

Abstract
Resmetirom (MGL‐3196), a selective thyroid hormone receptor‐β agonist, was evaluated in a 36‐week paired liver biopsy study (NCT02912260) in adults with biopsy‐confirmed nonalcoholic steatohepatitis (NASH). The primary endpoint was relative liver fat reduction as assessed by MRI–proton density fat fraction (MRI‐PDFF), and secondary endpoints included histopathology. Subsequently, a 36‐week active treatment open‐label extension (OLE) study was conducted in 31 consenting patients (including 14 former placebo patients) with persistently mild to markedly elevated liver enzymes at the end of the main study. In patients treated with resmetirom (80 or 100 mg orally per day), MRI‐PDFF reduction at OLE week 36 was −11.1% (1.5%) mean reduction (standard error [SE]; P < 0.0001) and −52.3% (4.4%) mean relative reduction, P < 0.0001. Low‐density lipoprotein (LDL) cholesterol (−26.1% [4.5%], P < 0.0001), apolipoprotein B (−23.8% [3.0%], P < 0.0001), and triglycerides (−19.6% [5.4%], P = 0.0012; −46.1 [14.5] mg/dL, P = 0.0031) were reduced from baseline. Markers of fibrosis were reduced, including liver stiffness assessed by transient elastography (−2.1 [0.8] mean kilopascals [SE], P = 0.015) and N‐terminal type III collagen pro‐peptide (PRO‐C3) (−9.8 [2.3] ng/mL, P = 0.0004 (baseline ≥ 10 ng/mL). In the main and OLE studies, PRO‐C3/C3M (matrix metalloproteinase‐degraded C3), a marker of net fibrosis formation, was reduced in resmetirom‐treated patients (−0.76 [−1.27, −0.24], P = 0.0044 and −0.68, P < 0.0001, respectively). Resmetirom was well tolerated, with few, nonserious adverse events. Conclusion: The results of this 36‐week OLE study support the efficacy and safety of resmetirom at daily doses of 80 mg and 100 mg, used in the ongoing phase 3 NASH study, MAESTRO‐NASH (NCT03900429). The OLE study demonstrates a potential for noninvasive assessments to monitor the response to resmetirom from an individual patient with NASH.
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- ANCOVA
analysis of covariance
- ApoB
apolipoprotein B
- ApoCIII
apolipoprotein CIII
- AST
aspartate aminotransferase
- BL
baseline
- C3M
metalloproteinase‐degraded collagen III
- CAP
controlled‐attenuation parameter
- CFB
change from baseline
- CT1
corrected T1
- FT3
free T3
- FT4
free thyroxine
- GGT
gamma‐glutamyl transpeptidase
- LDL‐C
low‐density lipoprotein–cholesterol
- NAFLD
nonalcoholic fatty liver disease
- NAS
NASH activity score
- NASH
nonalcoholic steatohepatitis
- OLE
open‐label extension
- Pbo
placebo
- PDFF
proton density fat fraction
- PRO‐C3
N‐terminal type III collagen propeptide
- Res
resmetirom
- RT3
reverse T3
- SHBG
sex hormone binding globulin
- THR‐β
thyroid hormone receptor‐β
- T3
triiodothyronine
- ULN
upper limit of normal
- VCTE
vibration‐controlled transient elastography
Nonalcoholic steatohepatitis (NASH) is a progressive form of nonalcoholic fatty liver disease (NAFLD), defined as the presence of ≥5% hepatic steatosis with inflammation and hepatocyte injury (e.g., ballooning), with or without fibrosis.( 1 , 2 ) NAFLD, including NASH, is associated with a constellation of comorbid conditions that include metabolic syndrome (obesity, type 2 diabetes mellitus, hypertension, dyslipidemia) and hypothyroidism and is associated with increased cardiovascular risk.( 3 ) Patients with more advanced NASH fibrosis have increased morbidity and mortality from both cardiovascular disease( 4 ) and from progression of their liver disease, including progression to cirrhosis, liver failure, and hepatocellular carcinoma.( 5 , 6 ) Diagnosis of NASH is complicated by the requirement for an invasive procedure, liver biopsy, to confirm diagnosis, and there is an unmet need for noninvasive biomarkers and imaging that can diagnose and stage advanced NASH with fibrosis( 7 , 8 ) and monitor a patient's response to treatment.
There is no approved therapy for NASH, and its prevalence has increased with increasing world‐wide prevalence of obesity.( 9 , 10 ) Obeticholic acid, a bile acid analog that activates farnesoid X receptors, improved fibrosis in a phase 3 clinical trial in patients with NASH and F2 or F3 fibrosis.( 11 ) For other agents assessed in patients with NASH, trials with serial liver biopsies did not meet the primary endpoints of fibrosis reduction or NASH resolution.( 12 , 13 , 14 )
Evidence suggests that NASH may be, in part, a condition of diminished liver thyroid hormone levels or hepatic hypothyroidism, and that the incidence of clinical and subclinical hypothyroidism is higher in patients with NAFLD/NASH relative to age‐matched controls.( 15 , 16 )
Resmetirom (MGL‐3196) is a liver‐directed, orally active agonist of thyroid hormone receptor (THR) that is about 28‐fold more selective than triiodothyronine (T3) for THR‐β versus THR‐α.( 17 , 18 ) It is highly protein bound (>99%), has poor tissue penetration outside the liver, and demonstrates specific uptake into the liver. In NASH, selectivity for THR‐β may provide metabolic benefits of thyroid hormone that are mediated by the liver, including reduction of excess hepatic fat, atherogenic lipids (low‐density lipoprotein–cholesterol [LDL‐C], triglycerides), and lipoproteins (apolipoprotein B [ApoB], lipoprotein[a] [Lp(a)], Apo CIII), while avoiding unwanted systemic actions of excess thyroid hormone in heart and bone that are largely mediated through THR‐α.( 15 )
In a 36‐week phase 2 serial liver biopsy NASH clinical trial, resmetirom demonstrated statistically significant reductions compared with placebo in MRI–proton density fat fraction (PDFF) (a measure of liver steatosis), liver enzymes, atherogenic lipids, and Lp(a), markers of inflammation and fibrosis.( 19 ) Resmetirom‐treated patients also had improvements in NASH on liver biopsy compared with placebo. Patients who were on higher doses of resmetirom (≥80 mg vs. 60 mg), or who had higher exposure to resmetirom and/or demonstrated greater reduction in hepatic fat on week 12 MRI‐PDFF, had a higher incidence of NASH resolution and liver fibrosis reduction. An active treatment open‐label extension (OLE) study was conducted in a subset of patients completing the 36‐week main study, who were predicted to have an incomplete response to either placebo or resmetirom treatment in the main study based on residual minimally to markedly elevated liver enzymes (alanine aminotransferase [ALT] and/or aspartate aminotransferase [AST]) at the end of the main study. The OLE study assessed the impact of 80 and 100 mg daily doses of resmetirom on safety and noninvasive assessments of NASH. Additionally, in both the main and OLE studies, the impact of resmetirom treatment was assessed on a biomarker of net fibrosis formation (N‐terminal type III collagen propeptide [PRO‐C3]/metalloproteinase‐degraded collagen III [C3M]) and a measure of hepatic inflammation (free T3/reverse T3).
Participants and Methods
Patients and Clinical Trial Design and Oversight
The design, eligibility, and oversight of the main 36‐week study have been described.( 19 ) The protocol was designed by Madrigal (R.T.), S.A.H., M.B., and Medpace (KM). All study data were available to Madrigal, Medpace (K.M.), and S.A.H. The statistical analyses were performed by K.M., and the MRI‐PDFF analyses were performed by M.B. Briefly, NCT02912260 was a 36‐week multicenter, randomized, double‐blind, placebo‐controlled study in adults with biopsy‐confirmed NASH (fibrosis stages 1‐3) and hepatic fat fraction of at least 10% at baseline when assessed by MRI‐PDFF. Patients were randomized 2:1 to receive resmetirom or matching placebo, orally once a day. Serial hepatic fat measurements were obtained at weeks 12 and 36, and a second liver biopsy was obtained at week 36. The primary efficacy endpoint, relative reduction in liver fat as determined on MRI‐PDFF, and key secondary liver biopsy endpoints were met.( 19 ) During the main 36‐week study, a protocol amendment was completed to allow patients to enroll in a 36‐week active treatment OLE study in which all patients received open‐label resmetirom treatment, and safety, serial imaging, and biomarker assessments were conducted.
