


Abstract
A significant portion of human cancers are due to viruses integrating into human genomes. Therefore, accurately predicting virus integrations can help uncover the mechanisms that lead to many devastating diseases. Virus integrations can be called by analysing second generation high-throughput sequencing datasets. Unfortunately, existing methods fail to report a significant portion of integrations, while predicting a large number of false positives. We observe that the inaccuracy is caused by incorrect alignment of reads in repetitive regions. False alignments create false positives, while missing alignments create false negatives. This paper proposes SurVirus, an improved virus integration caller that corrects the alignment of reads which are crucial for the discovery of integrations. We use publicly available datasets to show that existing methods predict hundreds of thousands of false positives; SurVirus, on the other hand, is significantly more precise while it also detects many novel integrations previously missed by other tools, most of which are in repetitive regions. We validate a subset of these novel integrations, and find that the majority are correct. Using SurVirus, we find that HPV and HBV integrations are enriched in LINE and Satellite regions which had been overlooked, as well as discover recurrent HBV and HPV breakpoints in human genome-virus fusion transcripts.
INTRODUCTION
Virus integration is a structural variation that inserts a virus segment into a host genome. In human, it is responsible for a significant portion of cancers. Hepatitis B (HBV) and Hepatitis C (HCV) viruses are known to cause hepatocellular carcinoma (HCC), a form of liver cancer (1), while Human papillomaviruses (HPVs) are present in virtually all cervical cancers, and are also associated with cancers of the anus, penis, vulva as well as oropharyngeal cancer (2,3). The Epstein-Barrow virus (EBV) infects ∼90% of adults and is linked to several cancers (4). Other well known oncoviruses are the Human T-lymphotropic virus (HTLV), Kaposi’s sarcoma-associated herpesvirus (HHV-8) and Merkel cell polyomavirus (MCV).
Second-generation sequencing technologies offer the opportunity to inexpensively and efficiently detect and characterise viral integrations in large numbers of samples. However, compared to other genomic structural variations such as deletions or transpositions, the problem of computationally detecting virus integrations has not been sufficiently tackled, and only a handful of solutions exist, which are often computationally very expensive, produce inaccurate results and are not able to simultaneously scan for integrations from a set of different viruses. Existing integration callers routinely employ one of two strategies: read subtraction and host+viruses mapping. The first strategy consists of mapping read pairs onto the host (resp. viruses) genome, and then remap the unmapped reads onto the viruses (resp. host) genome; finally, from the remaining unmaped reads, virus integrations are called. This strategy is used by SeqMap (5), Vy-PER (6), Virus-Clip (7), ViralFusionSeq (8), BatVI (9) and VIcaller (10). Methods following the second strategy build a customised genome by concatenating the host and the viruses, and then map the read pairs directly on it, and they include VirusSeq (11), ViFi (12) as well as general SV callers. Some tools such as VirusFinder (13,14) and SummonChimera (15) use a combination of the two strategies. See (16) for a more in-depth review and comparison of different software. No matter what strategy is used, the second step is usually to extract pairs mapping partially to the host and partially to a virus, and cluster such pairs into viral integrations.
We observed that existing callers struggle to correctly predict integrations in repeat regions of the host genome. When multiple similar copies of a region exist throughout a genome, aligners often fail to correctly align reads to them, as some of the reads can align to multiple possible loci. Therefore, if a virus integrates into such a region, many reads that are supposed to support its correct location will be aligned to incorrect locations and may create false negatives and false positives (Figure 1).
Figure 1.

(A) A region (yellow) is repeated three times in the host reference, in chromosomes 3, 5 and 8. (B) A virus integrates into chromosome 3 in the sample; four pairs supporting the integration are sequenced. (C) The chimeric host reads (host reads having their mate mapped to a virus sequence) have each three possible alignments, hence we call them ambiguous reads. In this situation, mappers such as BWA MEM choose one location at random, which confuses virus integration callers, leading them to either ignore such reads (and entirely miss the integration), or call many different integrations.
Existing callers deal with ambiguous reads (i.e. reads that align to multiple locations) by either (a) ignoring them, (b) trusting them only if supported by non-ambiguous reads or (c) by simply trusting the location provided by the aligner. Strategies (a) and (b) will result in false negatives, while (c) will produce many false positives. We used ViFi, BatVI and VIcaller as representatives of strategies (a), (b) and (c), respectively. (These three callers have been shown to be the currently most accurate methods.) We applied these three methods on liver cancer and cervical cancer datasets and found that all three methods predict hundreds of HBV and HPV integrations per sample, which are unlikely to be true. Furthermore, these methods miss known and validated integrations.
To solve this problem we borrow from techniques successfully used to predict transpositions (17), but we significantly adapt them to the problem of virus integration calling. Our algorithm iteratively clusters reads that are deemed to support the same integration, and for each cluster it finds the location that is most likely to be correct in both the host and the virus genomes.
The result is SurVirus, a sensitive, precise and fast virus integration caller. Given a second-generation paired-end dataset, a host genome and a database of viruses, SurVirus predicts the integration events that occurred, providing the precise integration loci on the host genome as well as which segments of which viruses integrated. We use well-studied HCC and cervical cancer datasets, both WGS and HIVID (targeted deep sequencing), as well as simulations, to show that SurVirus is able to predict many previously missed integrations, while being more precise than other published solutions. In particular, SurVirus is able to predict more known validated virus integrations compared to the other methods in both HCC and the cervical cancer patients. Twenty-eight percent of our HPV calls and 7% of our HBV calls were novel, and most of them are in repeat regions. We observe that HPV integrations in cervical cancer are enriched in LINE regions, which was previously observed for HPV-associated head and neck squamous cell carcinoma, but not in cervical cancer, to the best of our knowledge. We validated a subset of novel HPV integrations, and found that the majority of them was real. Our novel HBV integrations were mainly located in Satellite and LINE regions.
While the other methods call hundreds of false positives per sample, we are significantly more precise, and we call only a fraction of the unrealistic number of calls predicted by them (9 and 7 breakpoints per sample for HPV and HBV respectively). Similarly, when the virus integration callers are applied on DNA and RNA data from the same sample, SurVirus has the largest overlap among all the methods. Additionally, we found one recurrent HBV breakpoint in hg-HBV fusion transcripts and three recurrent HPV breakpoints in hg-HPV fusion transcripts. The HBV recurrent breakpoint and one of the HPV recurrent breakpoint have never been observed before, to the best of our knowledge.
METHODS
Overview of the algorithm
SurVirus requires a set of read pairs from the sample, a host reference genome and a database of virus genomes. It then operates in two steps (Figure 2). In the first step (Supplementary Section Chimeric pairs extraction), we retain the subset of pairs that are relevant for predicting virus integrations. Such pairs partially align to the host genome, and partially to the virus database. In the second step (Supplementary Section Candidate integration discovery), we identify ambiguously aligned reads and correct their alignments; then, we iteratively group and refine the clusters of read pairs that may support the same integration, and for each cluster we determine the location where the integration most likely happened. Supplementary Figure S9 illustrates how SurVirus works on a real example.
