

Abstract
Transcriptional enhancer factor domain family member 4 (TEAD4) is a downstream effector of the conserved Hippo signaling pathway, regulating the expression of genes involved in cell proliferation and differentiation. It is up‐regulated in several cancer types and is associated with metastasis and poor prognosis. However, its role in hepatocellular carcinoma (HCC) remains largely unexplored. Using data from The Cancer Genome Atlas, we found that TEAD4 was overexpressed in HCC and was associated with aggressive HCC features and worse outcome. Overexpression of TEAD4 significantly increased proliferation and migration rates in HCC cells in vitro as well as tumor growth in vivo. Additionally, RNA sequencing analysis of TEAD4‐overexpressing HCC cells demonstrated that TEAD4 overexpression was associated with the up‐regulation of genes involved in epithelial‐to‐mesenchymal transition, proliferation, and protein‐folding pathways. Among the most up‐regulated genes following TEAD4 overexpression were the 70‐kDa heat shock protein (HSP70) family members HSPA6 and HSPA1A. Chromatin immunoprecipitation–quantitative real‐time polymerase chain reaction experiments demonstrated that TEAD4 regulates HSPA6 and HSPA1A expression by directly binding to their promoter and enhancer regions. The pharmacologic inhibition of HSP70 expression in TEAD4‐overexpressing cells reduced the effect of TEAD4 on cell proliferation. Finally, by overexpressing TEAD4 in yes‐associated protein (YAP)/transcriptional coactivator with PDZ binding motif (TAZ)‐knockdown HCC cells, we showed that the effect of TEAD4 on cell proliferation and its regulation of HSP70 expression does not require YAP and TAZ, the main effectors of the Hippo signaling pathway. Conclusion: A novel Hippo‐independent mechanism for TEAD4 promotes cell proliferation and tumor growth in HCC by directly regulating HSP70 family members.
Abbreviations
- CAM
chorioallantoic membrane
- ChIP
chromatin Immunoprecipitation
- CREAM
Cognitive Reliability and Error Analysis Method
- CTGF
connective tissue growth factor
- CTR
control
- DMSO
dimethyl sulfoxide
- EMT
epithelial‐to‐mesenchymal transition
- GO
Gene Ontology
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- IgG
immunoglobulin
- IHC
immunochemistry
- kb
kilobase
- KNK437
heat shock protein family A inhibitor 1
- qPCR
quantitative real‐time polymerase chain reaction
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- seq
sequencing
- TAZ
transcriptional coactivator with PDZ binding motif
- TCGA
The Cancer Genome Atlas
- TEAD
transcriptional enhancer factor domain
- TEAD4
transcriptional enhancer factor domain family member 4
- YAP
yes‐associated protein
Transcriptional enhancer factor domain (TEAD) proteins are a family of transcription factors that bind the consensus 5′‐CATTCCA/T‐3′ sequence through their shared TEA DNA binding domain.( 1 ) TEAD‐regulated transcription strictly depends on the binding of TEAD transcription factors with various coactivators,( 2 , 3 ) among which are the yes‐associated protein (YAP) and transcriptional coactivator with PDZ binding motif (TAZ), two major effectors of the Hippo signaling pathway,( 4 , 5 ) and the vestigial‐like protein 1 (VGLL1).( 6 ) In mammals, TEADs are highly conserved and widely expressed( 7 , 8 ) and play a pivotal role in development by mediating cell proliferation and organ size control through the Hippo signaling pathway.( 9 ) In cancer cells, TEADs regulate proliferation, migration, differentiation, epithelial‐to‐mesenchymal transition (EMT), apoptosis, and invasion.( 10 ) In particular, TEAD factors regulate the expression of progrowth factors, such as connective tissue growth factor (CTGF),( 10 , 11 ) cysteine‐rich angiogenic inducer 61 (Cyr61),( 11 ) AXL receptor tyrosine kinase (AXL),( 12 ) MYC proto‐oncogene bHLH transcription factor (Myc),( 13 ) baculoviral inhibitor of apoptosis repeat‐containing 5 (survivin),( 13 ) and insulin‐like growth factor binding protein 5 (IGFBP5).( 6 ) Furthermore, overexpression of TEADs and their association with a poor clinical outcome have been reported in many cancer types, including breast, lung, prostate, colon, and gastric cancers as well as melanoma and glioblastoma.( 14 , 15 , 16 )
TEAD family member 4 (TEAD4), the gene encoding for the transcriptional enhancer factor (TEF)‐3, is overexpressed in several tumor types, such as breast and gastric cancers,( 16 ) as well as hepatoblastoma, the most common type of pediatric liver cancer.( 17 ) Notably, TEAD4 acts as an oncogene through Hippo signaling‐dependent( 18 ) and signaling‐independent mechanisms.( 6 , 15 ) In hepatocellular carcinoma (HCC), it is known that TEAD4 acts through the Hippo signaling pathway, wherein the TEAD4/YAP complex cooperates with forkhead box M1 (FOXM1) in inducing chromosome instability.( 19 ) It has also been shown that up‐regulation of the YAP2/TEAD4 axis by sirtuin 1 (SIRT1) deacetylation of YAP2 promotes HCC cell proliferation.( 20 ) Furthermore, TEAD4/YAP and hepatocyte nuclear factor 4α (HNF4α) regulate hepatocarcinogenesis by reciprocal repression in mice and rats.( 21 ) Thus, until now, TEAD4 was thought to play a YAP‐dependent role in HCC oncogenesis.
In this study, we show that TEAD4 is overexpressed in a subset of HCC and that it promotes cell proliferation in HCC cells both in vitro and in vivo. We further show that TEAD4 directly regulates HSP70 in a Hippo‐independent mechanism.
Materials and Methods
Cell Lines
HCC‐derived cell lines (HepG2, SNU449, HLE, and Huh7) were maintained in a 5% CO2‐humidified atmosphere at 37°C and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin (Bio‐Concept, Allschwil, Switzerland), and 1% minimal essential medium–nonessential amino acids (ThermoFisher Scientific, Basel, Switzerland). Stable YAP‐ and TAZ‐knockdown Huh7 and SNU449 cell lines (described in Tang et al.( 22 )) and their respective controls were maintained in complete medium supplemented with puromycin (1 μg/mL). All cell lines were confirmed negative for mycoplasma infection using the polymerase chain reaction (PCR)‐based Universal Mycoplasma Detection kit (American Type Culture Collection, Manassas, VA). For HSP70 inhibitor 1 (KNK437; CAS 218924‐25‐5; Calbiochem, Sigma‐Aldrich, St. Louis, MO), cells were incubated with 100 μM of the inhibitor and the corresponding dimethyl sulfoxide (DMSO) control.
