with dopaminergic abnormalities in the PFc of genetically vulnerable mice induces glutamatergic disturbances, which leads to behavioral deficits in the young adult stage.
Keywords: Adolescent stress, Genetic vulnerability, Glutamatergic neuron, Dopaminergic neuron, Prefrontal cortex
Introduction
Pathophysiology of psychiatric disorders including schizophrenia is associated with the disturbance of molecular and functional modifications in neurons (Goff and Coyle 2001; Hayashi-Takagi et al. 2010; McCullumsmith 2015; McCullumsmith et al. 2004; Moghaddam 2003; Tomoda et al. 2016; Xing et al. 2016). Numerous studies strongly support the involvement, at least in part, of disturbance in neuronal signaling pathways which results in the loss of proper brain activity, in the pathogenesis of schizophrenia and associated disorders. (Glantz and Lewis 2000; Glessner et al. 2010; Hayashi-Takagi 2017; Hayashi-Takagi et al. 2015; Kenny et al. 2014; Kirov et al. 2012; Lips et al. 2012; McGlashan and Hoffman 2000).
Deficits in sociability and impaired learning and memory are clinical features of psychiatric disorders including schizophrenia, and are hypothesized to result from a disturbance in neural systems (Gunaydin et al. 2014; Hayashi et al. 2016; Nagai et al. 2011; Takeuchi et al. 2016). Adult sociability, learning, and memory are altered by adolescent stress (Chaby et al. 2015; Li et al. 2015; McCormick et al. 2012; Niwa et al. 2011; Novick et al. 2016; Sarro et al. 2014). Adolescence is a critical time in development in which dramatic changes in hormone levels and fine-tuning of neurocircuitry facilitate an individual’s maturation into adulthood (Blakemore 2008). Therefore, adolescent brains are vulnerable to stress (Paus et al. 2008). Nonetheless, the biological mechanisms linking adolescent stress to adult behavioral changes are not well understood.
Psychological stress activates the hypothalamic-pituitary-adrenal (HPA) axis and induces production of glucocorticoids in the adrenals, which can trigger psychiatric conditions (Axelrod and Reisine 1984; Joels and Baram 2009; Sorrells et al. 2009). Patients with mood disorders, psychosis, and cocaine dependence show increased levels of corticotropin-releasing factor, adrenocorticotropic hormone, and glucocorticoids (Kreek et al. 2005). A glucocorticoid receptor (GR) antagonist, RU486 (mifepristone), is uniquely beneficial in human psychotic depression (Flores et al. 2006). In the previous study, our results suggested that an environmental stress during adolescence combined with an appropriate genetic risk can elicit elevated corticosterone, and result in the alterations of epigenetic modifications in mesocortical dopaminergic neurons (Niwa et al. 2013). Under this condition, molecular changes in dopaminergic neurons and associated behavioral alterations were blocked by administration of the GR antagonist RU486 (Niwa et al. 2013, 2016b). Several studies reported that use of RU486 may be effective in the treatment of psychiatric symptoms and may regulate the HPA axis (Belanoff et al. 2001, 2002; Flores et al. 2006; Young et al. 2004). Thus, overactivation of GR signaling might underlie neuronal dysfunction and behavioral deficits induced by psychological stress.
Alterations in the levels of glutamate and its metabolite N-acetylaspartate and a reduction in glutamate receptor binding in the prefrontal cortex (PFc) have been reported at clinical level in psychiatric disorders including schizophrenia (Hashimoto et al. 2005; Hayashi-Takagi et al. 2010; Pilowsky et al. 2006; Steen et al. 2005). In animal models, administration of N-methyl-d-aspartate (NMDA)-type glutamate receptor antagonists, such as phencyclidine (PCP) and ketamine, elicits various pathological alterations affecting physical and behavioral processes that are possibly relevant for psychiatric disorders including schizophrenia with an adult onset (Jaaro-Peled et al. 2009; Lu et al. 2011; Toriumi et al. 2012; Warden et al. 2012). Increasing evidence suggests that the glutamatergic system of PFc relies on mesocortical dopaminergic neurons (Novick et al. 2016; Owen et al. 2016; Swerdlow et al. 2001; Warden et al. 2012). Neuronal microcircuits in the PFc, consisting of glutamatergic pyramidal neurons and local inhibitory interneurons, are essential for the encoding and the maintenance of information in the PFc (Goldman-Rakic 1995; Kabanova et al. 2015; Tanaka et al. 2011). Midbrain dopaminergic neurons projecting to the PFc are predominantly located in the medial ventral tegmental area (VTA) and modulate cognitive functions including perseverative behavior and impulsivity (Goldman-Rakic 1995; Sandson and Albert 1984). The mesocortical dopaminergic neurons are indicative of ability to co-release dopamine and glutamate in their target areas. Dopaminergic inputs into the PFc optimize the signal-to-noise ratio in these local microcircuits and are thought to modulate their excitability through the alteration of input efficacy (Gorelova et al. 2002; Matsuda et al. 2006; Vijayraghavan et al. 2007; Yang and Seamans 1996). NMDA-mediated excitation was enhanced by a dopamine-D1 receptor agonist in prefrontal cortical pyramidal neurons through protein kinase A (PKA)-dependent mechanisms (Snyder et al. 1998; Tingley et al. 1997; Wang and O’Donnell 2001). Our previous study showed that dopaminergic function, especially dopamine-D1 receptor signaling, is critical for the regulation of NMDA-Ca2+/calmodulin kinase II (CaMK II) signaling in the PFc of PCP-treated mice (Aoyama et al. 2014; Mouri et al. 2007b). Thus, disturbance in the dopamine-glutamate system in the PFc induced by psychological stress may lead to adult behavioral deficits relevant to psychiatric disorders.
We previously reported that psychosocial stress imposed during adolescence in the presence of a genetic risk affects the mesocortical projection of dopaminergic neurons, which is associated with behavioral deficits, such as increased immobility in forced swim test, impairment of prepulse inhibition, and aberrant locomotor activity in adulthood (Niwa et al. 2013). Furthermore, we have recently reported that the first 1-week period during stress regimen in the adolescence may correspond specifically to the timing of maturation and function of mesocortical dopaminergic neurons and their sensitivity to glucocorticoids (Niwa et al. 2016b).
Nonetheless, there are at least three crucial but unanswered questions related to the model reported in our previous publications (Niwa et al. 2013, 2016b). First, it has not been determined whether adult sociability, learning, and memory are altered by adolescent stress in the model. Second, it also remains unknown how dopaminergic disturbance affects other neuronal systems such as glutamatergic neurons in the PFc in genetically vulnerable mice. Third, we have not yet identified which antipsychotic drugs can ameliorate the behavioral deficits and neurochemical changes in the model. The present study was designed to address these questions in order to enhance our understanding of neuronal, stress-associated disturbances, especially those affecting glutamatergic and dopaminergic neurons that have been frequently reported in psychiatric disorders. In the present study, we designated the model as disease model (DM).
Materials and methods
Animals
Disrupted-in-Schizophrenia 1 (DISC1) dominant-negative transgenic mice under control of the prion protein promoter (DISC1-DN-Tg-PrP) were generated at the Transgenic Core Laboratory of Johns Hopkins University. Heterozygous transgenic line 51 mice and wild-type littermates established by mating with C57BL6 mice maintaining the purity of the genetic background were compared in the present experiments. Our group and others previously reported that DISC1, one of the most popular leads to explore molecular pathways underlying the pathophysiology of psychiatric disorders, has crucial roles in brain maturation and adult behaviors (Brandon and Sawa 2011; Callicott et al. 2005; Furukubo-Tokunaga et al. 2016; Hamshere et al. 2005; Hashimoto et al. 2006; Hennah et al. 2003; Hodgkinson et al. 2004; Ibi et al. 2010; Ishizuka et al. 2011; Jaaro-Peled et al. 2016; Jaaro-Peled et al. 2013; Kamiya et al. 2005; Kilpinen et al. 2008; Nagai et al. 2011; Niwa et al. 2010, 2013, 2016b; O’Tuathaigh et al. 2017; Saito et al. 2016; Seshadri et al. 2015; Sullivan 2013; Trossbach et al. 2016). We selected further research on the bright promise of DISC1 protein as a molecular driver for the biology of the psychiatric disorders including schizophrenia (Niwa et al. 2016a).
For the control (CTL) group, wild-type littermate mice were housed in groups in wire-topped clear plastic cages (21 × 32 × 13 cm) until sampling after behavioral tests at 8 weeks of age, under a controlled environment (23 ± 1 °C; 50 ± 5% humidity; light and dark cycles starting at 8 am and 8 pm, respectively) with free access to food and water. For the DM group, DISC1-DN-Tg-PrP mice were isolated from 5 to 8 weeks of age in individual wire-topped opaque polypropylene cages (12.5 × 20 × 11 cm), and maintained until sampling after behavioral tests. Mild isolation stress during late adolescence causes no endocrinological, neurochemical, and behavioral changes (Niwa et al. 2013), although we acknowledge effects of adolescent isolation stress during different periods on behaviors (Hong et al. 2012; Kercmar et al. 2011; Leussis and Andersen 2008; Mathews et al. 2008; Niwa et al. 2011; Weiss et al. 2004). All animal procedures were in accordance with guidelines for the care and use of laboratory animals issued by the National Institutes of Health, Japanese Pharmacological Society, and Meijo University.
We reported similar changes in the phenotypes between male and female mice in a previously published study (Niwa et al. 2013), although we acknowledge sex-specific effects of DISC1 and adolescent stress in the other animal models (Abazyan et al. 2014; Ayhan et al. 2011; Holley et al. 2013; Hong et al. 2012; Kuroda et al. 2011; Leussis and Andersen 2008; Nakai et al. 2014; Pletnikov et al. 2008; Weiss et al. 2004). The other labs also reported no sex-specific findings in the DISC1 animal model (Clapcote et al. 2007; Koike et al. 2006). Furthermore, in our experimental conditions, we did not observe any significant differences among the unstressed wild-type, isolated wild-type, and unstressed DISC1 mutant mice in behavioral tests, neurochemical analyses, and assessment of corticosterone levels in our previous study (Niwa et al. 2013). We used the same mouse model and followed the same protocol in our previous published studies (Niwa et al. 2013, 2016b). Furthermore, we confirmed that there are no differences among unstressed wild-type, stressed wild-type, and unstressed DISC1 mutant mice under our current experimental conditions. Therefore, we used only male mice, and defined the CTL and CTL + Veh groups as “Control” in the present study.
Drug treatment
D-cycloserine (DCS; Sigma-Aldrich, MO, USA, 30 mg/kg, subcutaneously (s.c.)), methamphetamine (METH; 1 mg/kg, intraperitoneally (i.p.)), and SKF81297 (SKF; Sigma-Aldrich, 10 nmol/bilaterally) were dissolved in saline. DL-threo-β-benzyloxyaspartate (TBOA; Tocris, MO, USA, 10 nmol/unilaterally or bilaterally) was prepared as a stock solution of 100 mM in 50% dimethyl sulfoxide and 100 mM NaOH, and dissolved in phosphate-buffered saline (PBS) before the experiments. 5-Methylpyrazole-3-carboxylic acid (MPC; Sigma-Aldrich, 100 pmol/bilaterally) was dissolved in PBS (Ogaya et al. 2010). Clozapine (CLZ; Sigma-Aldrich, 3 mg/kg, i.p.) was dissolved in 0.3% carboxymethyl cellulose sodium salt-saline. The dose of CLZ (3 mg/kg, i.p.) used in this experiment was the maximal concentration that did not cause marked sedation in mice administered acutely (Niwa et al. 2010, 2011). RU486 (mifepristone: 17-hydroxy-11-(4-dimethylamino-phenyl)-17-(prop-1-ynyl)-estra-4, 9-dien-3-one) was dissolved in saline with 2% ethanol. Mice were administered RU486 (20 mg/kg, s.c., once per day) from 5 weeks until decapitation after the behavioral experiments (Niwa et al. 2013, 2016b).
The mice were administered DCS, TBOA, MPC, and CLZ or SKF 30 and 10 min before behavioral test and training, respectively. TBOA, MPC, and SKF were microinjected into the PFc (15° angle from anteroposterior (AP) +1.7 mm, mediolateral (ML) ±1.0 mm from the bregma, dorsoventral (DV) −2.0 mm from the dura) according to the atlas of Franklin and Paxinos (2007). Microinjection cannulas attached to tubing were inserted through the stainless pipe. The other end of the tubing was connected to a 10-μl Hamilton syringe. The volumes of 1 μl/unilaterally of TBOA, MPC, and SKF were injected over a period of approximately 30 s, and the injector was left in place for 1 min to allow diffusion. The compounds injected i.p. or s.c. were administered as a volume of 0.1 ml/10 g body weight.
Behavioral analyses
Two cohorts of mice were used. The first one was tested for locomotor activity, social interaction, novelty preference, and prepulse inhibition, while the second one was subjected to the forced swim test.
Locomotor activity test
To measure novel environment-induced locomotor activity, mice were placed in a transparent acrylic cage, and locomotion and rearing were measured every 5 min for 2 h by using digital counters with infrared sensors (Scanet SV-10; Merquest), as described previously (Coitinho et al. 2002; Huang et al. 2015; Mizoguchi et al. 2010; Nakajima et al. 2004; Niwa et al. 2007, 2011, 2013; Reynolds 2003; Takamatsu et al. 2006; Toriumi et al. 2014) with minor modifications. To measure novel environment-, saline-, and METH (1 mg/kg, i.p.)-induced locomotor activity, locomotion was measured every 5 min for 6 h (habituation session for 2 h; saline session for 2 h; METH session for 2 h) (Fig. S4). The respective locomotion over time graphs show that the mice were habituated at the end of the habituation session and recovered from the saline injection before the METH injection.
Forced swim test
Each mouse was placed in a transparent glass cylinder (8 cm in diameter × 20 cm high), containing water at 22–23 °C to a depth of 15 cm, and forced to swim for 10 min. The duration of immobility was measured using digital counters with infrared sensors (Mouri et al. 2012). The time spent in immobility was calculated as follows: 600 (s) − swimming time (s) = immobility time (s).