Figure 2.

SurVirus pipeline. The input to the algorithm is a host genome, a database of virus sequences and a set of read pairs. The algorithm then proceeds in two steps. Step 1: Chimeric pairs extraction. It extracts the subset of read pairs that are useful to the prediction of virus integrations, and maps them onto the human and viruses genomes using a standard aligner. Step 2: Candidate integration discovery. It corrects the alignment of the reads that were incorrectly aligned (for example, in the figure, the read represented as dotted arrow is realigned to the correct location), and clusters the pairs to predict virus integrations. Each cluster represents the breakpoint of an integration. (For example, in the figure, we discovered two clusters which are enclosed by green dotted lines.)
Details of software and datasets
A number of virus integration detection software exists, and testing them all was impractical. We selected three recent tools: BatVI (9), ViFi (12) and VIcaller (10). As described in the introduction, they represent the three main strategies for dealing with ambiguously aligned reads: ignoring them (ViFi), using them only when supported by uniquely aligned reads (BatVI) and trusting their original alignments (VIcaller). These software have already been tested and proven to be superior to older methods such as VirusSeq (11), ViralFusionSeq (8) and VirusFinder (13).
All the software were run with FASTQ files as input. ViFi is the only software that does not have the ability to deal with PCR duplicates, so we used FastUniq (18) to create a new set of FASTQ files with duplicates removed.
Simulated datasets
In order to determine if current callers fail in repetitive regions, we simulated two distinct genomes: one where integrations were inserted in random locations (RANDSIM), and one where they were inserted in repetitive regions (REPSIM).
For RANDSIM, we generated a genome by selecting 100 locations on hg19 at random, and simulating a HPV insertion for each location. For REPSIM, the 100 locations were selected to be in repeats (Supplementary Section REPSIM dataset in detail). Finally, for each simulated genome we generated four datasets using pIRS (19) with different coverages: 5 ×, 10 ×, 20 × and 50 ×. For all the datasets, the read length was 100 bp and the mean insert size 500 bp.
In the results section, we show that while the existing callers perform very well on RANDSIM, their performance degrade on REPSIM. We further show that the degradation of performance on REPSIM is due to the incorrect alignment of ambiguous reads.
Real datasets
We test SurVirus, BatVI, ViFi and VIcaller on three real datasets, which we name HPV HIVID, HBV HIVID and HBV WGS. This section describes each dataset, while Table 1 summarizes their features.
Table 1.
Summary of the three real-world datasets used to benchmark the performance of the callers
| Name | HPV HIVID | HBV HIVID | HBV WGS |
|---|---|---|---|
| Samples | 135 | 426 × 2 | 88 × 2 |
| Tissue | Cervical cancer | HCC (tumor/control) | HCC (tumor/control) |
| Sequencing method | HIVID | HIVID | WGS |
| Virus integrated | HPV | HBV | HBV |
| Sensitivity tested by | 75 Sanger-validated calls | 145 Sanger-validated calls | 246 HIVID calls |
| Precision tested by | RNA-seq | RNA-seq | 246 HIVID calls |
HPV HIVID dataset
In Hu et al. (20), 135 samples of cervical cancer were sequenced using HIVID, which is a targeted Illumina sequencing method that produces large numbers of read pairs from regions where the virus is integrated. The paper validated 211 candidate HPV integrations with Sanger sequencing, and 174 appear to be successful. Interestingly, we noticed that many of the Sanger sequences reported in (20) are not supported by the actual Illumina datasets (Supplementary Section Curating HPV Sanger validated calls, Supplementary Figures S1 and S2). We eventually retained 75 calls that showed the minimum required support, and we used them to assess the sensitivity of the callers.
Furthermore, for 10 patients, RNA sequencing is available. We use it to assess the precision of the methods, by computing the overlap between DNA and RNA calls.
In the results section, we run SurVirus, BatVI, ViFi and VIcaller on both HIVID and RNA-seq datasets to compare their performance. Unfortunately we failed to run VIcaller on the HIVID datasets for four cervical cancer samples (T2020, T2023, T2116, T2122), therefore we excluded these sample from the comparison.
HBV HIVID dataset
Zhao et al. (21) sequenced 426 HCC patients using HIVID. For each patient, two samples (control and tumor) were sequenced. One hundred forty-six integrations called by HIVID were reported as validated using Sanger sequencing. We exclude one because the Sanger sequence can be entirely mapped to the human genome, and we use the remaining 145 as a truth set for sensitivity. For 12/145 calls, the Sanger sequence can be aligned to multiple locations in the human genome other than the one predicted by HIVID. Therefore a single Sanger sequence can be used to validate multiple integration sites, and it is not guaranteed that the site predicted by HIVID is the correct one. We refer to these 12 calls as ambiguously validated.
RNA sequencing is available for 12 samples, and we repeat the analysis that was performed for HPV HIVID.
HBV WGS dataset
Sung et al. (22) produced WGS datasets for both tumor and adjacent normal tissue of 88 HCC patients, 81 of which are HBV-positive. Li et al. (23) predicted 246 HBV integrations on a subset of 28 patients using HIVID. We use the HIVID calls as a truth set to test the performance of the methods on the WGS datasets.
RESULTS
Performance on simulated datasets
In the introduction, we claim that ambiguous reads pose a challenge to existing callers, and we describe the different strategies used and the shortcomings of each strategy. In this section, we provide support for our claim by showing that ViFi, BatVI and VIcaller all work very well when the viruses are randomly integrated, but fail when viruses integrate in repetitive regions.
Supplementary Figure S4 shows that, when virus segments are randomly integrated into the human genome, the callers perform well. All methods predict nearly all the virus integrations correctly and produce very few false positives. However, when tested on REPSIM, the callers perform very differently (Figure 3A–C). Except for SurVirus, all the callers are lacking in either sensitivity, precision, or both. VIcaller demonstrates good sensitivity, but suffers from poor precision; furthermore, its precision decreases with increasing coverage. BatVI and ViFi show good precision, but opposite behaviours: BatVI is precise at low coverage, but becomes worse as the depth increases; ViFi starts with poor precision at 5×, but becomes more precise as the depth increases. SurVirus is both the most sensitive and the most precise caller in all the datasets. Remarkably, no other caller comes close in both sensitivity and precision, and this is reflected in the F1 score. It must be noted that the sole purpose of our simulated dataset is to demonstrate that existing callers struggle in detecting virus integrations in repetitive regions, while our algorithm better handles such integrations. It is not meant to be representative of general performance on biological dataset, which will be extensively tested in the following sections.