Human Samples
Ten human, unselected, nonconsecutive HCCs retrieved from the archives of the Institute of Medical Genetics and Pathology at the University Hospital Basel (Basel, Switzerland) were included in this study. These 10 samples had already been prescreened for HSP70 protein expression. The study was performed in accordance with the Helsinki Declaration and approved by the Ethics Committee of Basel. Data were collected retrospectively in a nonstratified and nonmatched manner, including patient age, tumor diameter, location, prognostic tumor/prognostic node stage, grade, histologic subtype, and vascular invasion.
Immunohistochemistry
Immunohistochemistry (IHC) was performed as described.( 23 ) Details are presented in the Supporting Information.
Plasmids and Transfection
For TEAD4 overexpression, pLV[Exp]‐enhanced green fluorescent protein (EGFP)/Neo‐EF1A>hTEAD4 was designed and ordered on the vector builder website (https://en.vectorbuilder.com/), and the empty control vector was plasmid cytomegalovirus (pCMV)‐mir EGFP. For TEAD4 silencing, pSuper‐retro‐puro empty vector and pSuper‐retro‐puro shTEAD1/3/4 were adapted from Zhao et al.( 11 ) The expression vectors were transiently transfected using the jetPRIME transfection reagent (Polyplus, Illkirch, France) following the manufacturer's instructions. The expression of the plasmids was evaluated by western blot and quantitative reverse‐transcription (qRT)‐PCR analysis. Cells were harvested 48 hours after transfection for further experiments.
Protein Extraction and Western Blot RNA Extraction and qRT‐PCR and RNA Sequencing
Protein extraction and western blot RNA extraction along with qRT‐PCR and RNA sequencing (RNA‐seq) are detailed in the Supporting Information.
Proliferation and Migration Assays
Proliferation and cell migration assays were performed using the xCELLigence Real‐Time Cell Analysis‐dual purpose (ACEA Biosciences, San Diego, CA) system. All experiments were performed in triplicate. Results are shown as mean ± SD. Statistical significance was assessed by the t test.
Wound‐Healing Assay
Twenty‐four hours after transfection, a 100‐μL micropipette tip was used to create a scratch in the cell monolayer in each well of the six‐well plate. To monitor the wound closure, representative phase‐contrast images of each well were taken at 0, 8, 12, and 24 hours after the scratch was made. Each experimental condition was evaluated in triplicate. Statistical significance was assessed by the t test.
Chorioallantoic Membrane Assay
Fertilized chicken eggs were obtained from a local hatchery (Gepro Geflügelzucht AG, Flawil, Switzerland) at day 1 of gestation and were incubated at 37°C with 60% humidity for 10 days. The cells were harvested 24 hours after transfection, suspended (2 × 106 cells per chorioallantoic membrane [CAM] assay) in 10 μL of medium (DMEM), mixed 1:1 with matrigel (Matrigel Matrix; Ref. 354234; Corning, Tewksbury, MA), and grafted onto the CAM of 9‐day‐old chicken embryos. The chicken embryos were maintained in ovo at 37°C for another 4 days, followed by removal of the tumors. Pictures of each tumor were taken with a Canon EOS 1100D digital camera. Tumor size measurements were performed by averaging the volume (height × width × width) of each tumor, using ImageJ as described.( 24 )
CellTiter‐Glo Cell Viability Assay
CellTiter‐Glo (G7573; Promega, Dübendorf, Switzerland) was used to determine the number of viable cells based on adenosine triphosphate content. Twenty‐four hours after transfection, cells were seeded in a 96‐well plate. After 8 hours, drug treatment was added to the cells and cell viability was measured by adding 100 μL of CellTiter‐Glo/well at 24, 48, and 72 hours posttreatment. Statistical significance was assessed by multiple t test. In sorafenib experiments, results were normalized to DMSO. Curve fitting was performed using Prism software (GraphPad Software, San Diego, CA) and the nonlinear regression equation.
Analysis of Chromatin Immunoprecipitation Sequencing Data
Chromatin immunoprecipitation (ChIP)‐seq data for TEAD4 in the HepG2 human liver cell line were produced using mouse monoclonal immunoglobulin G (IgG) raised against recombinant protein TEAD4 and were obtained from the Gene Expression Omnibus (accession number GSM1010875).( 25 ) Processed data as an hg19 bigWig file were loaded into Integrative Genomics Viewer,( 26 ) and the “Find Motif” tool was used to search for the most conserved bases of TEAD4 binding motif within regions of TEAD4 ChIP‐seq peaks. TEAD4 binding motif was obtained from the JASPAR 2018 database.( 27 ) TEAD4 enhancers were called from the TEAD4 broadPeak BED file using the Cognitive Reliability and Error Analysis Method (CREAM),( 28 ) with the options WScutoff = 1.5, Minlength = 1000, and peakNumMin = 2. CREAM calls enhancers based on clusters of peaks, taking into account the distribution of distances between peaks in ChIP‐seq data from a given sample.
ChIP
Cells from four 10‐cm Petri dishes at 70%‐80% confluence were crosslinked in 1% formaldehyde for 10 minutes with continuous shaking. The crosslinking was stopped by adding 0.15 M glycine while continuing shaking. After collecting the cells by scraping, the pellet was washed 3 times with cold phosphate‐buffered saline. The nuclei of the cells were isolated and lysed. Chromatin shearing was performed using Bioruptor Pico (Diagenode, Liège, Belgium). The number of cycles and the settings were as described.( 29 ) At the same time, the antibody was coupled with magnetic protein G beads (100‐03D; Invitrogen, Carlsbad, CA) by incubating 75 μL of protein G beads with 10 μg of TEAD4 antibody (TEF‐3, sc‐101184; Santa Cruz Biotechnology, Dallas, TX) or 10 μg of mouse IgG (sc‐2025; Santa Cruz Biotechnology) as a negative control for 1 hour with constant rotation. At the end of the sonication process, an aliquot of the chromatin was kept as input control for every sample and an equal amount of sonicated chromatin was incubated with the previously produced antibody‐coupled magnetic beads at 4°C overnight while rotating. The samples were washed and then eluted (all buffers are as described in the original protocol from Blecher‐Gonen et al.( 30 )). Ribonuclease treatment and then proteinase K treatment were performed on all samples, followed by overnight reverse crosslinking at 65°C with continuous shaking. DNA purification was performed using Agencourt AMPure XP (A63880; Beckman Coulter, Brea, CA). TEAD4 abundance on specific target gene promoters was quantified by qRT‐PCR compared to IgG negative control. Primer sequences are listed in Supporting Table S1.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism version 6.0 and R. For in vitro and in vivo studies, statistical significance was determined by the two‐tailed unpaired Student t test. Differences were considered statistically significant at P < 0.05. All experiments were performed at least twice. The statistical parameters (i.e., the exact value of n, P values) are noted in the figures and figure legends. Results are shown as mean ± SD.
