

Abstract
Biomimetic biomaterials are being actively explored in the context of cancer immunotherapy because of their ability to directly engage the immune system to generate anti-tumor responses. Unlike cellular therapies, biomaterial-based immunotherapies can be precisely engineered to exhibit defined characteristics including biodegradability, physical size, and tuned surface presentation of immunomodulatory signals. In particular, modulating the interface between the biomaterial surface and the target biological cell is key to enable biological function. Synthetic artificial antigen presenting cells (aAPCs) are promising as a cancer immunotherapy but are limited in clinical translation by the requirement of ex vivo cell manipulation and adoptive transfer of antigen-specific CD8+ T cells. To move towards acellular aAPC technology for in vivo use, we combine poly(lactic-co-glycolic acid) (PLGA) and cationic poly(beta-amino-ester) (PBAE) to form a biodegradable blend based on the hypothesis that therapeutic aAPCs fabricated from a cationic blend may have improved function. PLGA/PBAE aAPCs demonstrate enhanced surface interactions with antigen-specific CD8+ T cells that increase T cell activation and expansion ex vivo, associated with significantly increased conjugation efficiency of T cell stimulatory signals to the aAPCs. Critically, these PLGA/PBAE aAPCs also expand antigen-specific cytotoxic CD8+ T cells in vivo without the need of adoptive transfer. Treatment with PLGA/PBAE aAPCs in combination with checkpoint therapy decreases tumor growth and extends survival in a B16-F10 melanoma mouse model. These results demonstrate the potential of PLGA/PBAE aAPCs as a biocompatible, directly injectable acellular therapeutic for cancer immunotherapy.
Keywords: artificial antigen presenting cell, immunotherapy, cancer, immunoengineering, bioengineering, biomimetic, particle
Graphical Abstract
INTRODUCTION
Biotechnologies to harness and modulate the immune system have increasingly shown promise in diseases ranging from cancer and infectious diseases to transplantation and autoimmune diseases. Biomimicry is a fascinating area of materials research where bio-inspired properties enable new functionality for maintaining health and treating disease. A particularly promising approach in immunoengineering is the use of biodegradable particulate systems, ranging from the size of a virus to the size of a cell and mimicking the surfaces of the natural biologic materials, as agents of cellular programming. Such therapeutic agents have the potential to be used “off-the-shelf” as biological, but non-cellular, therapies with greatly reduced costs, manufacturing challenges, and regulatory hurdles compared to cellular therapies. In particular, biomimetic artificial antigen presenting cells (aAPC) have shown increasing promise to serve as a cancer immunotherapy.1 aAPC technology consists of at least three components: a biocompatible three-dimensional core material, a protein to serve as “Signal 1” or antigen recognition signal, and a protein to serve as “Signal 2” or effector direction signal.2 The two proteins are conjugated to the surface of the particle to engage corresponding receptors on the surface of T cells to initiate activation in a process that mimics natural antigen presentation to T cells by APCs. Signal 1 proteins can be non-specific T cell stimulators (e.g. anti-CD3) or they may consist of tumor-specific peptide-MHC complexes (pMHC) to activate T cells that will recognize and destroy the tumor. In the past decade, there have been great strides in the advancement of aAPC technology and engineered surfaces for T cell activation.3 Multiple platforms have been developed to enhance this stimulation including controlled release of a “Signal 3” protein to direct the T cell response,4–6 incorporation of magnetite to enable magnetic isolation of aAPC from T cells for therapeutic use,7 use of magnetite as the core material to enable magnetic clustering of T cell receptors,8–10 and modulation of the surface area available for T cell activation through modification of particle size, shape,11 or surface roughness.12 Many of these innovations have significantly increased aAPC efficacy. However, the promise of aAPC translation to the clinic has been hampered by the requirement of ex vivo manipulation of antigen specific T cells to achieve a therapeutic effect. This ex vivo culture step adds significant time and cost to the therapy, limiting its use in the clinic. An “off-the-shelf” enhanced aAPC therapy that could be administered directly in vivo and circumvent the need for ex vivo expansion of cells would provide greater patient access to T cell therapies. However, an improved aAPC formulation is necessary to achieve robust expansion of host T cells.
One promising area of research that has been recently investigated is the use of different biodegradable polymers for the plastic core of the aAPC. Primarily, these aAPC particle cores have been composed of poly(lactic-co-glycolic acid) (PLGA),4–5, 11, 13 a hydrolytically biodegradable polymer that has also been used in the construction of multiple FDA-approved devices. Although PLGA is well suited for various applications, it has several limitations as a material for aAPC. Firstly, it has been shown that the elastic modulus of a polymeric substrate for T cell activation can have a significant effect on T cell activation and subsequent phenotype.14 In addition, lateral rigidity of the surface proteins has been shown to impact the magnitude and direction of an artificially directed T cell response.15 In these cases, softer, more pliable materials have been found to be more beneficial than PLGA. In conjunction with the type of material, the surface density of signal proteins has been found to be an important parameter governing T cell activation.11 Ligand spacing has been found to be optimal when protein are spaced closely together as opposed to farther apart.16 However, protein coupling to PLGA particles can be highly inefficient and obtaining a sufficient amount of protein on the surface for T cell activation can be challenging.13 Not only can other polymers express these properties, but they have additional benefits, such a non-specific adjuvant effect during a biomaterials/immune cell interaction.17–18
Poly(beta-amino-ester) (PBAE) is a hydrophobic, cationic biodegradable polyester. PBAE particles have been shown to act directly as immunostimulatory agents, although this effect is attenuated as the material breaks down into lower molecular weight and free polymer.17 PLGA/PBAE blended particles support surface functionalization19 and encapsulation of nucleic acids, peptides, or small molecule drugs.20 Despite some work on PLGA/PBAE micro- and nanoparticles for intracellular delivery, no one has explored this combination of polymers in the context of aAPC technology. A blend of PLGA/PBAE as a core material for next generation aAPCs could potentially have enhanced biological efficacy and may promote particle/cell interactions due to the increased positive charge of the particles.
In this study, we have developed a hybrid PLGA/PBAE based aAPC for cancer immunotherapy (Figure 1). We have found that the particles made from this dual polymeric blend improve immune stimulation ex vivo and in vivo compared to traditional PLGA aAPC. This extra immune stimulatory capacity may be related to an increase in signal protein density on the surface of the particle compared to the PLGA particle. The enhanced activity seen in vitro also translated an enhanced ability to expand cells in vivo that were able to slow cancer progression in a murine melanoma model. A modest therapeutic effect was seen in the absence of adoptively transferred or ex vivo-manipulated T cells. Taken together, these results highlight the potential to use PLGA/PBAE aAPC as an acellular therapy for melanoma and other difficult to treat solid tumors.
Figure 1.
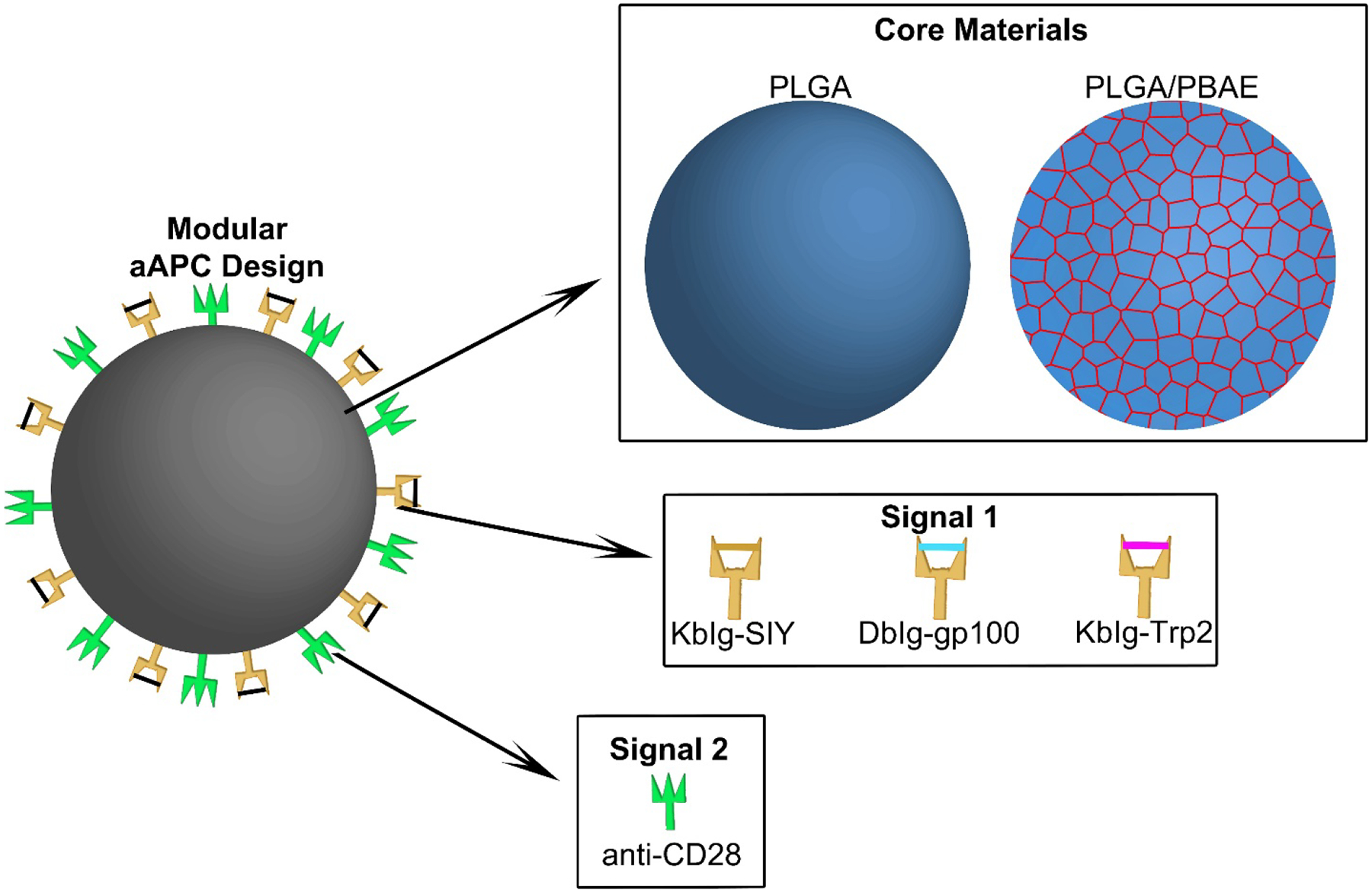
Schematic showing the modular design of a PLGA-based aAPC for melanoma immunotherapy. A PLGA/PBAE blend is explored as a novel core material for the aAPC to enhance presentation of surface-conjugated MHC Ig loaded with melanoma antigenic peptides (Signal 1) and Signal 2 molecules.