Figure 3.
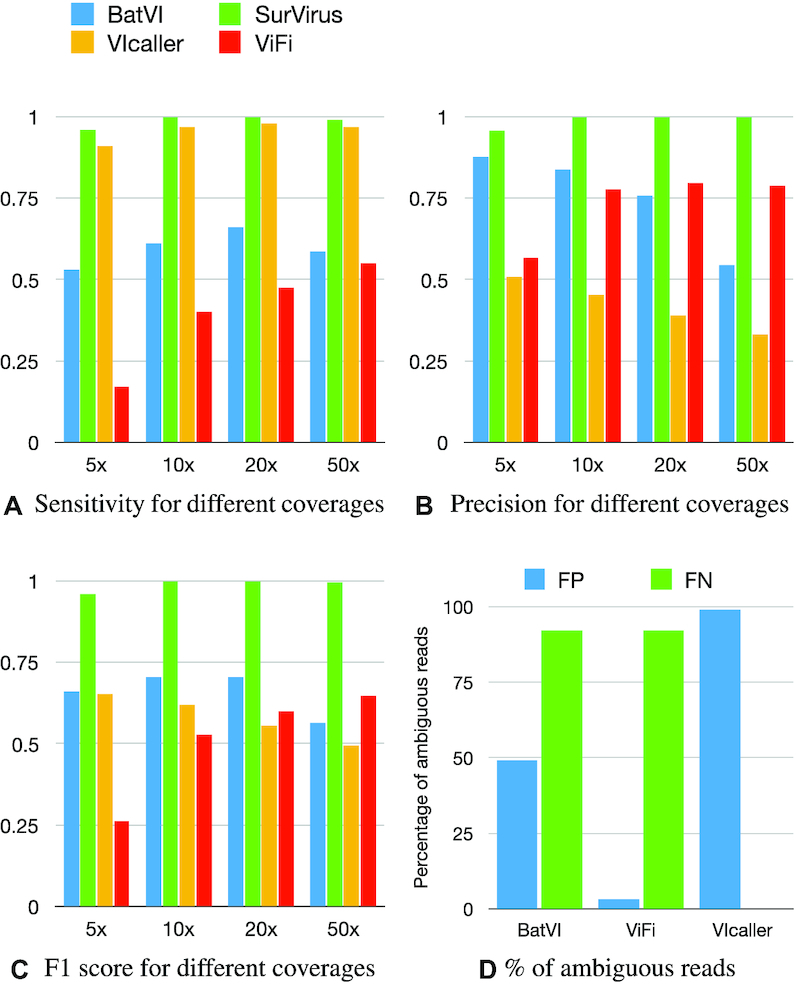
(A–C) The performance of the three tested callers plus SurVirus, on the REPSIM datasets at different coverage. A simulated integration is considered as predicted if the predictions is within 100 bp from it. (D) Percentage of ambiguous reads in false positives (blue) and false negatives (green) for each caller.
Figure 3D shows how ambiguous reads contribute to false positives and false negatives in REPSIM for each caller. We mark a read as ambiguous if its mapQ score is less than 10 when mapped to hg19. In BatVI, ambiguous reads constitute about half of all the reads used to call false positives. This is expected since BatVI uses ambiguous reads, as long as they are supported by a sufficient number of unambiguous reads. On the other hand, ViFi entirely discards ambiguous reads, therefore they do not contribute to false positives; by closer inspection, all false positives by ViFi are due to breakpoints imprecisely predicted (farther than 100 bp from the correct location). Finally, ambiguous reads contribute almost entirely to false positives by VIcaller, suggesting that it uses them incorrectly.
As for false negatives, it is immediately obvious that ambiguous reads contribute almost entirely to missed integrations in BatVI and ViFi. We did not consider VIcaller because it only has three false negatives, which would not be a significant number to infer any pattern. Supplementary Figure S5 breaks down the performance of the methods according to the difficulty of the repeats.
Performance on HIVID datasets
To benchmark the performance of SurVirus, BatVI, ViFi and VIcaller, we run these four callers on HPV HIVID and HBV HIVID. Their performance are compared using their Sanger sequencing and RNA-seq datasets.
Sensitivity on Sanger validated calls
Using the Sanger sequence, we are able to identify the host breakpoint at a single base pair resolution. Hence, we employ a very strict comparison criteria when comparing the calls of each caller to the benchmark validated calls (Supplementary Section Comparing integrations called by different methods, D = 10 bp). Figure 4A, C shows the percentage of validated calls that each method is able to detect, while Figure 4B, D shows the number of virus integrations called by each method.
Figure 4.
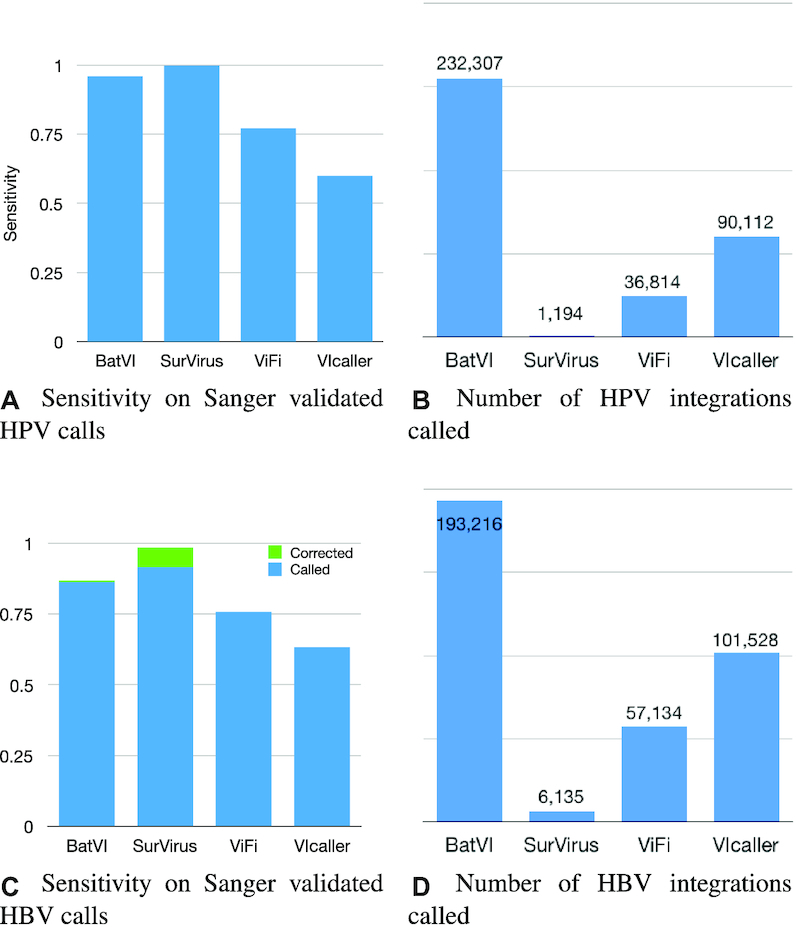
(A, B) Sensitivity and number of calls for the callers on the HPV HIVID dataset. Although the reliable ground truth is only a small set of Sanger validated integrations, it is easy to see that the number of calls BatVI, ViFi and VIcaller predict is unrealistic, and it is most likely the results of tens of thousands of false positives. (C, D) The same analysis is repeated on detected HBV integrations in HBV HIVID dataset. For a subset of validated calls (marked as ‘Corrected’), SurVirus actually selects a better location compared to what was reported by Zhao et al. (21).