Eligibility for the OLE study was defined in the protocol amendment dated November 6, 2017. Patients eligible for entry into the OLE study must have completed the main 36‐week study after the OLE study amendment was approved, undergone a liver biopsy and MRI‐PDFF assessment at week 36, and had ALT or AST levels that had not fully normalized during weeks 16‐30 of the main study (normal defined as ALT ≤ 30 IU/L for males and ≤19 IU/L for females; AST ≤ 30 IU/L). With respect to ALT or AST, eligible patients had to have:
Worsening ALT or AST ≥ 30% from baseline, and >upper limit of normal (ULN)
Improvement in ALT or AST, but levels remained elevated ≥1.5‐2‐fold ULN
ALT or AST ≥ 2‐fold ULN (whether improved or worsened)
Thirty‐one of a total 38 eligible patients (14 of 19 placebo and 17 of 19 resmetirom‐treated) from 12 sites from the main study signed an informed consent to participate in the OLE study (Fig. 1) and were enrolled from December 14, 2017, to May 8, 2018; all received active treatment in the OLE study (Fig. 1A). The OLE study enrollment occurred at week 38 of the main study, after the 2‐week follow‐up off the study drug, up to 2 months after completion of the main study (Fig. 1B). Treatment and dose were blinded at the time of entry into the OLE study, and an unblinded reviewer assigned dose until the main study was completed and had been unblinded. After unblinding of the main study and reviewing the data, all patients in the OLE study had a dose increase to at least 80 mg, the last increase in dose occurring at OLE week 24 (Fig. 1B).
Fig. 1.
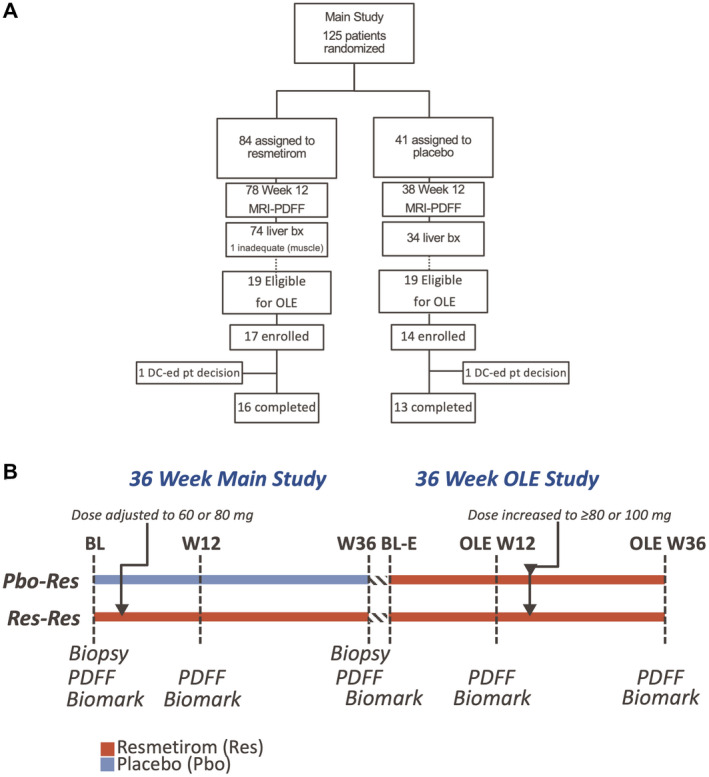
Disposition (A) of and treatment schematic (B) of patients in the MGL‐3196‐05 main and OLE studies. Abbreviations: BL, baseline; DC, discontinued.
Written, informed consent was obtained from all patients before enrollment in the main study, and a second informed consent was obtained for participation in the OLE study. The studies were performed in accordance with ethical principles of the Declaration of Helsinki and were consistent with the International Conference on Harmonization/Good Clinical Practice and applicable regulatory requirements. The institutional review board or independent ethics committee of each study center approved the study and all amendments.
Assessments
Study populations: All OLE study patients had completed the 36‐week main study, including week 36 liver biopsy and weeks 12 and 36 MRI‐PDFF. Two major groups were included in the OLE study: former resmetirom patients, many of whom were treated with a higher dose of resmetirom during the OLE (Res/Res), and former placebo patients who were treated with resmetirom during the OLE study (Pbo/Res).
Dose determination: Res/Res patients entering the OLE study initially continued on the dose of resmetirom that they were on at the end of the main study. Pbo/Res patients were started on an 80‐mg dose of resmetirom on day 1 of the OLE study. Based on a trough and 4‐hour post‐dose pharmacokinetic assessment at week 2, patients remained on the initial dose or were down‐titrated or up‐titrated by 20 mg at week 4, as determined by an unblinded reviewer. After the main study was unblinded, all patients in the OLE study had doses increased to at least 80 mg or 100 mg of resmetirom (the patients most advanced in the OLE study had the dose increase no later than week 24).
Procedures: The 36‐week OLE study followed the design of the 36‐week main study( 19 ) with a major exception that no additional liver biopsy was obtained after week 36 of the OLE study. In addition, most OLE patients had liver stiffness measurement by vibration‐controlled transient elastography (VCTE) to assess liver fibrosis, and controlled‐attenuation parameter (CAP) score to assess liver steatosis (FibroScan; Echosens, Paris, France), determined at OLE day 1 and OLE week 36. The main study week 36 PDFF was the baseline PDFF for the OLE study. NASH biomarkers and PDFF measurements were made at weeks 12 and 36 of the OLE study, and safety and lipid laboratory determinations were made at monthly visits throughout the study.
Statistical Methods
Outcomes
All OLE study endpoints were exploratory. For most parameters, baseline was defined as the time of initial treatment with resmetirom, the main study baseline for Res/Res and the OLE study baseline for Pbo/Res. The main efficacy outcomes included relative and absolute change in MRI‐PDFF at OLE week 36. Other key outcomes included safety assessments and change from baseline in liver enzymes (ALT, AST, and gamma‐glutamyl transpeptidase [GGT]), PRO‐C3 as a biomarker of liver fibrosis (Nordic Bioscience, Herlev, Denmark), C3M as biomarker of fibrosis regression,( 20 ) LDL‐C and other lipids, and FibroScan TE and CAP.
Statistical Analyses
In the statistical analysis plan, the prespecified main analyses population included week 36 completers (Pbo/Res, n = 13; Res/Res, n = 16), and the safety population included all 31 patients who enrolled in the OLE study and received at least one dose. Twenty nine of the 31 patients completed all 36 weeks of the OLE study; 2 discontinuations were patient decision. The primary week 36 MRI‐PDFF analysis population included Pbo/Res patients (n = 12) plus Res/Res patients with a dose increase in the OLE study (n = 11) receiving a dose of 80 mg or greater at week 36, and excluding patients with a weight loss or weight gain ≥9.5%. Completers by dose included patients on 80 mg (n = 21), 1 Pbo/Res patient on 60 mg (protocol deviation), and patients on ≥100 mg (n = 7).
Endpoints were summarized using descriptive statistics by key groups listed previously and continuous endpoints were analyzed using analysis of covariance (ANCOVA) or a paired t test to describe relative and/or absolute change from baseline to various time points including weeks 12, 32, and 36. Because of the absence of a placebo control group, within‐group analyses (t test) and paired group analyses (t test or least‐squared mean) were conducted where appropriate. For group analyses, the ANCOVA model included the specified group as a factor and baseline level as a covariate. The presentation of results included the estimated means overall and by group, their standard errors, and P values.
Results
Patient Characteristics
The baseline characteristics of patients enrolled in the OLE study are found in Table 1. The mean age was 48.2 (12.3) years, 51.6% were male, and 87.1% were White, with a mean body mass index of 35.3 (5.2) kg/m2. Diabetes was present in 45.2%, and hypertension was present in 51.6%. All patients met the liver enzyme entry criteria for the OLE study, including several patients in the Res/Res population who had demonstrated improvement at the end of the main study compared with baseline.
Table 1.