Analysis of The Cancer Genome Atlas Liver Data Set
Analysis of The Cancer Genome Atlas (TCGA) liver data set is described in the Supporting Information.
Results
TEAD4 Overexpression Promotes HCC Tumor Growth In Vitro and In Vivo
As TEAD4 overexpression has been associated with poor clinical outcome in a number of cancer entities, we asked whether TEAD4 overexpression plays a similar role in HCC. We first confirmed that TEAD4 was overexpressed in HCC using the TCGA data set. Compared to the nontumoral livers, HCCs showed TEAD4 overexpression (P < 0.001, Mann‐Whitney U test) in 21% (n = 73/371) of cases (Supporting Fig. S1A,B). Additionally, we observed that TEAD4 expression was associated with aggressive HCC features, such as high Edmondson grade, high stage, and histologic changes, more frequently observed in severe diseases, such as the presence of necrosis, pleomorphic, and multinucleated cells (P < 0.05; Supporting Fig. S1C). Additionally, TEAD4‐overexpressing tumors were more enriched in Hoshida molecular subtype S1, which is associated with higher proliferation and worse outcome (Supporting Fig. S1C). Furthermore, we observed that TEAD4 overexpression was associated with worse overall survival and was independently prognostic in patients with HCC in univariate and multivariate models (hazard ratio, 1.52; 95% confidence interval, 1.00‐2.29) (Supporting Fig. S1D; Supporting Table S2).
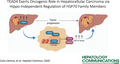
Having determined that TEAD4 is indeed overexpressed in HCC, we modulated the expression of TEAD4 in HCC cell lines, examined its effect on cell growth, and further exploited these models to dissect the possible oncogenic role of TEAD4. Using a complementary DNA construct, we overexpressed TEAD4 in two HCC cell lines with low endogenous levels of TEAD4 expression (Huh7 and SNU449; Supporting Fig. S2A,B). We further used short‐hairpin RNAs to silence TEAD4 in two HCC cell lines with high endogenous levels of TEAD4 expression (HLE and HepG2; Supporting Fig. S2A,B). We observed that TEAD4 overexpression significantly increased proliferation as well as cell migration in both Huh7 and SNU449 cells (Fig. 1A,B; Supporting Fig. S2C,D). On the contrary, TEAD4‐silenced HLE and HepG2 cells significantly proliferated and migrated less compared to control cells (Fig. 1A,B; Supporting Fig. S2C,D). The wound‐healing assay further confirmed the role of TEAD4 in regulating the migration potential of HCC cells. Indeed, forced expression of TEAD4 in Huh7 led to an increase in the gap closure rate, while TEAD4 silencing slowed the speed of gap closure in HLE cells (P < 0.01, Mann‐Whitney U test) (Fig. 1C). Finally, because sorafenib is still the primary choice for first‐line treatment of patients with HCC, we tested if the modulation of TEAD4 expression might affect the response to sorafenib. Our results showed that overexpression or silencing of TEAD4 did not alter the sensitivity of the cells to sorafenib (Supporting Fig. S2E).
FIG. 1.
Modulation of TEAD4 expression level impacts cell proliferation and migration. (A) Proliferation and (B) migration kinetics of TEAD4‐overexpressing (TEAD4ox) Huh7 and TEAD4‐silenced (shTEAD4) HLE cells compared to their respective controls. (C) Wound‐healing assay in TEAD4‐overexpressing Huh7 and TEAD4‐silenced HLE cells compared to their respective controls at specific time points. Representative pictures at time points 0 and 24 hours after scratch are shown for each condition. Scale bar in (C), 1 cm. Statistical significance was determined by the t test; *P < 0.05,**P < 0.01. Data are mean ± SD. Abbreviations: h, hours; sh, short hairpin.
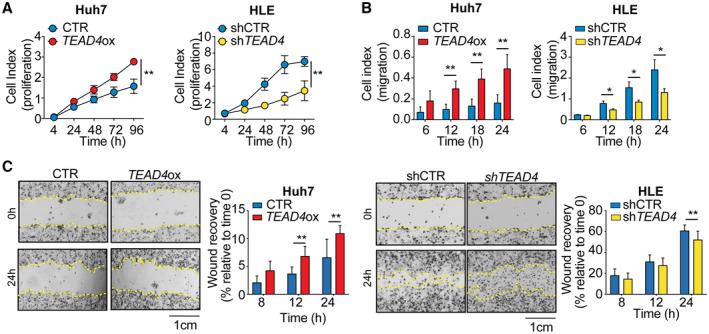
To further demonstrate the oncogenic role of TEAD4 in HCC, we xenotransplanted TEAD4‐overexpressing and control Huh7 cells into the chicken CAM and assessed tumor growth in vivo (Fig. 2A). Engraftment of tumor cells in the CAM has been successfully used as a model of tumorigenesis.( 31 ) Indeed, it has been shown that this densely vascularized extraembryonic tissue enables a fast, reproducible, and precise analysis of the principal steps of tumor progression and is a powerful tool for preclinical in vivo studies.( 32 ) Briefly, 24 hours posttransfection, cells were harvested and resuspended in matrigel before being seeded into the CAMs. After 4 days, eggs were screened for tumor formation and tumors were harvested for quantification and further analysis. In accordance with the results obtained in vitro, TEAD4‐overexpressing engrafted cells formed significantly larger tumors compared to control cells (Fig. 2B,C; P < 0.05, Mann‐Whitney U test). IHC and western blot analysis confirmed that the resected tumors were indeed of human origin and that TEAD4 overexpression could still be detected 5 days posttransfection (Fig. 2D).
FIG. 2.
TEAD4 overexpression increases tumor growth in vivo. (A) Schematic representation of the CAM assay. (B) Photographs of TEAD4‐overexpressing or control Huh7 cells implanted in CAMs and grown for 4 days postimplantation. (C) Volume of tumors derived from the CAM experiment (n ≥ 7 tumors over two independent experiments). Values are normalized to the mean volume of control. (D) TEAD4 expression in Huh7 tumors extracted 4 days postimplantation analyzed by western blot (left) and IHC (right). Tumoral cells were immunostained with the TEAD4 antibody. Scale bars, (B) 1 cm and (D) 50 μm and 20 μm. Statistical significance was determined by the Mann‐Whitney U test; *P < 0.05. Data are mean ± SD. Abbreviations: H&E, hematoxylin and eosin; ox, overexpression.