SurVirus is the only tool that precisely predicts all the validated HPV integrations. Remarkably, it does so while generating a fraction of the calls reported by other methods (Figure 4A, B). BatVI, the second most sensitive tool, predicts 196 × more integrations than SurVirus. ViFi and VIcaller predict 31× and 76× more calls than SurVirus, respectively, ant yet miss a significant portion of validated calls.
Similarly, SurVirus is also able to detect more validated HBV integrations while reporting far less calls than the other methods (Figure 4C, D). Interestingly, for the 12 HBV integrations which are ambiguously validated by Sanger sequences (i.e. these 12 Sanger sequences align to multiple locations on the human genome), SurVirus reports an alternative location compared to what was reported by HIVID. In 10/12 (=83.3%) cases, the location reported by SurVirus is better than that reported by Zhao et al. (21) (Supplementary Section Correcting the location of the HBV Sanger validated calls).
The number of integrations called by methods other than SurVirus are not realistic. ViFi, the method with the second least number of calls, predicts an average of over 350 HPV integrations per cervical sample, while VIcaller almost 700 and BatVI well over 1500 (Figure 4B). Such numbers point to an extremely high number of false positives. This is further reinforced by the fact that different methods call vastly different integrations, as shown in Figure 5A, C. In each figure there are seven subsets that are made of calls not predicted by SurVirus. We analyse them to determine the reason why SurVirus did not call them.
Figure 5.

(A) Intersection of the calls from the different callers on HPV HIVID datasets. (B) For each of the seven subsets of (A) not predicted by SurVirus, we randomly sampled 200 calls, and we classified them into six categories of false positives. Callers are abbreviated as BV (BatVI), VF (ViFi) and VC (VIcaller). (C) Intersection of the calls from the different callers on HPV HIVID datasets. (D) Classification of the seven subsets of (C) not predicted by SurVirus.
Most predictions not called by SurVirus are plausibly false positives
We examined the seven subsets of calls in Figure 5A, C that were not called by SurVirus but were called by at least one other tool. Given the large number of calls, 200 calls were randomly sampled for each subset. For each call, we analyse the reads that were used to call it. When multiple tools called the same integration, we analyse the supporting reads according to ViFi, if available (since it was shown in simulation to be the most precise); otherwise we analyse the supporting reads according to BatVI.
We classified these calls into seven categories, according to the reason why SurVirus would not trust the call. Namely:
1 or 2 unique support: Most callers fail at correctly handling PCR duplicates. For many calls, the tools report a large number of pairs supporting the call. However, upon closer inspection, the pairs are all PCR duplicates of one or two distinct pairs.
inconsistent reads: Callers sometimes cluster pairs that cannot support the same breakpoint. For example, the host reads may belong to opposite strands, or the virus reads may belong to different viruses, opposite strands or they may map too far from each other. See Supplementary Section Consistency of read pairs and Supplementary Figure S3.
low-quality unmapped: Some reads cannot be mapped by BWA MEM (32) onto either the human reference or to virus database, and have low average base quality (lower than 10); they are most likely artifacts. Some callers, in particular ViFi, employ custom algorithms that force their alignment against the virus database;
non-chimeric: The pairs used to support the integration can be properly aligned (as reported by BWA MEM) to either the host or a virus. They are not chimeric pairs;
short coverage: The host region next to the integration is expected to be covered by the chimeric host reads by >100 bp (since average insert size is >200 bp). Chimeric host reads are reads that support the existence of an integration, i.e. their mate is a virus read. If this is not true, we classify the call as short coverage. No constraint is given on virus coverage, since the virus segment which is integrated into the host may be short.
Figure 5B shows the classification of the sampled HPV calls into the six categories. The majority of the calls, after PCR duplicate removal, are supported by only 1 pair. Furthermore, a large number of calls were due to inconsistent virus reads and short coverage. Few cases were due to calls supported by two pairs, low-quality unmapped reads or non-chimeric. All of the calls could be categorised into these six categories.
For the HBV HIVID datasets, Figure 5D shows that all the calls except for three could be classified as false positives. Compared to HPV HIVID, the number of calls classified as short coverage was considerably less, and those classified as non-chimeric or low-quality unmapped were significantly more.
In total, out of these 1400 HPV integrations and 1400 HBV integrations that are not called by SurVirus, only three HBV integrations (0.1%) may potentially be true positives.
Validation of novel calls by SurVirus
For the HPV HIVID dataset, 385 integrations are only predicted by SurVirus. Unfortunately, only 21 samples were still available for validation. Out of the 385 calls, only 8 belonged to one of these samples. Five calls were successfully validated by Sanger validation (Supplementary Table S2). Two out of three integrations that failed to validate have split reads support, so there is a possibility that they are correct. It should be noted that those integrations seem to appear in low concentration. This, coupled with the fact that the organic samples maybe degraded and that they are in repeated regions, makes them more difficult to validate.
Given the limited number of DNA calls suitable for validation, in order to strengthen our confidence in the precision of the method, we validated the transcriptomic calls for two samples for which we possessed high quality RNA. 19 calls out of 19 successfully validated (Supplementary Table S4).
SurVirus predicts novel integrations in repetitive regions
For every SurVirus-unique call, we compute the percentage of reads that are not ambiguous (i.e. uniquely aligned). Figure 6 shows the frequency histograms of such percentages for the HPV and HBV HIVID datasets.
Figure 6.

Frequency histograms of the percentages of uniquely aligned reads for each SurVirus-unique call. For most calls, nearly all their reads are not uniquely aligned, which suggests they lie in repetitive regions, and they are missed by other methods for this reason.
The vast majority of SurVirus-unique HPV calls (81%) have <50% of their reads uniquely aligned. Furthermore, a large proportion of the calls (41%) have 20% or less of their reads uniquely aligned. This is even more evident in the HBV dataset, where 52% of the calls have <20% of their reads uniquely aligned, and 85% of them have <50% of uniquely aligned reads. This statistics indicate that SurVirus calls many novel integrations by using ambiguous reads.