Baseline Characteristics
| Pbo/Res (n = 14) | Res/Res (n = 17) | All (n = 31) | |
|---|---|---|---|
| Age, years (SD) | 42.4 (10.5) | 53.1 (11.8) | 48.2 (12.3) |
| Male, n (%) | 8 (57.1) | 8 (47.1) | 16 (51.6) |
| Race, White, n (%) | 14 (100.0) | 13 (76.5) | 27 (87.1) |
| Black | 0 (0) | 1 (5.9 ) | 1 (3.2) |
| Asian | 0 (0) | 2 (11.8) | 2 (6.4) |
| Other | 0 (0) | 1 (5.9) | 1 (3.2) |
| Hispanic, n (%) | 9 (64.3) | 7 (41.2) | 16 (51.6) |
| BMI, mean (SD) | 35.1 (5.2) | 34.5 (5.2) | 35.3 (5.2) |
| T2D, n (%) | 5 (35.7) | 9 (52.9) | 14 (45.2) |
| Hypertension, n (%) | 6 (42.9) | 10 (58.8) | 16 (51.6) |
| NAS, main BL mean (SD) | 4.7 (0.9) | 4.9 (1.1) | 4.8 (1.0) |
| NAS, main week 36 mean (SD) | 4.2 (1.5) | 3.9 (1.4) | 4.1 (1.4) |
| NAS, 2‐point decrease, n (%) | 2 (14.3) | 9 (52.9) | 11 (35.5) |
| Fibrosis stage, main BL mean (SD) | 1.6 (1.0) | 1.8 (1.0) | 1.7 (1.0) |
| Fibrosis stage, main week 36 mean (SD) | 1.8 (1.0) | 2.0 (0.8) | 1.8 (1.0) |
| F0 at week 36, n (%) | 0 | 3 (17.6) | 3 (9.7) |
| F2‐F3 at week 36, n (%) | 7 (50.0) | 13 (76.5) | 20 (64.6) |
| MRI‐PDFF main BL mean% (SD%) | 17.4 (7.6) | 21.0 (6.4) | 19.4 (7.1) |
| MRI‐PDFF main week 36 mean% (SD%) | 18.0 (7.0) | 14.2 (6.1) | 15.9 (6.7) |
| %CFB, main (BL to week 36) | 12.2 (46.6) | −27.9 (37.0) | −9.8 (45.6) |
| ALT (IU/L), main BL | 58.9 (27.4) | 68.3 (40.9) | 64.1 (35.2) |
| ALT (IU/L), OLE BL | 70.6 (51.7) | 58.5 (35.6) | 64.0 (43.2) |
| AST (IU/L), main BL | 36.0 (19.7) | 46.6 (19.5) | 41.8 (20.0) |
| AST (IU/L), OLE BL | 40.9 (24.8) | 43.8 16.4) | 42.5 (20.3) |
| GGT (IU/L), main BL | 70.3 (61.5) | 62.6 (33.1) | 66.1 (47.3) |
| GGT (IU/L), OLE BL | 76.6 (75.1) | 57.6 (30.8) | 66.2 (55.2) |
| Total bilirubin (mg/dL), OLE BL | 0.506 (0.17) | 0.574 (0.20) | 0.543 (0.19) |
| Direct bilirubin (mg/dL), OLE BL | 0.089 (0.035) | 0.106 (0.043) | 0.099 (0.040) |
| Alkaline phosphatase (IU/L), OLE BL | 83.4 (30.1) | 78.0 (20.3) | 79.6 (27.4) |
| PRO‐C3 ng/mL, main BL mean (SD) | 15.3 (8.7) | 23.2 (10.8) | 19.6 (10.5) |
| PRO‐C3 ng/mL, OLE BL mean (SD) | 19.6 (13.6) | 18.4 (6.3) | 19.0 (10.1) |
| C3M ng/mL, main BL, mean (SD) | 11.8 (2.9) | 10.9 (1.9) | 11.3 (2.4) |
| C3M ng/mL, OLE BL, mean (SD) | 12.2 (3.0) | 11.5 (2.3) | 11.8 (2.6) |
| PRO‐C3/C3M, main BL | 1.30 (0.63) | 2.10 (0.89) | 1.74 (0.87) |
| PRO‐C3/C3M, OLE BL | 1.70 (1.00) | 1.70 (0.80) | 1.67 (0.89) |
| FibroScan VCTE (kPa) OLE BL, mean (SD) | 8.3 (2.6) | 11.9 (5.0) | 10.3 (4.4) |
| FibroScan CAP, OLE BL, mean (SD) | 325.5 (77.5) | 317.8 (71.0) | 320.8 (72.5) |
| Direct LDL (mg/dL) OLE BL, mean (SD) | 121 (35.1) | 125.7 (42.9) | 123.7 (39.1) |
| TGs (mg/dL) OLE BL, mean (SD) | 176.1 (110.1) | 178.4 (72.0) | 177.4 (88.8) |
| HDL (mg/dL) OLE BL, mean (SD) | 43.5 (11.2) | 46.8 (14.2) | 45.1 (12.6) |
| ApoB (mg/dL) OLE BL, mean (SD) | 110 (29) | 112 (30) | 110.9 (29.5) |
| ApoCIII (mg/dL), main BL, mean (SD) | 10.3 (3.3) | 11.2 (3.8) | 10.8 (3.6) |
| ApoCIII (mg/dL), OLE BL, mean (SD) | 10.4 (5.3) | 10.6 (3.2) | 10.5 (4.1) |
| Common concomitant meds | |||
| NSAIDs | 4 (28.6) | 8 (47) | 12 (39) |
| Biguanides (metformin) | 4 (28.6) | 7 (41.2) | 11 (35.5) |
| Proton pump inhibitors | 4 (28.6) | 7 (41.2) | 11 (35.5) |
| Statins | 3 (21.4) | 6 (35.3) | 9 (29.0) |
| Angiotensin‐converting enzyme inhibitors | 4 (28.6) | 4 (23.5) | 8 (25.8) |
Note: Data are presented as n (%) or mean (SD). For FibroScan, n = 11; Pbo/Res and Res/Res, n = 14.
Abbreviations: BL, baseline; BMI, body mass index; %CFB, percent change from baseline; HDL, high density lipoprotein; NSAID, nonsteroidal anti‐inflammatory drug; TGs, triglycerides; T2D, type 2 diabetes.
The NASH activity score (NAS) improved at week 36 of the main study compared with baseline biopsy in Res/Res patients, in whom 53% of biopsies showed a 2‐point reduction of NAS at week 36, and 17.6% of biopsies showed a reduction in fibrosis as compared with 14.3% and 0% in Pbo/Res, respectively.
MRI‐PDFF Outcomes
In the Res/Res population, the average reduction in PDFF during the main study at week 36 was 27.9%, and 31.6% at OLE study week 12, and did not change significantly from week 36 of the main study. At OLE week 12, 10/16 Res/Res patients were on the same dose of resmetirom as in the main study; seven additional dose increases were made after week 12, and one additional increase to 100 mg after the initial increase to 80 mg at week 4. At OLE week 36, Pbo/Res patients and Res/Res patients with a dose increase during the OLE study (primary efficacy population) experienced a mean relative reduction of 52.3% (standard error = 4.4%, P < 0.0001) (Table 2 and Figure 2) and absolute reduction of 11.1% (1.5%) in PDFF. Patients receiving 100 mg experienced a mean relative reduction in PDFF of 58.8% (6.8%) and absolute reduction of −14.3% (1.9%). Most of the OLE patients (85%) experienced at least a 30% PDFF relative reduction, and all patients had at least 20% relative reduction. All patients (100%) receiving 100 mg had ≥5% absolute reduction. Two patients with >10% weight gain during the study experienced >20% and <30% relative PDFF reduction.
Table 2.
Change in MRI‐PDFF
| n | Pbo/Res | P Value | n | Res/Res | P Value | n | All | P Value | |
|---|---|---|---|---|---|---|---|---|---|
| Week 12 %CFB | 13 | −39.9 (4.2) | <0.0001 | 15 | −33.5 (5.6) | <0.0001 | 28 | −36.4 (3.6) | <0.0001 |
| Week 36 %CFB | 11 | −52.0 (7.1) | <0.0001 | 15 | −45.8 (5.1) | <0.0001 | 26 | −48.4 (4.2) | <0.0001 |
| Primary | 11 | −52.0 (7.1) | <0.0001 | 10 | −52.6 (5.2) | <0.0001 | 21 | −52.3 (4.4) | <0.0001 |
| 80 mg | 19 | −44.6 (4.9) | <0.0001 | ||||||
| 100 mg | 7 | −58.8 (6.8) | <0.0001 | ||||||
| Week 12 CFB | 13 | −7.4 (1.4) | 0.0002 | 15 | −7.8 (1.8) | 0.0006 | 28 | −7.6 (1.1) | <0.0001 |
| Week 36 CFB | 11 | −10.1 (2.0) | 0.0005 | 15 | −10.3 (1.7) | <0.0001 | 26 | −10.2 (1.3) | <0.0001 |
| Primary | 11 | −10.1 (2.0) | 0.0005 | 10 | −12.2 (2.2) | 0.0003 | 21 | −11.1 (1.5) | <0.0001 |
| 80 mg | 19 | −8.7 (1.5) | <0.0001 | ||||||
| 100 mg | 7 | −14.3 (1.9) | 0.0003 | ||||||
| Week 12 ≥ 30% PDFF reduction | 12 | 8 (66.7%) | 15 | 9 (60%) | 27 | 17 (63%) | |||
| Week 36 ≥ 30% PDFF reduction | 10 | 7 (70%) | 15 | 13 (86.7%) | 25 | 20 (80%) | |||
| Primary | 10 | 7 (70.0%) | 10 | 10 (100.0%) | 20 | 17 (85%) | |||
| 80 mg | 18 | 14 (77.8%) | |||||||
| 100 mg | 7 | 6 (85.7%) | |||||||
| Week 12 ≥ 5% PDFF reduction | 12 | 8 (66.7%) | 15 | 12 (80%) | 27 | 20 (74.1%) | |||
| Week 36 ≥ 5% PDFF reduction | 10 | 8 (80%) | 15 | 14 (93.3%) | 25 | 22 (88%) | |||
| Primary | 10 | 80% | 10 | 100% | 20 | 18 (90%) | |||
| 80 mg | 18 | 15 (83.3%) | |||||||
| 100 mg | 7 | 7 (100%) |
Note: Baseline is defined as the value at the main study screening visit for Res/Res patients and main study week 36 for former Pbo/Res patients. Week 12 and 36 are OLE week 12 and 36, respectively. Patients with >9.5% weight loss or gain from baseline to OLE weeks 12 and 36 and patients who are not compliant were excluded from the respective analyses. The means, standard errors, and P values come from a paired t test.