Taken together, our in vitro and in vivo data provide compelling evidence of the oncogenic role of TEAD4 in HCC. In particular, we demonstrate the role of TEAD4 in promoting proliferation and migration of liver cancer cells.
TEAD4 Regulates Cell Differentiation and Proliferation and the Expression of HSP70 Family Genes
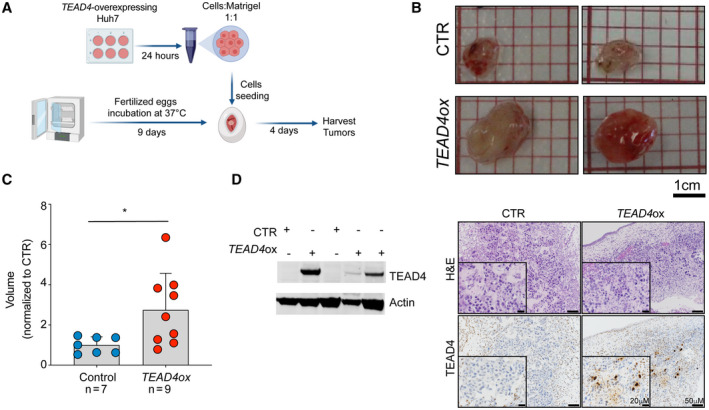
To define the transcriptional changes induced by TEAD4 overexpression, we performed RNA‐seq on Huh7 cells overexpressing TEAD4 (n = 4) or control vector (n = 4; Fig. 3A). Differential expression analysis identified 569 up‐regulated and 316 down‐regulated genes following TEAD4 overexpression (quasi‐likelihood F test, adjusted P < 0.05) (Fig. 3B; Supporting Table S3). Gene set enrichment analysis (GSEA)( 33 ) revealed the up‐regulated genes were involved in pathways, such as myogenesis, EMT, and the P53 pathway, all associated with cell differentiation and proliferation (Fig. 3C). Conversely, we observed a down‐regulation of gene sets involved in metabolism, such as protein secretion and bile acid metabolism (Fig. 3C). These results are consistent with the aggressive phenotype we observed in our in vitro and in vivo models following TEAD4 overexpression as well as with the well‐known role of TEAD proteins in regulating EMT and proliferation.( 10 )
FIG. 3.
TEAD4 regulates cell differentiation and proliferation and the expression of HSP70 family genes. (A) Heatmap showing the top 50 differentially expressed genes in TEAD4‐overexpressing Huh7 cells. (B) Volcano plot of –log10 adjusted P value against log2 fold change to illustrate the differentially expressed genes between TEAD4‐overexpressing Huh7 cells and control cells. Red dots indicate differentially expressed genes (adjusted P < 0.05). (C‐E) GSEA plots show selected significantly enriched gene sets in TEAD4‐overexpressing cells: Hallmark gene sets from (C) MSigDB database, (D) HSP family gene set, and (E) GO gene sets from the MSigDB database. (F) The Huh7 cell line was transiently transfected with a vector overexpressing TEAD4 and the corresponding control. RNA was extracted at different time points. mRNA expression levels of HSPA1A (left) and HSPA6 (right) were measured by qRT‐PCR using GAPDH as the internal control. Data are shown as fold change relative to control. Results represent three independent experiments. Statistical significance was determined by the Mann‐Whitney U test; *P < 0.05, **P < 0.01, ***P < 0.001. Data are mean ± SD. Abbreviations: E2F, E2 factor; FDR, false discovery rate; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; LATS1, large tumor suppressor kinase 1; mRNA, messenger RNA; mTORC1, mammalian target of rapamycin complex 1; NFKB, nuclear factor kappa B; ox, overexpression; TJP2, tight junction protein 2; TNFa, tumor necrosis factor alpha; UV, ultraviolet.
In‐depth analysis of the RNA‐seq data revealed altered expression of the HSP family members, including some small HSPs (HSPB9 and HSPB1), HSP40 (DNAJA4), and HSP70 (HSPA1A, HSPA1B, and HSPA6). Furthermore, the set of HSP70 genes defined by Klimczak et al.( 34 ) was found to be significantly enriched in the TEAD4‐overexpressing Huh7 cells (P < 0.05, GSEA; Fig. 3D), while other HSP gene sets (HSP40 and HSP90) were not significantly altered. In line with this, GSEA using Gene Ontology (GO) pathways showed enrichment of gene sets downstream of the HSP genes, such as those involved in HSP‐binding protein‐folding chaperones and unfolded protein binding (Fig. 3E). Surprisingly, the canonical Hippo targets seem not to be up‐regulated in TEAD4‐overexpressing cells, suggesting a potential Hippo‐independent role of TEAD4 in regulating HSP70 family members.
Among the HSP70 genes, HSPA1A and HSPA6 were the most significantly up‐regulated genes following TEAD4 overexpression (Fig. 3B). Using qPCR, we confirmed that overexpression of TEAD4 led to a significant increase in HSPA6 and HSPA1A expression in our in vitro models (Huh7 and SNU449; Fig. 3F). Our results suggested that HSP70 is a novel family of TEAD4 target genes in HCC.
TEAD4 Directly Regulates the Transcription of HSP70 Family Genes
The HSP70 family comprises 13 gene products that differ from each other by expression level, subcellular location, and amino acid composition. Following exposure to different stressors, HSP70s bind to misfolded proteins and prevent their aggregation. As HSP70 family members and their overexpression have been shown to play an oncogenic role in different cancer types,( 35 , 36 ) including HCC,( 37 , 38 ) we asked if HSP70 family genes were direct transcriptional targets of TEAD4. Because HSPA6 and HSPA1A were the most up‐regulated genes following TEAD4 overexpression (Fig. 3B), we focused on these two members of the family. We analyzed a set of peak calls from TEAD4 ChIP‐seq performed on the HepG2 cell line.( 25 ) Interestingly, we observed a TEAD4 peak within the HSPA6 promoter region, ~100 base pairs (bp) upstream of the transcription starting site (TSS), that contained two regions with the TEAD4 binding motif (chr1:161493541‐161493545 and chr1:161493648‐161493652; Fig. 4A). Additional analysis of the peak calls using CREAM( 28 ) identified a cluster of TEAD4 peaks ~50 kilobases (kb) upstream of the HSPA6 promoter (i.e., a TEAD4 enhancer, chr1:31,776,368‐31,832,367; Fig. 4A) and another enhancer ~47 kb downstream of the HSPA1A promoter (chr6:31,830,291‐38,133,978; Fig. 4A), both well within the 100‐kb range of most promoter‐enhancer interactions.( 39 ) Peaks in both enhancers and at the gene promoters were also confirmed to contain the TEAD4 binding motif (Fig. 4A).