We further classify the SurVirus-unique calls based on the classes of repeat regions they belong to (Figure 7); nearly all of them belong to at least one class of repeats. More than half of the SurVirus-unique HPV calls are located in LINE repeats. The fact that HPV tends to integrate in LINE repeats was observed by Hatano et al. (24) in HPV-associated head and neck squamous cell carcinoma, but it has been overlooked in cervical cancer, to the best of our knowledge, probably due to limitations of existing computational methods. The second and third most commonly missed repeats were Satellite (23%) and SINE (13%). Indeed, out of the five validated calls, two were located in LINEs repeat, one in Satellite, and one in SINE.
Figure 7.
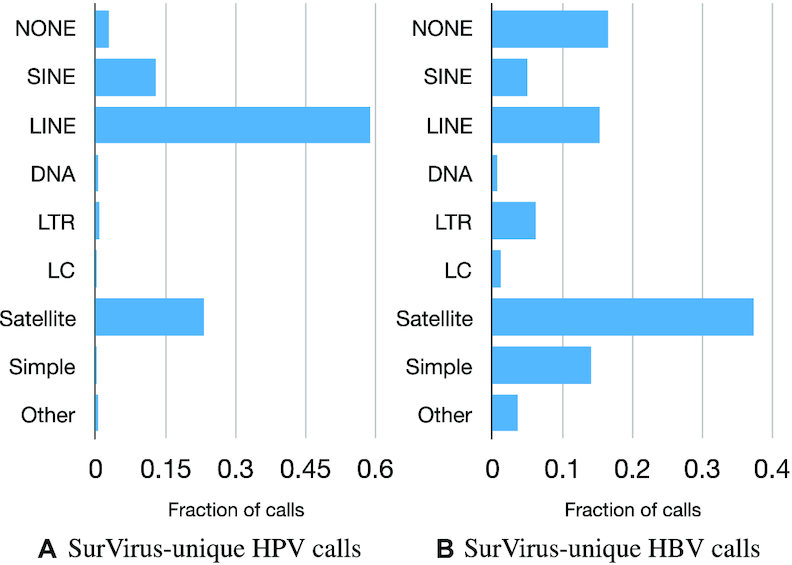
Classification of SurVirus-unique HPV calls (A) and HBV calls (B) according to which classes of repeats they belong to.
For the HBV HIVID dataset, most of the SurVirus-unique calls were in Satellite regions (40%), followed by LINE (22%). There are a few publications reporting that HBV is frequently integrated into Satellite and LINE (25–29).
RNA-sequencing
We ran SurVirus and the other methods on the available RNA data from cervical cancer and HCC patients. We expect that each RNA call is supported by a DNA call, either
directly: there is a DNA breakpoint that precisely matches in chromosome, strand and position of the RNA breakpoint;
as a potential alternative splicing: there is a DNA breakpoint that is within 100 000 bp of the RNA breakpoint, either downstream (if the RNA breakpoint is on the positive strand) or upstream (if the RNA breakpoint is on the negative strand).
Figure 8 illustrates the two cases: Transcript 1 in the figure represents case (i), while Transcript 2 represents case (ii).
Figure 8.
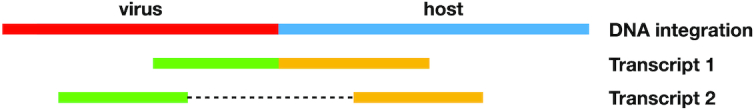
A virus is integrated into a genome. We show two cases: if Transcript 1 is expressed, then the breakpoint detected from RNA-seq will match the one detected from DNA-seq. If Transcript 2 is expressed, i.e. the genomic region containing the breakpoint is not transcribed, then the RNA call will not directly match the DNA call, and it will be classified as a potential alternative splicing.
SurVirus and ViFi display comparable precision and perform much better than BatVI and VIcaller (Figure 9). ViFi has less calls that are directly supported by DNA breakpoints. This is probably due to its low accuracy in determining the breakpoints (as we have demonstrated in Section Performance on simulated datasets).
Figure 9.
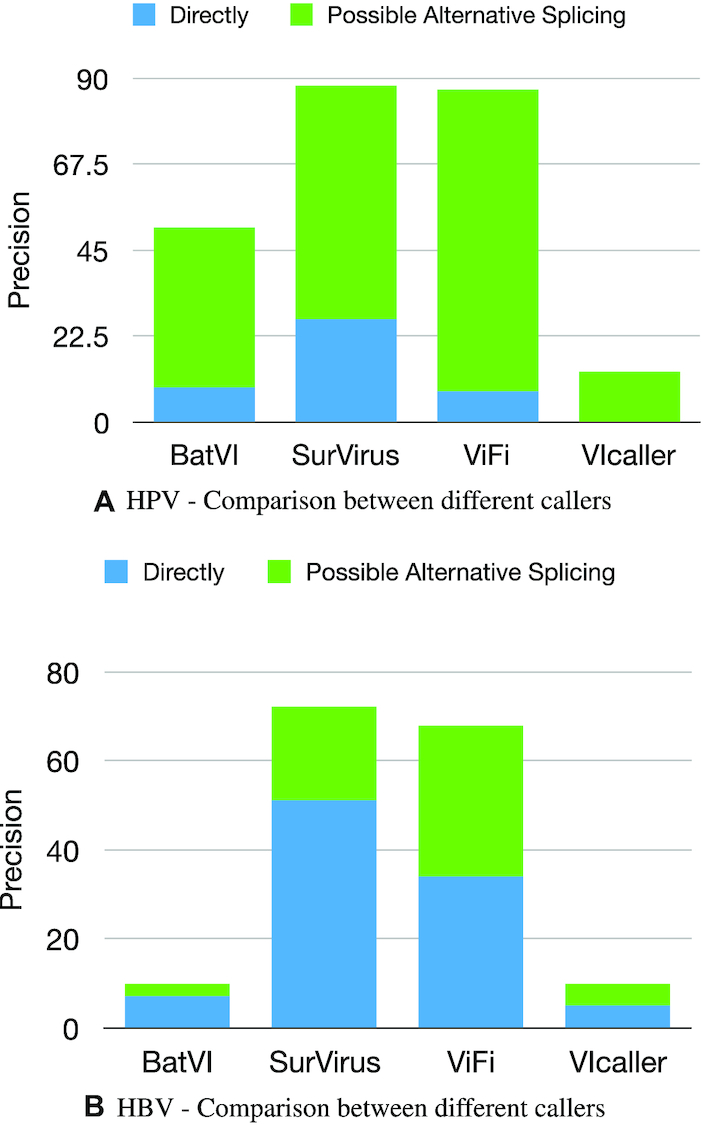
We consider an RNA call a FP if it is not supported by any DNA call on the same sample, neither directly or as a potential alternative splicing. The figure shows the precision on (A) the HPV HIVID and (B) the HBV HIVID datasets.