Fig. 2.
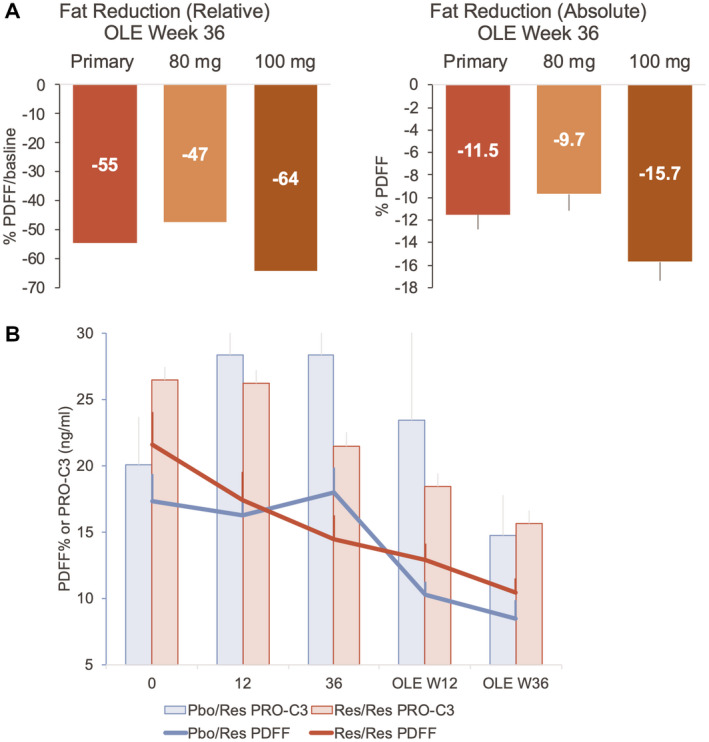
MRI‐PDFF results in the OLE study relative median (−54.6 [−35.6, −65.8]) (A) and absolute mean fat reduction (B) (Table 2) as determined by MRI‐PDFF at week 36 in the primary population (Pbo/Res and Res/Res patients with a dose increase during the OLE study) and by final dose. Time course of PDFF in the Pbo/Res (blue line) and primary Res/Res (red line) population compared with change in PRO‐C3.
The CAP score, a component of the FibroScan measurement that is considered a marker of hepatic steatosis,( 21 ) showed no significant correlation with PDFF, at either week 36 (main) or OLE week 36 (R < 0.02, non‐significant) and did not decrease significantly with treatment.
Lipids, Lipoproteins, and Liver Enzymes
Atherogenic lipids and lipoproteins were reduced in resmetirom‐treated patients, including statistically significant reductions in LDL‐C (−26.1% [4.5%], P < 0.0001), ApoB (−23.8% [3.0%], P < 0.0001), apolipoprotein CIII (ApoCIII) (−21.6% [3.7%], P < 0.0001), and triglycerides (−46.1 [14.5] mg/dL, P = 0.0036) (Table 3).
Table 3.
Effects on Lipids and Lipoproteins
| Lipids and Lipoproteins | n | CFB | P Value | %CFB | P Value |
|---|---|---|---|---|---|
| LDL‐C (mg/dL) | |||||
| Week 12 | 29 | −31.6 (5.2) | <0.0001 | −23.4 (2.9) | <0.0001 |
| Week 32 | 28 | −39.8 (8.4) | <0.0001 | −26.1 (4.5) | <0.0001 |
| 80 mg | 21 | −33.1 (5.7) | <0.0001 | −23.0 (4.1) | <0.0001 |
| 100 mg | 7 | −30.1 (9.8) | 0.0051 | −23.5 (7.2) | 0.0030 |
| No dose increase* | 5 | −17.9 (11.4) | 0.14 | −15.6 (7.6) | 0.057 |
| Dose increase* | 11 | −40.7 (7.7) | 0.0002 | −27.0 (5.1) | 0.0001 |
| Primary | 23 | −35.3 (7.8) | 0.0002 | −24.7 (4.2) | <0.0001 |
| LDL‐C (BL ≥ 100 mg/dL) | |||||
| BL, mg/dL (SD) | 21 | 139.7 (35.1) | |||
| Week 12 | 21 | −36.3 (6.6) | <0.0001 | −23.9 (3.5) | <0.0001 |
| Week 32 | 20 | −39.8 (8.4) | 0.0001 | −26.1 (4.5) | <0.0001 |
| 80 mg | 15 | −40.7 (7.9) | <0.0001 | −25.1 (5.3) | 0.0002 |
| 100 mg | 5 | −36.9 (13.7) | 0.016 | −29.4 (9.0) | 0.0047 |
| No dose increase | 3 | −15.4 (16.6) | 0.38 | −11.9 (9.8) | 0.25 |
| Dose increase | 8 | −53.9 (10.1) | 0.0007 | −33.2 (6.0) | 0.0004 |
| Primary | 19 | −43.5 (9.6) | 0.0002 | −28.7 (5.0) | <0.0001 |
| ApoB (mg/dL) | |||||
| Week 12 | 29 | −25.4 (3.8) | <0.0001 | −21.6 (2.5) | <0.0001 |
| Week 32 | 28 | −28.5 (5.0) | <0.0001 | −23.8 (3.0) | <0.0001 |
| 80 mg | 21 | −29.1 (4.3) | <0.0001 | −23.7 (3.6) | <0.0001 |
| 100 mg | 8 | −26.6 (7.6) | 0.0017 | −24.3 (6.1) | <0.0005 |
| No dose increase | 5 | −15.0 (8.5) | 0.10 | −15.1 (6.1) | 0.028 |
| Dose increase | 11 | −34.2 (5.7) | <0.0001 | −27.6 (4.1) | <0.0001 |
| Primary | 23 | −31.3 (5.9) | <0.0001 | −25.7 (3.4) | <0.0001 |
| ApoB (BL LDL‐C ≥ 100 mg/dL) | |||||
| Baseline | 21 | 122.7 (26.4) | |||
| Week 32 | 20 | −32.8 (6.6) | <0.0001 | −24.8 (3.9) | <0.0001 |
| TG (mg/dL) | |||||
| Week 12 | 29 | −33.0 (11.2) | 0.014 | −12.1 (6.3) | 0.065 |
| Week 32 | 28 | −46.1 (14.5) | 0.0036 | −19.6 (5.4) | 0.0012 |
| 80 mg | 21 | −44.2 (11.7) | 0.023 | −14.6 (6.0) | 0.023 |
| 100 mg | 7 | −51.7 (22.2) | 0.028 | −34.7 (10.4) | 0.0027 |
| No dose increase | 5 | −19.3 (22.5) | 0.41 | −9.0 (11.8) | 0.46 |
| Dose increase | 11 | −50.4 (15.1) | 0.0054 | −26.0 (8.0) | 0.0056 |
| Primary | 23 | −51.6 (16.7) | 0.0054 | −21.9 (5.7) | 0.0009 |
| TG (mg/dL) >150 | |||||
| BL mg/dL (SD) | 16 | 226.1 (89.7) | |||
| Week 32 | 16 | −70.8 (21.7) | 0.0052 | −24.8 (6.5) | 0.0016 |
| ApoCIII (mg/dL) | |||||
| Week 12 | 29 | −2.5 (0.46) | <0.0001 | −21.0 (3.8) | <0.0001 |
| Week 36 | 27 | −2.8 (0.62) | 0.0001 | −21.6 (3.7) | <0.0001 |
| 80 mg | 20 | −3.1 (0.42) | <0.0001 | −21.5 (4.4) | <0.0001 |
| 100 mg | 7 | −1.9 (0.73) | 0.018 | −21.9 (7.4) | 0.0067 |
| HDL‐C (mg/dL) | |||||
| Week 12 | 29 | −1.2 (1.1) | 0.25 | −1.4 (2.3) | 0.54 |
| Week 32 | 28 | −1.7 (1.2) | 0.15 | −1.1 (3.2) | 0.72 |
Note: Week 12 and 36 denote OLE weeks 12 and 36, respectively. Baseline is the OLE baseline for all indices except ApoCIII; baseline refers to main study baseline for Res/Res and OLE baseline for Pbo/Res. “n” refers to the number of patients with measurements at both extension baseline day 1 and this visit. The means, standard errors, and P values come from a paired t test.
No dose increase; refers only to Res/Res patients.
Liver enzymes, particularly ALT (−23.3 [6.7] IU/L, P = 00016) and GGT (−24.4 IU/L, P = 0.0006) (Table 4) declined over time, and sex hormone binding globulin (SHBG), a biomarker of resmetirom activity, increased (Supporting Table S1 and Fig. 3).
Table 4.