FIG. 4.
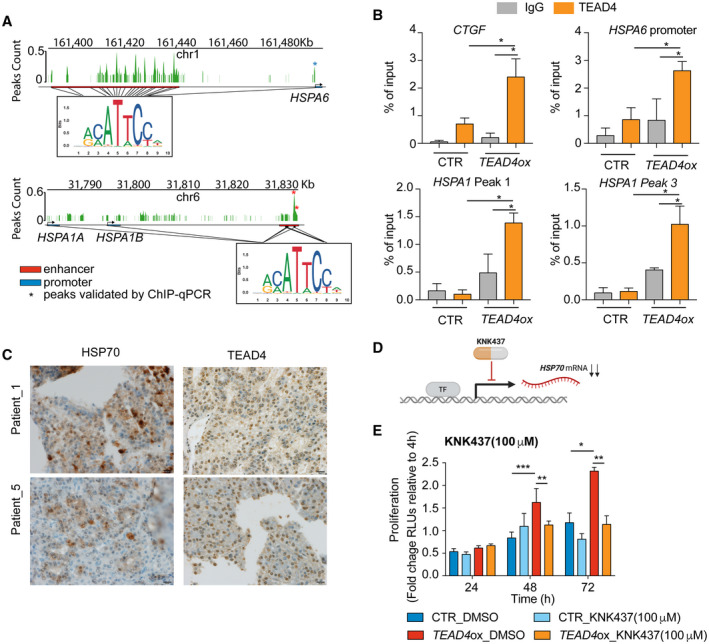
TEAD4 impacts cell proliferation by directly binding HSP70 promoters and associated enhancers. (A) TEAD4 ChIP‐seq peaks at the promoters of HSPA6 and HSPA1A, and regions called as TEAD4 enhancers within 100 kb of HSPA6 and HSPA1A, with TEAD4 binding motifs labeled. Enhancers and promoters are labeled as red and blue blocks, respectively. The TEAD4 binding motif logos are shown below the track, and positions in the HSPA6 and in HSPA1A enhancers harboring this motif are indicated (this motif was also found at the promoter of all three genes). Asterisks indicate peaks validated by ChIP‐qPCR. (B) ChIP‐qPCR showing enrichment for TEAD4 binding at the HSPA6 promoter and the HSPA1A enhancer. CTGF was used as a positive control. (C) Representative immunostain of TEAD4 and HSP70 on human HCCs. Magnification ×40; scale bar 20 μm. (D) Schematic representation of the mechanisms of action of KNK437 on HSP70. (E) Proliferation of TEAD4‐overexpressing Huh7 cells treated with 100 μM of KNK437 at various time points, relative to proliferation at 4 hours. DMSO was used as control. Results represent three independent experiments. Statistical significance was determined by the t test; *P < 0.05, **P < 0.01, ***P < 0.001. Data are mean ± SD. Abbreviations: chr, chromosome; h, hours; mRNA, messenger RNA; ox, overexpression; RLU, relative light unit; TF, transcription factor.
To confirm that TEAD4 directly binds to the promoter region of the HSPA6 gene and the enhancer region of HSPA1A, we performed ChIP in TEAD4‐overexpressing Huh7 cells, using anti‐TEAD4 antibody or IgG followed by qPCR. Using primers spanning the predicted TEAD4 binding region on the HSPA6 promoter, the HSPA1A enhancer region, and the known TEAD4 target CTGF promoter,( 7 ) we confirmed that TEAD4 indeed binds to the HSPA6 promoter and HSPA1A enhancer region (Fig. 4B).
To corroborate the results obtained in vitro we performed IHC staining on 10 HCC samples using TEAD4 and HSP70 antibodies. Analysis of this cohort revealed that samples positive for TEAD4 expression were also positive for HSP70 and vice versa (P = 0.06, Spearman correlation test; Fig. 4C). Of these 10 samples, eight were positive for HSP70 and two were negative. Six out of eight HCCs positive for HSP70 were also positive for TEAD4 (Fig. 4C), while the two HCCs negative for HSP70 were also negative for TEAD4.
Taken together, our results demonstrate that HSPA6 and HSPA1A are transcriptional targets of TEAD4 in HCC through both TEAD4‐promoter and TEAD4‐enhancer interactions.
Oncogenic Effects of TEAD4 Are Mediated Through Its Up‐Regulation of HSP70
Given that the HSP70 family genes are direct transcriptional targets of TEAD4 and that HSP70s are known to play an oncogenic role in various cancer types, including HCC,( 37 ) we hypothesized that the oncogenic effects of TEAD4 may be at least partially mediated by its up‐regulation of HSP70. We therefore inhibited HSP70 pharmacologically in TEAD4‐overexpressing cells with the benzylidene lactam compound KNK473 (Fig. 4C). KNK437 has been shown to inhibit the induction of HSPs, including HSP70, HSP40, and HSP105, in vitro.( 40 ) Additionally, KNK437 was shown to revert E2 factor (E2F) transcription factor 1‐mediated HSP40 induction, which plays a role in promoting colorectal cancer tumor growth and metastasis in vitro and in vivo.( 41 ) Supporting our hypothesis, treatment of TEAD4‐overexpressing Huh7 cells with 100 μM of KNK437 abolished the effect of TEAD4 overexpression on cell proliferation at 48 and 72 hours posttreatment (Fig. 4D). No significant reduction on cell proliferation was observed in control cells following treatment with KNK437.
Taken together, our results demonstrate that TEAD4 promotes tumor growth in HCC at least partially through direct regulation of the HSP70 family members. We further showed that the pharmacologic inhibition of HSP70 induction reverted the increased cell proliferation phenotype induced by TEAD4 overexpression.
TEAD4 Regulates HSP70 and Cell Proliferation Independent of Hippo Signaling
The transcriptional activity of TEAD proteins is highly dependent on the binding of their C‐terminal protein interaction domain to several coactivators.( 42 , 43 ) Among those cofactors, YAP and TAZ,( 44 , 45 ) two major Hippo signaling pathway transcriptional coactivators, are the most well studied. When the Hippo signaling pathway is active, YAP and TAZ proteins are located in the nucleus, thus binding to TEADs and inducing transcription of TEAD downstream targets (Fig. 5A). Conversely, when the Hippo pathway is inactive, phosphorylation of YAP and TAZ allows TEADs to bind to other cofactors and activate alternative transcriptional targets (Fig. 5A).( 46 ) The role of TEAD4 in HCC carcinogenesis has so far been linked to the Hippo signaling pathway.( 19 , 21 ) Interestingly, analysis of RNA‐seq data from TEAD4‐overexpressing Huh7 cells revealed no significant change in well‐known targets and/or effectors of the Hippo signaling pathway (e.g., YAP1, large tumor suppressor kinase 1 [LATS1], tight junction protein 2 [TJP2], and CTGF; Fig. 3B) or in the Hippo signaling pathway as a whole (q > 0.01 GO; Fig. 3E), suggesting that the oncogenic properties of TEAD4 in HCC are independent of the Hippo pathway.