Social interaction test
The apparatus used for the social interaction test comprised a square open arena (25 × 25 × 30 cm) with no top, made of gray non-reflecting acrylics, illuminated with lamps that were not directly visible to the mice. The light was diffused to minimize shadows in the arena. Each mouse was placed alone in the test box for 10 min for two consecutive days for habituation to the apparatus before the social interaction test. On the testing day, the mouse was randomly assigned to a same sex 8-week-old C57BL6 mouse in a different home cage as the unfamiliar partner. The mouse and its unfamiliar partner were left in this box for 10 min. The duration of social interaction (sniffing, grooming, following, mounting, and crawling except for aggressive behavior) were measured using a stopwatch after being recorded on a videotape. It must be emphasized that passive contact (sitting or lying with bodies in contact) was not considered as social interaction.
Novelty preference test
The novelty preference test was carried out as described previously (Ano et al. 2017; Mouri et al. 2007a; Nagai et al. 2003; Niwa et al. 2011; Sanna et al. 2017; Tang et al. 1999; Wang et al. 2017) with minor modifications. The experimental apparatus consisted of a Plexiglas open field box (30 × 30 × 35 cm), with the floor covered in sawdust. The test procedure consisted of three sessions: habituation, training, and retention. Each mouse was individually habituated to the box, with 10 min of exploration in the absence of objects each day for three consecutive days (habituation session). On day 4, two novel objects were symmetrically fixed to the floor of the box, 8 cm from the walls, and each animal was allowed to explore the box for 10 min (training session). The objects were different in shape and color, but similar in size. An animal was considered as exploring the object when its head was facing the object or it was touching or sniffing the object. After training sessions, the mice were immediately returned to their home cages. Even if we used the two separated objects in the training session, we did not see any differences in the exploratory preference (Fig. 1d, 6d, S1E, S1F, and S5D left-hand columns), suggesting that there was no biased exploratory preference in either group. Twenty-four hours following the training sessions, the animals were placed back into the same box with one of the familiar object from the training session and one novel object. The animals were allowed to explore freely for 10 min, and the time spent exploring each object was recorded on a videotape.
Fig. 1.
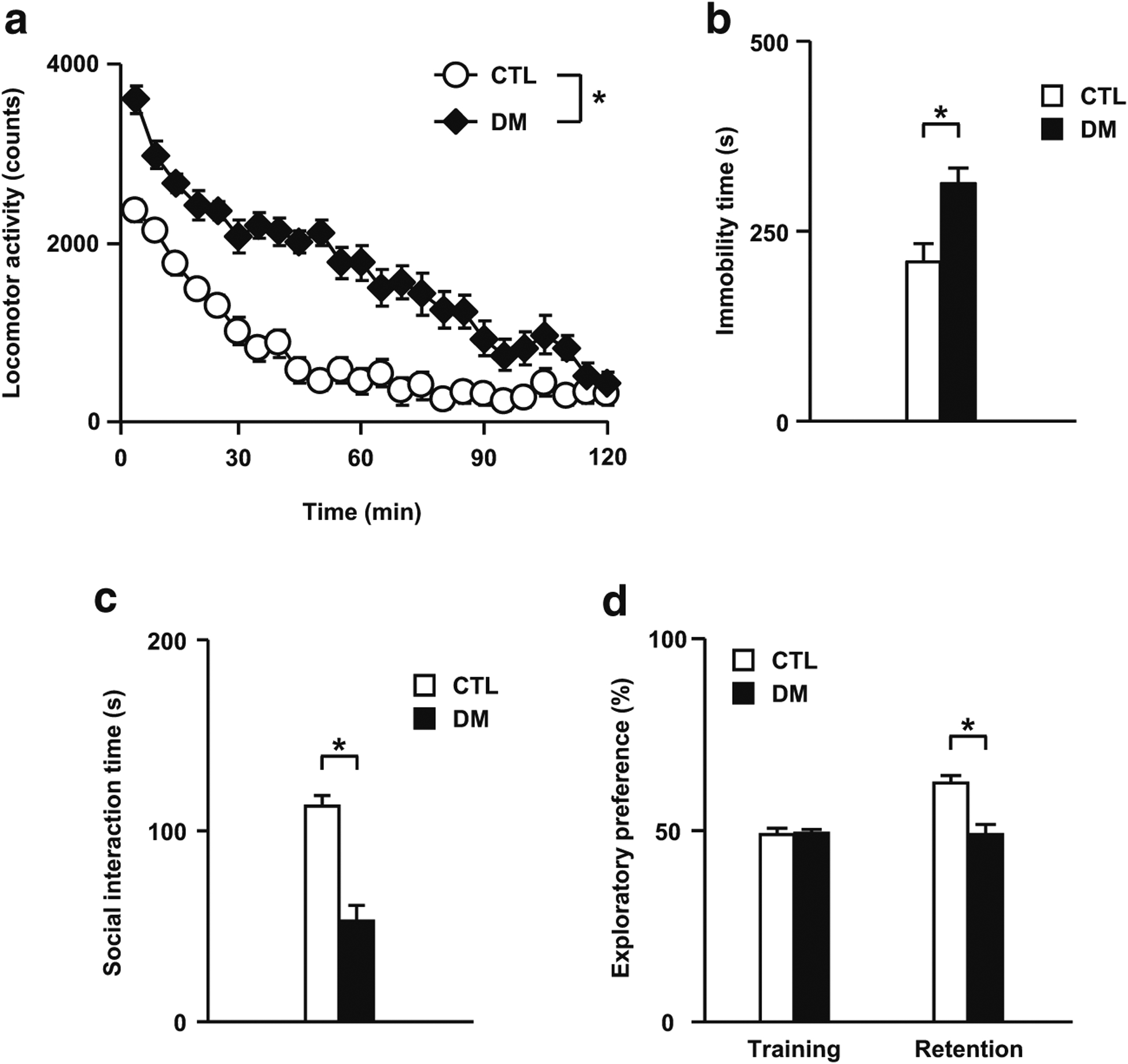
Behavioral abnormalities in the DM. a Aberrant locomotor activity. b Increased immobility in the forced swim test. c Deficits in sociability in the social interaction test. d Impaired visual memory in the novelty preference test. CTL wild-type mice without adolescent isolation, DM DISC1 mutant mice exposed to a 3-week period of isolation. N = 13–17 for a, N = 20 for b, N = 13–17 for c and d. Values are means ± SE. Statistical differences were determined using a two-way ANOVA with repeated measures (group, F(1, 28) = 68.15, P < 0.05 for a), and the t test for b to d (*P < 0.05)
Prepulse inhibition test
Mice were subjected to startle trials with three conditions: (i) a 120-dB noise burst presented alone; (ii) a prepulse that was 4, 8, or 16 dB above background noise (i.e., 74, 78, or 86 dB) followed by a 120 dB noise burst; or (iii) no stimulus (background noise alone), which was used to measure baseline movements in the startle chamber (SR-Laboratory Systems, San Diego Instruments). Percent prepulse inhibition of a startle response was calculated as:
Neurochemical and biochemical analyses
Western blotting
Western blotting was performed as described previously (Niwa et al. 2013) with a minor modification. The PFc was dissected out according to the atlas of Franklin and Paxinos (2007). Rabbit anti-phospho- NMDA receptor subunit 1 (p-NR1; 1:500, Millipore), goat anti-total-NR1 (t-NR1; 1:500, Santa Cruz Biotechnology), rabbit anti- phospho-CaMK II (p-CaMK II; 1:1000, Millipore), rabbit anti-total-CaMK II (t-CaMK II; 1:1000, Sigma-Aldrich), guinea-pig anti-GLAST (1:1000, Millipore), guinea-pig anti-GLT-1 (1:1000, Millipore), mouse anti-tyrosine hydroxylase (TH; 1:1000, Millipore), rabbit anti- dopamine D2 receptor (D2R; 1:500, Millipore), and goat anti-β-actin (1:500, Santa Cruz Biotechnology) antibodies were used as primary antibodies. Horseradish peroxidase-conjugated anti-rabbit, anti-goat, anti-guinea-pig, and anti-mouse IgG (1:2000, Kierkegaard & Perry Laboratories) were used as secondary antibodies. These biochemical assessments were made using the brains of mice isolated immediately after the forced swim test or 24 h after the last RU486 treatment.
In vivo microdialysis
Microdialysis was carried out as previously described (Murai et al. 2007; Niwa et al. 2013), with a minor modification. A guide cannula was implanted into the PFc (15° angle from AP +1.7 mm, ML −1.0 mm from the bregma, DV −2.0 mm from the dura) or nucleus accumbens (NAc; AP +1.7 mm, ML −0.8 mm from the bregma, DV −4.0 mm from the dura) according to the atlas of Franklin and Paxinos (2007). Artificial cerebrospinal fluid (147 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, and 1.2 mM MgCl2) was perfused at a flow rate of 1.0 μl/min. The dialysates were collected every 10 min and analyzed by a high-performance liquid chromatography (HPLC) system. Three or six samples were taken to establish the baseline levels of extracellular glutamate or dopamine, respectively.
The extracellular levels of glutamate upon potassium stimulation (50 mM KCl-containing Ringer solution was delivered through the dialysis probe for 30 min) or dopamine upon METH challenge (1 mg/kg, i.p.) were measured in the PFc and NAc, to clarify tonic and phasic responses in mesocortical and mesolimbic dopaminergic neurons, respectively (Grace 1991). For the rescue study with TBOA, a dialysis probe equipped with a microinjection tube was used. After the collection of baseline fractions, 10 nmol of TBOA dissolved in 1 μl of PBS were injected bilaterally during a 10-min period through the microinjection tube into the PFc.
High-performance liquid chromatography condition
Fluorescent derivatization of d-serine in rat microdialysis sample was carried out according to our previously published method with minor modifications (Fukushima et al. 2004). The HPLC system consists of an auto-sampler AS-2057 (Jasco Corporation, Tokyo, Japan); two pumps PU-2080 Plus (Jasco) and L-6200 (Hitachi, Tokyo, Japan); two fluorescence detectors L-7480 (Hitachi) and X-LCTM 3120FP (Jasco); two integrators, both Chromato-integrator D-7500s (Hitachi); and a six-port valve HV-2080–01 (Jasco) equipped with 100 μL of the sample loop. The column-switching HPLC method was carried out according to previously published studies with slight modifications (Fukushima et al. 2004). First, the peak of NBD-serine was separated from the peaks of other endogenous amino acids on the C18 column. The time program of mobile phase 1 was as follows: [CH3CN-0.1% TFA in H2O (10:90)]: [CH3CN-0.1% TFA in H2O (90:10)] (92:8) was isocratically eluted from 0 to 25 min at a flow rate of 0.8 mL/min, then CH3CN-0.1% TFA in H2O (90:10) from 25.1 to 35 min at 0.8 mL/min, and finally, [CH3CN-0.1% TFA in H2O (10:90)]: [CH3CN-0.1% TFA in H2O (90:10)] (92:8) from 35.1 to 45 min at 0.8 mL/min to start the C18 column. The fluorescence detector wavelength was set at 540 nm with an excitation wavelength of 470 nm. Both C18 (TSKgel ODS-80Ts) with a guard column (TSKguardgel ODS-80Ts) and a tandem series of two chiral columns (Sumichiral OA-2500(S)) were kept at 40 °C in a column oven CO-2065 Plus (Jasco).
Real-time reverse transcription polymerase chain reaction
Total RNA was isolated using an RNeasy Kit (Qiagen, Hilden, Germany) and converted into cDNA using a SuperScript™ III First-Strand System for reverse transcription polymerase chain reaction (RT-PCR) Kit (Invitrogen). The levels of serine racemase and d-amino acid oxidase mRNA were determined by real-time RT-PCR using a TaqMan probe. β-Actin mRNA was used as the internal control. The serine racemase primers used for real-time RT-PCR were as follows: 5′-CTGG ACAAGGAACAATTG-3′ (forward) and 5′-GGGC CTTAATTGTAATGG-3′ (reverse), and the TaqMan probe was 5′-CAACCATTCCTCCTCCTCCTACTG-3′ (probe). The d-amino acid oxidase primers used for real-time RT-PCR were as follows: 5′-CTGAGAGGTTAACTGAGA-3′ (forward) and 5′-GTGCAGTTGATAATCACA-3′ (reverse), and the TaqMan probe was 5′-ACTCCTCTTGCCAC CTCTTCG-3′ (probe). The β-actin primers used for real-time RT-PCR were as follows: 5′-GGGCTATGCTCTCC CTCACG-3′ (forward) and 5′-GTCACGCACGATTT CCCTCTC-3′ (reverse), and the TaqMan probe was 5′-CCTGCGTCTGGACCTGGCTGGC-3′ (probe). The amplification consisted of an initial step (95 °C for 3 min) followed by 40 cycles of denaturation for 1 min at 95 °C, annealing for 1 min at 55 °C, and the extension time for 1 min at 72 °C in an iCycle iQ Detection System (Bio-Rad Laboratories, Inc., CA, USA). The expression levels were calculated as described previously (Niwa et al. 2007).
Statistical analyses
All data were expressed as means ± SE. Statistical analyses were performed using the commercial software (IBM SPSS statistics 23, IBM). Statistical differences between two groups were calculated using Student’s t test. Statistical differences among three groups/factors or more were determined using a one-way analysis of variance (ANOVA) and a two-way ANOVA with repeated measures, followed by the Bonferroni post hoc tests. Corrections for multiple comparisons were made when appropriate. Statistical significance was *P < 0.05.
Results
Behavioral deficits in the DM
The DM animals showed robust deficits in the novelty-induced locomotor activity (Fig. 1a), forced swim (Fig. 1b), and prepulse inhibition (Fig. 6e) tests, which was consistent with our previous findings (Niwa et al. 2013, 2016b). In this study, to examine the roles of psychosocial stress in sociability, learning, and memory in our model, we performed social interaction and novelty preference tests. The DM animals showed significant decreases in social interaction than the CTL did (Fig. 1c), suggesting that adolescent stress leads to deficits in sociability in adulthood. As shown in Fig. 1d, there were no significant differences in the exploratory preference between two objects during the training session (Fig. 1d, two left-hand columns), suggesting that there was no biased exploratory preference in either group. However, for the retention session, the levels of exploratory preference for the novel objects in the DM significantly decreased compared to those in CTL (Fig. 1d, two right-hand columns), suggesting impaired learning and memory in DM animals.
Fig. 6.