Liver Enzymes and Biomarkers at OLE Week 12 and 36
| n | Pbo/Res | P Value | n | Res/Res | P‐Value | n | All | P Value | |
|---|---|---|---|---|---|---|---|---|---|
| Liver enzymes | |||||||||
| ALT (IU/L) | |||||||||
| Week 12, CFB | 14 | −16.8 (4.7) | 0.0014 | 16 | −14.4 (4.4) | 0.0029 | 30 | −15.5 (4.8) | 0.0028 |
| Week 36, CFB | 13 | −31.7 (4.6) | <0.0001 | 16 | −16.4 (4.1) | 0.0005 | 29 | −23.3 (6.7) | 0.0016 |
| 80 mg | 21 | −24.4 (4.1) | <0.0001 | ||||||
| 100 mg | 7 | −20.3 (7.4) | 0.011 | ||||||
| Dose increase | 11 | −18.1 (5.8) | 0.0085 | ||||||
| No dose increase | 5 | −17.5 (8.8) | 0.067 | ||||||
| AST (IU/L) | |||||||||
| Week 12, CFB | 14 | −5.7 (4.2) | 0.19 | 16 | −4.1 (3.9) | 0.30 | 30 | −4.9 (3.5) | 0.17 |
| Week 36, CFB | 13 | −16.6 (3.1) | <0.0001 | 16 | −1.2 (2.8) | 0.68 | 29 | −8.1 (4.1) | 0.061 |
| 80 mg | 21 | −7.2 (3.0) | 0.025 | ||||||
| 100 mg | 7 | −10.2 (5.4) | 0.068 | ||||||
| Dose increase | 11 | −9.1 (3.5) | 0.020 | ||||||
| No dose increase | 5 | 4.7 (5.2) | 0.39 | ||||||
| GGT (IU/L) | |||||||||
| Week 12, CFB | 14 | −15.4 (4.0) | 0.0007 | 16 | −14.4 (3.8) | 0.0007 | 30 | −14.9 (3.4) | 0.0002 |
| Week 36, CFB | 13 | −26.5 (5.5) | <0.0001 | 16 | −22.2 (4.9) | 0.0001 | 29 | −24.1 (6.2) | 0.0006 |
| 80 mg | 21 | −22.2 (4.4) | <0.0001 | ||||||
| 100 mg | 7 | −28.8 (7.5) | 0.0008 | ||||||
| Dose increase | 11 | −22.2 (3.4) | <0.0001 | ||||||
| No dose increase | 5 | −13.1 (5.1) | 0.023 | ||||||
| PRO‐C3 (ng/mL) | |||||||||
| Week 12, CFB | 14 | −2.9 (1.8) | 0.13 | 16 | −4.85 (1.7) | 0.0091 | 30 | −3.94 (1.4) | 0.0098 |
| Week 36, CFB | 13 | −7.8 (1.0) | <0.0001 | 16 | −6.9 (0.93) | <0.0001 | 29 | −7.32 (1.9) | 0.0005 |
| 80 mg | 21 | −6.8 (0.78) | <0.0001 | ||||||
| 100 mg | 7 | −7.8 (1.3) | <0.0001 | ||||||
| BL ≥ 10.0 | |||||||||
| BL | 9 | 12.3 (79.0) | 14 | 26.1 (9.5) | 23 | 24.7 (12.0) | |||
| Week 12, CFB | 9 | −3.2 (2.6) | 0.24 | 13 | −6.2 (2.2) | 0.010 | 22 | −4.97 (1.9) | 0.014 |
| Week 36, CFB | 8 | −9.9 (1.5) | <0.0001 | 13 | −9.8 (1.2) | <0.0001 | 21 | −9.8 (2.3) | 0.0004 |
| 80 mg | 14 | −9.2 (1.1) | <0.0001 | ||||||
| 100 mg | 6 | −10.6 (1.6) | <0.0001 | ||||||
| BL ≥ 14.0 | |||||||||
| BL | 7 | 25.1 (17.2) | 12 | 26.5 (9.2) | 19 | 26.0 (12.3) | |||
| Week 12, CFB | 7 | −2.5 (3.1) | 0.44 | 12 | −7.0 (2.4) | 0.01 | 19 | −5.3 (2.1) | 0.02 |
| Week 36, CFB | 6 | −10.8 (1.9) | <0.0001 | 12 | −10.7 (1.3) | <0.0001 | 18 | −10.7 (2.6) | 0.0008 |
| 80 mg | 12 | −10.1 (1.2) | <0.0001 | ||||||
| 100 mg | 5 | −11.5 (1.9) | <0.0001 | ||||||
| C3M (ng/mL) | |||||||||
| Week 12, CFB | 14 | 0.66 (0.46) | 0.23 | 16 | 0.61 (0.49) | 0.16 | 30 | 0.64 (0.33) | <0.0001 |
| Week 36, CFB | 13 | 0.22 (0.51) | 0.68 | 16 | 0.81 (0.46) | 0.086 | 29 | 0.54 (0.37) | <0.0001 |
| 80 mg | 21 | 0.82 (0.40) | 0.05 | ||||||
| 100 mg | 7 | −0.16 (0.69) | 0.82 | ||||||
| PRO‐C3/C3M | |||||||||
| Week 12, CFB | 14 | −0.41 (0.14) | 0.005 | 16 | −0.46 (0.13) | 0.0015 | 30 | −0.44 (0.13) | <0.0001 |
| Week 36, CFB | 13 | −0.68 (0.09) | <0.0001 | 16 | 0.68 (0.08) | <0.0001 | 29 | −0.68 (0.15) | <0.0001 |
| 80 mg | 21 | −0.67 (0.07) | <0.0001 | ||||||
| 100 mg | 7 | −0.65 (0.12) | <0.0001 | ||||||
| FibroScan VCTE (kPa) | |||||||||
| Week 36, CFB | 11 | −2.0 (0.66) | 0.0064 | 14 | −2.2 (0.58) | 0.0012 | 25 | −2.1 (0.8) | 0.015 |
| 80 mg | 19 | −1.8 (0.45) | 0.0007 | ||||||
| 100 mg | 5 | −3.4 (0.88) | 0.0008 | ||||||
| FibroScan CAP | |||||||||
| BL | 11 | 325.5 (77.5) | 13 | 316.8 (71.0) | 24 | 320.8 (72.5) | |||
| Week 36, CFB | 11 | −14.5 (12.4) | 0.26 | 13 | −10.7 (11.4) | 0.36 | 24 | −12.4 (11.3) | 0.29 |
| 80 mg | 19 | −8.3 (9.3) | 0.39 | ||||||
| 100 mg | 4 | −33.2 (20.3) | 0.12 | ||||||
| Adiponectin (mg/L) | |||||||||
| BL | 14 | 3.9 (1.4) | 16 | 4.5 (2.2) | 4.25 (1.9) | ||||
| Week 12, CFB | 14 | 0.84 (0.30) | 0.79 | 16 | −0.039 (0.29) | 30 | 0.018 (0.22) | 0.93 | |
| Week 36, CFB | 13 | 1.3 (0.30) | 0.0002 | 16 | 0.95 (0.27) | 0.0017 | 29 | 1.1 (0.20) | <0.0001 |
| Week 36 (%CFB) | 13 | 32.7 (6.6) | <0.0001 | 16 | 23.4 (6.0) | 0.0006 | 27.5 (4.4) | <0.0001 | |
| 80 mg | 21 | 0.98 (0.21) | 0.0001 | ||||||
| 100 mg | 7 | 1.1 (0.39) | 0.0082 | ||||||
| Reverse T3 (ng/dL) | |||||||||
| Baseline | 13 | 19.3 (4.6) | 16 | 17.6 (5.1) | 29 | 18.33 (4.9) | |||
| Week 12, CFB | 13 | −4.22 (0.81) | <0.0001 | 16 | −4.61 (0.73) | <0.0001 | 29 | −4.4 (0.59) | <0.0001 |
| Week 36, CFB | 13 | −2.6 (0.99) | 0.014 | 16 | −4.02 (0.89) | 0.0001 | 29 | −3.4 (0.73) | <0.0001 |
| 80 mg | 21 | −3.4 (0.81) | 0.0002 | ||||||
| 100 mg | 7 | −3.2 (1.4) | 0.030 |
Note: Baseline is defined as the value at the main study baseline visit for Res/Res patients and OLE baseline for Pbo/Res patients except for FibroScan kilopascals and CAP, where baseline is OLE baseline. Week 12 and 36 are OLE weeks 12 and 36, respectively. “n” refers to the number of patients with measurements at both baseline and this visit. All, means, standard errors, and P values come from a paired t test. For Pbo/Res and Res/Res, 80 mg and 100 mg, the liver stiffness means, standard errors, and P values come from an ANCOVA model with percent change or change from baseline as the dependent variable and former treatment group as a factor. For the analysis of change from baseline, baseline is also included as a covariate.
Fig. 3.