FIG. 5.
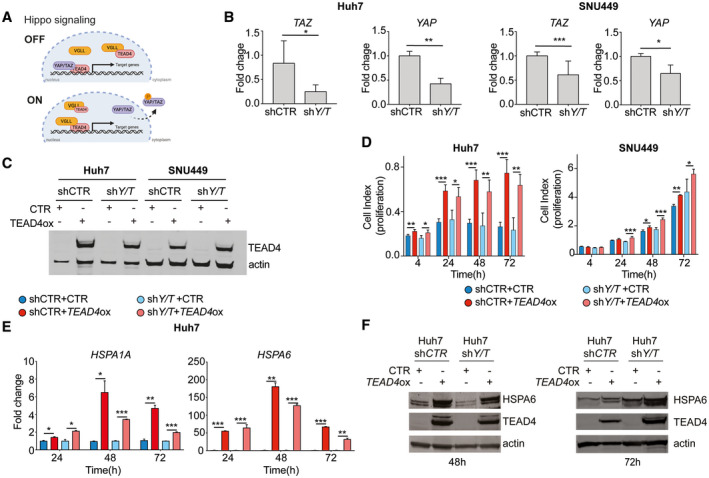
The effect of TEAD4 on cell proliferation is independent of expression of cofactors YAP and TAZ. (A) Schematic representation of TEAD4 transcriptional activity associated with YAP/TAZ cofactor expression. (B) Expression levels of YAP and TAZ were measured in stable YAP/TAZ‐knockdown (shY/T) and control (shCTR) Huh7 and SNU449 cell lines by qRT‐PCR normalized to GAPDH as internal control. (C) TEAD4 expression in stably TEAD4‐overexpressing and control Huh7 and SNU449 cell lines with knockdown of YAP/TAZ or control (shCTR). (D) Proliferation kinetics of stable TEAD4‐overexpressing and control Huh7 cell lines with YAP/TAZ knockdown or control (shCTR) (left) and in TEAD4‐overexpressing and control SNU449 stable cell lines with YAP/TAZ knockdown and control (shCTR) (right). (E) HSPA6 and HSPA1A expression levels in TEAD4‐overexpressing and control Huh7 cell lines with knockdown of YAP/TAZ or control (shCTR) were measured by qRT‐PCR using GAPDH as reference. (F) Expression levels of HSPA6 in TEAD4‐overexpressing and control Huh7 cell lines with knockdown of YAP/TAZ or control (shCTR) were measured by western blot at 48 and 72 hours posttransfection. Data are shown as fold change to control. Results represent three independent experiments. Statistical significance was determined by t test; *P < 0.05, **P < 0.01, ***P < 0.001. Data are mean ± SD. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; h, hours; ox, overexpression; sh, short hairpin; VGLL, vestigial‐like protein 1.
To test the hypothesis that TEAD4 does not require Hippo signaling to promote cell proliferation in liver cancer cells, we overexpressed TEAD4 in stable YAP/TAZ‐knockdown and control liver cancer cell lines (Supporting Fig. S3A,B; Fig. 5B,C). We first confirmed that the stable YAP/TAZ‐knockdown cells had impaired Hippo signaling, as indicated by reduced Cyr61, CTGF, and vimentin expression (Supporting Fig. S3A,B). When TEAD4 was overexpressed (Supporting Fig. S3C), we observed that the proliferation rate was significantly increased in both stable YAP/TAZ‐knockdown and control Huh7 and SNU449 cells (P < 0.001 and P < 0.05, respectively; Fig. 5D), suggesting that the presence or absence of the cofactors YAP and TAZ did not alter how TEAD4 modulates cell proliferation in vitro.
Finally, to confirm that TEAD4 regulates HSP70 expression independent of Hippo signaling, we measured HSPA1A and HSPA6 expression in the stable YAP/TAZ‐knockdown and control cells. In line with our RNA‐seq analysis, HSPA1A and HSPA6 were up‐regulated following TEAD4 overexpression in both the stable YAP/TAZ‐knockdown and the control cells (Fig. 5E; Supporting Fig. S3D), demonstrating that TEAD4 regulates HSP70 expression independent of its YAP and TAZ coactivators. Additionally, we also performed western blot analysis to evaluate if the changes observed at the RNA level were also observed at the protein level. Indeed, we observed that the overexpression of TEAD4 in cells with knockdown of YAP/TAZ led to an increased level of HSP70 at the protein level (Fig. 5F).
Our results suggest that the oncogenic effects of TEAD4 overexpression in HCC are, at least in part, independent of the expression of the Hippo signaling YAP and TAZ coactivators.
Discussion
TEAD4 is a member of the TEAD transcriptional enhancer factor family composed of four members (TEAD1‐4). Deregulation of TEAD4 expression, as with other members of the family, has been extensively reported in several tumor types.( 16 , 47 , 48 ) In the present study, we showed that TEAD4 promotes cell proliferation and tumor growth in HCC by directly regulating HSP70 family members through, at least in part, a Hippo‐independent mechanism.
After confirming the overexpression of TEAD4 in a subset of HCC in the TCGA data set, we evaluated the functional relevance of TEAD4 up‐regulation in HCCs by performing a series of in vitro and in vivo experiments. We demonstrated that TEAD4 overexpression promoted tumor growth, cell proliferation, and migration in liver cancer cells while TEAD4 silencing had the opposite effect. Our observations are consistent with the oncogenic role played by TEAD4 in other tumor types. Indeed, it has been shown that down‐regulation of TEAD4 expression hampers cancer cell proliferation and invasiveness in in vitro and in vivo models of colorectal and gastric cancers.( 48 , 49 ) Our findings demonstrate that TEAD4 also acts as an oncogene in HCC.