MATERIALS AND METHODS
2.1. PBAE synthesis and characterization
Poly(beta-amino ester) (PBAE) was synthesized using a two-step procedure as previously described.21 An acrylate-terminated base polymer was prepared through the Michael addition of 4,4’-trimethylenedipiperidine to 1,4-butanediol diacrylate at stoichiometric ratios of 1.05, 1.1 or 1.2:1 acrylate: amine monomers. Monomers were reacted at 90°C for 24 hours to generate the base PBAE polymer. The base polymer was dissolved in anhydrous tetrahydrofuran (THF) and reacted with an excess of end-capping monomer 1-(3-aminopropyl)-4-methylpiperazine at room temperature for 1 hour. The resulting end-capped PBAE polymer was purified with hexane, dried under vacuum, and stored at −20°C under nitrogen gas. The molecular weight of the PBAE was determined through gel permeation chromatography using an Ultrastyragel column with a molecular weight range of 500–30 kDa (Waters; Milford, MA) and a mobile phase of THF with 5% DMSO and 1% piperidine.
2.2. Artificial antigen presenting cell synthesis and characterization
aAPCs were synthesized in a two-step core particle formation and functionalization. Particle cores were synthesized from poly (lactic-co-glycolic acid) (PLGA 50:50 lactic acid to glycolic acid ratio, MW 34,000–58,000 Da) that was purchased commercially (Sigma Aldrich; St. Louis, MO) or a blend of PLGA and PBAE. For a typical microparticle synthesis, 100 mg of PLGA or 75 mg/ 25 mg PLGA/ PBAE were dissolved in 5 mL dichloromethane (DCM) and homogenized into a 50 mL, 1% poly vinyl alcohol (PVA) solution by a T-25 digital ULTRA-TURRAX IKA tissue homogenizer (IKA Works; Wilmington, NC) at a speed of 15,000 rpm (IKA Works; Wilmington, NC). The resulting microemulsion was further diluted in 100 mL of 0.5% PVA solution and stirred for 4 hours to allow the DCM to evaporate. After particle hardening, microparticles were washed three times in water through centrifugation at 3,000 x g for 5 min. The washed microparticle solution was frozen and lyophilized prior to characterization and use. Particle imaging was conducted with a Leo FESEM scanning electron microscope. To prepare samples for analysis, lyophilized particles were mounted onto carbon tape (Nisshin EM Co.; Tokyo, Japan) and placed upon aluminum tacks (Electron Microscopy Services; Hatfield, PA). Samples were sputter coated with a 20 nm thick layer of gold-palladium prior to imaging. Images were processed in ImageJ to obtain size information. Zeta potential measurements were conducted using a Malvern Panalytical Zetasizer. Particles were resuspended at 0.2 mg/mL in 0.1X PBS at pH 7.4 or 6.0 for zeta potential measurements.
Functionalization was achieved through EDC/NHS chemistry to conjugate carboxylic acid-terminated PLGA to amines on proteins of interest. Lyophilized particles were dissolved at a concentration of 2 mg/mL in 0.1 M MES buffer at pH 6.0. 100 μL of EDC/NHS (Sigma Aldrich; St. Louis, MO) stock solution consisting of 40 mg/mL EDC and 48 mg/mL NHS was added to each sample and the particles were activated for 30 minutes at room temperature. Surface-activated particles were washed in PBS through centrifugation at 5,000 × g for 5 min. The particles were resuspended in PBS at 2 mg/mL. 8 μg MHC IgG dimer, synthesized as described elsewhere22 and loaded with the antigen of choice, and 10 μg anti-CD28 monoclonal antibody (mAb) (BD Biosciences; San Jose, CA) was added to each sample and the particles were reacted with the proteins overnight at 4 °C. The resulting aAPCs were washed in PBS through centrifugation at 5000 × g and resuspended in 100 mg/mL endotoxin-free sucrose, then frozen and lyophilized prior to use. aAPC size was measured via dynamic light scattering using a Malvern Panalytical Zetasizer Pro.
To determine the amount of protein on the surface, PLGA aAPCs and PLGA/PBAE aAPCs (PLGA/PBAE aAPCs) were conjugated with fluorescently labelled MHC DbIg-gp100 dimer and anti-CD28 (Biolegend; San Diego, CA). For conjugation optimization experiments, aAPCs were conjugated with an Alexa Fluor 546 goat anti-hamster IgG (Life Technologies; Grand Island, NY). The particles were washed with PBS three times and evaluated for fluorescence with a BioTek Synergy 2 plate reader (Biotek; Winooski, VT). For protein retention studies, conjugated particles were incubated in PBS at 37 °C for up to 10 days and conjugation efficiency was measured. The mass of protein on the particle was calculated from a standard curve to evaluate conjugation efficiency. Conjugation efficiency was calculated as (mass of protein calculated on particle surface)/ (mass of protein added during conjugation) *100%.
2.3. In vitro artificial antigen presenting cell binding studies
To assess the interaction of PLGA and PLGA/PBAE aAPC with T cells, confocal imaging and flow cytometry was used to evaluate aAPC/T cell binding. PLGA and PLGA/PBAE aAPC were synthesized and conjugated as previously described with the addition of 10 μg of Vybrant DiD Cell-Labeling Solution (Thermo Fisher Scientific; Waltham, MA) into the polymer solution during synthesis. All mice were maintained according to Johns Hopkins University’s Institutional Review Board. PMEL CD8+ transgenic mice were sacrificed and CD8+ T cells were isolated from the spleens using the Miltenyi CD8a+ Isolation Kit (Miltenyi; Auburn, CA). The cells were then stained with Vybrant Cell Tracker carboxyfluorescein succinyl ester (CFSE) (Life Technologies; Grand Island, NY) dye following the manufacturer’s protocol. Prior to confocal imaging, T cells were incubated with aAPCs at 37 °C for 1 hour at a ratio of 1 mg of particles to 500,000 T cells in a volume of 100 μL. The mixture was then imaged under confocal microscopy using a Zeiss 710 LSM (Carl Zeiss Microscopy; Jena, Germany). To quantitatively assess binding events, T cells were incubated with aAPCs at a ratio of 0.01 mg of particles to 50,000 T cells in a volume of 100 μL. Samples were acquired on an Accuri C6 Flow Cytometer (BD Biosciences; San Jose, CA). Geometric mean fluorescence intensity (MFI) of DiD-labelled aAPC was normalized to samples with T cells and blank particles.
2.4. In vitro artificial antigen presenting cell/ T cell stimulation
To compare PLGA/PBAE aAPC to PLGA aAPC in their ability to stimulate antigen-specific CD8+ T cells, we performed a functional efficacy assay with primary CD8+ T cells isolated from 2C mice. 2C CD8+ T cells were isolated from mouse spleens and labelled with CFSE. Cells were then incubated with aAPCs bearing cognate antigens at titrated concentrations of aAPC (polymer weight) / 100,000 CD8+ T cells in RPMI supplemented with L-glutamine, non-essential amino acids, vitamin solution, sodium pyruvate, β-mercaptoethanol, 10% FBS, ciproflaxin, and a cocktail of T cell growth factors. After three days of incubation, CFSE dilution was assessed through flow cytometry analysis on a BD FACSCalibur. Each generation is defined as a distinct peak of the flow cytometry CFSE histogram, as the CFSE dye is diluted in half with each cell division. Total proliferation was assessed through manual counting of the cell sample seven days after stimulation using trypan blue to visually isolate viable cells.
2.5. In vivo antigen specific T cell expansion
One day prior to treatment, C57BL/6 (B6) mice were irradiated with a central dose of 500 cGy, a sublethal dose to induce transient lymphopenia.13 On the day of treatment, CD8+ T cells were isolated from the spleens of PMEL mice and labelled with CellTrace™ Violet (Life Technologies) according to the manufacturer’s protocol. Then, 1×106 labelled cells were adoptively transferred per B6 mouse either with or without PLGA or PLGA/PBAE aAPCs. For those groups who received co-injection of cells with aAPCs, the CD8+ T cells were incubated with aAPCs for 1 hour at 4 °C prior to co-injection. 30,000 units of IL-2 (Prometheus Laboratories) were administered intraperitoneally on the day of and the day after treatment. Tail vein bleeds were performed on day 3, and on day 7 mice were sacrificed and spleens and lymph nodes were harvested for analysis by flow cytometry. Blood samples were treated with 1 mL of ACK lysis buffer for 4 minutes and splenocytes were treated with 4 mL of ACK lysis buffer for 1 minute to lyse red blood cells. Total immune cells from the spleen and lymph nodes were measured with manual counting on a hemocytometer, using Trypan blue staining to exclude dead cells. Immune cells were stained with surface staining antibodies, PerCP anti-mouse CD90.1 (Thy1.1), clone OX-7 (Biolegend) and APC anti-mouse CD8a, clone 53–6.7 (Biolegend) for 15 minutes at 4 °C. All samples were then washed and analyzed by flow cytometry on a BD FACSCelesta Instrument. CellTrace™ Violet dilution peaks were used to ascertain the percent of divided Thy1.1+ PMEL CD8+ T cells on Day 3 and Thy1.1 staining was used to quantify the frequency of Thy1.1+ PMEL CD8+ T cells on Day 7.
