


Abstract
Various in vivo mutagenesis methods have been developed to facilitate fast and efficient continuous evolution of proteins in cells. However, they either modify the DNA region that does not match the target gene, or suffer from low mutation rates. Here, we report a mutator, eMutaT7 (enhanced MutaT7), with very fast in vivo mutation rate and high gene-specificity in Escherichia coli. eMutaT7, a cytidine deaminase fused to an orthogonal RNA polymerase, can introduce up to ∼4 mutations per 1 kb per day, rivalling the rate in typical in vitro mutagenesis for directed evolution of proteins, and promotes rapid continuous evolution of model proteins for antibiotic resistance and allosteric activation. eMutaT7 provides a very simple and tunable method for continuous directed evolution of proteins, and suggests that the fusion of new DNA-modifying enzymes to the orthogonal RNA polymerase is a promising strategy to explore the expanded sequence space without compromising gene specificity.
Graphical Abstract
Graphical Abstract.
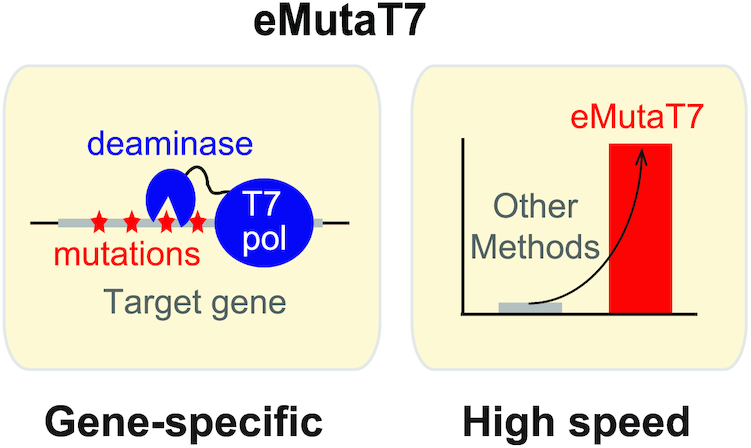
eMutaT7: a rapid in vivo mutagenesis method for continuous directed evolution of proteins.
INTRODUCTION
Directed evolution is a powerful tool for studying protein evolution and obtaining new functional biomolecules for research, biotechnology, and medicine (1). Traditional directed evolution methods usually rely on in vitro diversification of a target gene, followed by selection or screen of variants exhibiting better function. Despite their broad application, these methods consist of labour-intensive discrete steps and generate limited sequence diversity due to relatively low efficiency of cellular DNA uptake. To explore the expanded sequence space with minimal manual labour, various in vivo mutagenesis methods have been recently developed and enable continuous directed evolution (CDE), in which gene diversification and selection are simultaneously performed inside of cells (2).
The ideal in vivo mutagenesis method for CDE of proteins should have high gene specificity to reduce off-target mutagenesis and should make evenly distributed mutations within targets to explore wider sequence space. In most methods, however, the target region is not well correlated with the specific gene region (2). For example, several Cas9-based methods such as base editor (3), CRISPR-X (4), and EvolvR (5), target 3–350 nucleotides from gRNA-binding sites without distinguishing gene boundary. Methods using an orthogonal error-prone DNA polymerase target the whole or part of a plasmid with neither gene specificity nor temporal control of mutagenesis (6,7). A virus-based method, PACE (8), is a pioneering example of CDE of a target gene with many successful examples of protein evolution (2), and has recently been extended to mammalian cells (9,10). However, PACE is inherently based on random mutagenesis without positional specificity and requires specialized equipment for continuous supply of fresh host cells (8). The truly gene-specific in vivo mutagenesis was reported in systems using a fusion protein of a cytidine deaminase and T7 RNA polymerase (T7RNAP), MutaT7 in bacteria (11) and TRACE in human cells (12). T7RNAP allows specific targeting to T7 promoter-controlled genes, upon which a cytidine deaminase introduces the C→T and G→A mutations on the target and non-target strands, respectively. Despite the high gene-specificity, they showed low mutation rates (e.g. MutaT7, 0.34 mutations day−1 kb−1; TRACE, 0.1–0.8 mutations day−1 kb−1), undermining its advantage over the typical in vitro mutagenesis-based methods.