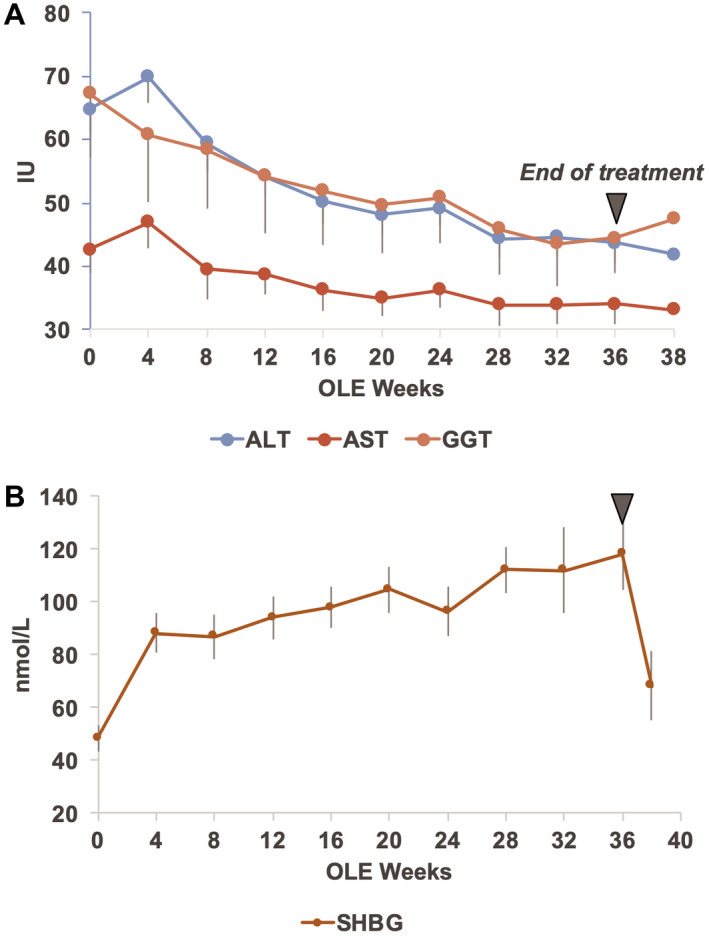
Time course in OLE study patients of liver enzymes ALT, AST, and GGT (A) and SHBG (B). The baseline (week 0) is the OLE baseline for both Res/Res and Pbo/Res patients.
Markers of Fibrosis
The biomarker C3M, a marker of fibrosis regression, and the ratio, PRO‐C3/C3M, a proposed measure of net fibrosis formation, had not been assessed in the main study samples and were evaluated in a post hoc analysis of the main study. Baseline main study PRO‐C3/C3M ratio levels were correlated significantly with baseline fibrosis stage on liver biopsy (r = 0.24, P = 0.001), ballooning (r = 0.29, P = 0.003), and not with other NAS parameters. In the main study, C3M levels increased in the resmetirom treatment group, with no change in the placebo group, and PRO‐C3/C3M showed a significant decrease with resmetirom treatment (P = 0.0044) (Supporting Table S2 and Fig. 4).
Fig. 4.
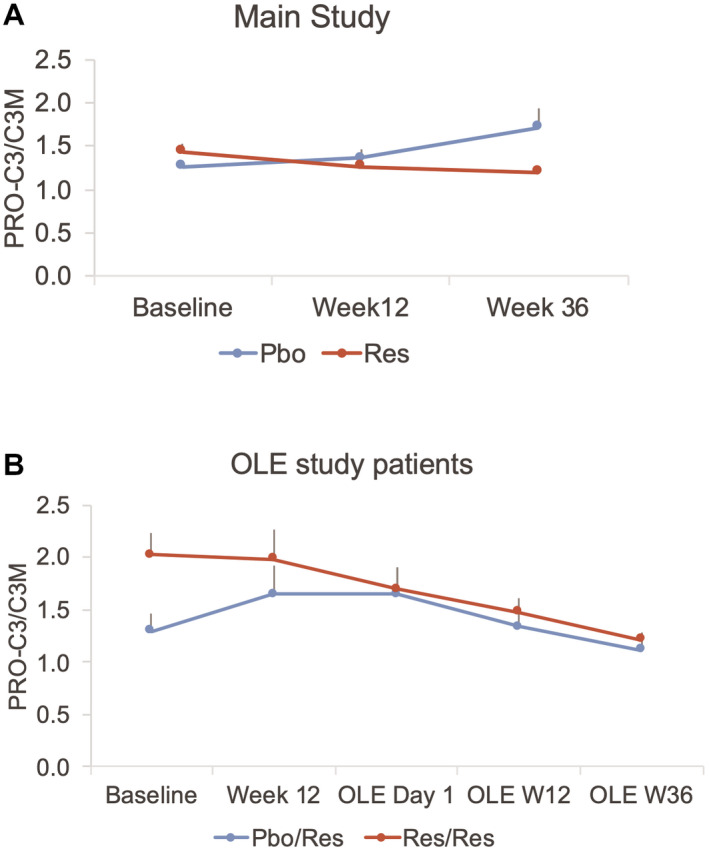
Time course of PRO‐C3/C3M in the main (A) and OLE (B) studies. Both the main and OLE times are shown in (B). Res/Res patients were on resmetirom for both the main and OLE 36‐week studies, and Pbo/Res were on placebo during the main study and started on resmetirom on OLE day 1 for 36 weeks.
In the OLE study, similar to the main study, PRO‐C3 was reduced significantly with resmetirom treatment (P = 0.0005), with greatest reduction at OLE week 36, coincident with decrease in PDFF (Fig. 2B and Table 4). The magnitude of reduction in PRO‐C3 was greater with higher baseline PRO‐C3. Similarly, in the OLE, C3M increased and PRO‐C3/C3M decreased significantly with treatment (P < 0.0001 for each measurement) (Fig. 4 and Table 4).
In the OLE, other potential fibrosis markers such as adiponectin, which has been proposed as inversely correlated with fibrosis in the liver,( 22 ) increased significantly (Table 4). Liver stiffness (VCTE) on FibroScan, which is predictive of liver fibrosis stage,( 21 , 23 ) decreased with resmetirom treatment (−2.1 [0.8] kPa, P = 0.015). In Res/Res and Pbo/Res patients, another potential marker of inflammation/fibrosis, corrected T1 (CT1) measured on MRI, showed normalization with time on resmetirom treatment (Fig. 5).
Fig. 5.
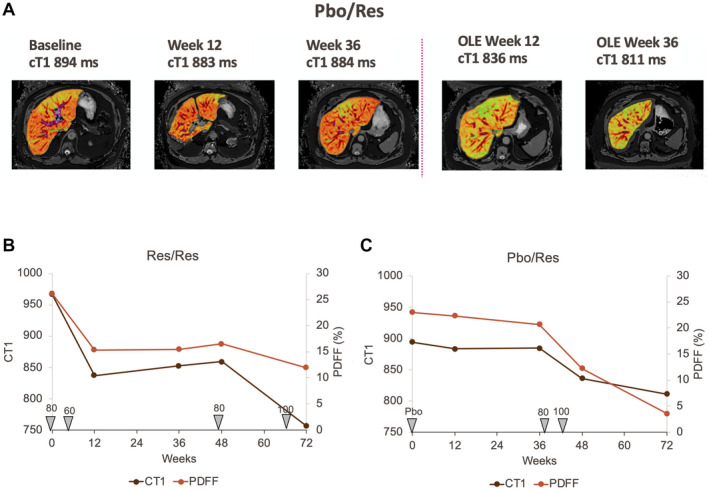
Time course of CT1 and PDFF and dose in individual Pbo/Res and Res/Res patients. (A) CT1 images at indicated assessment times of a Pbo/Res patient. (B,C) The Res/Res and Pbo/Res patient, respectively, time course of CT1, PDFF, and dose administered over time from the start of the main study to the end of the OLE.
Thyroid Parameters
Thyroid axis hormones may be altered in NASH, which is associated with subclinical and clinical hypothyroidism.( 15 , 16 ) In a post hoc analysis, evidence for intrahepatic hypothyroidism at baseline was assessed in randomized main and OLE study patients. Compared with a data set of non‐NASH patients of similar age, there were no differences in baseline free T4 (FT4) or thyroid stimulating hormone (TSH) in patients with NASH (Supporting Table S3). Reverse T3 (rT3), a marker of hepatic inflammation, was statistically higher (P < 0.0001), and free T3(FT3)/rT3 was lower in the NASH compared with the non‐NASH population; the ratio of FT3/rT3 declined with fibrosis state (Fig. 6A). Treatment with resmetirom significantly reduced rT3 and increased the ratio of FT3/rT3 in both the main and OLE studies (Supporting Table S3 and Fig. 6B).
Fig. 6.