2.6. In vivo T cell killing assay
PMEL CD8+ T cells were adoptively transferred into irradiated B6 mice, either alone or with PLGA or PLGA/PBAE aAPCs, as described above. Six days after treatment, splenocytes from naive B6 mice were labelled with either 5 μM or 0.5 μM CFSE (Invitrogen), to generate CFSEhi and CFSElo populations, respectively. After labelling, CFSEhi splenocytes were loaded for 1 hour at 37°C with 1μg gp100 (GenScript) peptide per 107 cells, washed twice in PBS, counted, and then mixed 1:1 with unloaded CFSElo splenocytes. 107 cells of a 1:1 mixture of gp100 loaded CFSEhi and unloaded CFSElo cells were injected per recipient mouse from the various treatment groups. The following day, the mice were sacrificed, and their spleens were harvested and processed as described above. Percent specific killing was calculated as 100% *(1-[(CFSElo,control/CFSEhi,control)/(CFSElo,treatment/CFSEhi,treatment)]) .
2.7. In vivo tumor treatment study
To assess the efficacy of treatment with PLGA/PBAE aAPC in vivo, we utilized an immunotherapy murine melanoma treatment model. C57BL/6 mice (Jackson Laboratories; Bar Harbor, ME) were inoculated with 3 × 105 B16-F10 melanoma cells subcutaneously on the right flank. On days 4 and 11 after tumor inoculation, PLGA/PBAE aAPCs were administered intravenously at a 2 mg dose to half of the mice, and the other half received no particle treatment. Anti-PD1 clone RMP-14 (BioXCell; West Lebanon, NH) was administered intraperitoneally at 200 μg on the day of particle treatment and 100 μg on the following day. Beginning eleven days after tumor inoculation, tumor size was assessed with digital calipers every other day until the tumor area reached 200 mm2, after which the mice were sacrificed by CO2 asphyxiation and cervical dislocation.
2.8. Statistical methods
Multiple comparisons were made using ANOVAs with Sidak’s or Tukey’s post-tests when comparing across groups. Two-tailed Student’s t tests were used when comparing two groups. All graphs show mean and error bars that represent SEM. All n values are present within figure legends. All statistical analysis was performed using GraphPad Prism software version 8.1.
RESULTS AND DISCUSSION
3.1. aAPC Synthesis and Characterization
Artificial antigen presenting cells (aAPCs) are promising for cancer immunotherapy, but their clinical use has so far been limited to ex vivo expansion of CD8+ T cells prior to adoptive transfer. Cell-based therapies, though promising, are expensive to manufacture and have limited availability due to costs and the personalized nature of the therapeutic.23 However, improvements to aAPC technology would allow for them to be used acellularly, making it easier for the therapy to be mass-produced and stably stored over long periods of time. Therefore, we wanted to improve the efficacy of PLGA-based aAPCs beyond previous capacities that require adoptive transfer.13 Our strategy was to reformulate the particle core by incorporating a cationic hydrophobic polyester, PBAE 1-(3-aminopropyl)-4-methylpiperazine end-capped poly(1,4-butanediol diacrylate-co-4,4-trimethylenedipiperidine). PBAE is a softer material that alone has demonstrated immunostimulatory effects. PLGA/PBAE blended particles have shown improved material properties and cellular interactions in other contexts such as encapsulation of cargo for intracellular delivery and controlled release kinetics. They have not been previously evaluated for protein coupling to their surface or been investigated for their potential to function as aAPCs. The addition of PBAE may alter the physical properties of the particle, including its hydrophobicity and stiffness, which could enhance aAPC functioning.
Briefly, a linear biodegradable PBAE was synthesized from two sequential Michael addition reactions (Figure 2A). Monomers were reacted at stoichiometric ratios of 1.05, 1.1, and 1.2 methacrylate: amine monomers to prepare PBAEs. Resulting number average molecular weights were 18,322; 18,813; and 10,693 Da, respectively (Table S1). Microparticles were fabricated from PLGA or a blend of PLGA and PBAE, and subsequently functionalized with MHC Ig dimer and anti-CD28 to generate PLGA and PLGA/PBAE aAPCs.
Figure 2.
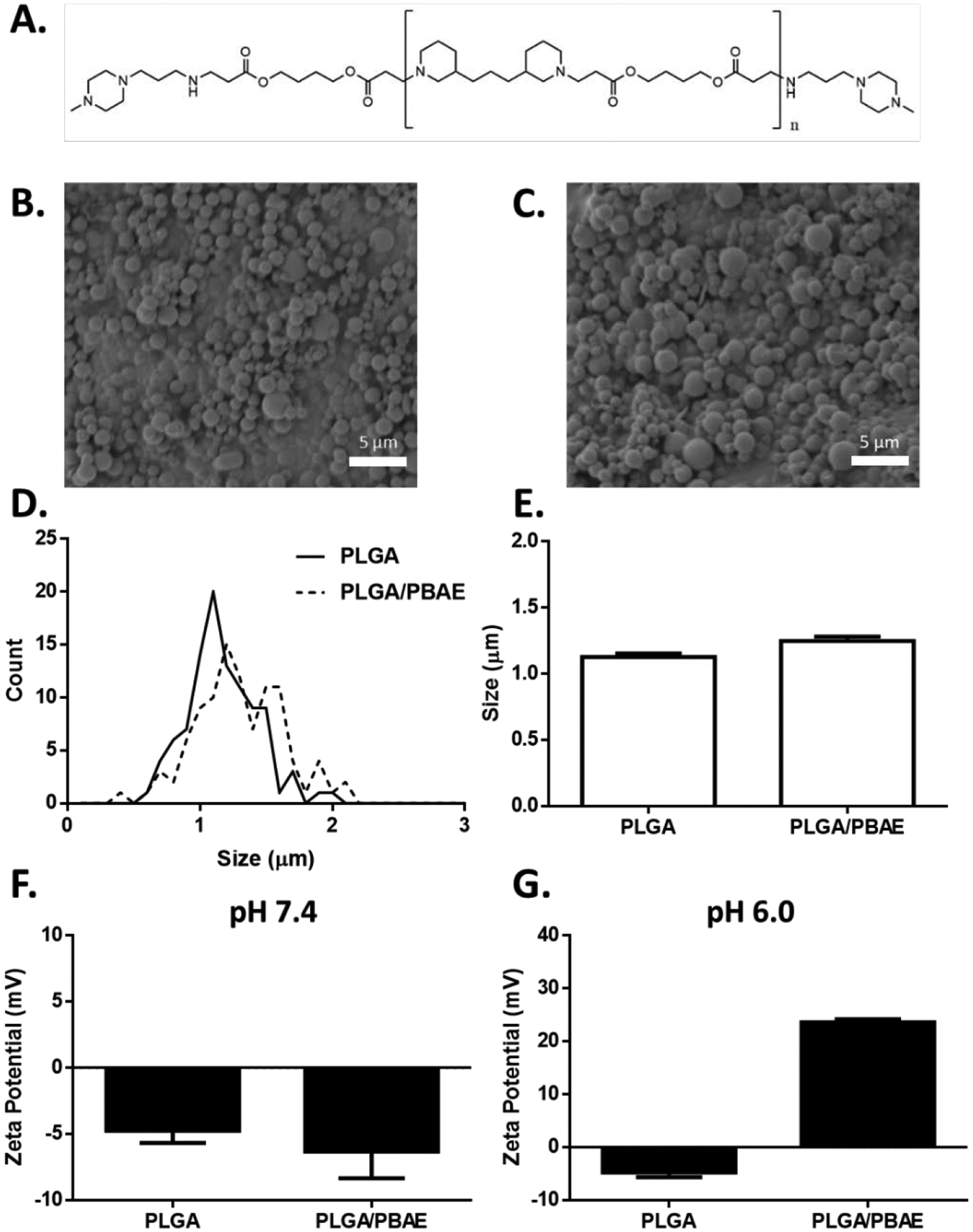
aAPC characterization and functional assessment. A) Chemical structure of PBAE synthesized via Michael addition of 1,4-butanediol diacrylate and 4,4-trimethylenedipiperidine and endcapped with 1-(3-aminopropyl)-4-methylpiperazine. SEM micrographs of B) PLGA and C) PLGA/PBAE microparticles. D) Particle size distribution of PLGA and PLGA/PBAE microparticles as evaluated by image analysis of SEM micrographs. E) Mean diameter of PLGA and PLGA/PBAE microparticles. Error bars represent SEM of 100 particles. F) PLGA and PLGA/PBAE have similar zeta potentials at pH 7.4. G) At pH 6.0, PLGA/PBAE particles exhibit a positive shift in zeta potential. Error bars in F) and G) represent SEM of 3 replicates.
We evaluated the physical properties of PLGA and PLGA/PBAE microparticles by imaging lyophilized particles using scanning electron microscopy (SEM) to determine size and morphology. The spherical morphology and topology of the surfaces of the PLGA and PLGA/PBAE particles were found to be similar between the two samples (Figure 2B–C). Upon sizing by ImageJ, it was found that the both particles had statistically similar sizes and size distributions (Figure 2D–E). PLGA particles were measured as 1.1 μm ± 0.3 μm and the PLGA/PBAE particles were 1.3 μm ± 0.3 μm, within 1 standard deviation of each other. This size was determined to be sufficient for T cell activation based on previous results with aAPC technology.24–25 In addition, based on our previous work it was found that the particles would be safe to administer intravenously as larger particles were determined to be pharmacokinetically safe.13 PLGA and PLGA/PBAE particles both possess a slightly negative surface charge at pH 7.4 (Figure 2F). To highlight a chemical difference between the particles, at pH 6, which is more representative of acidic environments, such as the tumor microenvironment, the zeta potential of PLGA/PBAE particles rises to 24 ± 1 mV, while PLGA particles retain a slight negative surface charge of −5 ± 2 mV (Figure 2G).