More interestingly, we found that out of the 23 RNA integrations that are potentially due to alternative splicing, 20 splicing loci in the viruses belong to three recurring categories: type16:880 (eight calls), type16:226 (five calls) and type18:929 (seven calls). Figure 10 shows an example for each category. Two of the three locations (type16:880 and type18:929) have been reported as splicing sites in HPV-human fusion transcripts by Brant et al. (30), and type16:226 is reported as a known splice site in HPV transcripts, although to the best of our knowledge, splicing in this location has never been observed in HPV-human fusion transcripts. All of our calls in cell-lines successfully validated. We could not validate the calls in patients since the RNA samples are degraded.
Figure 10.
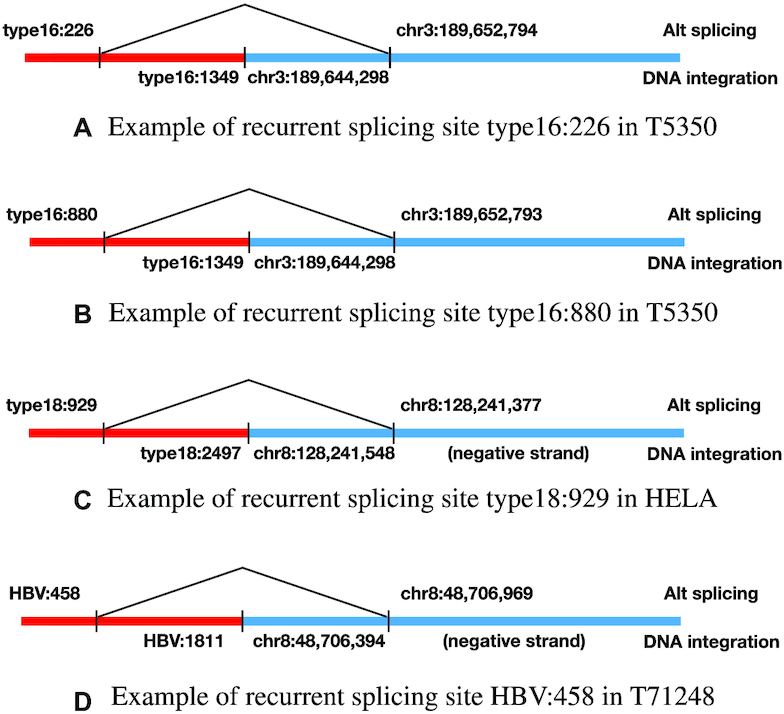
Examples for the three recurring categories of alternative splicing locations in HPV and HBV.
For HBV, we detected one recurrent splicing site HBV:458 in HBV-human fusion transcripts, which appeared in four out of 12 patients. This is the first time we discovered this recurrent splicing site in HBV-human fusion transcripts, though this splicing site is known to occur in HBV transcripts (31).
Performance on the HBV WGS dataset
Figure 11A, B shows the sensitivity and precision for different callers on the HBV WGS datasets, when using the HIVID calls as benchmark. Note that HIVID uses deep sequencing and as such, it is very sensitive but also prone to noise, hence the sensitivities of the methods are likely underestimated. We failed to run VIcaller on these datasets, as the software required >100GB of RAM for every dataset we tried. Therefore, we used the HBV integrations of VIcaller provided by Chen et al. (10).
Figure 11.
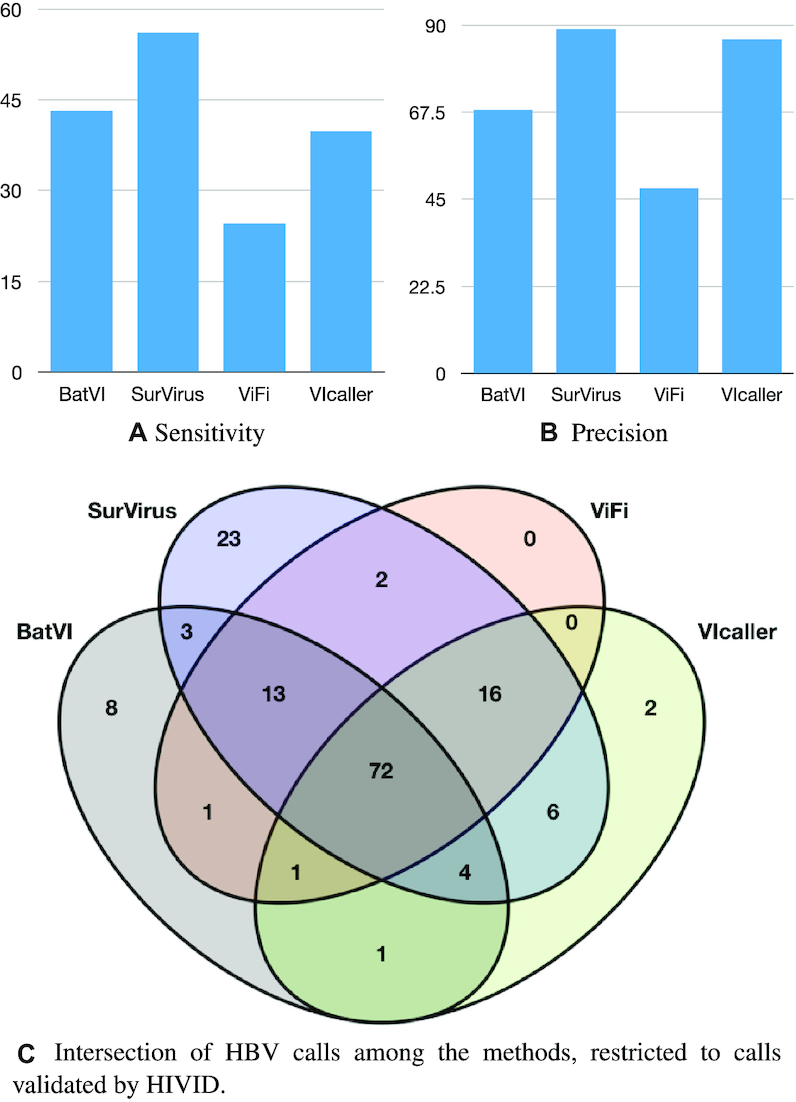
(A, B) Sensitivity and precision for the callers on the HCC WGS datasets. The HIVID calls are used as ground truth. (C) Intersection between the calls from different callers, restricted to the HIVID calls.
SurVirus has better performance on this dataset as well. BatVI has the second best sensitivity, yet SurVirus is 35% more sensitive while having sensibly higher precision. VIcaller almost matches SurVirus in precision, but it is noticeably less sensitive. ViFi performs the worst in both sensitivity and precision, with SurVirus calling more than twice the number of true positives while nearly doubling the precision.