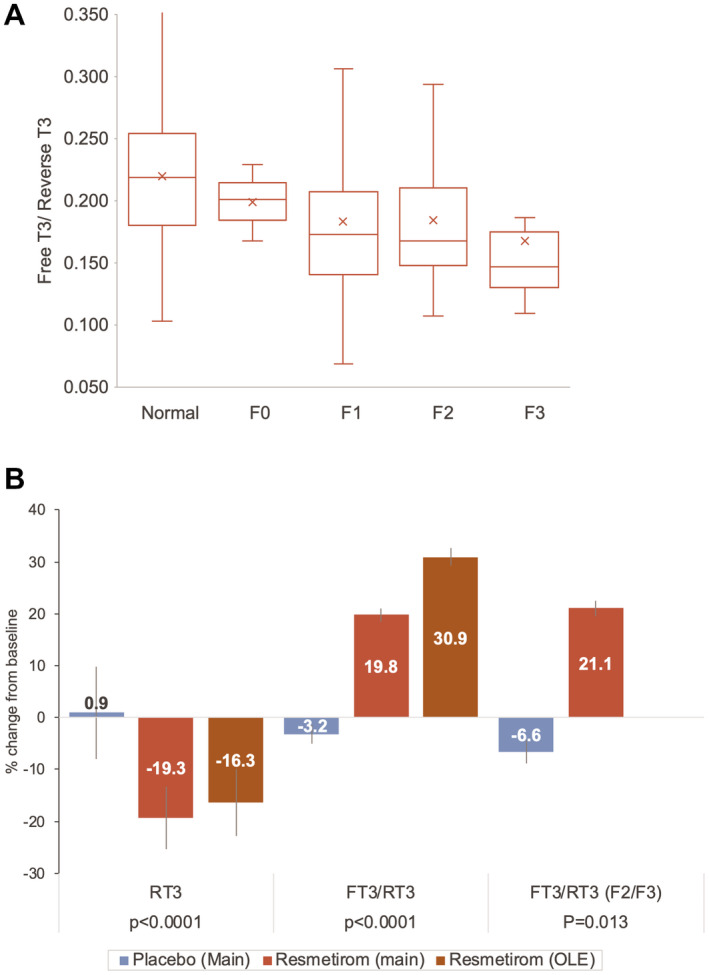
Effects of resmetirom on RT3 and FT3/RT3 in patients with NASH. (A) Baseline FT3/RT3 in normal (non‐NASH) and NASH according to liver biopsy fibrosis stage. Shown as a box and whisker plot with box defined by quartile 1, 3, and median, with quartile line shown; “x” indicates the mean, SD, and error bars. (B) Effect of resmetirom on thyroid pathway hormones at week 36 in the main study or OLE. Compared with placebo in the main study, within‐group comparison over time in the OLE. Abbreviation: F2/F3, patients with baseline NASH fibrosis stage of F2 or F3.
As in the main study, in OLE study patients, resmetirom mildly (−10.9%, P < 0.05) decreased FT4, a prohormone that is converted to the active hormone FT3 in the liver and other tissues. There were no significant effects on active thyroid pathway hormones TSH and only 4% increase in FT3 (Supporting Table S1). There were no adverse events (AEs) consistent with hypothyroidism or hyperthyroidism.
Safety
Comparison was made between the OLE study and the placebo group population from the main study (Table 5). Resmetirom at doses of 80 mg and 100 mg in the OLE study was well‐tolerated with no serious or severe AEs. There was no increase in gastrointestinal AEs in the OLE study compared with the placebo group in the main study. No changes in vital signs, including body weight, heart rate or blood pressure, were seen (Supporting Table S1).
Table 5.
Safety
| Main Study | OLE Study | |
|---|---|---|
| Placebo,* n = 41 | Resmetirom, n = 31 | |
| Patients with AEs, n (%) | 28 (68) | 18 (58) |
| Severe | 2 (5) | 0 |
| Moderate | 13 (32) | 10 (32) |
| Mild | 13 (32) | 8 (26) |
| Patients with severe AEs | 2 (5) | 0 |
| Most common AEs, n (%) | ||
| Diarrhea | 4 (10) | 3 (10) |
| Nausea | 3 (7) | 1 (3) |
| Headache | 6 (15) | 0 |
| Urinary tract infection | 4 (10) | 1 (3) |
| Dizziness | 4 (10) | 1 (3) |
| Grade 3 Common terminology criteria, n (%) | ||
| ALT > 5 × ULN | 3 (7) | 0 |
| GGT > 5 × ULN | 5 (12) | 0 |
Reported in Harrison et al.( 19 )
Discussion
The 36‐week OLE study of MGL‐3196‐05 was an exploratory study conducted in patients with NASH predicted to have an incomplete response to either placebo or resmetirom treatment in the main 36‐week study based on residual minimally to markedly elevated liver enzymes (ALT and/or AST) at the end of the main study. The study examined whether an increase in resmetirom dose and/or 72 weeks of treatment in Res/Res patients or 36 weeks of treatment with ≥80 mg resmetirom in Pbo/Res patients could lead to improvement in noninvasive measures of NASH and fibrosis. At the time the patients entered the OLE study, their treatment code and treatment response on liver biopsy, MRI‐PDFF, lipids, and other pharmacodynamic biomarkers (e.g., SHBG, FT4) were blinded to the study team. Eligible patients included an equal number from the placebo and resmetirom treatment groups, and because the study was randomized 2:1 resmetirom, placebo, indicated a higher percentage of placebo patients qualified for the OLE (55.9%, placebo; 25.7% resmetirom). Some patients in the OLE study, particularly in the resmetirom treatment group, demonstrated NASH and fibrosis reduction on liver biopsy, PDFF, and biomarkers at week 36 of the main study (Table 1). Thus, liver enzyme response did not always predict biopsy or MRI‐PDFF response.
In the main study, about half of the patients on resmetirom received 60 mg, which was shown to be a less effective dose than 80 mg or 100 mg in reducing PDFF or achieving NASH resolution.( 19 ) The OLE study provided an opportunity to determine whether increasing the dose of resmetirom from 60 mg to at least 80 mg in Res/Res patients and/or switching to resmetirom treatment in patients who were on placebo in the main study would improve the biomarker and PDFF responses. Predefined treatment groups (e.g., Pbo/Res vs. Res/Res; Res/Res with a dose increase vs. Res/Res no dose increase; 80 mg vs. 100 mg dose group) were assessed.
Effect on noninvasive efficacy endpoints that had been observed during the main study were confirmed during the OLE study. These included a MRI‐PDFF reduction of >50% relative fat reduction compared with baseline, and a high percentage of patients achieving >30% fat reduction on MRI‐PDFF, which was shown to be associated with increased NASH reduction and resolution in the main study.( 19 , 24 ) Atherogenic lipid and lipoprotein lowering of >20 to >25% were observed for LDL‐C, ApoB, triglycerides, and ApoCIII.
The change in PDFF was −26.8% (11.4) and −52.6% (5.2) at the end of the main and OLE studies, respectively, in Res/Res patients with a dose increase during the OLE study, who also showed an improvement in lipid lowering, liver enzymes, and other biomarkers relative to patients who did not receive a dose increase. There was little change in the PDFF or other biomarkers in placebo patients during the main study, and a convincing improvement in several imaging and biomarker responses in the Pbo/Res group during the 36‐week OLE study. At the end of the OLE study, Pbo/Res patients, who were dosed at 80 mg or 100 mg, showed improvement in PDFF, biomarkers, and lipid endpoints relative to the main study (Tables and Fig. 2B). An increase to 100 mg from an 80 mg dose appeared to improve the PDFF and CT1 responses in 2 patients (Table 2, Figs. 2A and 5). However, given the small number of patients treated with 100 mg compared with 80 mg, no statistical comparison was possible between the two doses. Notably, there were no safety findings associated with higher doses of resmetirom used in the OLE, as compared with the main study.
Type III collagen is a key component of liver fibrosis in patients with NASH, and identifying reliable noninvasive measures of fibrosis progression or regression will be critical in the long‐term treatment of NASH. PRO‐C3 and C3M, serum markers reflecting type III collagen formation and degradation, respectively, were assessed to determine the net effect on collagen. The observed reduction in PRO‐C3 along with increase or no change in C3M levels as reflected by the PRO‐C3/C3M level may indicate an overall decrease in fibrosis.( 8 ) Similarly, liver stiffness (VCTE) on FibroScan showed statistically significant improvement during the 36‐week OLE study and, as an increasingly validated measure of liver fibrosis, represents an important noninvasive in‐office test that has potential utility to monitor individual NASH patient response to treatment.( 21 )
Notably, CAP, another parameter measured by FibroScan, was uncorrelated with main week 36 PDFF or OLE week 36 PDFF; the change in CAP did not show a relationship to the change in PDFF. The CAP score may provide a categorical measure of liver fat that approximately compares with the steatosis score on liver biopsy, but unlike MRI‐PDFF, does not represent a precise measure of liver fat.( 25 )
Mechanistically, as a targeted hepatic therapeutic acting through THR‐β, resmetirom may restore components of liver function that are defective in NASH. Thyroid hormone acting through THR‐β is vital to maintain normal lipid regulation and mitochondrial function in the liver.( 15 ) In a large National Health and Nutrition Education Survey database and median follow‐up of 23 years, individuals with low thyroid function demonstrated an association with NAFLD.( 26 ) Low thyroid function was associated with a higher risk for all‐cause and cardiovascular mortality in individuals with NAFLD and not in those without NAFLD. Another study reports intrahepatic hypothyroidism in human NASH livers, hypothesized as caused by depressed conversion of prohormone T4 to active hormone T3.( 16 ) This conversion mediated by deiodinase 1 and is depressed in NASH livers, while the level of thyroid hormone degradative enzyme deiodinase 3 made in stellate cells is increased. Decreased RT3 and an increase in the FT3/RT3 ratio by resmetirom may reflect a correction in endogenous hepatic thyroid hormone activity and improvement in hepatic function through increased direct THR‐β activity. The improvements in atherogenic lipids and lipoproteins, coupled with the improvement in hepatic thyroid function and reduction in lipotoxic fat, support the potential for reduced atherosclerotic risk in patients with NASH treated with resmetirom.