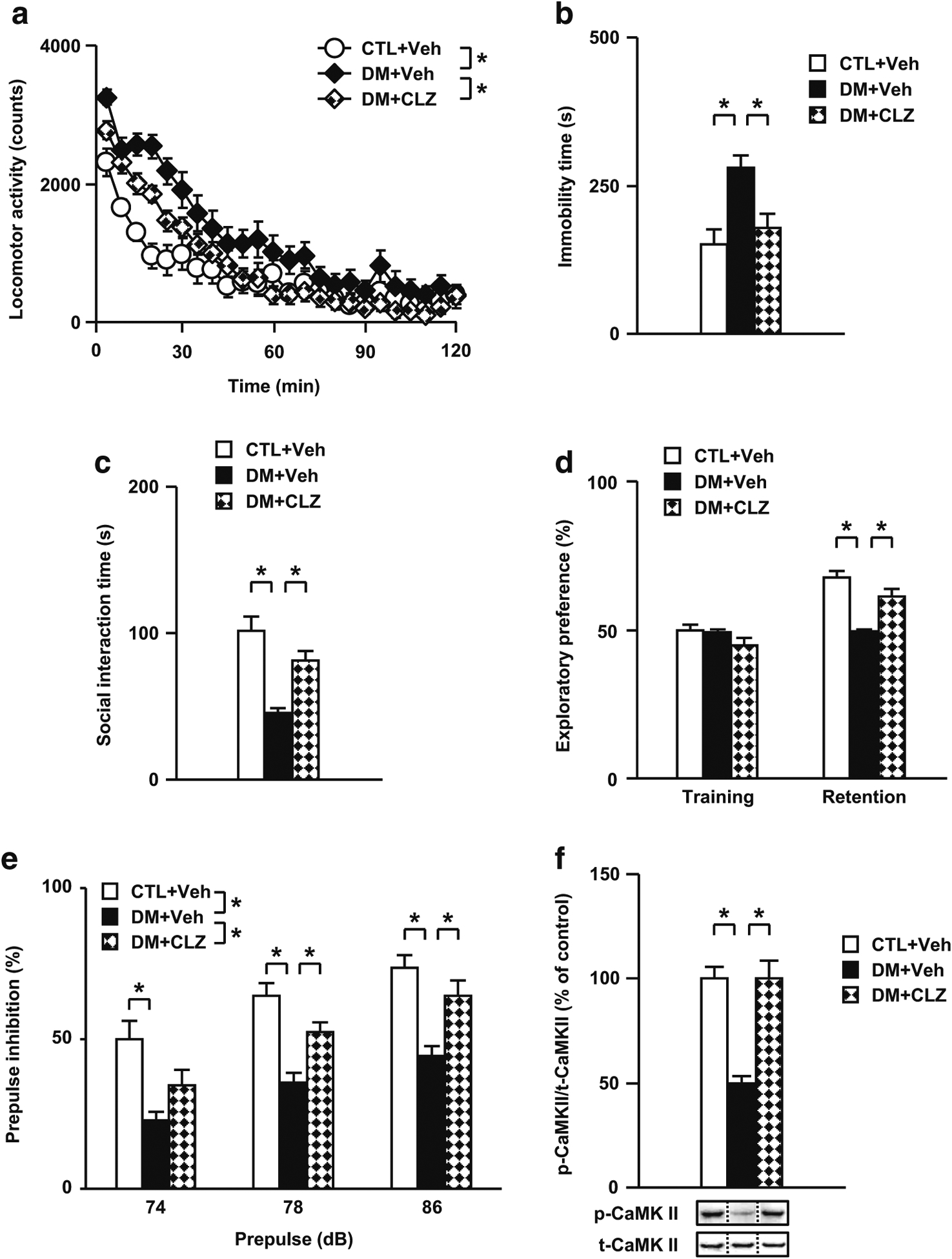
Effects of an atypical antipsychotic drug CLZ on behavioral and neurochemical abnormalities in the DM. a Effects of CLZ on aberrant locomotor activity. b Effects of CLZ on the impaired performance in the forced swim test. c Effects of CLZ on the impaired performance in the social interaction test. d Effects of CLZ on the impaired performance in the novelty preference test. e Effects of CLZ on the impaired performance in the prepulse inhibition test. f Normalization effect of CLZ on the decreased rate of CaMK II phosphorylation in the PFc of the DM after the forced swim test. p- phospho-, t- total-, Veh treated with vehicle, CLZ treated with clozapine. N = 11–12 for a, N = 9–11 for b, N = 11–12 for c to e, N = 7 for f. Values are means ± SE. Statistical differences were determined using a two-way ANOVA with repeated measures (group, F(2, 32) = 7.31, P < 0.05 for a; F(2, 32) = 15.68, P < 0.05 for e), and one-way ANOVA (group, F(2, 27) = 7.92, P < 0.05 for b; F(2, 32) = 16.70, P < 0.05 for c; F(2, 32) = 25.41, P < 0.05 for retention of d; F(2, 32) = 7.93, P < 0.05 for prepulse 74 in e; F(2, 32) = 16.91, P < 0.05 for prepulse 78 in e; F(2, 32) = 14.49, P < 0.05 for prepulse 86 in e; F(2, 18) = 21.79, P < 0.05 for f), followed by Bonferroni post hoc tests (*P < 0.05)
Role of glucocorticoids in the behavioral deficits observed in the DM
In our previous studies, the DM showed a significant increase in levels of corticosterone (Niwa et al. 2013, 2016b). A glucocorticoid receptor antagonist, RU486, normalized mesocortical dopaminergic disturbance and ameliorated behavioral deficits observed in the DM (Niwa et al. 2013, 2016b). Thus, we next investigated whether RU486 also ameliorates the behavioral deficits observed in the present experiments. We first confirmed our previous data that RU486 significantly ameliorates the impaired performance in locomotor activity in the DM animals (Fig. S1A). We examined whether RU486 ameliorates the impairment of performance in the social interaction and novelty preference tests. Treatment with RU486 significantly ameliorated the decrease in the exploratory preference during the novelty preference test in the DM (Fig. S1E). Moreover, the treatment with RU486 tended to ameliorate the impaired performance in the social interaction test in the DM (Fig. S1C). However, RU486 had no effect on behavioral scores in CTL (Fig. S1B, D, and F). These results suggest that elevation in the levels of corticosterone, a rodent glucocorticoid, could underlie the behavioral deficits in the DM.
Alterations of glutamatergic neurons in the PFc of the DM
We have previously reported a hypofunction of the mesocortical dopaminergic neuron in the DM (Niwa et al. 2013). Increasing evidence suggests that dopaminergic neurons regulate glutamatergic neurons in the PFc (Aoyama et al. 2014; Mouri et al. 2007b; Swerdlow et al. 2001; Warden et al. 2012). Thus, we examined glutamatergic alterations underlying the behavioral deficits in the DM. We investigated the changes in the activation of CaMK II signaling via NMDA receptors in the PFc after the forced swim test, since we had previously reported that the phosphorylation levels of NR1, a subunit of NMDA receptors, and CaMK II in the PFc decreased in PCP-treated mice, showing increased immobility after the forced swim test (Murai et al. 2007). The phosphorylation levels of NR1 and CaMK II in the DM significantly decreased compared with those of CTL (Fig. 2a, b). No difference in the expression levels of total NR1 and CaMK II was observed between the two groups (data not shown).
Fig. 2.
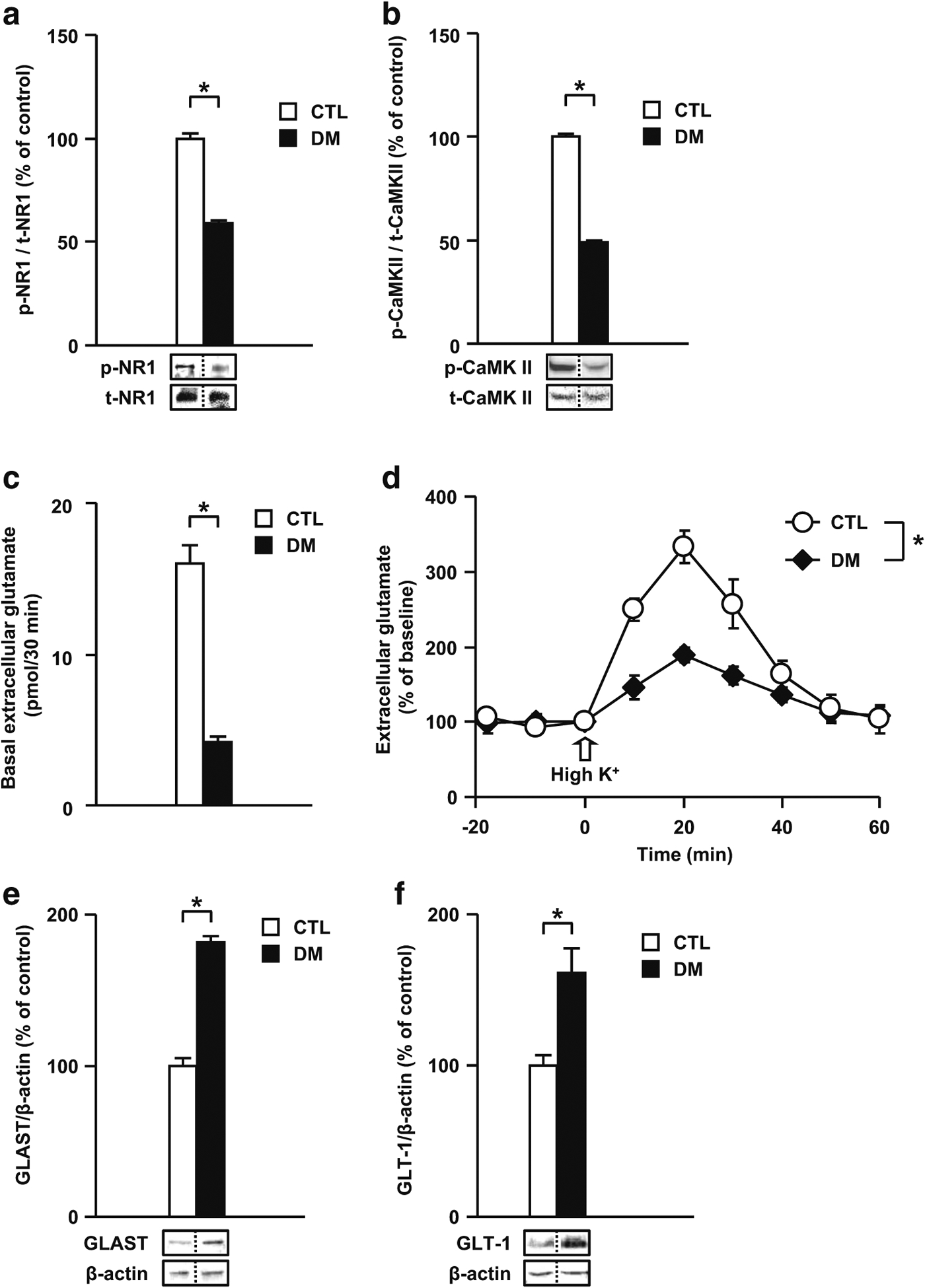
Glutamatergic disturbances in the DM. a Levels of phospho-NR1/total-NR1 in the PFc. b Levels of phospho-CaMK II/total-CaMK II in the PFc. c Extracellular levels of glutamate at baseline in the PFc. d Extracellular levels of glutamate upon high K+ stimulation in the PFc. Extracellular levels of glutamate were measured by in vivo microdialysis. e Levels of GLAST in the PFc. f Levels of GLT-1 in the PFc. p- phospho-, t- total-. N = 7 for a, b, e, and f; N = 6–7 for c and d. Values are means ± SE. Statistical differences were determined using the t test for a, b, c, e, and f, and the two-way ANOVA with repeated measures (group, F(1, 12) = 12.39, P < 0.05 for d), (*P < 0.05)
To investigate the mechanisms underlying the decrease in the levels of p-NR1 and p-CaMK II in the DM, we examined the extracellular levels of glutamate in the PFc of the DM by using an in vivo microdialysis technique. After the basal extracellular levels of glutamate reached a steady state, they were monitored during 30 min of dialysis. The DM animals showed dramatically decreased extracellular levels of glutamate at baseline in the PFc (DM 4.21 ± 0.35 pmol/30 min, CTL 16.03 ± 1.22 pmol/30 min) (Fig. 2c). We investigated the effect of high potassium (high K+) (50 mM) stimulation in the PFc on the extracellular levels of glutamate in the DM. The extracellular levels of glutamate in the PFc of the DM were significantly lower than those in CTL (Fig. 2d).
Glial glutamate transporters, i.e., GLAST and GLT-1, play an important role in regulating glutamate transmission by rapidly clearing glutamate from extracellular fluid. To examine whether the decrease in the extracellular levels of glutamate was due to changes in the expression levels of glutamate transporters, we investigated the levels of glial glutamate transporters in the PFc of the DM by immuno-blotting. The levels of GLAST and GLT-1 significantly increased in the DM compared with those in CTL (Fig. 2e, f). These results suggest that glutamatergic signaling is altered in the DM and that the increase in the expression levels of glutamate transporters may be responsible for decreased extracellular levels of glutamate, both at baseline and upon K+-stimulation, and decreased phosphorylation of NR1 and CaMK II.
Role of glutamate transporters in the behavioral deficits and impairment of CaMK II activation in the DM
GLAST and GLT-1 mediate glutamate uptake by astrocytes (Pines et al. 1992; Storck et al. 1992), and the ability of excitatory amino acid transporters to rapidly and efficiently clear synaptic glutamate is integral to physiological excitatory neurotransmission (Chawla et al. 2017; Haydon and Carmignoto 2006; Rosenberg and Aizenman 1989). It has been suggested that the disturbance of the glutamatergic system shown in this experiment is due to an increased expression of glial glutamate transporters (Murai et al. 2007). We investigated whether a competitive glutamate transport inhibitor TBOA would ameliorate the behavioral deficits and glutamatergic disturbance in the DM. As shown in Fig. 3a, the treatment with TBOA in the PFc significantly ameliorated the increased immobility time in the DM. TBOA also significantly ameliorated the decreased basal extracellular levels of glutamate in the PFc of the DM (Fig. 3b). Furthermore, TBOA significantly ameliorated the decreased phosphorylation levels of CaMK II in the PFc of the DM (Fig. 3c). However, TBOA had no effect on behavioral and neurochemical measurements in CTL (Fig. S2). These results suggest that the increase in the expression levels of glial glutamate transporter may be related to a decrease in the extracellular levels of glutamate in the PFc of the DM.