We reasoned that the method using an orthogonal RNA polymerase could be an ideal solution for gene-specific in vivo mutagenesis, and could substantially boost its potential by accelerating the mutation rate. We also thought that the fast doubling time could make bacteria a better host to explore the higher clonal diversity. Here, we report a new gene-specific in vivo mutagenesis system that has significantly higher mutation rate in E. coli. Our results also demonstrate that the systematic search for new DNA-modifying enzymes for fusion to an orthogonal RNA polymerase may greatly broaden the sequence space to explore for directed evolution of proteins.
MATERIALS AND METHODS
Plasmid cloning and materials
Escherichia coli strains, plasmids, and primers used in this study are listed in Supplementary Tables S1–S3, respectively. The mutator plasmids expressing eMutaT7 (pHyo094) and MutaT7 (pHyo099) were obtained as follows: the deaminase gene that expresses Petromyzon marinus cytidine deaminase (PmCDA1) and rat apolipoprotein B mRNA editing catalytic polypeptide-like (rApobec1) were amplified from AIDv2 (Addgene #79620) and pCMV-BE3 (Addgene #73021), respectively (3,13). T7 RNA polymerase gene, in which NdeI site was deleted using inverse PCR (14), was amplified from pNL001 (15). Two DNA fragments were linked by overlapping PCR with a linker gene for the 16-amino acid XTEN linker (3) between two genes. The fused DNA expressing PmCDA1-XTEN-T7RNAP was inserted into pSK633 (16) between the NdeI and PstI sites. The XTEN linker of pHyo099 was exchanged into the linker of MutaT7 (pHyo287) and the araBAD promoter of pHyo287 was exchanged into the A1lacO promoter (pHyo288) by inverse PCR.
All target plasmids (pHyo182, pheS_A294G; pHyo245, pheS_A294G with the dual promoter system; pHyo249, degP_A184S with the dual promoter system; pHyo253, TEM-1 with the dual promoter system) were constructed in a low copy-number plasmid (one or two copy per a single cell; the parent plasmid is pNL001 (15)). To make pheS_A294G gene transcribed with T7 RNA polymerase (pHyo175, pET28b-pheS_A294G), pheS_A294G gene was amplified from the SK324 strain (17) and inserted into pET28b vector using NdeI and XhoI sites. DNA fragment from lacI to pheS_A294G gene containing T7-lac promoter and T7 terminator (lacI-T7-lac promoter-pheS_A294G-T7 terminator) was amplified from pHyo175, and inserted in the pNL001 vector between BamHI and EcoRI sites to make pHyo182 (pVS133-pheS_A294G). To make dual promoter system (pHyo245), a new T7-lac promoter and T7 terminator sequences were inserted in front of T7 terminator and between T7 promoter and lacI gene region, respectively, in the reverse direction.
For degP evolution experiments, pheS_A294G was replaced with degP_A184S amplified from pSK735 (16) and wild-type degP amplified from p7 (18) to make pHyo249 (degP_A294G) and pHyo246 (degP), respectively, using NEBuilder Hifi DNA assembly master Mix (New England Biolabs, USA).
For TEM-1 evolution experiments, ampicillin resistance gene in the target plasmid was replaced with tetracycline resistance gene amplified from pREMCM3 (19) and TEM-1 gene was replaced with pheS_A294G gene to make pHyo253, using NEBuilder Hifi DNA assembly master Mix.
All plasmids harboring variants of the mutator or targets (mutation, deletion, and insertion) were constructed using inverse PCR. eMutaT7 variants (low processive variants or linker length variants) were made on pHyo094. The degP variants were made on pHyo246 and pHyo249 to make pHyo254 (degP_P231L) and pHyo255 (degP_A184S, P231L), respectively, for in vivo viability assay, and on p7 and pSK735 to make pHyo256 (degP_P231L) and pHyo257 (degP_A184S_P231L), respectively, for in vitro enzymatic assay.
All PCR experiments were conducted with KOD Plus neo DNA polymerase (Toyobo, Japan). Restriction enzymes, T4 polynucleotide kinase and T4 DNA ligases were purchased from Enzynomics (South Korea). Plasmids and DNA fragments were prepped with LaboPass™ plasmid DNA purification kit mini, LaboPass™ PCR purification kit, and LaboPass™ Gel extraction kit (Cosmogenetech, South Korea). Sequences of genes in plasmids constructed in this study were confirmed by Sanger sequencing (Macrogen, South Korea). Antibiotics (ampicillin, chloramphenicol, kanamycin, and tetracycline), arabinose, and IPTG were purchased from LPS solution (South Korea). Cefotaxime and ceftazidime were purchased from Tokyo chemical industry (Japan). H-p-Chloro-dl-Phe-OH (p-Cl-Phe) was purchased from Bachem (Switzerland).