As a transcription factor, TEAD4 regulates the transcription of many genes, activating or repressing several downstream pathways, such as cell growth, differentiation, and apoptosis.( 11 ) Consistent with the role of TEAD4 in cell differentiation, the gene expression profile of TEAD4‐overexpressing Huh7 cells showed up‐regulation of pathways, such as spermatogenesis, EMT, and myogenesis. Surprisingly, chaperone folding and HSP pathways were also found to be significantly enriched in TEAD4‐overexpressing cells. Specifically, we identified two members of the HSP70 family (HSPA6 and HSPA1A) as among the most up‐regulated genes. HSP70B and HSP70‐1, the protein products of HSPA6 and HSPA1A, respectively, are members of the ubiquitous and highly conserved HSP70 family of molecular chaperones. In normal cell conditions, chaperones are expressed at very low levels, ensuring the correct folding and transport of newly synthesized proteins.( 50 ) However, chaperone levels increase during cell cycle and development( 51 ) as well as in response to cellular stress as occurs, for example, in tumors. Overexpression of HSP70 family members has been shown to play an oncogenic role in different cancer types,( 35 , 36 ) including HCC.( 37 , 38 ) In HCC, HSP70 has been reported to drive cell migration,( 52 ) and genetic ablation of HSP70 was able to markedly impair chemically induced liver tumorigenesis and tumor progression.( 38 ) Notably, HSP70 is a clinically important marker in HCC diagnosis; HSP70 expression, together with that of glutamine synthetase and glypican 3, is commonly used to differentiate early and low‐grade tumors from dysplastic nodules.( 53 )
Our reanalysis of the ChIP‐seq data from the HepG2 cell line( 25 ) revealed two mechanisms by which TEAD4 can regulate the expression of HSP70 family genes; first by binding sites mapped less than 1 kb from the TSS of HSPA6 and second through TEAD4 enhancer regions ~50 kb upstream of the HSPA6 promoter and ~47 kb downstream of HSPA1A. Both putative TEAD4 enhancers and the HSP70 promoter regions were found to harbor TEAD4 binding motif, and we confirmed the direct binding of TEAD4 at these loci by ChIP‐qPCR in TEAD4‐overexpressing Huh7 cells. In fact, we demonstrated that the increased cell proliferation resulting from TEAD4 overexpression was reversed by the pharmacologic inhibition of HSP70. We note that although KNK437 is a nonspecific HSP70 inhibitor, it has been shown to inhibit only inducible HSPs but has no effect on the expression of constitutively expressed HSP family members, including HSC70 and HSP90.( 40 ) Therefore, although KNK437 is not specific to HSP70, it would only inhibit HSPs induced following TEAD4 overexpression. Our results thus indicate that the oncogenic effect of TEAD4 in HCC acts through its regulation of HSP70.
The transcription factor TEAD4 and its coactivators YAP and TAZ are considered major downstream effectors of the conserved Hippo signaling pathway.( 18 ) However, in colorectal and prostate cancers,( 6 , 15 ) TEAD4 has been found to play a role in EMT and cell proliferation independent of YAP/TAZ expression. Here, we demonstrated that TEAD4 overexpression increased liver cancer cell proliferation independent of the expression of YAP/TAZ. Supporting our YAP/TAZ‐knockdown experiments, our RNA‐seq data from TEAD4‐overexpressing cells revealed no significant changes on Hippo signaling following TEAD4 overexpression. Furthermore, we showed that TEAD4 overexpression induced HSPA1A and HSPA6 expression in the context of YAP/TAZ knockdown. Together, our results indicate that TEAD4 regulates HSP70 expression and liver cancer cell proliferation independent of Hippo signaling.
In conclusion, we showed that TEAD4 plays an oncogenic role in HCC and that its oncogenic effect is mediated in part by its regulation of HSP70 genes. More importantly, our results unveil a novel role of TEAD4 outside its canonical Hippo‐dependent mechanism.
Supporting information
Supplementary Material
Supported by the Swiss Cancer League (KLS‐3639‐02‐2015 to L.M.T., KFS‐4543‐08‐2018 to C.K.Y.N., and KFS‐4988‐02‐2020‐R to S.P.), Swiss National Science Foundation (PZ00P3_168165 to S.P.), University of Basel (Research Fund Junior Researchers to S.P.), and Krebsliga Beider Basel (KLbB‐4473‐03‐2018 to S.P.).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Bürglin TR. The TEA domain: a novel, highly conserved DNA‐binding motif. Cell 1991;66:11‐12. [DOI] [PubMed] [Google Scholar]
- 2. Pobbati AV, Hong W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol Ther 2013;14:390‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiao JH, Davidson I, Matthes H, Garnier JM, Chambon P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF‐1. Cell 1991;65:551‐568. [DOI] [PubMed] [Google Scholar]
- 4. Chen L, Chan SW, Zhang X, Walsh M, Lim CJ, Hong W, et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev 2010;24:290‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, et al. Structural insights into the YAP and TEAD complex. Genes Dev 2010;24:235‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pobbati AV, Chan SW, Lee I, Song H, Hong W. Structural and functional similarity between the Vgll1‐TEAD and the YAP‐TEAD complexes. Structure 2012;20:1135‐1140. [DOI] [PubMed] [Google Scholar]
- 7. Benhaddou A, Keime C, Ye T, Morlon A, Michel I, Jost B, et al. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ 2012;19:220‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joshi S, Davidson G, Le Gras S, Watanabe S, Braun T, Mengus G, et al. TEAD transcription factors are required for normal primary myoblast differentiation in vitro and muscle regeneration in vivo. PLoS Genet 2017;13:e1006600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao B, Li L, Lei Q, Guan K‐L.. The Hippo‐YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24:862‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang H, Liu C‐Y, Zha Z‐Y, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial‐mesenchymal transition. J Biol Chem 2009;284:13355‐13362. Erratum in: J Biol Chem 2019;294:5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP‐dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011;30:1229‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 2010;107:1437‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knight JF, Shepherd CJ, Rizzo S, Brewer D, Jhavar S, Dodson AR, et al. TEAD1 and c‐Cbl are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br J Cancer 2008;99:1849‐1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y, Wang G, Yang Y, Mei Z, Liang Z, Cui A, et al. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial–mesenchymal transition and metastasis in a YAP‐independent manner. Oncogene 2016;35:2789‐2800. [DOI] [PubMed] [Google Scholar]