3.2. PBAE enhances surface protein presentation on aAPC
Blending PBAE with PLGA up to 25% PBAE did not alter particle size or morphology, but we found that increasing the relative amount of PBAE in the particle composition led to a corresponding increase in protein conjugation efficiency. We first sought to identify the optimal ratio of PLGA to PBAE to maximize protein conjugation to the particle surface. Microparticles were fabricated by blending 0, 5, 10, 15, or 25% PBAE by weight with PLGA. Particles were subsequently coupled with fluorescent monoclonal IgG antibody using EDC/NHS chemistry, which links proteins of interest to carboxylic acid groups on PLGA via amide bonds. Interestingly, the addition of PBAE dramatically increased the protein conjugation efficiency to the particle surface. The highest conjugation efficiency was achieved with the 25% PBAE blend (Figure 3A) at 8.7% compared to 0.06% for PLGA. As previously reported with a related PBAE, we found that increasing the amount of PBAE past 25% led to particle aggregation.19 Thus a 75%: 25% PLGA: PBAE blend was used to synthesize particles for all additional experiments.
Figure 3.
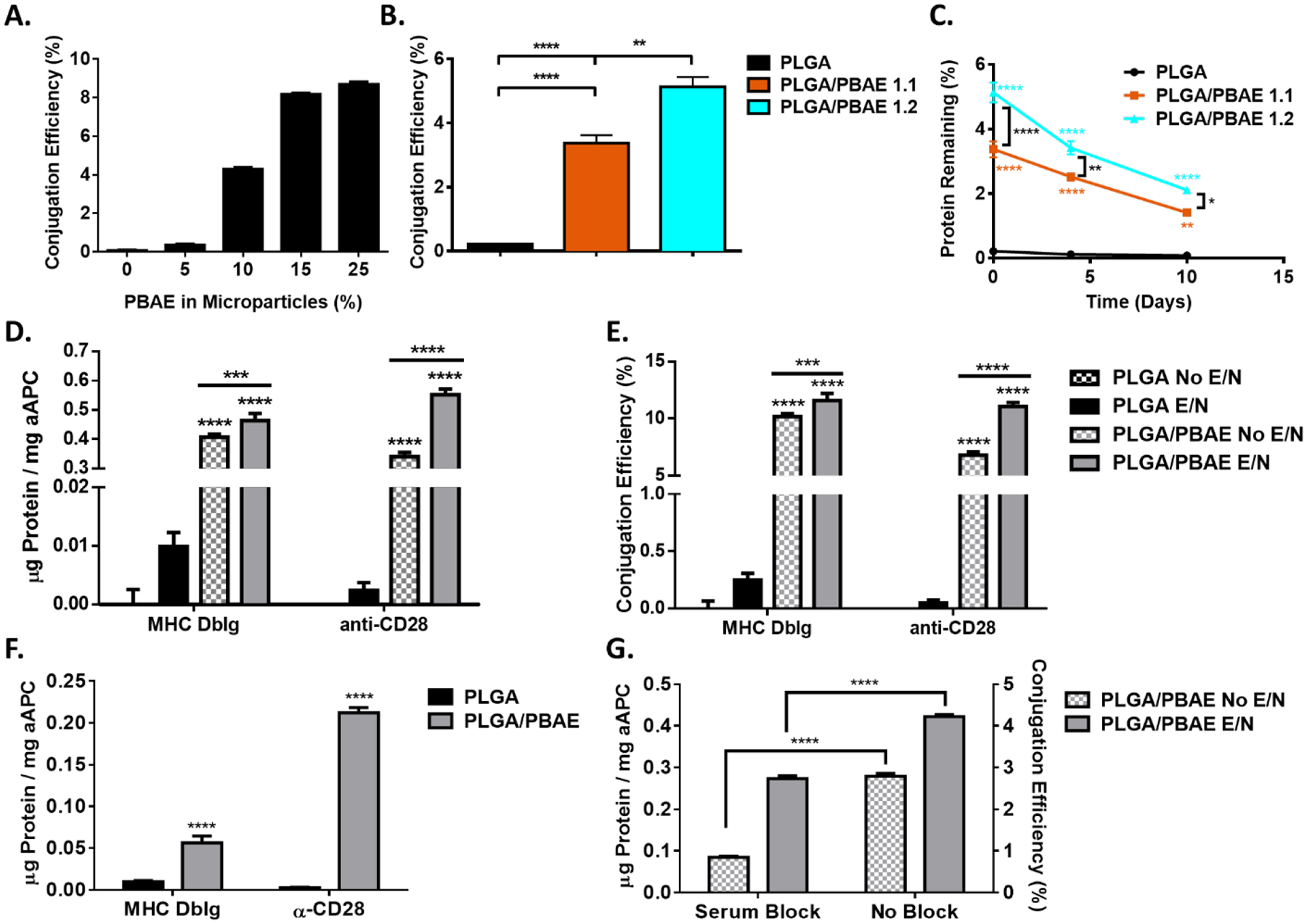
Characterization of PLGA and PLGA/PBAE aAPC. A) Increasing the PBAE content in microparticles improves protein conjugation efficiency. B) PLGA/PBAE particles were formed with PBAE synthesized with either 1.1:1 or 1.2:1 methacrylate: amine stoichiometric ratios. Particles were then conjugated with a fluorescent MHC DbIg and conjugation efficiency was measured using a fluorescent microplate reader. C) Particles were incubated at 37 °C and protein retention on the particle surface was measured at different timepoints using a fluorescent microplate reader. Error bars may be smaller than symbols. D-E) PLGA/PBAE particles conjugate signal 1 and 2 proteins with higher efficiency than PLGA in the presence and absence of EDC/NHS (E/N) conjugation reagents F) Non-covalently attached protein was subtracted from total conjugated protein. G) PLGA/PBAE particles were incubated with FBS prior to protein conjugation with or without EDC/NHS. Error bars in A) – C) represent the SEM of 3 replicates, error bars in D) – G) represent the SEM of 4 replicates, (* = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
We further looked to optimize protein conjugation by varying the stoichiometric ratio of monomers used to synthesize the PBAE. We analyzed the effect of varying the methacrylate: amine stoichiometric ratio during PBAE synthesis and measured the resulting molecular weight of the PBAE using gel permeation chromatography (Table S1). Increasing the stoichiometric ratio generally corresponded with a decrease in the molecular weight of the PBAE polymer. PLGA and PLGA/PBAE microparticles were synthesized and conjugated with a fluorescent antibody, and surface protein content was analyzed immediately after conjugation (Figure 3B) and after incubation at 37 °C over the course of 10 days (Figure 3C). There is a 1.5-fold increase in conjugation efficiency for particles made with PBAE synthesized from a 1.2:1 stoichiometric ratio compared to a 1.1:1 ratio. Protein stability over time was similar across all particles, with approximately 40% of protein remaining on the aAPC surface after 10 days. aAPCs were also incubated for one hour in PBS or media containing 10% serum and dynamic light scattering was utilized to assess particle size stability over time. PLGA aAPCs and PLGA/PBAE aAPCs did not increase in size due to the presence of serum for the duration of the study (Figure S1). The presence of high salt may lead to interactions between aAPCs that increases their measured hydrodynamic diameter at the initial time point compared to SEM particle size measurements, but the initial size did not significantly increase over time for either aAPC type in either incubation condition.
Following PBAE optimization we sought to compare conjugation efficiency of MHC Ig dimer and anti-CD28 to the surface of PLGA and PLGA/PBAE aAPC in the presence or absence of chemical conjugation reagents EDC and NHS (Figure 3D–E). PLGA/PBAE aAPC conjugated 46 times more signal 1 and 231 times more signal 2 protein mass compared to PLGA aAPC when EDC/NHS chemistry was used. The MHC Ig dimer and anti-CD28 were conjugated with similar efficiency to the surface of PLGA/PBAE particles (Figure 3E), although a larger amount of MHC Ig dimer is adsorbed compared to anti-CD28, which could potentially be due to its larger molecular weight26 or characteristic differences of the proteins. Some variation in conjugation efficiency and the ratio of MHC Ig dimer to anti-CD28 on the surface of the particle is typically observed.13, 27
When protein was incubated with particles in the absence of chemical conjugation reagents, the resulting protein on the surface of PLGA/PBAE particles was approximately 62–88% of the total amount measured after conjugation with EDC and NHS. This suggests that non-covalently attached protein mainly contributes to the enhanced conjugation efficiency of PLGA/PBAE over PLGA. When the amount of non-covalently attached surface protein was subtracted from the total measured amount of surface protein, PLGA/PBAE aAPC were found to conjugate 5.6 times more signal 1 and 89 times more signal 2 than PLGA aAPC as a direct result of EDC/NHS conjugation (Figure 3F). Enhanced chemical conjugation efficiency may be due in part to hydrolysis of PBAE at the surface of the particle, which could increase the presence of carboxylic acid groups available for protein conjugation.