The OLE had significant limitations. Although the completion rate in the OLE study was high, the sample size was relatively small. Differences between doses could not be adequately explored because of the relatively small numbers of patients on a 100‐mg dose. Nonetheless, the 36‐week OLE study and additional post hoc assessments from the main study (PRO‐C3/C3M, FT3/RT3) demonstrated that resmetirom has a positive impact on several noninvasive markers of liver fat, inflammation, and fibrosis. Furthermore, these noninvasive biomarkers may be useful to monitor response to treatment over time. Taken as a whole, this study demonstrates that a series of noninvasive biomarkers, including imaging, may be useful in monitoring response to resmetirom in individual patients with NASH.
Based in part on the safety and efficacy of the OLE study, a resmetirom phase 3 clinical trial MAESTRO‐NASH (NCT03900429) was initiated in patients with NASH and stage F2 or F3 fibrosis, to test whether resmetirom at daily doses of 80 mg and 100 mg compared with placebo will resolve NASH, reduce liver fibrosis, and reduce LDL‐C after 52 weeks of treatment. MAESTRO‐NAFLD‐1 (NCT04197479), a 52‐week phase 3 “real‐life NASH study” that enrolls patients based on NASH diagnosed using noninvasive assessments is also being conducted to assess safety and the effects of 80 mg and 100 mg of resmetirom on serial biomarkers, lipids, MRI‐PDFF, and FibroScan.
Supporting information
Table S1‐S3
Acknowledgments
We thank the investigators, trial participants, and their families.
Supported by Madrigal Pharmaceuticals, West Conshohocken, Pennsylvania.
Potential conflict of interest: Dr. Harrison owns stock in, advises, consults, and received grants from Akero, Galectin, Genfit, Hepion, Metacrine, NGM, and Northsea. He advises, consults for, and received grants from Axcella, Civi, Cymabay, Galmed, Gilead, Hightide, Intercept, Madrigal, Novartis, Novo Nordisk, Poxel, and Sagimet. He owns stock in, advises, and consults for Histoindex. He owns stock in and advises Pathai. He owns stock in and received grants from Cirus. He advises and consults for Altimmune, Echosens, Foresite Labs, Indalo, Medpace, Prometic, Ridgeline, and Terns. He consults and received grants from Enyo and Viking. He advises 89 Bio, Arrowhead, and Theratechnologies. He consults for Boston, B Riley FBR, Canfite, Fibronostics, Fortress, GNS Healthcare, Inipharm, Ionis, Kowa Research Institute, Microba PTY, Piper Sandler & Co., and Sonic Incytes Medical Corp. Dr. Frias consults for, is on the speakers’ bureau for, and received grants from Merck and Sanofi. He consults and received grants from Boerhinger Ingelheim, Eli Lilly, and Novo Nordisk. He consults for Altimmune, Axcella, Coherus, Gilead, and Intercept. He received grants from Allergan, AstraZeneca, BMS, Janseen, Madrigal, Novartis, Pfizer, and Theracos. Dr. Alkhouri consults for and is on the speakers’ bureau for Gilead, Intercept, and Echosens. He consults for and received grants from Pfizer. He consults for Allergan and Perspectum. He is on the speakers’ bureau for AbbVie and Alexion. He received grants from Akero, NGM, NorthSea, Genfit, Madrigal, Novo Nordisk, and Zydus. Dr. Bashir consults for ICON and PLC. He received grants from Carmot, CymaBay, Madrigal, Metacrine, NGM, Pinnacle Clinical Research, and ProSciento. Dr. Taub owns stock and is employed by Madrigal.
References
Author names in bold designate shared co‐first authorship.
- 1. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 2. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 3. Stahl EP, Dhindsa DS, Lee SK, Sandesara PB, Chalasani NP, Sperling LS. Nonalcoholic fatty liver disease and the heart: JACC state‐of‐the‐art review. J Am Coll Cardiol 2019;73:948‐963. [DOI] [PubMed] [Google Scholar]
- 4. Henson JB, Simon TG, Kaplan A, Osganian S, Masia R, Corey KE. Advanced fibrosis is associated with incident cardiovascular disease in patients with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther 2020;51:728‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piazzolla VA, Mangia A. Noninvasive diagnosis of NAFLD and NASH. Cells 2020;9:1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyle M, Tiniakos D, Schattenberg JM, Ratziu V, Bugianessi E, Petta S, et al. Performance of the PRO‐C3 collagen neo‐epitope biomarker in non‐alcoholic fatty liver disease. JHEP Rep 2019;1:188‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: current and emerging. J Hepatol 2018;68:362‐375. [DOI] [PubMed] [Google Scholar]
- 10. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11‐20. [DOI] [PubMed] [Google Scholar]
- 11. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non‐alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo‐controlled phase 3 trial. Lancet 2019;394:2184‐2196. [DOI] [PubMed] [Google Scholar]
- 12. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J Hepatol 2020;73:26‐39. [DOI] [PubMed] [Google Scholar]
- 13. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC‐0602K in non‐alcoholic steatohepatitis: a randomized, double‐blind, placebo‐controlled phase IIb study. J Hepatol 2020;72:613‐626. [DOI] [PubMed] [Google Scholar]
- 14. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo‐controlled trial of emricasan in patients with NASH and F1–F3 fibrosis. J Hepatol 2020;72:816‐827. [DOI] [PubMed] [Google Scholar]
- 15. Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 2019;29:1173‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bohinc BN, Michelotti G, Xie G, Pang H, Suzuki A, Guy CD, et al. Repair‐related activation of hedgehog signaling in stromal cells promotes intrahepatic hypothyroidism. Endocrinology 2014;155:4591‐4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelly MJ, Pietranico‐Cole S, Larigan JD, Haynes NE, Reynolds CH, Scott N, et al. Discovery of 2‐[3,5‐dichloro‐4‐(5‐isopropyl‐6‐oxo‐1,6‐dihydropyridazin‐3‐yloxy)phenyl]‐3,5‐dio xo‐2,3,4,5‐tetrahydro[1,2,4]triazine‐6‐carbonitrile (MGL‐3196), a highly selective thyroid hormone receptor beta agonist in clinical trials for the treatment of dyslipidemia. J Med Chem 2014;57:3912‐3923. [DOI] [PubMed] [Google Scholar]
- 18. Taub R, Chiang E, Chabot‐Blanchet M, Kelly MJ, Reeves RA, Guertin MC, et al. Lipid lowering in healthy volunteers treated with multiple doses of MGL‐3196, a liver‐targeted thyroid hormone receptor‐beta agonist. Atherosclerosis 2013;230:373‐380. [DOI] [PubMed] [Google Scholar]
- 19. Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL‐3196) for the treatment of non‐alcoholic steatohepatitis: a multicentre, randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet 2019;394:2012‐2024. [DOI] [PubMed] [Google Scholar]
- 20. Barascuk N, Veidal SS, Larsen L, Larsen DV, Larsen MR, Wang J, et al. A novel assay for extracellular matrix remodeling associated with liver fibrosis: an enzyme‐linked immunosorbent assay (ELISA) for a MMP‐9 proteolytically revealed neo‐epitope of type III collagen. Clin Biochem 2010;43:899‐904. [DOI] [PubMed] [Google Scholar]
- 21. Eddowes PJ, Sasso M, Allison M, Tsochatzis E, Anstee QM, Sheridan D, et al. Accuracy of fibroscan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2019;156:1717‐1730. [DOI] [PubMed] [Google Scholar]
- 22. Park PH, Sanz‐Garcia C, Nagy LE. Adiponectin as an anti‐fibrotic and anti‐inflammatory adipokine in the liver. Curr Pathobiol Rep 2015;3:243‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hashemi SA, Alavian SM, Gholami‐Fesharaki M. Assessment of transient elastography (FibroScan) for diagnosis of fibrosis in non‐alcoholic fatty liver disease: a systematic review and meta‐analysis. Caspian J Intern Med 2016;7:242‐252. [PMC free article] [PubMed] [Google Scholar]
- 24. Patel J, Bettencourt R, Cui J, Salotti J, Hooker J, Bhatt A, et al. Association of noninvasive quantitative decline in liver fat content on MRI with histologic response in nonalcoholic steatohepatitis. Therap Adv Gastroenterol 2016;9:692‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caussy C, Reeder SB, Sirlin CB, Loomba R. Noninvasive, quantitative assessment of liver fat by MRI‐PDFF as an endpoint in NASH trials. Hepatology 2018;68:763‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D, Vazquez‐Montesino LM, Escober JA, Fernandes CT, Cholankeril G, Loomba R, et al. Low thyroid function in nonalcoholic fatty liver disease is an independent predictor of all‐cause and cardiovascular mortality. Am J Gastroenterol 2020;115:1496‐1504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S3