E. coli strain construction
All the strains in this study were constructed by λ-Red-mediated recombineering with the pSIM6 plasmid (20). To construct the Δung strain (cHYO057), kanamycin resistance gene region including 100-bp upstream and 100-bp downstream was amplified with primers 180 and 181, and the open reading frame (ORF) of wild-type ung in W3110 was replaced by the kanamycin resistance gene. To construct the ΔdegP Δung strain (cHYO059), SK345 (ΔdegP) (17) was used as a recipient cell. Proper gene deletion or insertion were confirmed by colony PCR using 2X TOP Simple™ DyeMIX-Tenuto (Enzynomics), and 30 μg/ml of kanamycin was used for selection.
Western blot
Cells expressing N-terminal FLAG-tagged pheS_A924G were grown to OD600 ∼0.6 in LB at 37°C. 30-μl samples of the cultures were mixed with 4× SDS sample buffer, heated at 95°C for 15 min, and centrifuged at 13 000 × g for 10 min. Samples (7 μl) were separated by SDS/PAGE and transferred to PVDF membrane (ATTO, Japan). The membrane was sequentially incubated in Ezblock protein-free Blocking buffer (ATTO), anti-OctA (FLAG octapeptide) antibody (2000:1 dilution; SantaCruz, USA), and anti-rabbit IgG-HRP (2000:1 dilution; SantaCruz), and then blotted with the WestSaveUp ECL solution (AbFrontier, South Korea). Image was obtained using Amersham imager 600 (GE Healthcare, USA).
Mutation cycle
The Δung cells (cHYO057) harboring a mutator plasmid (pHyo094) and a target plasmid (pHyo182 for single promoter and pHyo245 for dual promoter) were grown overnight in LB broth supplemented with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol (cycle #0). The overnight cultures (3.5 μl) were mixed with 350 μl of LB broth supplemented with 100 μg/ml ampicillin, 35 μg/ml chloramphenicol, 0.2% arabinose and 0.1 mM IPTG in a 96-deep well plate (Bioneer, South Korea) and incubated at 37°C for 4 h (cycle #1). The dilution–incubation cycle was repeated up to 27 times. At the end of each cycle, a fraction of cells were stored at −80°C as 15% glycerol stocks.
To determine the number of accumulated mutations on the pheS_A294G gene, cells at the selected mutagenesis cycles (cycle #3, #6, #9, #18 and #27) were streaked on LB-Agar plate containing 100 μg/ml ampicillin. Five colonies were randomly chosen for plasmid isolation and the sequence determination. Mutations were counted in the region from 80-bp upstream to 35-bp downstream of the pheS gene (total 1099 bp). Primers 507 and 529 was used for sequencing of target plasmids that have dual promoter system, and universal sequencing primers were used for sequencing of plasmids that have single promoter system (primers T7 promoter and T7 terminator in Supplementary Table S3).
PheS_A294G suppression assay
Samples obtained at the endpoint of each cycle (overnight culture for cycle #0) were diluted to OD600 ∼0.2. Serial 10-fold dilutions of cells (5μl) were spotted on YEG-agar plates and grown on either selective (1.6 mM p-Cl-Phe, 0.2% arabinose and 0.1 mM IPTG) or nonselective (no additives) YEG-agar plates at 37°C overnight. Viable cells on each condition were counted and the suppressor frequency was calculated as N1/N0 (N1: colony forming unit (CFU) in selective plates and N0: CFU in nonselective plates).
Viability assay
Overnight cultures expressing eMutaT7, unfused PmCDA1 and T7RNAP, no mutator, or MP6, were diluted 100-fold in LB broth supplemented with 35 μg/mL chloramphenicol, grown to a log phase (OD600 = 0.2–0.5) at 37°C, and diluted to OD600 ∼0.2. Serial 10-fold dilutions of cells (5 μl) were spotted on LB-agar plates supplemented with 0.2% arabinose and 35 μg/ml chloramphenicol and grown at 37°C for 16 h. Viable cells were counted to calculate CFU/ml (Figure 2E and Supplementary Figure S4).