Fig. 3.
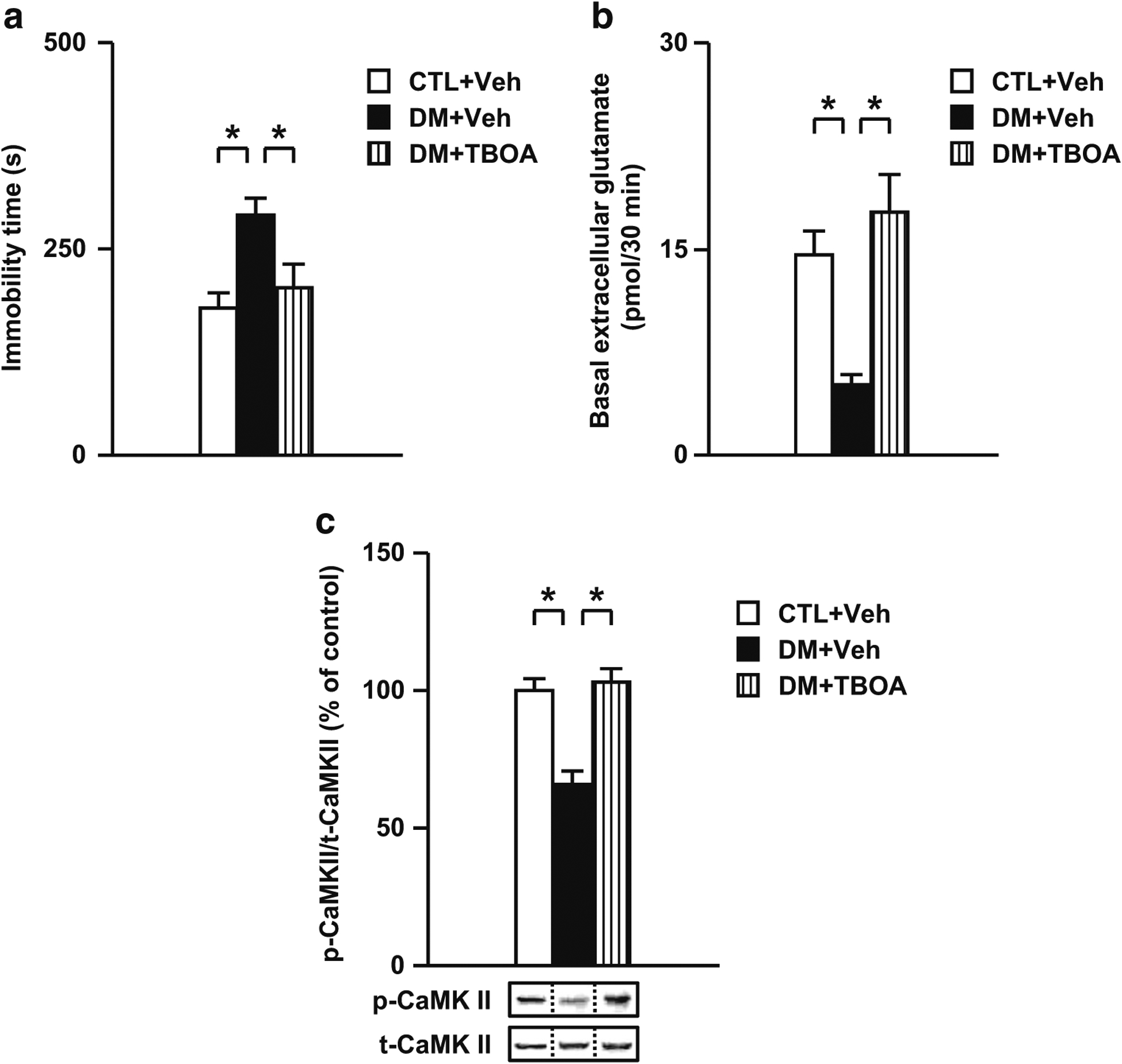
Effects of a competitive glutamate transport inhibitor TBOA on behavioral and neurochemical abnormalities in the DM. a Effects of TBOA on the impaired performances in the forced swim test. b Normalization effect of TBOA on the decreased extracellular levels of glutamate at baseline in the PFc of the DM. c Normalization effect of TBOA on the decreased rate of CaMK II phosphorylation in the PFc of the DM after the forced swim test. p- phospho-, t- total-, Veh treated with vehicle, TBOA treated with DL-threo-β-benzyloxyaspartate. N = 12–14 for a, N = 7–9 for b, N = 5 for c. Values are means ± SE. Statistical differences were determined using a one-way ANOVA (group, F(2, 37) = 6.45, P < 0.05 for a; F(2, 23) = 10.87, P < 0.05 for b; F(2, 12) = 17.35, P < 0.05 for c), followed by Bonferroni post hoc tests (*P < 0.05)
Changes of the serine system in the PFc of the DM
D-serine modulates glutamatergic neuronal function as an endogenous co-agonist of NMDA receptors. Therefore, we investigated whether levels of d-serine and its synthetizing/degradation enzymes were altered in the DM. As shown in Fig. 4, the concentration of d-serine significantly decreased in the DM (Fig. 4a). Further, in the DM animals, the mRNA expression levels of serine racemase, a synthetizing enzyme of d-serine, significantly decreased (Fig. 4b), whereas the mRNA expression levels of its degradation enzyme d-amino acid oxidase significantly increased (Fig. 4c). These results suggest that the aberrant d-serine system may, at least in part, decrease glutamatergic neuronal activity in the PFc of the DM.
Fig. 4.
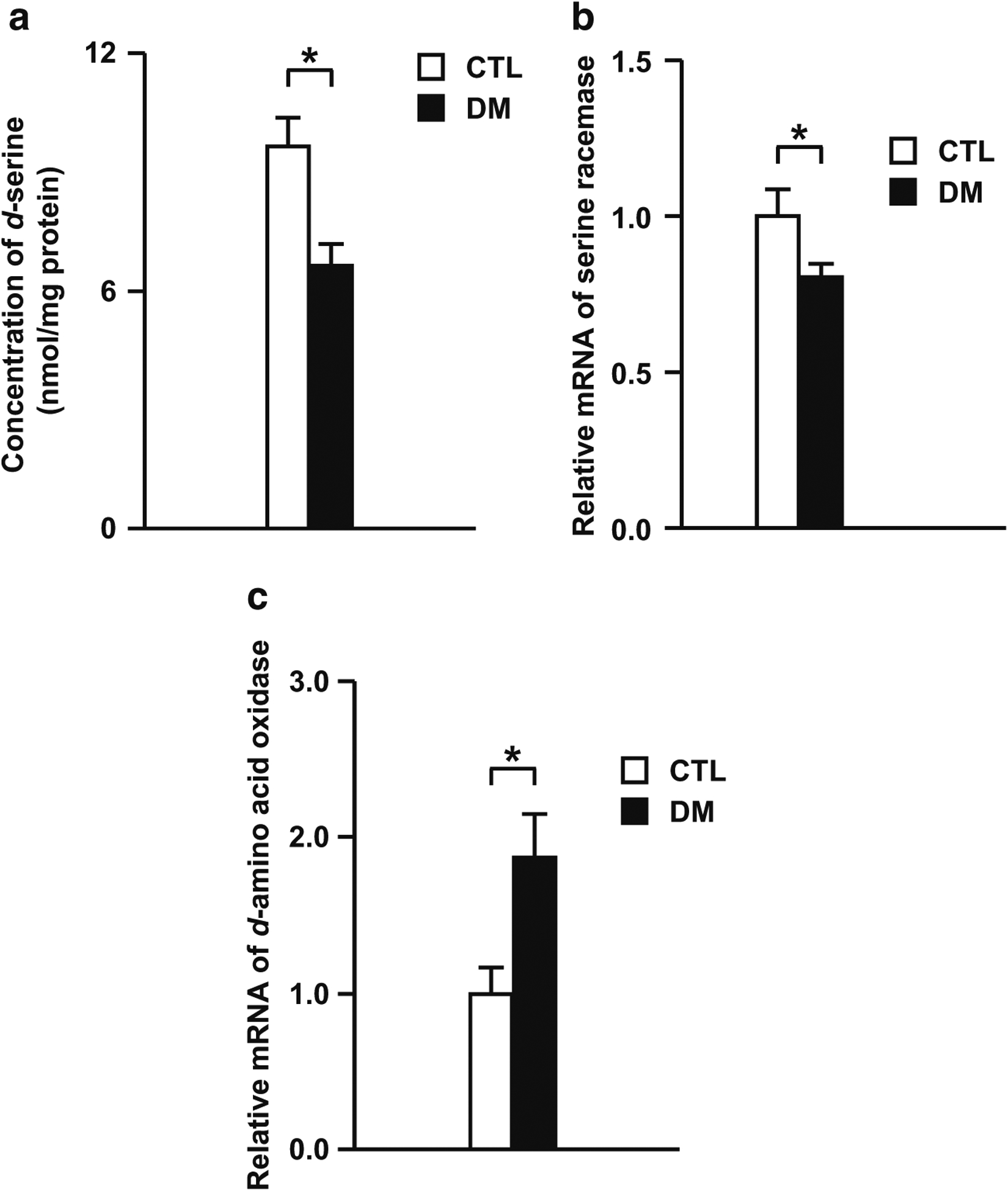
Serine system disturbance in the DM. a Concentration of d-serine in the PFc of the DM. b The mRNA expression levels of serine racemase in the PFc of the DM. c The mRNA expression levels of d-amino acid oxidase in the PFc of the DM. N = 8 for a, N = 14 for b, N = 13–14 for c. Values are means ± SE. Statistical differences between two groups were analyzed with the t test (*P < 0.05)
Roles of the NMDA receptor and serine system in the behavioral deficits and impairment of CaMK II activation in the DM
To investigate the relationship between behavioral deficits and glutamatergic disturbance, the effects of DCS, a partial NMDA receptor glycine-site agonist, on impaired performance in the forced swim test and changes in CaMK II activation in the DM were examined. As shown in Fig. 5a, acute treatment with DCS in the PFc significantly ameliorated the increased immobility time in the forced swim test in the DM. Moreover, the treatment with DCS significantly ameliorated the decreased phosphorylation levels of CaMK II in the PFc of the DM (Fig. 5b). However, DCS had no effect on behavioral and neurochemical measurements in CTL (Fig. S3A, B). These results suggest that the impairment of NMDA-CaMK II signaling in the PFc may be, at least in part, involved in the behavioral deficits in the DM.
Fig. 5.
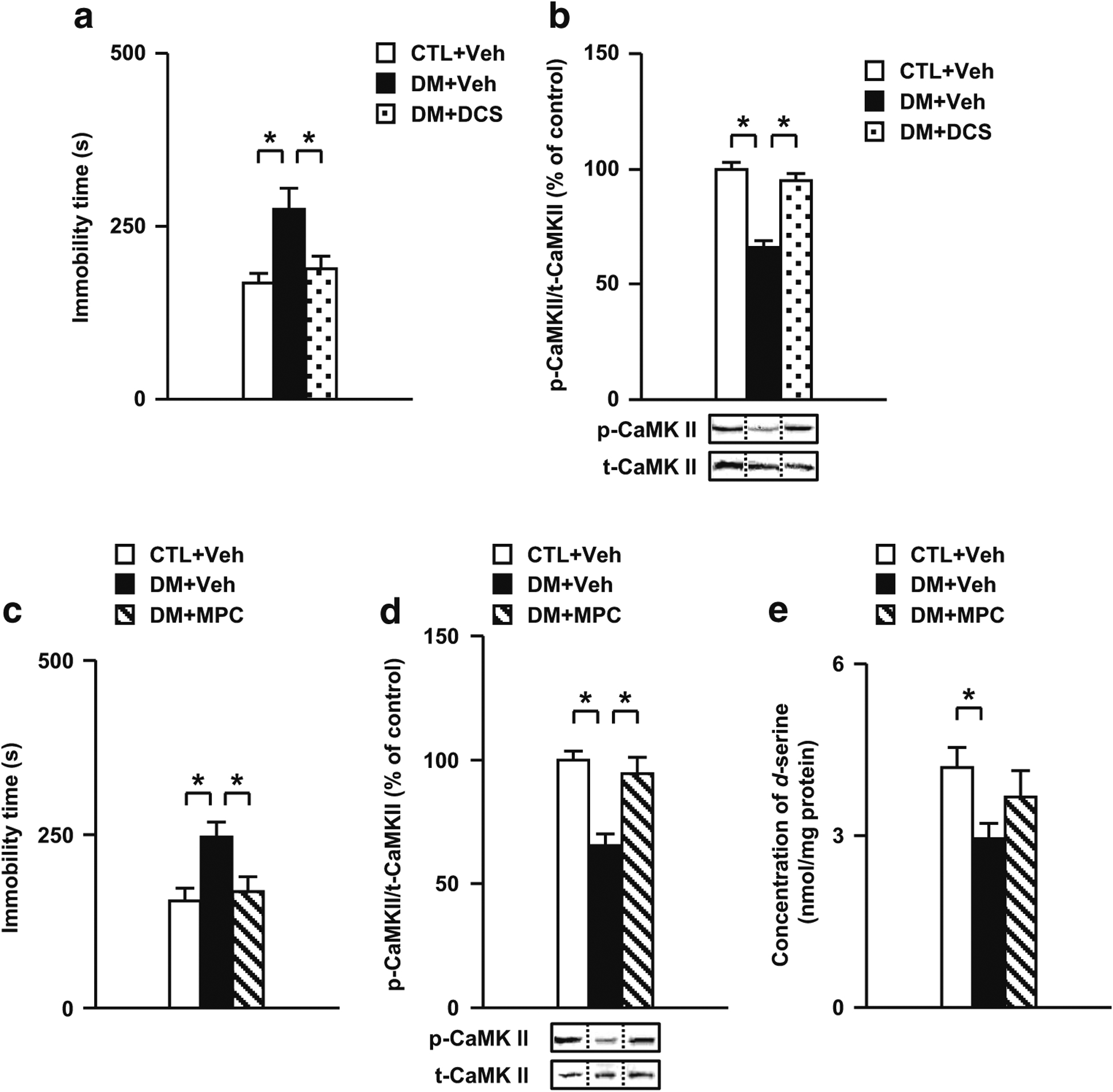
Effects of a partial NMDA receptor glycine-site agonist DCS and an inhibitor of d-amino acid oxidase MPC on behavioral and neurochemical abnormalities in the DM. a Effects of DCS on the impaired performances in the forced swim test. b Normalization effect of DCS on the decreased rate of CaMK II phosphorylation in the PFc of the DM after the forced swim test. c Effects of MPC on the impaired performances in the forced swim test. d Normalization effect of MPC on the decreased rate of CaMK II phosphorylation in the PFc of the DM after the forced swim test. e Effects of MPC on the decreased concentration of d-serine in the PFc of the DM. p- phospho-, t- total-, Veh treated with vehicle, DCS treated with D-cycloserine, MPC treated with 5-methylpyrazole-3-carboxylic acid. N = 12 for a, N = 10 for b, N = 17–19 for c, N = 6 for d, N = 10–13 for e. Values are means ± SE. Statistical differences were determined using one-way ANOVA (group, F(2, 33) = 6.54, P < 0.05 for a; F(2, 27) = 42.24, P < 0.05 for b; F(2, 51) = 6.33, P < 0.05 for c; F(2, 15) = 13.32, P < 0.05 for d; F(2, 33) = 3.50, P < 0.05 for e), followed by Bonferroni post hoc tests (*P < 0.05)
To investigate the relationship between behavioral deficits and abnormalities in the serine system, the effects of MPC, an inhibitor of d-amino acid oxidase, on the forced swim test, impairments of CaMK II activation, and the serine system in the DM were examined. As shown in Fig. 5c, acute treatment with MPC in the PFc significantly ameliorated the increased immobility time during the forced swim test in the DM. Moreover, the treatment with MPC significantly ameliorated decreased phosphorylation levels of CaMK II in the PFc of the DM (Fig. 5d). Furthermore, MPC tended to normalize the decreased concentration of d-serine in the PFc of the DM (Fig. 5e). However, MPC had no effect on behavioral and neurochemical measurements in CTL (Fig. S3C to E). These results suggest that abnormalities in the serine system in the PFc may be, at least in part, involved in behavioral deficits in the DM via changes in NMDA-CaMK II signaling.