Figure 11C shows the intersection of the methods, restricted to the calls predicted by HIVID. 23 HIVID calls are called only by SurVirus. For comparison, the total number of calls predicted by any of the other three methods and missed by SurVirus was 13, less than half. Interestingly, the majority (8/13) are predicted only by BatVI.
Runtime comparison
We compare the running time (Table 2) and the memory usage (Table 3) of the software on two datasets: an HIVID dataset (SiHa) and a WGS dataset (260T).
Table 2.
Running time of the different virus integration callers, on an HIVID dataset (SiHa) and a WGS dataset (260T)
| HIVID | WGS | |
|---|---|---|
| BatVI | 7h 56m | 3h 44m |
| SurVirus | 4m | 2h 4m |
| ViFi | 15m | 2d 7h 30m |
| VIcaller | 30m | N.A. |
Table 3.
Memory usage of the different virus integration callers, on an HIVID dataset (SiHa) and a WGS dataset (260T)
| HIVID | WGS | |
|---|---|---|
| BatVI | 31GB | 31GB |
| SurVirus | 8GB | 9GB |
| ViFi | 6.5GB | 7GB |
| VIcaller | 11GB | >120GB |
The main difference between the two is that WGS datasets are much larger than HIVID datasets, but HIVID datasets often have very high numbers of chimeric pairs.
SurVirus is much faster than the other methods on both the WGS and the HIVID datasets. Remarkably, it is the only software that performs well on both datasets. Although BatVI is relatively fast on WGS, SurVirus takes nearly half the time, and it is more than a 100 times faster in processing the HIVID dataset. ViFi, on the other hand, was fast on the HIVID dataset, and yet SurVirus was nearly four times faster, and over 25 times faster on WGS. VIcaller was relatively slow on HIVID, and for every WGS dataset we tried, it required more than 120 GB of memory. The other methods required reasonable amounts of memory to process both the HIVID and the WGS datasets.
DISCUSSION
In this study, we tackled the problem of detecting virus integrations in a host genome. Integrations in repeat regions are difficult to predict, and existing solutions fail to call integrations in repetitive regions; the difficulty is due to the fact that reads are often aligned incorrectly to such repeat regions. We developed SurVirus, an algorithm that corrects the mapping of the reads by a technique similar to multiple sequence alignment, but does so efficiently and it is able to deal with large HTS datasets.
We used simulated and published biological dataset to demonstrate that SurVirus predicts novel integrations compared to the state-of-the-art methods, and we were able to validate them. Such novel integrations are mostly in repeat regions, especially in LINE and Satellite repeats, and the literature supports our finding. In particular, a study by Hatano et al. (24) combined Next-Generation Sequencing and Sanger sequencing to accurately detect the HPV integrations in HPV-associated head and neck squamous cell carcinoma, and found significant enrichment of integrations in LINE regions, compared to random expectation. Among the methods we tested, only SurVirus observed a similar enrichment for HPV integrations in cervical cancer datasets; this leads us to believe that revisiting available datasets using SurVirus may uncover a substantial number of integrations that were previously missed.
Furthermore, tested methods predict up to hundreds of thousands of false positives, which SurVirus successfully filters. DNA and RNA concordance is higher for SurVirus compared to other methods. These facts strongly suggest that SurVirus is significantly more precise than the state of the art.
SurVirus can be used with an arbitrarily large database of viruses, and it can quickly scan large datasets starting from a BAM file as well as from raw FASTQ files. This, plus the high sensitivity and precision, as well as the fact that SurVirus detects the breakpoints with extreme accuracy (Supplementary Figure S6), make SurVirus suitable for scanning for integrations in large populations. Higher sensitivity means SurVirus will detect integrations that would be otherwise missed by other callers. Higher precision means that less time will be wasted analysing false positives, as well as less noise as a confounding factor in the analysis. Higher accuracy in determining the breakpoint allows for a better characterisation of the breakpoints, e.g. determining the presence of inserted sequences in between the host and the virus sequences, or determining microhomologies. Indeed, we observed that the breakpoint predicted by SurVirus have significantly more microhomologies than what would be expected at random. Finally, SurVirus is open-source and free to use, it can be used on any organism, and its output is very clear and easy to process. For these reasons, we believe it represents a significant step forward in the study of how virus integrations affect their host.
DATA AVAILABILITY
Supplementary Table S10 reports the Accession number for every dataset we used. SurVirus calls on HPV and HBV HIVID datasets are reported in Supplementary Tables S1 and S6, respectively. SurVirus calls on HPV and HBV RNA-seq datasets are reported in Supplementary Tables S3 and S8, respectively. SurVirus calls on HBV WGS datasets are reported in Supplementary Table S9. Curated benchmark HPV integrations, as detailed in Section Curating HPV Sanger validated calls, are reported in Supplementary Table S5. Reads count for HBV integrations having disagreeing location between HIVID and SurVirus are listed in Supplementary Table S7.
The source code, along with instructions, can be found at https://github.com/kensung-lab/SurVirus.
Supplementary Material
ACKNOWLEDGEMENTS
We would to acknowledge to thank Chen Xun for its help in the usage of VIcaller.
Contributor Information
Ramesh Rajaby, School of Computing, National University of Singapore, 13 Computing Drive, 117417, Singapore; NUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, 28 Medical Drive, 117456, Singapore.