PLGA/PBAE aAPC also show significant levels of non-covalently coupled, surface coated protein in the absence of EDC and NHS (Figure 3E). It has previously been shown that proteins can coat the surface of hydrophobic polystyrene particles in the absence of a chemically active reagent.28 Blending a cationic polymer, PBAE, with PLGA may increase the positive surface charge of the particle, and regions of positive charge and of hydrophobicity can favor antibody adsorption at a neutral pH.29 To determine whether non-covalently attached protein was being physically absorbed to the surface of PLGA/PBAE particles, we added a blocking step with 50% FBS for 1 hour at room temperature prior to conjugation with or without EDC and NHS. We found that the blocking step led to about a 3-fold decrease of non-covalently coupled protein on the surface of PB particles and a 1.5-fold decrease in total surface protein after conjugation using EDC/NHS chemistry (Figure 3G). The ability of PLGA/PBAE particles to conjugate protein with higher efficiency can thus be attributed to both enhanced physical protein adsorption and improved chemical conjugation efficiency, with the data indicating that physical adsorption is the main contributor. This increased conjugation efficiency can be critical to aAPC activity, as it has previously been found that the surface density of proteins on the aAPCs is a critical parameter in T cell activation and must be optimized.16 Continued optimization of protein conjugation to increase ligand surface density on PLGA/PBAE particles may further improve biological activity.
3.3. PLGA/PBAE aAPC bind more effectively to cognate CD8+ T cells
For optimal activity of the hybrid particles, it is important for the aAPC to have high specificity for cognate T cells recognizing the correct tumor-associated antigen/ MHC I complex. Thus, to further study how PLGA/PBAE particles interact with cells of interest, we performed aAPC/cell binding studies using confocal microscopy and flow cytometry. The ability of PLGA and PLGA/PBAE aAPC encapsulating a fluorescent dye to bind to CFSE-labelled CD8+ T cells was analyzed and compared to noncognate controls. Confocal microscopy showed that the cognate Db-gp100 PLGA and PLGA/PBAE aAPC exhibited increased interaction with PMEL T cells compared to non-cognate Kb-SIY aAPC (Figure 4A–D). Visually, more cells were bound to at least one aAPC and the average number of aAPC bound per cell was larger for PLGA/PBAE aAPC samples (Figure 4C–D). This trend was reinforced by quantitative flow cytometry results that showed a 53-fold increase in mean fluorescence intensity (MFI) for bound cognate PLGA/PBAE aAPCs compared to PLGA aAPC (Figure 4E). T cells showed a minimal increase in aAPC MFI when incubated with cognate PLGA aAPC compared to noncognate, suggesting a weak targeted interaction with PLGA aAPC. However, T cells exhibited a 35-fold increase in aAPC MFI when incubated with cognate PLGA/PBAE aAPC compared to the non-cognate PLGA/PBAE aAPC, showing robust antigen-specific interactions. PLGA/PBAE aAPC exhibited higher levels of binding to cognate T cells than PLGA aAPC, which agrees with previously reported findings that increased density of targeting protein correlates to increased binding and subsequently stronger immune stimulation.30
Figure 4.
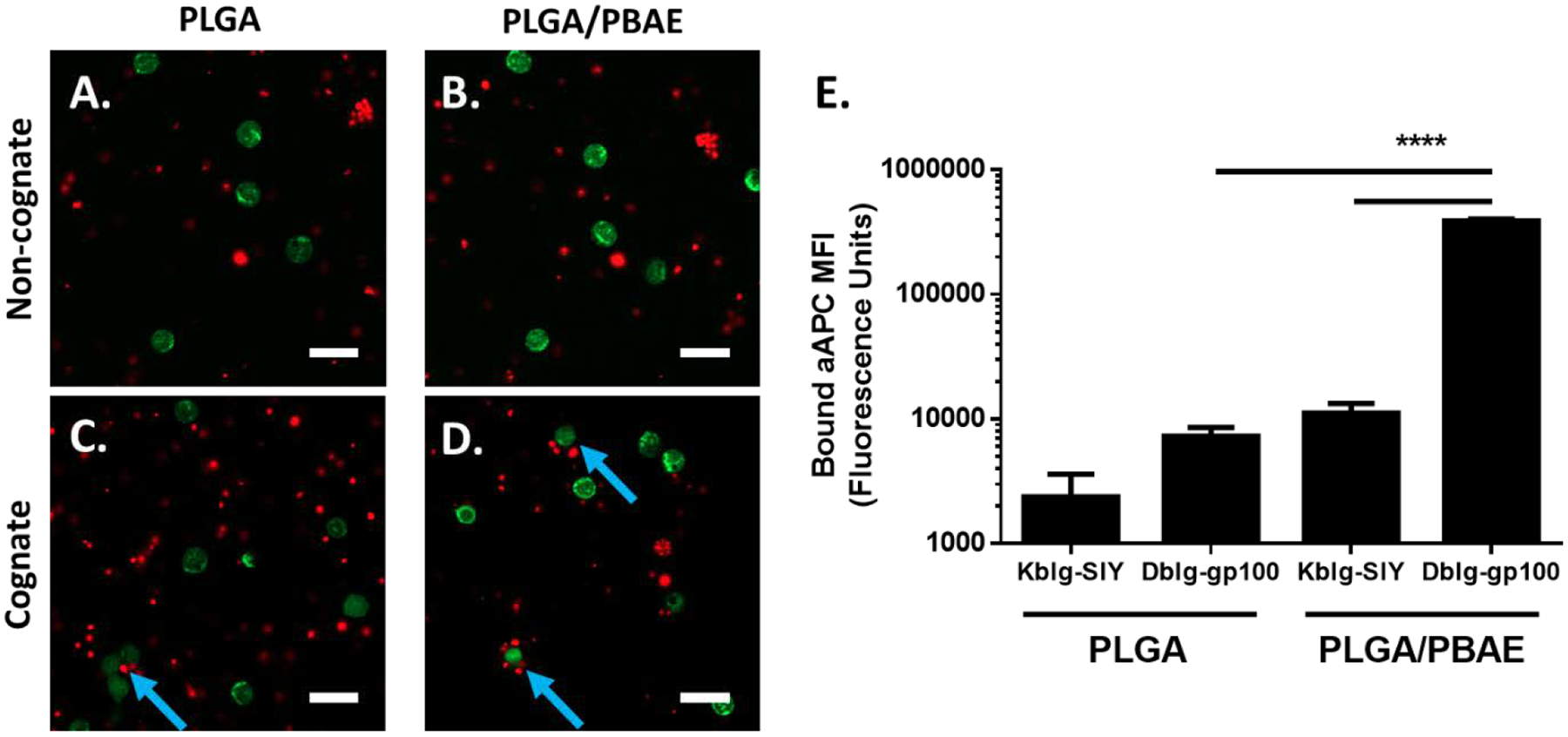
PLGA/PBAE aAPC (red) show higher levels of binding to cognate CD8+ T cells (green) compared to PLGA aAPC. DiD-labelled MHC DbIg-gp100 (cognate) or MHC KbIg-SIY (noncognate) aAPC were incubated with CFSE-labelled PMEL CD8+ T cells for 1 hour at 37 °C. Representative confocal images illustrate A) PLGA and B) PLGA/PBAE aAPC exhibit low levels of binding to non-cognate CD8+ T cells. C) PLGA aAPC show relatively low levels of binding to cognate CD8+ T cells. Blue arrows indicate binding events. D) PLGA/PBAE aAPC bind more frequently and in higher numbers to cognate-CD8+ T cells. Scale bars = 20 μm. E) After incubation of CD8+ T cells with particles, cells were washed and analyzed using flow cytometry. DiD fluorescence intensity was normalized to control samples with blank particles. PLGA/PBAE aAPC demonstrate enhanced binding to cognate CD8+ T cells, as evidenced by a more than 50-fold increase in DiD fluorescence intensity over CD8+ T cells incubated with PLGA aAPC. Error bars represent the SEM of 4 replicates, (* = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001).
3.4. aAPC stimulate peptide-specific CD8+ T cells in vitro
We next evaluated the ability of PLGA and PLGA/PBAE aAPC to stimulate CD8+ T cells from 2C and PMEL mice, two primary transgenic strains with CD8+ T cells recognizing a single peptide/MHC complex. To generate aAPCs, particles were functionalized with MHC DbIg-gp100 or KbIg-SIY, cognate to PMEL and 2C CD8+ T cells, respectively, and an anti-CD28 monoclonal antibody. aAPCs functionalized with each peptide-MHC complex were incubated at titrated doses with cognate CFSE-labelled 2C CD8+ T cells. Proliferation was assessed at 3 days by CFSE dilution and 7 days by counting.
CFSE dilution analyses revealed almost complete T cell activation by PLGA/PBAE aAPC at a dose of 1 μg per 100,000 T cells compared to 100 μg per 100,000 T cells for PLGA aAPC (Figure 5A). The limiting dose of activation was found to be 0.01 μg for PLGA/PBAE compared to 1 μg for PLGA. T cell stimulation by PLGA aAPC was similar in potency to our previously reported results.11, 13 Generational analysis shows the dose dependency of T cell expansion (Figure 5B). 45% of 2C cells divided four or more times at a 1 μg dose of PLGA/PBAE aAPC compared to only 15% at a 1 μg dose of PLGA aAPC. PLGA/PBAE aAPC also induced more cell division overall; at the 1 μg dose nearly 90% of cells had divided by Day 3 compared to about 43% of cells stimulated with PLGA aAPC. (Figure 5C).
Figure 5.