Effects of CLZ on the behavioral deficits and impairment of CaMK II activation in the DM
We have reported that CLZ, an atypical antipsychotic drug, ameliorated the behavioral deficits and impairment of CaMK II activation in the PCP animal model (Aoyama et al. 2014; Mouri et al. 2007b). Therefore, we investigated whether CLZ ameliorates the behavioral deficits and glutamatergic disturbance in the DM. The treatment with CLZ significantly ameliorated the increased locomotor activity, especially the first 30 min of the test session, in the DM (Fig. 6a). We next examined the effects of CLZ on novelty-, saline-, and METH-induced hyperactivity observed in the DM. Although the DM displayed hyperlocomotion compared to CTL, the treatment with CLZ significantly improved their hyperactivity (Fig. S4A). CLZ also significantly attenuated METH-induced hyperactivity in the DM. Furthermore, treatment with CLZ significantly ameliorated the increased immobility in the forced swim test in the DM (Fig. 6b). Moreover, CLZ significantly ameliorated not only the decrease of social interaction in the social interaction test, but also the decrease of exploratory preference in the novelty preference test in the DM (Fig. 6c, d). We also examined whether the deficit in sensory motor gating in the DM was reversed by CLZ. Although the DM showed decreased prepulse inhibition compared to CTL, the treatment with CLZ significantly improved the decreased prepulse inhibition in these animals (Fig. 6e). There were no significant differences in startle responses (data not shown). Further, CLZ significantly ameliorated the decreased phosphorylation levels of CaMK II in the PFc of the DM (Fig. 6f). However, CLZ had no effect on behavioral and neurochemical measurements in CTL (Fig. S4B, S5). These results suggest that CLZ may ameliorate behavioral deficits in the DM by normalizing altered NMDA-CaMK II signaling in the PFc.
The effects of CLZ on the dopaminergic disturbances in the DM
The following data from the present study support our previous findings regarding dopaminergic disturbance in the DM (Niwa et al. 2013, 2016b): (1) The extracellular levels of dopamine at baseline significantly decreased in the PFc but not in the NAc of the DM (Fig. 7a, c) compared with the CTL group. (2) The DM animals showed increased extracellular levels of dopamine upon METH challenge in the NAc but not in the PFc (Fig. 7b, d). (3) The DM animals showed changes in the expression levels of TH and D2R in the PFc. We investigated whether CLZ ameliorates these dopaminergic disturbances. CLZ normalized the dopamine-related abnormalities including basal and METH-induced extracellular levels of dopamine, but not the expression levels of TH and D2R in the DM (Fig. 7b, c, e, f). However, CLZ had no effect on neurochemical measurements in CTL (Fig. S6). These results suggest that CLZ may normalize dopaminergic disturbances and subsequent behavioral deficits by increasing basal extracellular levels of dopamine in the DM.
Fig. 7.
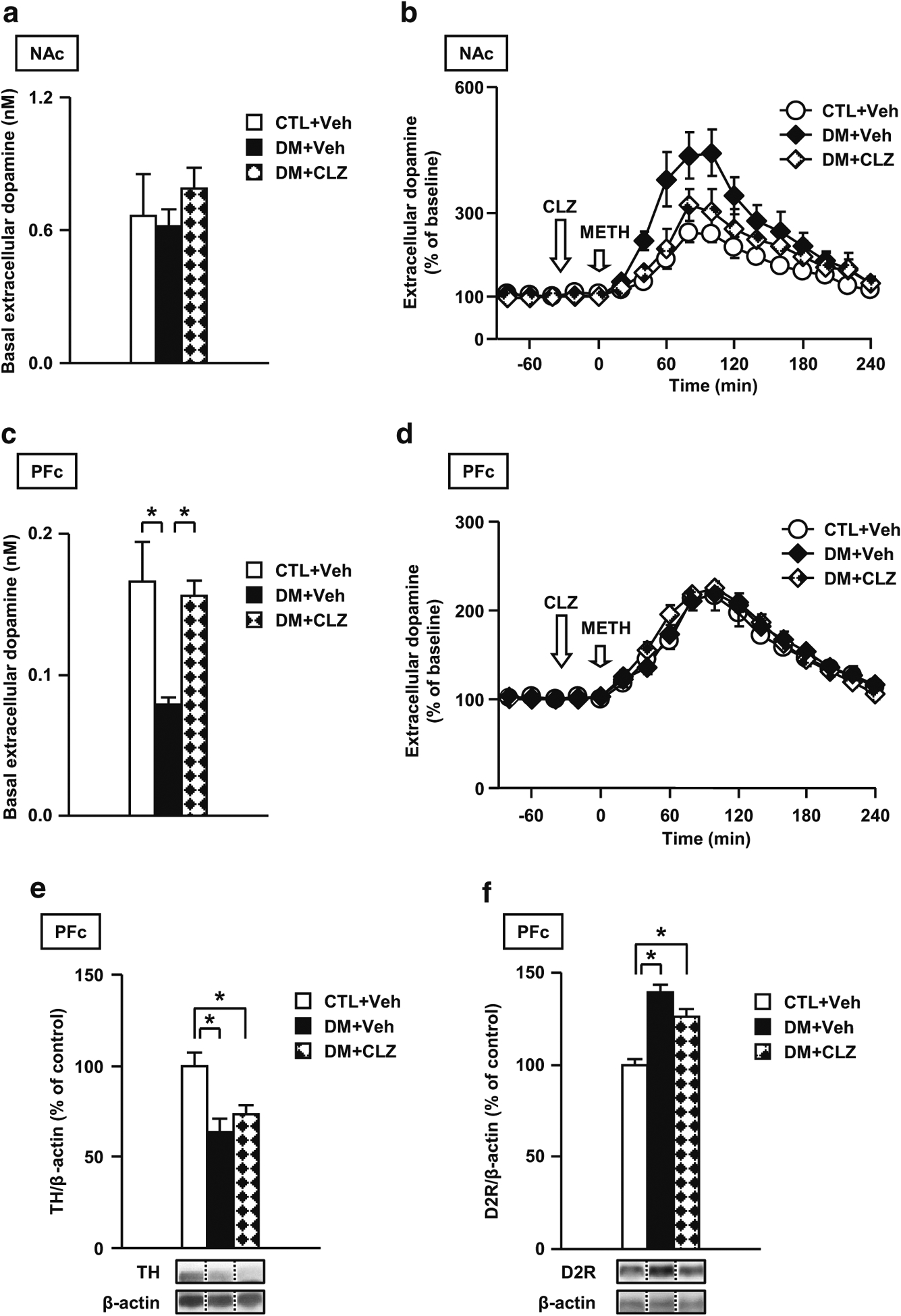
Effects of an atypical antipsychotic drug CLZ on dopaminergic disturbance in the DM. a Effects of CLZ on the extracellular levels of dopamine at baseline in the NAc. b Effects of CLZ on the levels of dopamine upon METH challenge in the NAc. c Effects of CLZ on the extracellular levels of dopamine at baseline in the PFc. d Effects of CLZ on the levels of dopamine upon METH challenge in the PFc. Effects of CLZ on the decreased levels of TH (e) and increased levels of D2R (f) in the PFc of the DM. Veh treated with vehicle, CLZ treated with clozapine. N = 7 for a and b, N = 7–8 for c and d, N = 6 for e and f. Values are means ± SE. Statistical differences were determined using a two-way ANOVA with repeated measures (group, F(2, 18) = 3.25, P > 0.05 for b; F(2, 19) = 0.39, P > 0.05 for d), and a one-way ANOVA (group, F(2, 18) = 0.50, P > 0.05 for a; group, F(2, 19) = 6.25, P < 0.05 for c; F(2, 15) = 8.49, P < 0.05 for e; F(2, 15) = 26.83, P < 0.05 for f), followed by Bonferroni post hoc tests (*P < 0.05)
Role of D1 receptor in the behavioral deficits and impairment of NR1 and CaMK II activation in the DM
We have reported that CLZ ameliorated the behavioral deficits and impairment of CaMK II activation through the activation of the dopamine D1 receptor in the PCP animal model (Aoyama et al. 2014; Mouri et al. 2007b). In addition, the infusion of a dopamine-D1 receptor agonist into the PFc attenuates the impairment of latent learning and the decrease of learning-associated NR1 phosphorylation, suggesting a functional linkage between glutamatergic and dopaminergic signaling (Aoyama et al. 2014; Hida et al. 2015; Mouri et al. 2007b). Thus, we investigated whether SKF, a dopamine-D1 receptor agonist, ameliorates the behavioral deficits and impairment of NR1 and CaMK II activation in the PFc of the DM. As shown in Fig. 8a, acute treatment with SKF in the PFc significantly ameliorated the increased immobility time in the DM. Moreover, treatment with SKF significantly ameliorated the decreased levels of phospho-NR1 and phospho-CaMK II in the DM (Fig. 8b, c). However, SKF had no effect on behavioral and neurochemical measurements in CTL (Fig. S7). These results suggest that abnormal behaviors in the DM resulting from the alterations of NMDA-CaMK II signaling in the PFc may be associated with a disturbance in dopaminergic function.
Fig. 8.
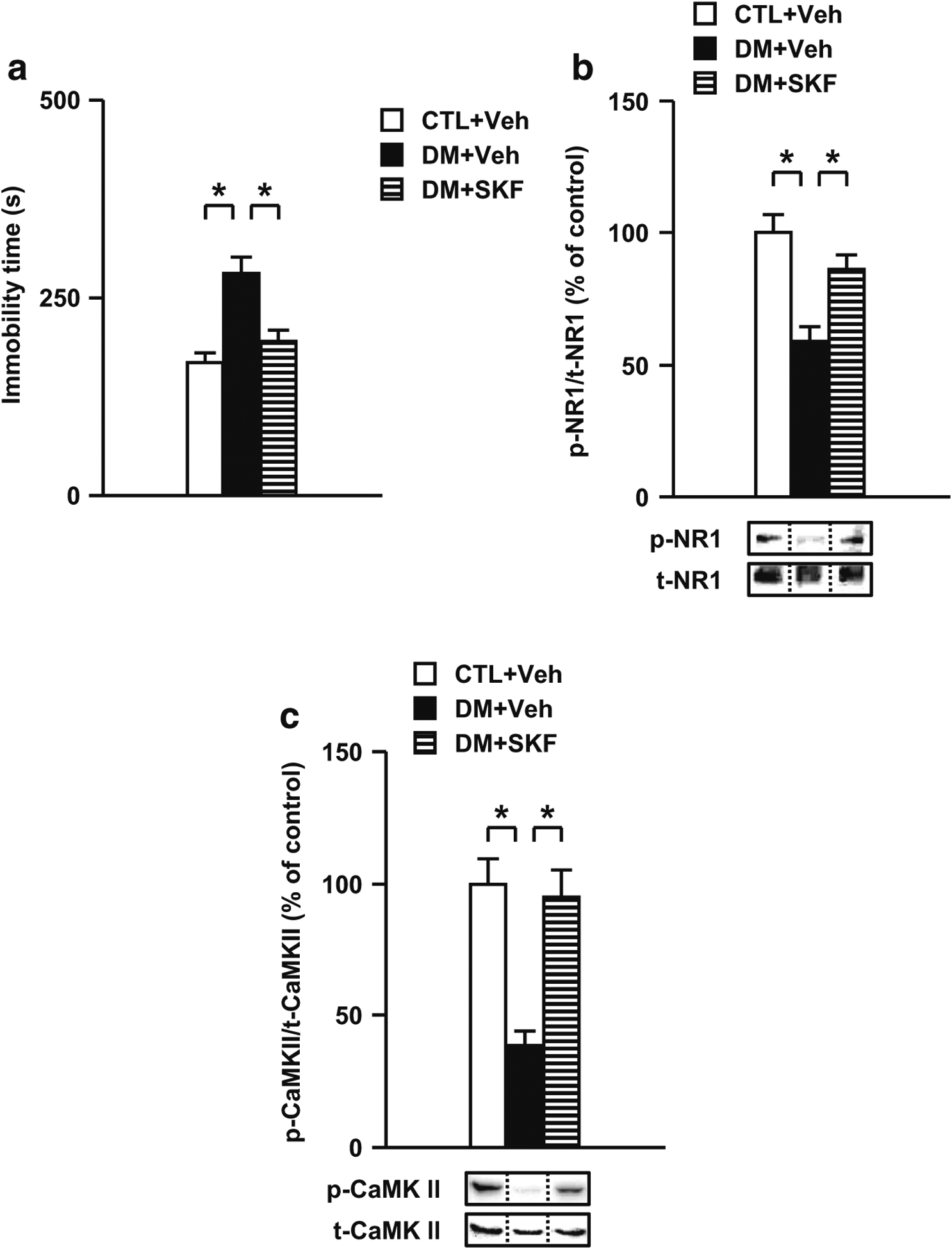
Effects of a dopamine-D1 receptor agonist SKF on behavioral and neurochemical abnormalities in the DM. a Effects of SKF on impaired performance in the forced swim test. b Effects of SKF on the decreased NR1 phosphorylation in the PFc of the DM after the forced swim test. c Normalization effect of SKF on the decreased CaMK II phosphorylation in the PFc of the DM after the forced swim test. Veh treated with vehicle, SKF treated with SKF81297. N = 8–10 for a, N = 7 for b, N = 8 for c. Values are means ± SE. Statistical differences were determined using a one-way ANOVA (group, F(2, 27) = 12.75, P < 0.05 for a; F(2, 18) = 13.94, P < 0.05 for b; F(2, 21) = 15.85, P < 0.05 for c), followed by Bonferroni post hoc tests (*P < 0.05)
Discussion
The ultimate goal of our research is to determine how neuroendocrinological changes induced by psychosocial stress alter neuronal function and connectivity in the brain, and dissect the mechanisms from the cellular level to circuitry and behavior. In this exploratory study, we aimed to address three unanswered questions related to the use of previously described genetically vulnerable mice exposed to late adolescent stress (Hayashi et al. 2016; Niwa et al. 2013, 2016b).