Figure 2.
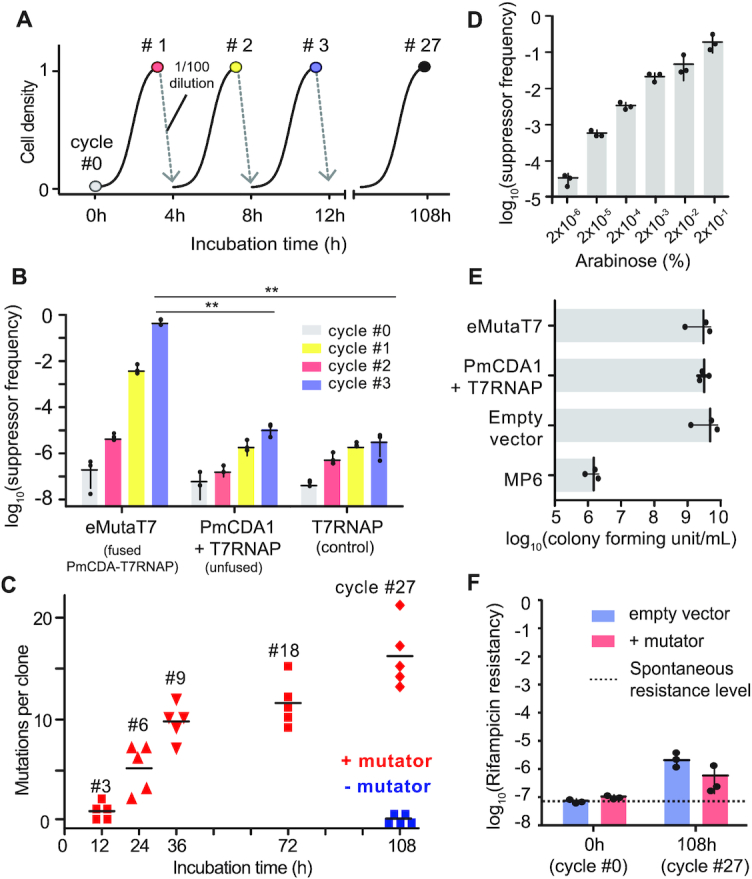
eMutaT7 induces rapid in vivo mutagenesis in the target gene. (A) General scheme of the mutagenesis cycle. (B) PheS_A194G suppressor frequency at each mutagenesis cycle for cells expressing PmCDA1-T7RNAP (eMutaT7), unfused PmCDA1 and T7RNAP, or T7RNAP only. (C) The number of mutations found in five clones at each mutagenesis cycle. (D) PheS_A194G suppressor frequency with different concentration of arabinose. (E) Viability of cells expressing eMutaT7, unfused proteins, no protein, or MP6. (F) Off-target mutation level estimated by rifampicin resistance frequency. The dotted line represents spontaneous rifampicin resistance level. Data are presented as dot plots with averages (short black lines) ± 1 SD; n = 3 (B, D–F) or 5 (C); **P < 0.01 by Student's t-test.
To determine the rifampicin resistance, samples after cycle #0 and #27 were grown to log phase in LB broth supplemented with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol, and subjected to viability assay on selective (80 μg/ml rifampicin) or nonselective (no rifampicin) plates.
High-throughput sequencing and data analysis
Cells taken at cycle 27 were grown in 15 ml of LB broth without arabinose and IPTG, and the target plasmids were extracted with Plasmid Miniprep Kit. The 3289-bp DNA fragments containing the pheS_A924G gene were amplified using primers 512 and 513 covering a region ranging from 1016 bp upstream from T7 promoter to 1069 bp downstream including T7 terminator. Samples were then prepared with TruSeq Nano DNA Kit and sequenced on Illumina HiSeq 2500 (Illumina, USA; operated by Macrogen) in 2 × 101 paired-end runs using the manufacturer's reagents following the manufacturer's protocol to determine the mutation pattern occurred on target plasmids.