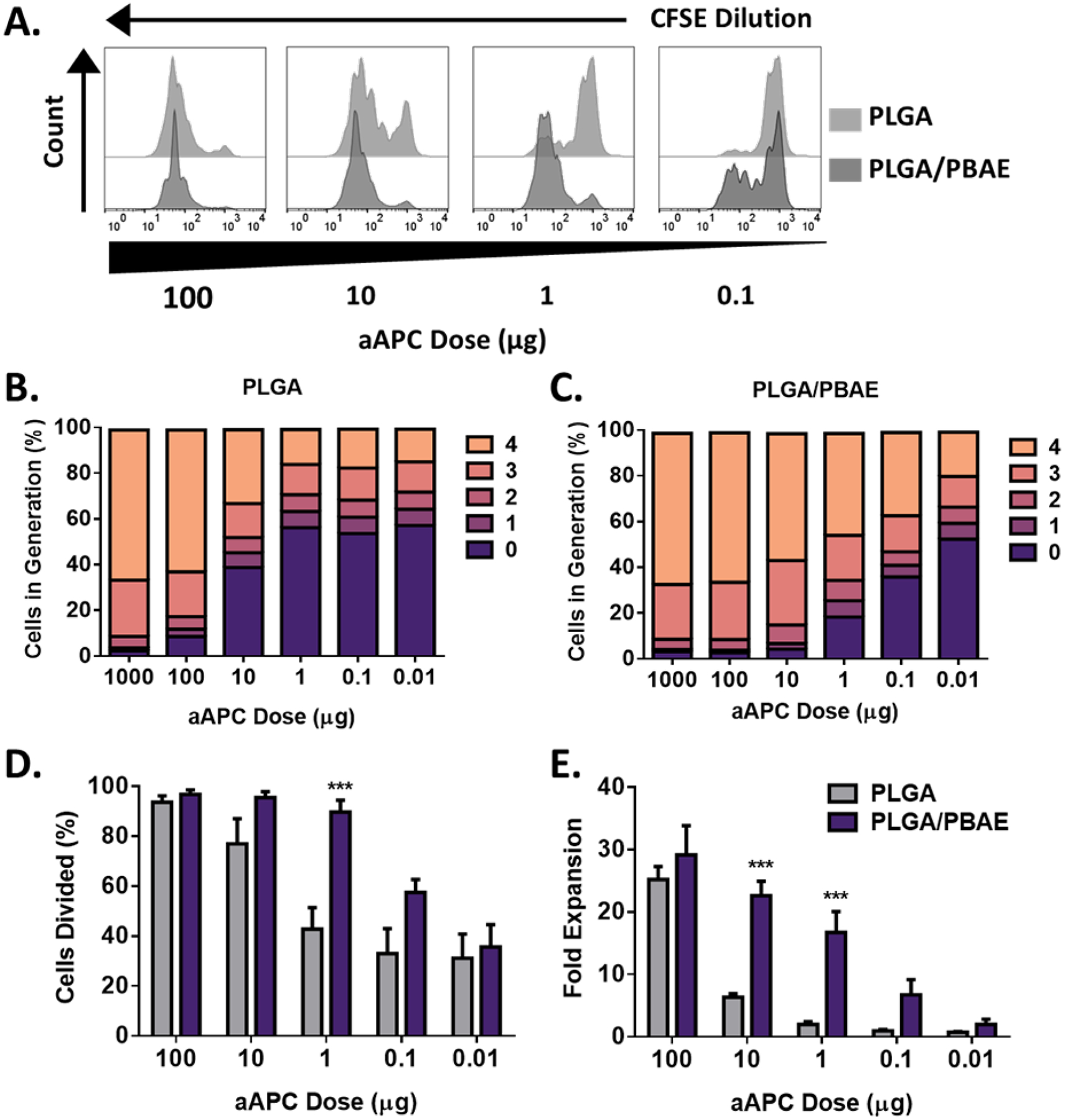
PLGA/PBAE aAPC mediate higher levels of antigen-specific 2C CD8+ T cell expansion ex vivo compared to PLGA aAPC, particularly at mid-range doses. A) Representative CFSE dilution peaks over a range of PLGA aAPC and PLGA/PBAE aAPC doses. Generation analyses of B) PLGA and C) PLGA/PBAE aAPC-treated CD8+ T cells. Generations were determined by analysis of CFSE dilution peaks. D) Percent of cells divided on day 3, quantified from CFSE dilution analysis. E) Fold expansion of CD8+ T cells on day 7. Error bars in D) and E) represent the SEM of 3 replicates, (*** = p<0.001).
After 7 days, CD8+ T cell counts confirmed dose-dependent expansion. PLGA/PBAE aAPC led to roughly a 15-fold increase in expansion compared to PLGA aAPC at 10 and 1 μg doses (Figure 5D). PLGA/PBAE aAPC were often nearly as effective as PLGA aAPC at 100-fold lower doses. There was also no adverse effect of the aAPC on the viability based on trypan blue analysis. These results suggest that the PLGA/PBAE aAPC are significantly stronger than PLGA aAPC for immune stimulation and could be used as an enhanced aAPC for immunotherapy.
3.5. aAPC expand peptide-specific CD8+ T cells in vivo
To be effective as an acellular therapy, aAPC must be able to activate and expand antigen specific CD8+ T cells in vivo. We next sought to determine the effectiveness of PLGA and PLGA/PBAE aAPC to expand anti-tumor CD8+ T cells in vivo, to find a leading candidate to be administered therapeutically. CellTrace Violet (CTV)-labelled PMEL CD8+ T cells and PLGA or PLGA/PBAE aAPC functionalized with Db-gp100 and anti-CD28 were co-administered to irradiated B6 mice. Mice received IL-2 intraperitoneally on days 2 and 4 post-treatment (Figure 6A). On day 3, blood samples were collected and analyzed for CTV-labelled CD8+ T cells (Figure 6B), and on day 7, spleens were harvested and analyzed for CTV-labelled CD8+ T cells (Figure 6C). The fraction of dividing PMEL cells in the blood on Day 3 was significantly increased in both PLGA and PLGA/PBAE aAPC-treated mice compared to control mice that received only PMEL cells, with 55% and 61% of cells divided, respectively, compared to 11% in control mice (Figure 6D). However, the mean CTV intensity of the PMEL cells was significantly reduced only in PLGA/PBAE aAPC-treated mice, indicative of cells in higher generations (Figure 6E). PLGA/PBAE aAPC treated mice had a 2.56-fold increase in PMEL cells within CD8+ T cell populations of the spleen compared to control mice (Figure 6F). Only PLGA/PBAE aAPC significantly expanded antigen specific CD8+ T cells in secondary lymphoid organs after 7 days.
Figure 6.
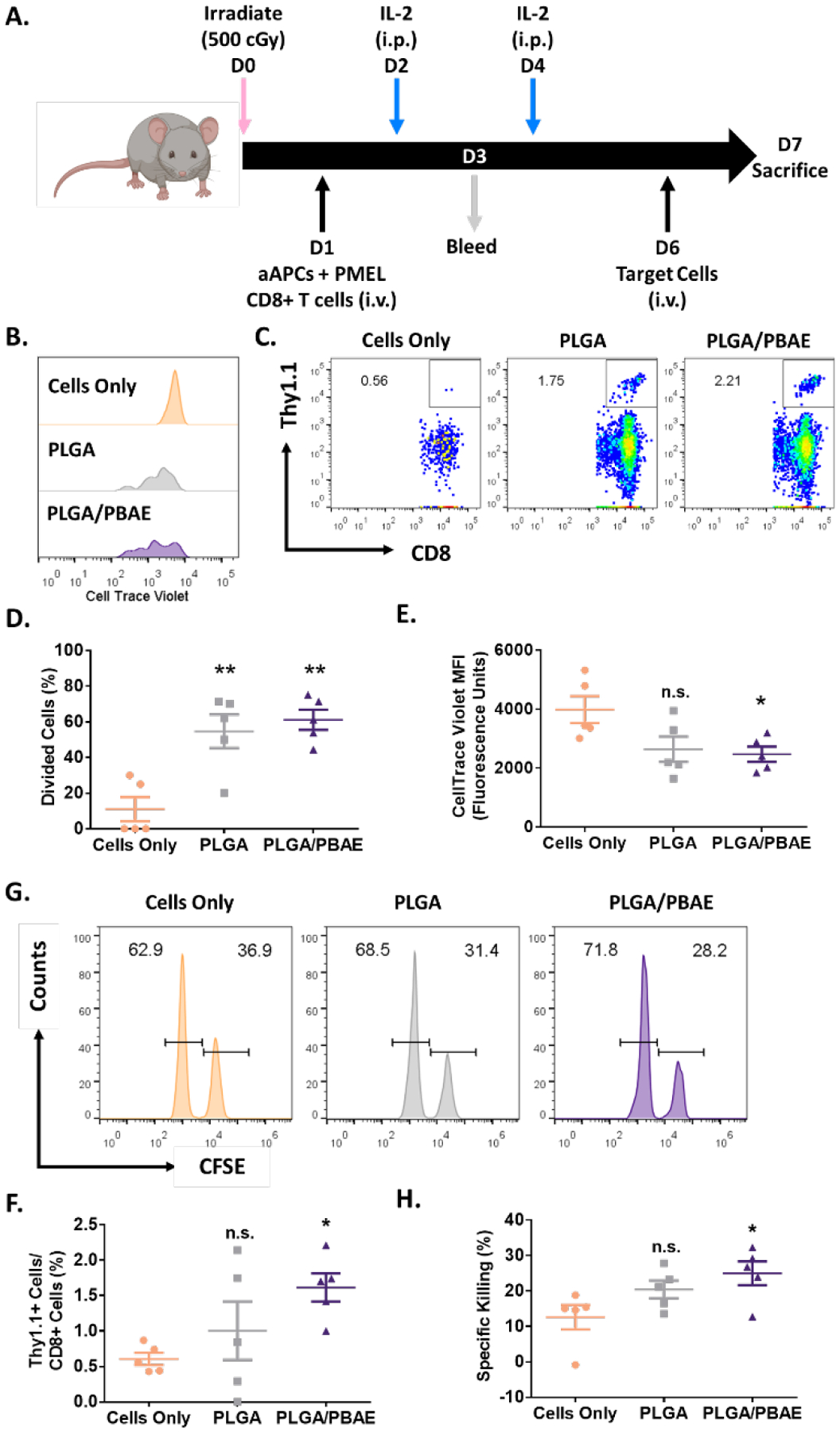
A) Schematic of in vivo antigen-specific CD8+ T cell expansion and killing assay timeline. Representative B) CTV dilution in PMEL cells in the blood on day 3 and C) FACS plots showing the percentage of Thy1.1+ CD8+ cells in the spleen on day 7. D) Percent of divided PMEL CD8+ T cells in the blood on day 3, quantified from analysis of CTV dilution in PMEL cells. E) MFI of CTV-labelled PMEL cells in the blood on day 3. F) Percent of Thy1.1+ PMEL cells within the CD8+ T cell population in the spleen of mice on day 7. G) Representative fractions of CFSEhi (peptide-pulsed) and CFSElo (control) target splenocyte populations in the spleen 24 hours after injection. H) Percent specific killing of target splenocytes on day 7 after aAPC treatment and 24 hours after administration of target cells. Error bars in E) – H) represent the SEM of 5 replicates, (* = p<0.05, ** = p<0.01, n.s.= not significant).