First, we aimed to determine whether sociability, learning, and memory were altered in the DM. Deficits in sociability in addition to impaired learning and memory are the key clinical features of several mental disorders including schizophrenia, and are attributed to alterations in neural systems (Gunaydin et al. 2014; Hayashi et al. 2016; Nagai et al. 2011; Tomoda et al. 2016). We observed impaired social interaction and novelty preference for object recognition memory in addition to dopaminergic and glutamatergic disturbances in the DM. These behavioral and neurochemical deficits were prevented by the treatment with CLZ, one of the atypical antipsychotics, which is coincident with clinical reports of psychiatric disorders including schizophrenia (Lee et al. 1999; Owen et al. 2016). It has been reported that dopaminergic hypofunction in the PFc is, at least in part, related to deficits in sociability, and learning and memory impairments in psychiatric disorders including schizophrenia (Coura et al. 2013; Hultman et al. 2016; Niwa et al. 2010, 2011). CLZ preferentially increases dopamine release in the PFc compared with that in the NAc (Kuroki et al. 2008; Nagai et al. 2011; Youngren et al. 1999). Our results have therefore confirmed previously published findings. In addition, a D1R agonist SKF also ameliorated both behavioral and neurochemical abnormalities. Therefore, we propose that the beneficial effect of CLZ on behavioral deficits may be related to the enhancement of baseline dopamine transmission in the PFc (Fig. 9).
Fig. 9.
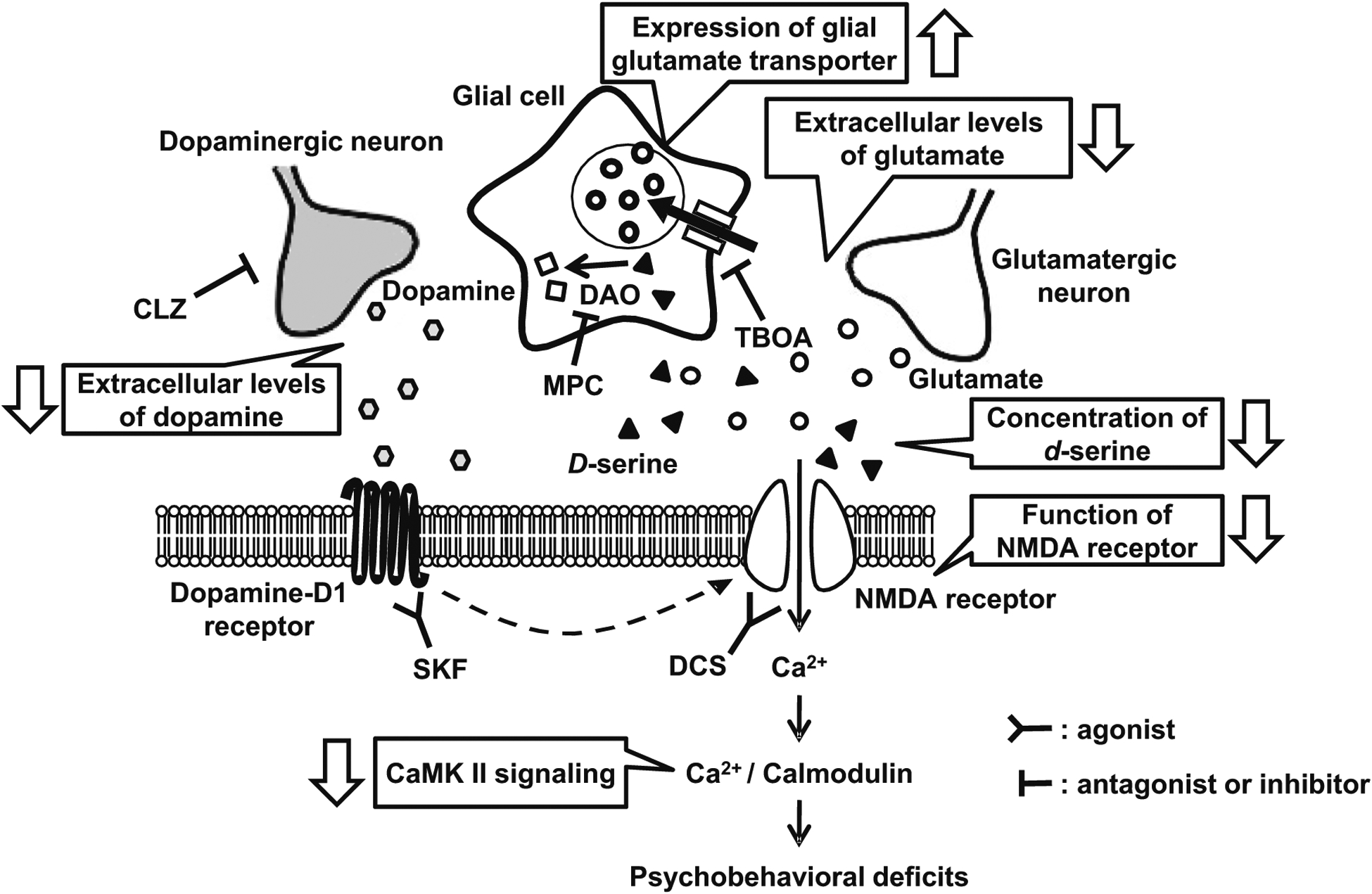
Adolescent stress leads to glutamatergic disturbance through alterations in dopaminergic signaling in the prefrontal cortex of the DM. Our results suggest that presynaptic and postsynaptic glutamate and dopamine transmissions in the PFc are impaired by adolescent stress in genetically susceptible animals. In the presynaptic glutamate transmission in the PFc, (1) the upregulation of glial glutamate transporters (GLAST, GLT-1), and (2) the decreased extracellular levels of dopamine and glutamate are observed. In the postsynaptic glutamate transmission in the PFc, (3) the functions of the NMDA receptor and serine system are impaired. These abnormalities of glutamate and dopamine transmissions, and a disturbance of the serine system induced (4) the abnormality of NMDA-CaMK II signaling via dopamine D1 receptor and (5) behavioral deficits. TBOA DL-threo-β-benzyloxyaspartate (a competitive glutamate transport inhibitor), DCS D-cycloserine (a partial NMDA receptor glycine-site agonist), CLZ clozapine (an atypical antipsychotic drug), SKF SKF81297 (a dopamine-D1 receptor agonist), MPC 5-methylpyrazole-3-carboxylic acid (an inhibitor of d-amino acid oxidase), DAO d-amino acid oxidase
Second, we aimed to elucidate whether the glutamatergic system was altered by the disturbance of dopaminergic signaling in the PFc of the DM. Disturbances in the glutamatergic neuronal system, such as the reduction in the numbers of dendritic spines observed in the autopsied brains and morphological changes of the dendritic spine that directly correlate with its functional deficits, underlie psychiatric disorders including schizophrenia (Goff and Coyle 2001; Hayashi-Takagi et al. 2010; McCullumsmith et al. 2004; Moghaddam 2003). D-serine, an NMDA receptor co-agonist, plays a crucial role in NMDA-dependent neurotransmission and is associated with a range of neuro-psychiatric disorders (Onozato et al. 2016). The levels of d-serine were lower in the plasma and cerebrospinal fluid of patients with psychiatric disease including schizophrenia (Fukushima et al. 2014; Hashimoto et al. 2005; Van Horn et al. 2013). The genes encoding serine racemase, the synthetizing enzyme of d-serine, and d-amino acid oxidase, the degradation enzyme of d-serine, are some of the promising candidates that may underlie the susceptibility to psychiatric disorders including schizophrenia and bipolar affective disorder (Schumacher et al. 2004). These results are coincident with clinical reports of psychiatric disease in which alterations in the levels of glutamate and d-serine and a reduction in NMDA activation were observed (Pilowsky et al. 2006; Steen et al. 2005). Our results showed an upregulation of glial transporters, a reduction in the basal levels of extracellular glutamate and dopamine, and changes in the levels of serine racemase and d-amino acid oxidase, which lead to altered glutamatergic abnormalities via NMDA-CaMK II signaling in the PFc and subsequent behavioral deficits (Fig. 9). Furthermore, these neuronal and behavioral deficits were ameliorated by a partial NMDA receptor glycine-site agonist DSC, an inhibitor of d-amino acid oxidase MPC, a competitive glutamate transport inhibitor TBOA, and a dopamine-D1 receptor agonist SKF (Fig. 9). We speculate that prolonged exposure to stress affects glutamatergic neurotransmission through dopaminergic alterations in the PFc and subsequently contributes to behavioral changes, since SKF ameliorated both dopaminergic and glutamatergic disturbance.
Third, we aimed to determine the effects of antipsychotics on behavioral deficits and neurochemical abnormalities in the DM. We showed that CLZ significantly ameliorated the impaired performance in locomotor activity, forced swim, social interaction, novelty preference, and prepulse inhibition tests in the DM. CLZ also normalized the glutamatergic and dopaminergic abnormalities studied in the DM, including activation of CaMK II and levels of basal and METH-induced extracellular dopamine. Although it has been reported that administration of CLZ increases the extracellular levels of dopamine in PFc and NAc of rats (Bortolozzi et al. 2010; Kuroki et al. 1999; Li et al. 2005; Moghaddam and Bunney 1990; Tanda et al. 2015), our data are compatible with the previous reports that CLZ restores the changes in the overflow of dopamine in the PFc of mice and phosphorylated CaMK II in a dopamine D1 receptor-dependent manner (Aoyama et al. 2014; Mouri et al. 2007b; Niwa et al. 2010). These findings suggest that dopamine modulates the increase of NMDA-mediated excitability in the prefrontal cortical neurons. Therefore, we propose that behavioral deficits in the DM implicate glutamatergic neuron alterations via NMDA-CaMK II signaling through a dopaminergic disturbance in the PFc.
Many epidemiological studies have indicated that initial and major risks for the psychiatric disease occur during neurodevelopment, although the onset of the disease occurs in juveniles and young adults. Therefore, it is important to address the mechanisms that can explain such onset of psychiatric disorders. Glutamatergic neurons undergo dynamic changes during postnatal brain maturation, particularly in late childhood and early adolescence (Jaaro-Peled et al. 2009; Owen et al. 2016). Deficits in postnatal modifications of glutamatergic neurons that occur just before the onset of the disease have been hypothesized as a potential mechanism (Hayashi-Takagi et al. 2010; Owen et al. 2016). We induced adverse experiences (e.g., social isolation in combination with a genetic risk in this case) during the period at which neuronal development was actively taking place (Niwa et al. 2013, 2016b). It is therefore possible that disturbances in the prefrontal glutamatergic and dopaminergic neurotransmission elicited by adolescent stress may contribute to the juvenile onset of the diseases. Further studies of the effects of psychosocial stress at different time points, including adulthood, are needed to enhance the importance of adolescence vulnerability.
In conclusion, we provide a potential mechanism underlying the link between adolescent stress and the onset of the disease during young adulthood by showing that (1) adolescent stress in combination with genetic vulnerability induces deficits in sociability and impaired learning and memory in adulthood, (2) prolonged exposure to stress affects glutamatergic neurotransmission through dopaminergic disturbance in the PFc and subsequently contributes to the behavioral deficits, and (3) antipsychotic drugs significantly ameliorate the behavioral deficits and neurochemical abnormalities (Fig. 9). The DM model may be useful to study the biology of the psychiatric disorders including schizophrenia. Further elucidation of such mechanisms would allow us to explore novel therapeutic strategies and provide a good template for prophylactic environmental readjustment, which is crucially important in clinical psychiatry and public health.
Supplementary Material
Acknowledgements
We thank Prof. A. Sawa, Johns Hopkins University, for critical reading and discussion of the manuscript; Prof. T. Yoshio, T. Izawa, and K. Sakaki, Toho University, for their technical assistance on HPLC analyses; and Editage (www.editage.jp) for English language editing.
This work was supported by the “Academic Frontier” Project for Private Universities from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), Research on Regulatory Science of Pharmaceuticals and Medical Devices and Risk of Chemical Substances from the Ministry of Health, Labour and Welfare of Japan (MHLW), JSPS grants 19659292, 20390073, 22659213, 22248033, 26460240, and 17H04252, and Research Grant from the SRF (T.N.); by NIH grants DA-040127 and K99MH-094408, NARSAD, JSPS grants 20007152 and 23-639, and JST PRESTO JPMJPR14M6 (M.N.); by Strategic Research Program for Brain Sciences from Japan Agency for Medical Research and Development (AMED) and Scientific Research on Innovative Areas, “Glial assembly: a new regulatory machinery of brain function and disorders” (N.O.); by JSPS grant 16K10195, and Research Grant from the SRF (A.M.).