Raw reads were trimmed to remove adapter and low quality end sequences using Trimmomatic v.0.36 palindrome mode with followed option; ILLUMINACLIP:Adapter.fa:2:30:10:8:true LEADING:15 TRAILING:15 SLIDINGWINDOW:4:15 MINLEN:36 (21). Trimmed reads were aligned against the target sequence using Burrows-Wheeler Alignment tool (BWA) v0.7.17 with mem mode and BAM files generated by mapping were sorted using SAMtools v.1.6 (22,23). Sorted bam files were subject to perform mpileup using SAMtools mpileup with maximum depth option, which was set as total number of trimmed reads, and output tag list option consisting of DP, DP4 and AD. BCFtools v1.6, which was a set of utilities of SAMtools package, was used to call allele for each locus with multiallelic-caller option. Allele count for each allele and ratio (each allele count/total allele count) were calculated based on AD information of VCF files.
Mutation rate calculation
Mutation rate caused by eMutaT7 was calculated and presented as two styles: (i) mutations per base per generation or (ii) mutations per day per 1 kb. Mutations were counted in the 1099-bp region of pheS for Sanger Sequencing. The number of generations per one cycle was calculated as 6.6 (log2100) because cell density increases 100-fold during one cycle (4 h). To present in the first style, the number of mutations was averaged for chosen clones and divided by the number of nucleotides (1099) and generations (6.6 × cycle number). To present in the second style, the number of mutations was averaged and divided by incubation days (6 cycles·day−1) and the length of the gene region (1.099 kb). To compare the values with those of MutaT7, the number of mutations were counted from Supplementary Figure S10 of the Shoulders group's report (11). Mutation rates of TRACE were estimated from Figure 2A and Supplementary Figure S3b of the Chen group's report (12).
Statistical analysis of high-throughput sequencing data
For high-throughput sequencing data (Figure 3), Mann–Whitney test (unpaired Wilcoxon test) was used to assess the significance of the mutation frequency caused by eMutaT7 system. Calculation was conducted using Stata (USA). Statistically significance were determined with P values defined as *P < 0.05, **P < 0.0001 for this experiment. For other data, we assumed the data will follow normal distribution and performed Student's t-test.
Figure 3.
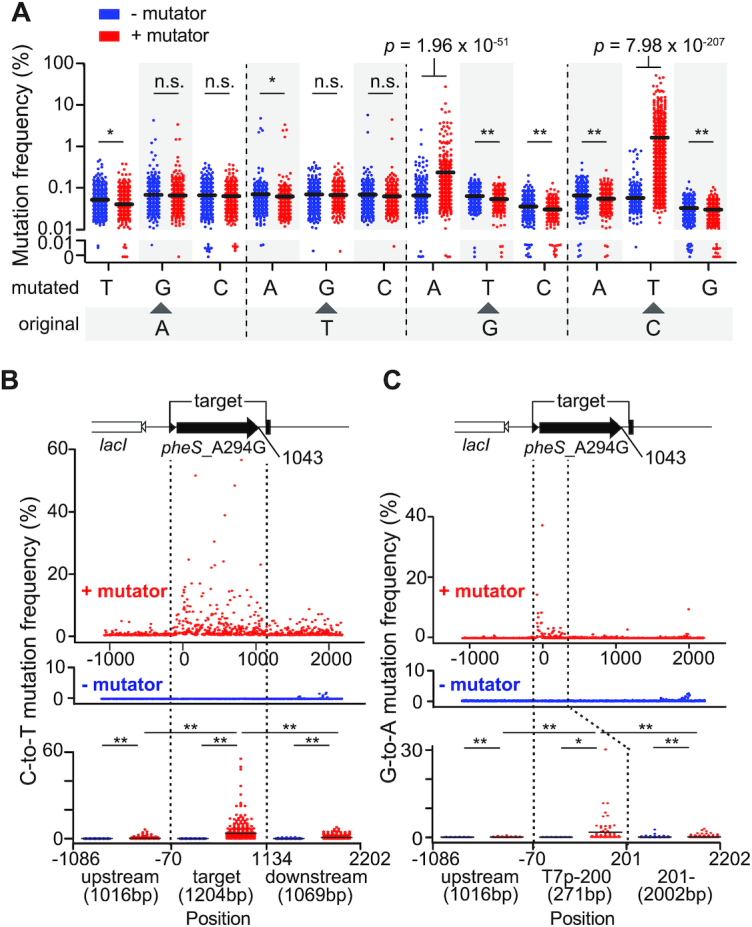
Illumina sequencing demonstrates gene specific mutagenesis of eMutaT7. (A) Mutation frequency of all possible substitution types with or without eMutaT7. Y-axes above and below 0.01% are in log- and linear-scale, respectively. (B, C) Frequency of all C→T (B) and G→A (C) mutations in ∼3.3 kb DNA around the target gene with or without eMutaT7. Collective mutation frequencies in upstream, target (C→T) or –70–200 (G→A), and downstream regions are shown as dot plots with averages (short black lines). *P < 0.05, **P < 0.0001 by Mann–Whitney's t-test.