3.6. PLGA/PBAE aAPC treatment results in superior killing compared to PLGA aAPC treatment by peptide-specific CD8+ T cells in vivo
CD8+ T cells expanded by aAPCs must have antigen-specific cytotoxic capabilities to function therapeutically. To evaluate the cytotoxicity of aAPC-activated CD8+ T cells, PMEL CD8+ T cells and PLGA or PLGA/PBAE-aAPC were co-administered to irradiated B6 mice, followed by treatment with IL-2 according to the dosing schedule shown in Figure 6A. Six days later, splenocytes were harvested from B6 mice and labelled with high (hi) or low (lo) concentrations of CFSE. CFSEhi target B6 splenocytes were pulsed with gp100 and administered along with non-pulsed CFSElo B6 splenocytes intravenously. On the following day, spleens were harvested and analyzed for relative CFSElo and CFSEhi populations to evaluate antigen-specific killing (Figure 6G). CD8+ T cells from PLGA/PBAE aAPC-treated mice showed the highest level of antigen-specific killing, nearly twice the amount of killing by CD8+ T cells from mice that did not receive aAPCs, while PLGA aAPC-expanded CD8+ T cells demonstrated weaker cytotoxicity (Figure 6H).
3.7. Biodegradable PLGA/PBAE aAPCs administered intravenously delays tumor growth and extends survival in a B16-F10 melanoma mouse model
Finally, we wanted to evaluate our leading aAPC formulation in an endogenous B16-F10 melanoma tumor model in combination with immune checkpoint blockade, anti-PD1, an emerging standard of care. B16-F10 is a particularly aggressive tumor line and it has been shown that checkpoint blockade enhances the therapeutic effect of adoptive T cell transfer in this model.31 We have also previously shown that checkpoint blockade synergizes with aAPC treatment and adoptive T cell therapy to treat B16-F10 melanoma in C57BL/6 mice.13 To demonstrate proof-of-concept, we tested whether PLGA/PBAE aAPC along with anti-PD1 could slow the growth of established tumors without adoptive transfer of CD8+ T cells. PLGA/PBAE aAPCs were synthesized as above and conjugated with a 1:1 ratio of Kb-trp2 and Db-gp100 as signal 1, and anti-CD28 mAb as signal 2. Wild type C57BL/6 mice (n=6/ group) were injected with 3 × 105 B16-F10 cells, which constitutively express Kb-trp2 and Db-gp100, subcutaneously on the flank on day 0. On days 4 and 11, half of the mice were treated with 2 mg aAPC intravenously and half the mice were control mice that did not receive particle treatment (Figure 7A). All mice, including the control group, received 200 μg and 100 μg of anti-PD-1 monoclonal antibody on days 4 and 5, respectively, doses shown to have no effect on tumor growth in this mouse model when given as a monotherapy.13 Throughout the experiment, tumors in the PLGA/PBAE aAPC-treated group trended smaller than the untreated group, and by 11 days post-treatment, tumor growth was significantly reduced in the treatment group. PLGA/PBAE aAPC treatment reduced tumor growth up to 33% and led to a corresponding 31% increase in median survival time compared to anti-PD1 monoclonal antibody therapy alone (Figure 7B–C). This result is noteworthy for the development of aAPC technology as it has not previously been shown that a similar biodegradable particle-based, cell-free therapy has the potential be used as an in vivo treatment for melanoma. The B16-F10 melanoma model is immunosuppressive and it typically requires multiple particle injections32 or multiple biologically engineered therapies to treat effectively.33 Here, we have shown that injection of PLGA/PBAE aAPC in context of an immune checkpoint blockade can significantly increase the survival in a murine melanoma model, thus validating the PLGA/PBAE particle as an enhanced aAPC for cancer immunotherapy.
Figure 7.
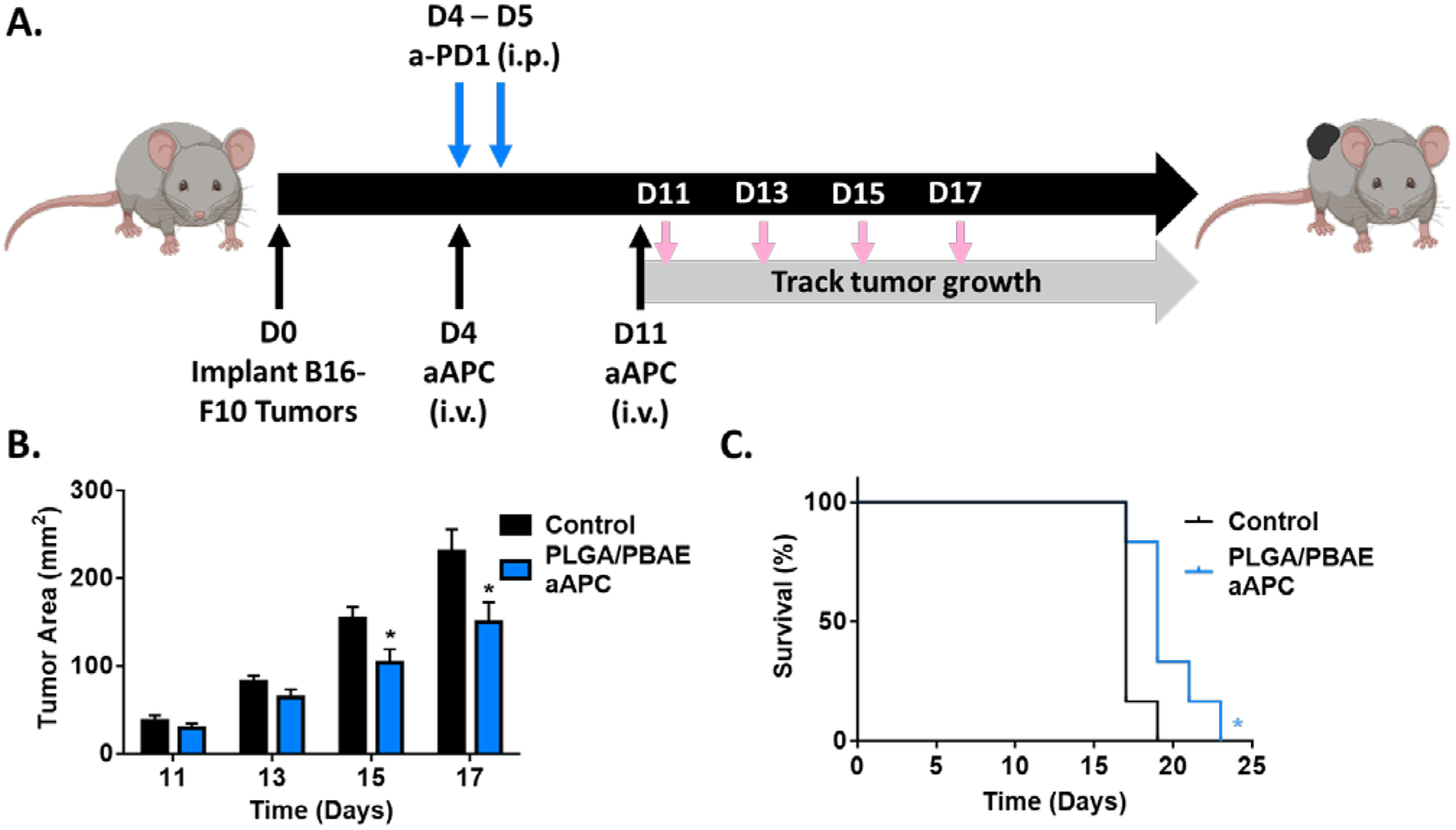
A) Schematic depicting dosing schedule for B16F10 tumor treatment model. Both the PLGA/PBAE aAPC and the control group received doses of anti-PD-1 monoclonal antibody i.p. on D4 and D5 and received no adoptive transfer of cells. B) Intravenous PLGA/PBAE aAPC treatment led to a reduction in tumor burden compared to untreated mice. C) Treatment with PLGA/PBAE aAPC extended survival compared to the control group. Error bars in B) and C) represent SEM of 6 replicates, (* = p<0.05).