- 16. Zhou Y, Huang T, Cheng ASL, Yu J, Kang W, To KF. The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci 2016;17:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J, Liu P, Tao J, Wang P, Zhang Y, Song X, et al. TEA domain transcription factor 4 is the major mediator of yes‐associated protein oncogenic activity in mouse and human hepatoblastoma. Am J Pathol 2019;189:1077‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu F‐X, Zhao B, Guan K‐L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015;163:811‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weiler SME, Pinna F, Wolf T, Lutz T, Geldiyev A, Sticht C, et al. Induction of chromosome instability by activation of yes‐associated protein and forkhead box M1 in liver cancer. Gastroenterology 2017;152:2037‐2051.e22. [DOI] [PubMed] [Google Scholar]
- 20. Mao B, Hu F, Cheng J, Wang P, Xu M, Yuan F, et al. SIRT1 regulates YAP2‐mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014;33:1468‐1474. [DOI] [PubMed] [Google Scholar]
- 21. Cai W‐Y, Lin L‐Y, Hao H, Zhang S‐M, Ma F, Hong X‐X, et al. Yes‐associated protein/TEA domain family member and hepatocyte nuclear factor 4‐alpha (HNF4α) repress reciprocally to regulate hepatocarcinogenesis in rats and mice. Hepatology 2017;65:1206‐1221. [DOI] [PubMed] [Google Scholar]
- 22. Tang F, Gao R, Jeevan‐Raj B, Wyss CB, Kalathur RKR, Piscuoglio S, et al. LATS1 but not LATS2 represses autophagy by a kinase‐independent scaffold function. Nat Commun 2019;10:5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coto‐Llerena M, Ercan C, Kancherla V, Taha‐Mehlitz S, Eppenberger‐Castori S, Soysal SD, et al. High expression of FAP in colorectal cancer is associated with angiogenesis and immunoregulation processes. Front Oncol 2020;10:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lauzier A, Normandeau‐Guimond J, Vaillancourt‐Lavigueur V, Boivin V, Charbonneau M, Rivard N, et al. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci Rep 2019;9:11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 2013;14:178‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khan A, Fornes O, Stigliani A, Gheorghe M, Castro‐Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open‐access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018;46:D260‐D266. Erratum in: Nucleic Acids Res 2018;46:D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tonekaboni SAM, Mazrooei P, Kofia V, Haibe‐Kains B, Lupien M. CREAM: Clustering of genomic REgions analysis method. bioRxiv 2017; 10.1101/222562. [DOI] [Google Scholar]
- 29. Dimitrova Y, Gruber AJ, Mittal N, Ghosh S, Dimitriades B, Mathow D, et al. TFAP2A is a component of the ZEB1/2 network that regulates TGFB1‐induced epithelial to mesenchymal transition. Biol Direct 2017;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blecher‐Gonen R, Barnett‐Itzhaki Z, Jaitin D, Amann‐Zalcenstein D, Lara‐Astiaso D, Amit I. High‐throughput chromatin immunoprecipitation for genome‐wide mapping of in vivo protein‐DNA interactions and epigenomic states. Nat Protoc 2013;8:539‐554. [DOI] [PubMed] [Google Scholar]
- 31. Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase‐8. Nature 2006;439:95‐99. [DOI] [PubMed] [Google Scholar]
- 32. Azoitei N, Hoffmann CM, Ellegast JM, Ball CR, Obermayer K, Gößele U, et al. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J Exp Med 2012;209:697‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sergushichev A. Algorithm for cumulative calculation of gene set enrichment statistic. Sci Tech J Inf Technol Mech Opt 2016;16:956‐959. [Google Scholar]
- 34. Klimczak M, Biecek P, Zylicz A, Zylicz M. Heat shock proteins create a signature to predict the clinical outcome in breast cancer. Sci Rep 2019;9:7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jäättelä M. Members of the heat‐shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev 2005;19:570‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fuller KJ, Issels RD, Slosman DO, Guillet JG, Soussi T, Polla BS. Cancer and the heat shock response. Eur J Cancer 1994;30A:1884‐1891. [DOI] [PubMed] [Google Scholar]
- 37. Xiong J, Jiang X‐M, Mao S‐S, Yu X‐N, Huang X‐X. Heat shock protein 70 downregulation inhibits proliferation, migration and tumorigenicity in hepatocellular carcinoma cells. Oncol Lett 2017;14:2703‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cho W, Jin X, Pang J, Wang Y, Mivechi NF, Moskophidis D. The molecular chaperone heat shock protein 70 controls liver cancer initiation and progression by regulating adaptive DNA damage and mitogen‐activated protein kinase/extracellular signal‐regulated kinase signaling pathways. Mol Cell Biol 2019;39:e00391‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of genome‐wide enhancer‐promoter interactions reveals co‐expression of interacting genes and modes of higher order chromatin organization. Cell Res 2012;22:490‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yokota S, Kitahara M, Nagata K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res 2000;60:2942‐2948. [PubMed] [Google Scholar]
- 41. Yang S, Ren X, Liang Y, Yan Y, Zhou Y, Hu J, et al. KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene 2020;39:249‐261. [DOI] [PubMed] [Google Scholar]
- 42. Anbanandam A, Albarado DC, Nguyen CT, Halder G, Gao X, Veeraraghavan S. Insights into transcription enhancer factor 1 (TEF‐1) activity from the solution structure of the TEA domain. Proc Natl Acad Sci U S A 2006;103:17225‐17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes‐associated protein localized in the cytoplasm. Genes Dev 2001;15:1229‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiao S, Li C, Hao Q, Miao H, Zhang L, Li L, et al. VGLL4 targets a TCF4‐TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat Commun 2017;8:14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Belandia B, Parker MG. Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem 2000;275:30801‐30805. [DOI] [PubMed] [Google Scholar]
- 46. Meng Z, Moroishi T, Guan K‐L. Mechanisms of Hippo pathway regulation. Genes Dev 2016;30:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget 2015;6:17658‐17697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Y, Huang T, Zhang J, Wong CC, Zhang B, Dong Y, et al. TEAD1/4 exerts oncogenic role and is negatively regulated by miR‐4269 in gastric tumorigenesis. Oncogene 2017;36:6518‐6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang J‐Y, Yu C‐Y, Bao Y‐J, Chen L, Chen J, Yang S‐L, et al. TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1. Cell Cycle 2018;17:102‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin J, Hartl FU. Molecular chaperones in cellular protein folding. BioEssays 1994;16:689‐692. [DOI] [PubMed] [Google Scholar]
- 51. Kiang JG, Tsokos GC. Heat shock protein 70 kDa: molecular biology, biochemistry, and physiology. Pharmacol Ther 1998;80:183‐201. [DOI] [PubMed] [Google Scholar]
- 52. Liu C‐C, Jan Y‐J, Ko B‐S, Wu Y‐M, Liang S‐M, Chen S‐C, et al. 14‐3‐3σ induces heat shock protein 70 expression in hepatocellular carcinoma. BMC Cancer 2014;14:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tommaso LD, Di Tommaso L, Franchi G, Park YN, Fiamengo B, Destro A, et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007;45:725‐734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material