Antibiotic resistance gene evolution
For TEM-1 and AmpC evolution, all the experiments were performed with LB supplemented with 6 μg/ml tetracycline, 35 μg/ml chloramphenicol, 0.2% arabinose and 0.1mM IPTG. We tested multiple antibiotic concentrations at the same time, of which the cells grown at the highest concentration were subjected to the next round of growth in fresh media with equal or higher amounts of antibiotics. The concentration of each antibiotic (cefotaxime, ceftazidime, or carbenicillin) was gradually increased as indicated in Figure 5A, Supplementary Figure S10. All the experiments were conducted in triplicate and if two out of three samples did not grow, one sample that was grown was inoculated into three samples for the next cycle. After the evolution, plasmids were extracted in bulk from evolution mixture grown in LB media supplemented only with 6 μg/ml tetracycline and inserted into fresh Δung cells (cHYO057) harboring the T7 RNA polymerase-expressing plasmid (pHyo183). Ten colonies were chosen to determine MICs, and plasmids from the five colonies with high MIC values (400–800 μg/ml for CTX, 2000–4000 μg/ml for CAZ and CB) were analyzed by Sanger sequencing. At the end of each cycle, cells were stored at −80°C as 15% glycerol stocks.
Figure 5.

eMutaT7 promotes rapid continuous directed evolution of model proteins. (A) TEM-1 evolution workflow. (B, C) Evolutionary pathway of TEM-1 for antibiotic resistance. Each number indicates a CTX (B) or CAZ (C) concentration in a culture. Strikethrough indicates no growth. Grey indicates that two cultures out of three did not grow. (D, E) Structure of TEM-1 (PDB, 1axb) with a covalent inhibitor (yellow), the active site (green) and mutations found in the evolved TEM-1 (blue and pink).
MIC determination
Each strain was grown overnight without selection pressure, inoculated at 1/10 000 dilution into fresh LB broth with increasing concentrations of antibiotics in 96-well culture plates (SPL, South Korea), and grown at 37°C with shaking (150 rpm) overnight with aluminum foil cover to prevent too much evaporation. Cell density (OD600) was measured with M200 microplate reader (TECAN, Switzerland).
DegP_A184S evolution
For degP evolution, all the experiments were performed with LB supplemented with 100 μg/ml ampicillin, 35 μg/ml chloramphenicol, 0.2% arabinose and 0.1 mM IPTG. At the end of each cycle, cells were stored at −80°C as 15% glycerol stock. We grew ΔdegP cells carrying pHyo249 (DegP_A184S-expressing plasmid) and pHyo094 (eMutaT7-expressing plasmid) at 37°C. The growth temperature increased by 1°C every 4-hr cycle and no cell death was detected while increasing to 44°C (Figure 6A). After the evolution was over, the plasmids were extracted and retransformed into a fresh cells (cHYO059). Ten colonies were chosen and the cell viability was tested on LB-Agar plates at 44°C, where originally degP_A184S could not grow. Plasmids from five colonies grown at 44°C were analyzed by Sanger sequencing (Supplementary Figure S11).
Figure 6.

Identification of an activating mutation in the DegP protease by eMutaT7. (A) Workflow of the DegP_A184S evolution. Mutations found in five clones are listed and a common mutation, P231L, is represented as red. ssA18V* and ssP21L* are mutations in the signal sequence, which is required for protein translocation to the periplasm and cleaved after translocation. (B) Addition of P231L in DegP_A184S restores the cell viability at high temperature (44°C), while P231L alone reduces cell viability. (C) Basal activities of various DegP mutants (10 μM) were determined by cleavage of the reporter peptide (100 μM). Data are presented as dot plots with averages (short black lines) ± 1 SD (n = 3). (D) Structure of the DegP trimer (PDB, 3otp) with active sites (blue), substrates (green), and the P231 residue (magenta).
The degP_P231L and degP_A184S P231L variants were freshly constructed (pHyo254 and 255 for cell viability test and pHyo256 and 257 for in vitro enzymatic assay) to remove possible mutations that could have occurred in other regions of the evolved plasmid.