CONCLUSION
We have designed, fabricated, and characterized a new polymer blend composed of a biodegradable anionic polymer, poly(lactic-co-glycolic acid), and a biodegradable cationic polymer, 1-(3-aminopropyl)-4-methylpiperazine end-capped poly(1,4-butanediol diacrylate-co-4,4-trimethylenedipiperidine) (PLGA/PBAE) and evaluated its utility as a core material for particulate aAPCs. These PLGA/PBAE aAPCs demonstrated enhanced binding, activation, and expansion of antigen-specific CD8+ T cells ex vivo compared to PLGA aAPCs using two different transgenic cell models. PLGA/PBAE aAPCs were observed to expand antigen-specific CD8+ T cells with specific cell-killing abilities in vivo. When evaluated therapeutically, PLGA/PBAE aAPCs were found to modestly stimulate endogenous cancer-targeting CD8+ T cells in an antigen-specific fashion. Intravenous therapeutic injection of PLGA/PBAE-aAPC can generate a cytotoxic T cell response in vivo and for the first time, has been shown to reduce tumor burden and increase median survival time in a tumor treatment model when used in conjunction with checkpoint blockade. Additional optimization of the PLGA/PBAE aAPC construct and dosing regimen may further enhance their therapeutic efficacy. These enhanced aAPCs can be constructed in a modular fashion to target different tumor antigens and may be promising for the treatment of a variety of solid tumors.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by grants from the National Institute of Biomedical Imaging and Bioengineering (P41EB028239 and R01EB029341), the National Cancer Institute (R01CA228133, R25CA153952, R33CA229042, and T32CA153952), the Troper Wojcicki Foundation, the Bloomberg~Kimmel Institute for Cancer Immunotherapy at Johns Hopkins, and the JHU Catalyst and Discovery award programs.
Footnotes
Supporting Information
Table S1
Figure S1
REFERENCES
- 1.Oelke M; Krueger C; Giuntoli RL 2nd; Schneck JP, Artificial Antigen-Presenting Cells: Artificial Solutions for Real Diseases. Trends Mol Med 2005, 11 (9), 412–420. [DOI] [PubMed] [Google Scholar]
- 2.Meyer RA; Green JJ, Artificial Antigen-Presenting Cells: Biomimetic Strategies for Directing the Immune Response. In Biomaterials in Regenerative Medicine and the Immune System, Santambrogio L, Ed. Springer International Publishing: Cham, 2015; pp 257–277. [Google Scholar]
- 3.Ben-Akiva E; Meyer RA; Wilson DR; Green JJ, Surface Engineering for Lymphocyte Programming. Adv Drug Deliv Rev 2017, 114, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steenblock ER; Fahmy TM, A Comprehensive Platform for ex vivo T-Cell Expansion based on Biodegradable Polymeric Artificial Antigen-Presenting Cells. Molecular Therapy 2008, 16 (4), 765–772. [DOI] [PubMed] [Google Scholar]
- 5.Steenblock ER; Fadel T; Labowsky M; Pober JS; Fahmy TM, An Artificial Antigen-Presenting Cell with Paracrine Delivery of IL-2 impacts the Magnitude and Direction of the T Cell Response. The Journal of Biological Chemistry 2011, 286 (40), 34883–34892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L; Wang L; Shahzad KA; Xu T; Wan X; Pei W; Shen C, Paracrine Release of IL-2 and anti-CTLA-4 Enhances the ability of Artificial Polymer Antigen-Presenting Cells to expand Antigen-Specific T Cells and Inhibit Tumor Growth in a Mouse Model. Cancer Immunology, Immunotherapy 2017, 66 (9), 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fadel TR; Sharp FA; Vudattu N; Ragheb R; Garyu J; Kim D; Hong E; Li N; Haller GL; Pfefferle LD; Justesen S; Herold KC; Fahmy TM, A Carbon Nanotube-Polymer Composite for T-Cell Therapy. Nat Nanotechnol 2014, 9 (8), 639–647. [DOI] [PubMed] [Google Scholar]
- 8.Perica K; De Leon Medero A; Durai M; Chiu YL; Bieler JG; Sibener L; Niemoller M; Assenmacher M; Richter A; Edidin M; Oelke M; Schneck J, Nanoscale Artificial Antigen Presenting Cells for T Cell Immunotherapy. Nanomedicine 2014, 10 (1), 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perica K; Tu A; Richter A; Bieler JG; Edidin M; Schneck JP, Magnetic Field-Induced T Cell Receptor Clustering by Nanoparticles enhances T Cell Activation and Stimulates Antitumor Activity. ACS Nano 2014, 8 (3), 2252–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perica K; Bieler JG; Schutz C; Varela JC; Douglass J; Skora A; Chiu YL; Oelke M; Kinzler K; Zhou S; Vogelstein B; Schneck JP, Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano 2015, 9 (7), 6861–6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunshine JC; Perica K; Schneck JP; Green JJ, Particle Shape Dependence of CD8+ T Cell Activation by Artificial Antigen Presenting Cells. Biomaterials 2014, 35 (1), 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fadel TR; Fahmy TM, Immunotherapy Applications of Carbon Nanotubes: from Design to Safe Applications. Trends Biotechnol 2014, 32 (4), 198–209. [DOI] [PubMed] [Google Scholar]
- 13.Kosmides AK; Meyer RA; Hickey JW; Aje K; Cheung KN; Green JJ; Schneck JP, Biomimetic Biodegradable Artificial Antigen Presenting Cells Synergize with PD-1 Blockade to Treat Melanoma. Biomaterials 2017, 118, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor RS; Hao X; Shen K; Bashour K; Akimova T; Hancock WW; Kam LC; Milone MC, Substrate Rigidity Regulates Human T Cell Activation and Proliferation. J Immunol 2012, 189 (3), 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu CJ; Hsieh WT; Waldman A; Clarke F; Huseby ES; Burkhardt JK; Baumgart T, Ligand Mobility Modulates Immunological Synapse Formation and T Cell Activation. PLoS One 2012, 7 (2), e32398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delcassian D; Depoil D; Rudnicka D; Liu M; Davis DM; Dustin ML; Dunlop IE, Nanoscale Ligand Spacing Influences Receptor Triggering in T Cells and NK Cells. Nano Lett 2013, 13 (11), 5608–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andorko JI; Hess KL; Pineault KG; Jewell CM, Intrinsic Immunogenicity of Rapidly-Degradable Polymers evolves during Degradation. Acta Biomater 2016, 32, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dold NM; Zeng Q; Zeng X; Jewell CM, A poly(Beta-Amino Ester) Activates Macrophages independent of NF-kappaB Signaling. Acta Biomater 2018, 68, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fields RJ; Cheng CJ; Quijano E; Weller C; Kristofik N; Duong N; Hoimes C; Egan ME; Saltzman WM, Surface Modified poly(Beta Amino Ester)-containing Nanoparticles for Plasmid DNA Delivery. J Control Release 2012, 164 (1), 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Vlerken LE; Duan Z; Little SR; Seiden MV; Amiji MM, Biodistribution and Pharmacokinetic Analysis of Paclitaxel and Ceramide administered in Multifunctional Polymer-Blend Nanoparticles in Drug Resistant Breast Cancer Model. Mol Pharm 2008, 5 (4), 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson DG; Peng W; Akinc A; Hossain N; Kohn A; Padera R; Langer R; Sawicki JA, A Polymer Library Approach to Suicide Gene Therapy for Cancer. Proc Natl Acad Sci U S A 2004, 101 (45), 16028–16033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneck JP; Slansky JE; O’Herrin SM; Greten TF, Monitoring Antigen-Specific T Cells using MHC-Ig Dimers. Curr Protoc Immunol 2001, Chapter 17, Unit 17 2. [DOI] [PubMed] [Google Scholar]
- 23.de Sa Silva F; Almeida PN; Rettore JV; Maranduba CP; de Souza CM; de Souza GT; Zanette Rde S; Miyagi SP; Santos Mde O; Marques MM; Maranduba CM, Toward Personalized Cell Therapies by using Stem Cells: Seven Relevant Topics for Safety and Success in Stem Cell Therapy. J Biomed Biotechnol 2012, 2012, 758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mescher MF, Surface Contact Requirements for Activation of Cytotoxic T Lymphocytes. The Journal of Immunology 1992, 149 (7), 2402–2405. [PubMed] [Google Scholar]
- 25.Hickey JW; Vicente FP; Howard GP; Mao HQ; Schneck JP, Biologically Inspired Design of Nanoparticle Artificial Antigen-Presenting Cells for Immunomodulation. Nano Lett 2017, 17 (11), 7045–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler JE, 9 - Solid Phases in Immunoassay. 1996, 205–225. [Google Scholar]
- 27.Meyer RA; Sunshine JC; Perica K; Kosmides AK; Aje K; Schneck JP; Green JJ, Biodegradable Nanoellipsoidal Artificial Antigen Presenting Cells for Antigen Specific T-Cell Activation. Small 2015, 11 (13), 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Champion JA; Mitragotri S, Role of Target Geometry in Phagocytosis. Proc Natl Acad Sci U S A 2006, 103 (13), 4930–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai TM; Mehta RC; DeLuca PP, Adsorption of Peptides to poly(D,L-lactide-co-glycolide) .2. Effect of Solution Properties on the Adsorption. Int J Pharm 1996, 127 (1), 43–52. [Google Scholar]
- 30.Cheung AS; Zhang DKY; Koshy ST; Mooney DJ, Scaffolds that Mimic Antigen-Presenting Cells enable ex vivo Expansion of Primary T Cells. Nat Biotechnol 2018, 36 (2), 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahvi DA; Meyers JV; Tatar AJ; Contreras A; Suresh M; Leverson GE; Sen S; Cho CS, CTLA-4 Blockade plus Adoptive T-Cell Transfer promotes Optimal Melanoma Immunity in Mice. J Immunother 2015, 38 (2), 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosmides AK; Sidhom JW; Fraser A; Bessell CA; Schneck JP, Dual Targeting Nanoparticle Stimulates the Immune System To Inhibit Tumor Growth. ACS Nano 2017, 11 (6), 5417–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moynihan KD; Opel CF; Szeto GL; Tzeng A; Zhu EF; Engreitz JM; Williams RT; Rakhra K; Zhang MH; Rothschilds AM; Kumari S; Kelly RL; Kwan BH; Abraham W; Hu K; Mehta NK; Kauke MJ; Suh H; Cochran JR; Lauffenburger DA; Wittrup KD; Irvine DJ, Eradication of Large Established Tumors in Mice by Combination Immunotherapy that Engages Innate and Adaptive Immune Responses. Nat Med 2016, 22 (12), 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.