Abbreviations
- CTL
Control
- DM
Disease model
- DCS
D-cycloserine
- METH
Methamphetamine
- SKF
SKF81297
- TBOA
DL-threo-β-benzyloxyaspartate
- MPC
5-Methylpyrazole-3-carboxylic acid
- CLZ
Clozapine
- PFc
Prefrontal cortex
- NMDA
N-methyl-d-aspartate
- NR1
NMDA receptor subunit 1
- CaMK II
Ca2+/calmodulin kinase II
- TH
Tyrosine hydroxylase
- D2R
Dopamine D2 receptor
- NAc
Nucleus accumbens
- PCP
Phencyclidine
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00213-017-4704-8) contains supplementary material, which is available to authorized users.
Compliance with ethical standards All animal procedures were in accordance with guidelines for the care and use of laboratory animals issued by the National Institutes of Health, Japanese Pharmacological Society, and Meijo University.
Conflict of interest The authors declare that they have no conflict of interest.
References
- Abazyan B, Dziedzic J, Hua K, Abazyan S, Yang C, Mori S, Pletnikov MV, Guilarte TR (2014) Chronic exposure of mutant DISC1 mice to lead produces sex-dependent abnormalities consistent with schizophrenia and related mental disorders: a gene-environment interaction study. Schizophr Bull 40:575–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano Y, Dhata A, Taniguchi Y, Hoshi A, Uchida K, Takashima A, Nakayama H (2017) Iso-Alpha-acids, bitter components of beer, prevent inflammation and cognitive decline induced in a mouse model of Alzheimer’s disease. J Biol Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama Y, Mouri A, Toriumi K, Koseki T, Narusawa S, Ikawa N, Mamiya T, Nagai T, Yamada K, Nabeshima T (2014) Clozapine ameliorates epigenetic and behavioral abnormalities induced by phencyclidine through activation of dopamine D1 receptor. Int J Neuropsychopharmacol 17:723–737 [DOI] [PubMed] [Google Scholar]
- Axelrod J, Reisine TD (1984) Stress hormones: their interaction and regulation. Science 224:452–459 [DOI] [PubMed] [Google Scholar]
- Ayhan Y, Abazyan B, Nomura J, Kim R, Ladenheim B, Krasnova IN, Sawa A, Margolis RL, Cadet JL, Mori S, Vogel MW, Ross CA, Pletnikov MV (2011) Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry 16:293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF (2001) Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol 21:516–521 [DOI] [PubMed] [Google Scholar]
- Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C, Schatzberg AF (2002) An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry 52: 386–392 [DOI] [PubMed] [Google Scholar]
- Blakemore SJ (2008) The social brain in adolescence. Nat Rev Neurosci 9:267–277 [DOI] [PubMed] [Google Scholar]
- Bortolozzi A, Masana M, Diaz-Mataix L, Cortes R, Scorza MC, Gingrich JA, Toth M, Artigas F (2010) Dopamine release induced by atypical antipsychotics in prefrontal cortex requires 5-HT1A receptors but not 5-HT2A receptors. Int J Neuropsychopharmacol 13:1299–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Sawa A (2011) Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci 12: 707–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, Verchinski BA, Meyer-Lindenberg A, Balkissoon R, Kolachana B, Goldberg TE, Weinberger DR (2005) Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci U S A 102:8627–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaby LE, Cavigelli SA, Hirrlinger AM, Lim J, Warg KM, Braithwaite VA (2015) Chronic stress during adolescence impairs and improves learning and memory in adulthood. Front Behav Neurosci 9:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla AR, Johnson DE, Zybura AS, Leeds BP, Nelson RM, Hudmon A (2017) Constitutive regulation of the glutamate/aspartate transporter EAAT1 by calcium-calmodulin-dependent protein kinase II. J Neurochem 140:421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ, Roder JC (2007) Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54:387–402 [DOI] [PubMed] [Google Scholar]
- Coitinho AS, Dietrich MO, Hoffmann A, Dall’Igna OP, Souza DO, Martins VR, Brentani RR, Izquierdo I, Lara DR (2002) Decreased hyperlocomotion induced by MK-801, but not amphetamine and caffeine in mice lacking cellular prion protein (PrPC). Brain Res Mol Brain Res 107:190–194 [DOI] [PubMed] [Google Scholar]
- Coura RS, Cressant A, Xia J, de Chaumont F, Olivo-Marin JC, Pelloux Y, Dalley JW, Granon S (2013) Nonaggressive and adapted social cognition is controlled by the interplay between noradrenergic and nicotinic receptor mechanisms in the prefrontal cortex. FASEB J 27: 4343–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF (2006) Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology 31:628–636 [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (2007) The mouse brain in stereotaxic coordinates. Academic Press; San Diego, ed. 3 [Google Scholar]
- Fukushima T, Kawai J, Imai K, Toyo’oka T (2004) Simultaneous determination of D- and L-serine in rat brain microdialysis sample using a column-switching HPLC with fluorimetric detection. Biomed Chromatogr BMC 18:813–819 [DOI] [PubMed] [Google Scholar]
- Fukushima T, Iizuka H, Yokota A, Suzuki T, Ohno C, Kono Y, Nishikiori M, Seki A, Ichiba H, Watanabe Y, Hongo S, Utsunomiya M, Nakatani M, Sadamoto K, Yoshio T (2014) Quantitative analyses of schizophrenia-associated metabolites in serum: serum D-lactate levels are negatively correlated with gamma-glutamylcysteine in medicated schizophrenia patients. PLoS One 9:e101652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukubo-Tokunaga K, Kurita K, Honjo K, Pandey H, Ando T, Takayama K, Arai Y, Mochizuki H, Ando M, Kamiya A, Sawa A (2016) DISC1 causes associative memory and neurodevelopmental defects in fruit flies. Mol Psychiatry 21:1232–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57:65–73 [DOI] [PubMed] [Google Scholar]
- Glessner JT, Reilly MP, Kim CE, Takahashi N, Albano A, Hou C, Bradfield JP, Zhang H, Sleiman PM, Flory JH, Imielinski M, Frackelton EC, Chiavacci R, Thomas KA, Garris M, Otieno FG, Davidson M, Weiser M, Reichenberg A, Davis KL, Friedman JI, Cappola TP, Margulies KB, Rader DJ, Grant SF, Buxbaum JD, Gur RE, Hakonarson H (2010) Strong synaptic transmission impact by copy number variations in schizophrenia. Proc Natl Acad Sci U S A 107:10584–10589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Coyle JT (2001) The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158: 1367–1377 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS (1995) Cellular basis of working memory. Neuron 14:477–485 [DOI] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR (2002) Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol 88:3150–3166 [DOI] [PubMed] [Google Scholar]
- Grace AA (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41:1–24 [DOI] [PubMed] [Google Scholar]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, Tye KM, Anikeeva P, Malenka RC, Deisseroth K (2014) Natural neural projection dynamics underlying social behavior. Cell 157: 1535–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamshere ML, Bennett P, Williams N, Segurado R, Cardno A, Norton N, Lambert D, Williams H, Kirov G, Corvin A, Holmans P, Jones L, Jones I, Gill M, O’Donovan MC, Owen MJ, Craddock N (2005) Genomewide linkage scan in schizoaffective disorder: significant evidence for linkage at 1q42 close to DISC1, and suggestive evidence at 22q11 and 19p13. Arch Gen Psychiatry 62:1081–1088 [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindstrom LH, Iyo M (2005) Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuro-Psychopharmacol Biol Psychiatry 29:767–769 [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Numakawa T, Ohnishi T, Kumamaru E, Yagasaki Y, Ishimoto T, Mori T, Nemoto K, Adachi N, Izumi A, Chiba S, Noguchi H, Suzuki T, Iwata N, Ozaki N, Taguchi T, Kamiya A, Kosuga A, Tatsumi M, Kamijima K, Weinberger DR, Sawa A, Kunugi H (2006) Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet 15:3024–3033 [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sawa A, Hikida T (2016) Impaired hippocampal activity at the goal zone on the place preference task in a DISC1 mouse model. Neurosci Res 106:70–73 [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A (2017) Synapse pathology and translational applications for schizophrenia. Neurosci Res 114:3–8 [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A (2010) Disrupted-in-schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 13:327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525:333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–1031 [DOI] [PubMed] [Google Scholar]
- Hennah W, Varilo T, Kestila M, Paunio T, Arajarvi R, Haukka J, Parker A, Martin R, Levitzky S, Partonen T, Meyer J, Lonnqvist J, Peltonen L, Ekelund J (2003) Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet 12:3151–3159 [DOI] [PubMed] [Google Scholar]
- Hida H, Mouri A, Mori K, Matsumoto Y, Seki T, Taniguchi M, Yamada K, Iwamoto K, Ozaki N, Nabeshima T, Noda Y (2015) Blonanserin ameliorates phencyclidine-induced visual-recognition memory deficits: the complex mechanism of blonanserin action involving D3-5-HT2A and D1-NMDA receptors in the mPFC. Neuropsychopharmacology 40:601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK (2004) Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet 75:862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Wang EA, Cepeda C, Jentsch JD, Ross CA, Pletnikov MV, Levine MS (2013) Frontal cortical synaptic communication is abnormal in Disc1 genetic mouse models of schizophrenia. Schizophr Res 146:264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Flashner B, Chiu M, ver Hoeve E, Luz S, Bhatnagar S (2012) Social isolation in adolescence alters behaviors in the forced swim and sucrose preference tests in female but not in male rats. Physiol Behav 105:269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W-J, Lee H-J, Chen H-L, Fan P-C, Ku Y-L, Chiou L-C (2015) Hispidulin, a constituent of Clerodendrum inerme that remitted motor tics, alleviated methamphetamine-induced hyperlocomotion without motor impairment in mice. J Ethnopharmacol 166:18–22 [DOI] [PubMed] [Google Scholar]
- Hultman R, Mague SD, Li Q, Katz BM, Michel N, Lin L, Wang J, David LK, Blount C, Chandy R, Carlson D, Ulrich K, Carin L, Dunson D, Kumar S, Deisseroth K, Moore SD, Dzirasa K (2016) Dysregulation of prefrontal cortex-mediated slow-evolving limbic dynamics drives stress-induced emotional pathology. Neuron 91:439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K (2010) Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res 206:32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka K, Kamiya A, Oh EC, Kanki H, Seshadri S, Robinson JF, Murdoch H, Dunlop AJ, Kubo K, Furukori K, Huang B, Zeledon M, Hayashi-Takagi A, Okano H, Nakajima K, Houslay MD, Katsanis N, Sawa A (2011) DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473: 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A (2009) Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci 32:485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Niwa M, Foss CA, Murai R, de Los RS, Kamiya A, Mateo Y, O’Donnell P, Cascella NG, Nabeshima T, Guilarte TR, Pomper MG, Sawa A (2013) Subcortical dopaminergic deficits in a DISC1 mutant model: a study in direct reference to human molecular brain imaging. Hum Mol Genet 22:1574–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Altimus C, LeGates T, Cash-Padgett T, Zoubovsky S, Hikida T, Ishizuka K, Hattar S, Mongrain V, Sawa A (2016) Abnormal wake/sleep pattern in a novel gain-of-function model of DISC1. Neurosci Res 112:63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joels M, Baram TZ (2009) The neuro-symphony of stress. Nat Rev Neurosci 10:459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabanova A, Pabst M, Lorkowski M, Braganza O, Boehlen A, Nikbakht N, Pothmann L, Vaswani AR, Musgrove R, Di Monte DA, Sauvage M, Beck H, Blaess S (2015) Function and developmental origin of a mesocortical inhibitory circuit. Nat Neurosci 18:872–882 [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, Ross CA, Hatten ME, Nakajima K, Sawa A (2005) A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 7:1167–1178 [DOI] [PubMed] [Google Scholar]
- Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, Kelleher E, Ennis S, Tropea D, Anney R, Corvin AP, Donohoe G, Gallagher L, Gill M, Morris DW (2014) Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry 19:872–879 [DOI] [PubMed] [Google Scholar]
- Kercmar J, Budefeld T, Grgurevic N, Tobet SA, Majdic G (2011) Adolescent social isolation changes social recognition in adult mice. Behav Brain Res 216:647–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen-von Wendt T, von Wendt L, Paunio T, Peltonen L (2008) Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry 13:187–196 [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayes A, Fernandez E, Olason PI, Bottcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ (2012) De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 17:142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA (2006) Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A 103:3693–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreek MJ, Nielsen DA, Butelman ER, LaForge KS (2005) Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nat Neurosci 8:1450–1457 [DOI] [PubMed] [Google Scholar]
- Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, Tsuboi D, Nishioka T, Namba T, Iizuka Y, Kubota S, Nagai T, Ibi D, Wang R, Enomoto A, Isotani-Sakakibara M, Asai N, Kimura K, Kiyonari H, Abe T, Mizoguchi A, Sokabe M, Takahashi M, Yamada K, Kaibuchi K (2011) Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 20:4666–4683 [DOI] [PubMed] [Google Scholar]
- Kuroki T, Meltzer HY, Ichikawa J (1999) Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther 288:774–781 [PubMed] [Google Scholar]
- Kuroki T, Nagao N, Nakahara T (2008) Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. Prog Brain Res 172:199–212 [DOI] [PubMed] [Google Scholar]
- Lee MA, Jayathilake K, Meltzer HY (1999) A comparison of the effect of clozapine with typical neuroleptics on cognitive function in neuroleptic-responsive schizophrenia. Schizophr Res 37:1–11 [DOI] [PubMed] [Google Scholar]
- Leussis MP, Andersen SL (2008) Is adolescence a sensitive period for depression? Behavioral and neuroanatomical findings from a social stress model. Synapse (New York, NY) 62:22–30 [DOI] [PubMed] [Google Scholar]
- Li Z, Huang M, Ichikawa J, Dai J, Meltzer HY (2005) N-desmethylclozapine, a major metabolite of clozapine, increases cortical acetylcholine and dopamine release in vivo via stimulation of M1 muscarinic receptors. Neuropsychopharmacology 30:1986–1995 [DOI] [PubMed] [Google Scholar]
- Li C, Liu Y, Yin S, Lu C, Liu D, Jiang H, Pan F (2015) Long-term effects of early adolescent stress: dysregulation of hypothalamic-pituitary-adrenal axis and central corticotropin releasing factor receptor 1 expression in adult male rats. Behav Brain Res 288:39–49 [DOI] [PubMed] [Google Scholar]