Yi Zhou, Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Yifan Meng, Department of Gynecologic Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China; Cancer Biology Research Center (Key laboratory of the ministry of education), Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Xi Zeng, Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Guoliang Li, Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Peng Wu, Department of Gynecologic Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China; Cancer Biology Research Center (Key laboratory of the ministry of education), Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Wing-Kin Sung, School of Computing, National University of Singapore, 13 Computing Drive, 117417, Singapore; Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China; Genome Institute of Singapore, 60 Biopolis Street, Genome 138672, Singapore.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Singapore Ministry of Education [Tier2 grant R-252-000-664-112]. Funding for open access charge: MoE Tier2 [R-252-000-664-112].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kao J.-H., Chen D.-S.. Global control of hepatitis B virus infection. Lancet Infect. Dis. 2002; 2:395–403. [DOI] [PubMed] [Google Scholar]
- 2. Schiffman M., Castle P.E., Jeronimo J., Rodriguez A.C., Wacholder S.. Human papillomavirus and cervical cancer. Lancet. 2007; 370:890–907. [DOI] [PubMed] [Google Scholar]
- 3. Parkin D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer. 2006; 118:3030–44. [DOI] [PubMed] [Google Scholar]
- 4. Xu M., Zhang W.-L., Zhu Q., Zhang S., Yao Y.-Y., Xiang T., Feng Q.-S., Zhang Z., Peng R.-J., Jia W.-H.et al.. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics. 2019; 9:1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hawkins T.B., Dantzer J., Peters B., Dinauer M., Mockaitis K., Mooney S., Cornetta K.. Identifying viral integration sites using SeqMap 2.0. Bioinformatics. 2011; 27:720–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forster M., Szymczak S., Ellinghaus D., Hemmrich G., Rühlemann M., Kraemer L., Mucha S., Wienbrandt L., Stanulla M.UFO Sequencing Consortium within I-BFM Study Group et al.. Vy-PER: eliminating false positive detection of virus integration events in next generation sequencing data. Sci. Rep. 2015; 5:11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ho D. W.H., Sze K. M.F., Ng I. O.L.. Virus-Clip: a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability. Oncotarget. 2015; 6:20959–20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J.-W., Wan R., Yu C.-S., Co N.N., Wong N., Chan T.-F.. ViralFusionSeq: accurately discover viral integration events and reconstruct fusion transcripts at single-base resolution. Bioinformatics. 2013; 29:649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tennakoon C., Sung W.K.. BATVI: Fast, sensitive and accurate detection of virus integrations. BMC Bioinformatics. 2017; 18:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X., Kost J., Sulovari A., Wong N., Liang W.S., Cao J., Li D.. A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome Res. 2019; 29:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y., Yao H., Thompson E.J., Tannir N.M., Weinstein J.N., Su X.. VirusSeq: software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics. 2013; 29:266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen N.-P.D., Deshpande V., Luebeck J., Mischel P.S., Bafna V.. ViFi: accurate detection of viral integration and mRNA fusion reveals indiscriminate and unregulated transcription in proximal genomic regions in cervical cancer. Nucleic Acids Res. 2018; 46:3309–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q., Jia P., Zhao Z.. VirusFinder: software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS One. 2013; 8:e64465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q., Jia P., Zhao Z.. VERSE: a novel approach to detect virus integration in host genomes through reference genome customization. Genome Med. 2015; 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katz J.P., Pipas J.M.. SummonChimera infers integrated viral genomes with nucleotide precision from NGS data. BMC Bioinformatics. 2014; 15:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen X., Kost J., Li D.. Comprehensive comparative analysis of methods and software for identifying viral integrations. Brief Bioinform. 2018; 20:2088–2097. [DOI] [PubMed] [Google Scholar]
- 17. Rajaby R., Sung W.-K.. TranSurVeyor: an improved database-free algorithm for finding non-reference transpositions in high-throughput sequencing data. Nucleic Acids Res. 2018; 46:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu H., Luo X., Qian J., Pang X., Song J., Qian G., Chen J., Chen S.. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 2012; 7:e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu X., Yuan J., Shi Y., Lu J., Liu B., Li Z., Chen Y., Mu D., Zhang H., Li N.et al.. pIRS: profile-based Illumina pair-end reads simulator. Bioinformatics (Oxford, England). 2012; 28:1533–1535. [DOI] [PubMed] [Google Scholar]
- 20. Hu Z., Zhu D., Wang W., Li W., Jia W., Zeng X., Ding W., Yu L., Wang X., Wang L.et al.. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015; 47:158–63. [DOI] [PubMed] [Google Scholar]
- 21. Zhao L.-H., Liu X., Yan H.-X., Li W.-Y., Zeng X., Yang Y., Zhao J., Liu S.-P., Zhuang X.-H., Lin C.et al.. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016; 7:12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung W.-K., Zheng H., Li S., Chen R., Liu X., Li Y., Lee N.P., Lee W.H., Ariyaratne P.N., Tennakoon C.et al.. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012; 44:765–769. [DOI] [PubMed] [Google Scholar]
- 23. Li W., Zeng X., Lee N.P., Liu X., Chen S., Guo B., Yi S., Zhuang X., Chen F., Wang G.et al.. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013; 102:338–344. [DOI] [PubMed] [Google Scholar]
- 24. Hatano T., Sano D., Takahashi H., Hyakusoku H., Isono Y., Shimada S., Sawakuma K., Takada K., Oikawa R., Watanabe Y.et al.. Identification of human papillomavirus (HPV) 16 DNA integration and the ensuing patterns of methylation in HPV-associated head and neck squamous cell carcinoma cell lines. Int. J. Cancer. 2017; 140:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ogata N., Tokino T., Kamimura T., Asakura H.. A comparison of the molecular structure of integrated hepatitis B virus genomes in hepatocellular carcinoma cells and hepatocytes derived from the same patient. Hepatology. 1990; 11:1017–1023. [DOI] [PubMed] [Google Scholar]
- 26. Tokino T., Fukushige S., Nakamura T., Nagaya T., Murotsu T., Shiga K., Aoki N., Matsubara K.. Chromosomal translocation and inverted duplication associated with integrated hepatitis B virus in hepatocellular carcinomas. J. Virol. 1987; 61:3848–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaul Y., Garcia P.D., Schonberg S., Rutter W.J.. Integration of hepatitis B virus DNA in chromosome-specific satellite sequences. J. Virol. 1986; 59:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Houck C.M., Rinehart F.P., Schmid C.W.. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 1979; 132:289–306. [DOI] [PubMed] [Google Scholar]
- 29. Tsuei D.-J., Chang M.-H., Chen P.-J., Hsu T.-Y., Ni Y.-H.. Characterization of integration patterns and flanking cellular sequences of hepatitis B virus in childhood hepatocellular carcinomas. J. Med. Virol. 2002; 68:513–521. [DOI] [PubMed] [Google Scholar]
- 30. Brant A.C., Menezes A.N., Felix S.P., de Almeida L.M., Sammeth M., Moreira M.A.M.. Characterization of HPV integration, viral gene expression and E6E7 alternative transcripts by RNA-Seq: A descriptive study in invasive cervical cancer. Genomics. 2019; 111:1853–1861. [DOI] [PubMed] [Google Scholar]
- 31. Hass M., Hannoun C., Kalinina T., Sommer G., Manegold C., Günther S.. Functional analysis of hepatitis B virus reactivating in hepatitis B surface antigen-negative individuals. Hepatology. 2005; 42:93–103. [DOI] [PubMed] [Google Scholar]
- 32. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary Table S10 reports the Accession number for every dataset we used. SurVirus calls on HPV and HBV HIVID datasets are reported in Supplementary Tables S1 and S6, respectively. SurVirus calls on HPV and HBV RNA-seq datasets are reported in Supplementary Tables S3 and S8, respectively. SurVirus calls on HBV WGS datasets are reported in Supplementary Table S9. Curated benchmark HPV integrations, as detailed in Section Curating HPV Sanger validated calls, are reported in Supplementary Table S5. Reads count for HBV integrations having disagreeing location between HIVID and SurVirus are listed in Supplementary Table S7.
The source code, along with instructions, can be found at https://github.com/kensung-lab/SurVirus.