- Lips ES, Cornelisse LN, Toonen RF, Min JL, Hultman CM, International Schizophrenia C, Holmans PA, O’Donovan MC, Purcell SM, Smit AB, Verhage M, Sullivan PF, Visscher PM, Posthuma D (2012) Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry 17:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Mamiya T, Lu P, Toriumi K, Mouri A, Hiramatsu M, Zou LB, Nabeshima T (2011) Prenatal exposure to PCP produces behavioral deficits accompanied by the overexpression of GLAST in the prefrontal cortex of postpubertal mice. Behav Brain Res 220:132–139 [DOI] [PubMed] [Google Scholar]
- Mathews IZ, Mills RG, McCormick CM (2008) Chronic social stress in adolescence influenced both amphetamine conditioned place preference and locomotor sensitization. Dev Psychobiol 50:451–459 [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Marzo A, Otani S (2006) The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J Neurosci 26:4803–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick BP, Snethen G, Lysaker PH (2012) Emotional episodes in the everyday lives of people with schizophrenia: the role of intrinsic motivation and negative symptoms. Schizophr Res 142:46–51 [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE (2015) Evidence for schizophrenia as a disorder of neuroplasticity. Am J Psychiatry 172:312–313 [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE, Clinton SM, Meador-Woodruff JH (2004) Schizophrenia as a disorder of neuroplasticity. Int Rev Neurobiol 59:19–45 [DOI] [PubMed] [Google Scholar]
- McGlashan TH, Hoffman RE (2000) Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry 57:637–648 [DOI] [PubMed] [Google Scholar]
- Mizoguchi H, Watanabe C, Osada S, Yoshioka M, Aoki Y, Natsui S, Yonezawa A, Kanno S, Ishikawa M, Sakurada T, Sakurada S (2010) Lack of a rewarding effect and a locomotor-enhancing effect of the selective mu-opioid receptor agonist amidino-TAPA. Psychopharmacology 212:215–225 [DOI] [PubMed] [Google Scholar]
- Moghaddam B (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40:881–884 [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Bunney BS (1990) Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem 54:1755–1760 [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Hara H, Mizoguchi H, Tabira T, Nabeshima T (2007a) Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J 21: 2135–2148 [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, Nitta A, Furukawa H, Nabeshima T (2007b) Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol 71:1598–1609 [DOI] [PubMed] [Google Scholar]
- Mouri A, Sasaki A, Watanabe K, Sogawa C, Kitayama S, Mamiya T, Miyamoto Y, Yamada K, Noda Y, Nabeshima T (2012) MAGE-D1 regulates expression of depression-like behavior through serotonin transporter ubiquitylation. J Neurosci 32:4562–4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai R, Noda Y, Matsui K, Kamei H, Mouri A, Matsuba K, Nitta A, Furukawa H, Nabeshima T (2007) Hypofunctional glutamatergic neurotransmission in the prefrontal cortex is involved in the emotional deficit induced by repeated treatment with phencyclidine in mice: implications for abnormalities of glutamate release and NMDA-CaMKII signaling. Behav Brain Res 180:152–160 [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y, Nabeshima T (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J 17:50–52 [DOI] [PubMed] [Google Scholar]
- Nagai T, Kitahara Y, Ibi D, Nabeshima T, Sawa A, Yamada K (2011) Effects of antipsychotics on the behavioral deficits in human dominant-negative DISC1 transgenic mice with neonatal polyI:C treatment. Behav Brain Res 225:305–310 [DOI] [PubMed] [Google Scholar]
- Nakai T, Nagai T, Wang R, Yamada S, Kuroda K, Kaibuchi K, Yamada K (2014) Alterations of GABAergic and dopaminergic systems in mutant mice with disruption of exons 2 and 3 of the Disc1 gene. Neurochem Int 74:74–83 [DOI] [PubMed] [Google Scholar]
- Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T, He J, Nitta A, Mizuno M, Tran MH, Seto A, Yoshimura M, Kitaichi K, Hasegawa T, Saito K, Yamada Y, Seishima M, Sekikawa K, Kim HC, Nabeshima T (2004) Role of tumor necrosis factor-alpha in methamphetamine-induced drug dependence and neurotoxicity. J Neurosci 24:2212–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Mizoguchi H, Ito Y, Noda Y, Nagai T, Nabeshima T (2007) A novel molecule “shati” is involved in methamphetamine-induced hyperlocomotion, sensitization, and conditioned place preference. J Neurosci 27:7604–7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Kamiya A, Murai R, Kubo K, Gruber AJ, Tomita K, Lu L, Tomisato S, Jaaro-Peled H, Seshadri S, Hiyama H, Huang B, Kohda K, Noda Y, O’Donnell P, Nakajima K, Sawa A, Nabeshima T (2010) Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65:480–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Matsumoto Y, Mouri A, Ozaki N, Nabeshima T (2011) Vulnerability in early life to changes in the rearing environment plays a crucial role in the aetiopathology of psychiatric disorders. Int J Neuropsychopharmacol 14:459–477 [DOI] [PubMed] [Google Scholar]
- Niwa M, Jaaro-Peled H, Tankou S, Seshadri S, Hikida T, Matsumoto Y, Cascella NG, Kano S, Ozaki N, Nabeshima T, Sawa A (2013) Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science 339:335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Cash-Padgett T, Kubo KI, Saito A, Ishii K, Sumitomo A, Taniguchi Y, Ishizuka K, Jaaro-Peled H, Tomoda T, Nakajima K, Sawa A, Kamiya A (2016a) DISC1 a key molecular lead in psychiatry and neurodevelopment: no-more disrupted-in-schizophrenia 1. Mol Psychiatry 21:1488–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Lee RS, Tanaka T, Okada K, Kano S, Sawa A (2016b) A critical period of vulnerability to adolescent stress: epigenetic mediators in mesocortical dopaminergic neurons. Hum Mol Genet 25:1370–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick AM, Mears M, Forster GL, Lei Y, Tejani-Butt SM, Watt MJ (2016) Adolescent social defeat alters N-methyl-D-aspartic acid receptor expression and impairs fear learning in adulthood. Behav Brain Res 304:51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogaya T, Song Z, Ishii K, Fukushima T (2010) Changes in extracellular kynurenic acid concentrations in rat prefrontal cortex after D-kynurenine infusion: an in vivo microdialysis study. Neurochem Res 35:559–563 [DOI] [PubMed] [Google Scholar]
- Onozato M, Nakazawa H, Hakariya H, Shishikura M, Nagashima C, Ishimaru K, Sakamoto T, Iizuka H, Ichiba H, Fukushima T (2016) Effect of risperidone on plasma d-serine concentration in rats post-administered with d-serine. Life Sci 158:98–103 [DOI] [PubMed] [Google Scholar]
- O’Tuathaigh CM, Fumagalli F, Desbonnet L, Perez-Branguli F, Moloney G, Loftus S, O’Leary C, Petit E, Cox R, Tighe O, Clarke G, Lai D, Harvey RP, Cryan JF, Mitchell KJ, Dinan TG, Riva MA, Waddington JL (2017) Epistatic and independent effects on schizophrenia-related phenotypes following co-disruption of the risk factors Neuregulin-1 × DISC1. Schizophr Bull 43:214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, Sawa A, Mortensen PB (2016) Schizophrenia. Lancet 388:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus T, Keshavan M, Giedd JN (2008) Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci 9:947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, Ell PJ (2006) First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry 11:118–119 [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360:464–467 [DOI] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA (2008) Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 13(173–86):115. [DOI] [PubMed] [Google Scholar]
- Reynolds D (2003) GABAA α1 subunit knock-out mice do not show a hyperlocomotor response following amphetamine or cocaine treatment. Neuropharmacology 44:190–198 [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E (1989) Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett 103:162–168 [DOI] [PubMed] [Google Scholar]
- Saito A, Taniguchi Y, Rannals MD, Merfeld EB, Ballinger MD, Koga M, Ohtani Y, Gurley DA, Sedlak TW, Cross A, Moss SJ, Brandon NJ, Maher BJ, Kamiya A (2016) Early postnatal GABAA receptor modulation reverses deficits in neuronal maturation in a conditional neurodevelopmental mouse model of DISC1. Mol Psychiatry 21: 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandson J, Albert ML (1984) Varieties of perseveration. Neuropsychologia 22:715–732 [DOI] [PubMed] [Google Scholar]
- Sanna MD, Ghelardini C, Thurmond RL, Masini E, Galeotti N (2017) Behavioural phenotype of histamine H4 receptor knockout mice: focus on central neuronal functions. Neuropharmacology 114:48–57 [DOI] [PubMed] [Google Scholar]
- Sarro EC, Sullivan RM, Barr G (2014) Unpredictable neonatal stress enhances adult anxiety and alters amygdala gene expression related to serotonin and GABA. Neuroscience 258:147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Jamra RA, Freudenberg J, Becker T, Ohlraun S, Otte AC, Tullius M, Kovalenko S, Bogaert AV, Maier W, Rietschel M, Propping P, Nothen MM, Cichon S (2004) Examination of G72 and D-amino-acid oxidase as genetic risk factors for schizophrenia and bipolar affective disorder. Mol Psychiatry 9:203–207 [DOI] [PubMed] [Google Scholar]
- Seshadri S, Faust T, Ishizuka K, Delevich K, Chung Y, Kim SH, Cowles M, Niwa M, Jaaro-Peled H, Tomoda T, Lai C, Anton ES, Li B, Sawa A (2015) Interneuronal DISC1 regulates NRG1-ErbB4 signalling and excitatory-inhibitory synapse formation in the mature cortex. Nat Commun 6:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P (1998) A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 18:10297–10303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM (2009) The stressed CNS: when glucocorticoids aggravate inflammation. Neuron 64: 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen RG, Hamer RM, Lieberman JA (2005) Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology 30:1949–1962 [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W (1992) Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A 89:10955–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF (2013) Questions about DISC1 as a genetic risk factor for schizophrenia. Mol Psychiatry 18:1050–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL (2001) Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology 156:194–215 [DOI] [PubMed] [Google Scholar]
- Takamatsu Y, Yamamoto H, Ogai Y, Hagino Y, Markou A, Ikeda K (2006) Fluoxetine as a potential pharmacotherapy for methamphetamine dependence: studies in mice. Ann N Y Acad Sci 1074:295–302 [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Sonneborn A, Spooner PA, Yamasaki M, Watanabe M, Smith CC, Fernandez G, Deisseroth K, Greene RW, Morris RG (2016) Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 537:357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka DH, Toriumi K, Kubo K, Nabeshima T, Nakajima K (2011) GABAergic precursor transplantation into the prefrontal cortex prevents phencyclidine-induced cognitive deficits. J Neurosci 31: 14116–14125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Valentini V, De Luca MA, Perra V, Serra GP, Di Chiara G (2015) A systematic microdialysis study of dopamine transmission in the accumbens shell/core and prefrontal cortex after acute antipsychotics. Psychopharmacology 232:1427–1440 [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999) Genetic enhancement of learning and memory in mice. Nature 401:63–69 [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272:5157–5166 [DOI] [PubMed] [Google Scholar]
- Tomoda T, Sumitomo A, Jaaro-Peled H, Sawa A (2016) Utility and validity of DISC1 mouse models in biological psychiatry. Neuroscience 321:99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriumi K, Mouri A, Narusawa S, Aoyama Y, Ikawa N, Lu L, Nagai T, Mamiya T, Kim HC, Nabeshima T (2012) Prenatal NMDA receptor antagonism impaired proliferation of neuronal progenitor, leading to fewer glutamatergic neurons in the prefrontal cortex. Neuropsychopharmacology 37:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriumi K, Kondo M, Nagai T, Hashimoto R, Ohi K, Song Z, Tanaka J, Mouri A, Koseki T, Yamamori H, Furukawa-Hibi Y, Mamiya T, Fukushima T, Takeda M, Nitta A, Yamada K, Nabeshima T (2014) Deletion of SHATI/NAT8L increases dopamine D1 receptor on the cell surface in the nucleus accumbens, accelerating methamphetamine dependence. Int J Neuropsychopharmacol 17:443–453 [DOI] [PubMed] [Google Scholar]
- Trossbach SV, Bader V, Hecher L, Pum ME, Masoud ST, Prikulis I, Schable S, de Souza Silva MA, Su P, Boulat B, Chwiesko C, Poschmann G, Stuhler K, Lohr KM, Stout KA, Oskamp A, Godsave SF, Muller-Schiffmann A, Bilzer T, Steiner H, Peters PJ, Bauer A, Sauvage M, Ramsey AJ, Miller GW, Liu F, Seeman P, Brandon NJ, Huston JP, Korth C (2016) Misassembly of full-length disrupted-in-schizophrenia 1 protein is linked to altered dopamine homeostasis and behavioral deficits. Mol Psychiatry 21:1561–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Horn MR, Sild M, Ruthazer ES (2013) D-serine as a gliotransmitter and its roles in brain development and disease. Front Cell Neurosci 7:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AFT (2007) Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10:376–384 [DOI] [PubMed] [Google Scholar]
- Wang J, O’Donnell P (2001) D1 dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cerebral cortex (New York, NY: 1991) 11:452–462 [DOI] [PubMed] [Google Scholar]
- Wang YT, Huang CC, Lin YS, Huang WF, Yang CY, Lee CC, Yeh CM, Hsu KS (2017) Conditional deletion of Eps8 reduces hippocampal synaptic plasticity and impairs cognitive function. Neuropharmacology 112:113–123 [DOI] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM, Deisseroth K (2012) A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492:428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss IC, Pryce CR, Jongen-Relo AL, Nanz-Bahr NI, Feldon J (2004) Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav Brain Res 152:279–295 [DOI] [PubMed] [Google Scholar]
- Xing J, Kimura H, Wang C, Ishizuka K, Kushima I, Arioka Y, Yoshimi A, Nakamura Y, Shiino T, Oya-Ito T, Takasaki Y, Uno Y, Okada T, Iidaka T, Aleksic B, Mori D, Ozaki N (2016) Resequencing and association analysis of six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci Rep 6:27491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK (1996) Dopamine D1 receptor actions in layers VVI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci 16:1922–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN (2004) Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology 29:1538–1545 [DOI] [PubMed] [Google Scholar]
- Youngren KD, Inglis FM, Pivirotto PJ, Jedema HP, Bradberry CW, Goldman-Rakic PS, Roth RH, Moghaddam B (1999) Clozapine preferentially increases dopamine release in the rhesus monkey prefrontal cortex compared with the caudate nucleus. Neuropsychopharmacology 20:403–412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.