To test viability of the ΔdegP cells (cHYO059) expressing degP_A184S variants, cells were grown on selective (44°C) and nonselective (37°C) LB-agar plates for 16 h.
DegP protein preparation and in vitro enzyme activity assay
DegP and its variants (P231L, A184S, P231L/A184S) were expressed and purified as previously described (18). In vitro enzymatic assays were performed as previously described (18,24). In vitro enzymatic assays were performed at 37°C in 50 mM sodium phosphate (pH 8) and 100 mM NaCl. Basal activities of DegP variants (10 μM) were monitored by increase of fluorescence after cleavage of the reporter peptide (100 μM; excitation, 320 nm; emission, 430 nm) using Infinite F200Pro microplate reader (TECAN).
RESULTS AND DISCUSSION
Design of the gene-specific in vivo mutagenesis system
Previously, Petromyzon marinus cytidine deaminase (PmCDA1) was reported to be the most efficient deaminase in E. coli (25) and was successfully used for genome editing (13). Therefore, instead of rAPOBEC1 in MutaT7, we fused PmCDA1 to the N-terminus of T7RNAP. We expected that our mutator, eMutaT7, would mainly generate the C→T mutations on the coding strand, because transcription process exposes the coding strand as a single-stranded DNA that is the main target of PmCDA1 (Figure 1) (26,27). To control mutator production and promote utilization in diverse E. coli strains, we cloned the mutator gene into a plasmid under control of an arabinose-inducible promoter. This plasmid also expresses UGI, an inhibitor of uracil–DNA glycosylase (UNG), which increases mutation efficiency in the ung+ strain (28).
Figure 1.
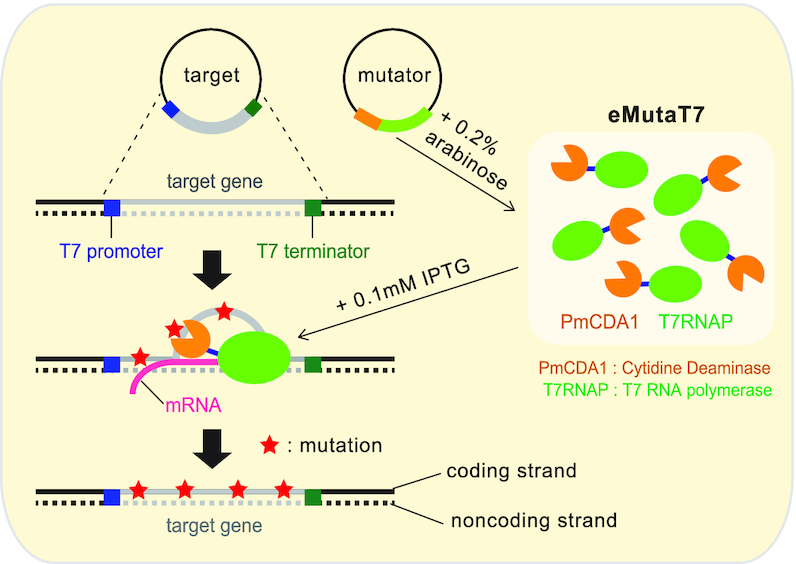
Schematic illustration of the eMutaT7 system. The mutator and the target gene are induced with 0.2% arabinose and 0.1 mM IPTG, respectively. T7 RNA polymerase in the mutator transcribes the target gene between T7 promoter and T7 terminator and, during transcription, exposes a short region of the coding strand as single strand DNA. The cytidine deaminase in the mutator may convert cytosine to thymine mainly on the exposed coding strand.
To sensitively detect the gene-specific mutagenesis, we chose as a model target a conditionally toxic allele of pheS (pheS_A294G), which enables toxic incorporation of p-chloro-phenylalanine (p-Cl-Phe) into the cognate tRNAPhe (29). Because p-Cl-Phe inhibits the growth of the pheS_A294G strain, we reasoned that the mutational inactivation of pheS_A294G could be monitored by counting viable cells in the presence of p-Cl-Phe, while the in vivo mutagenesis is performed without selection pressure in the absence of p-Cl-Phe. In particular, the C→T transition induced by PmCDA1 can generate 13 stop codons (CAG→TAG) throughout the pheS gene as well as other potentially inactivating mutations. To minimize the heterogeneity of the pheS variants in a single cell, we used a low-copy-number plasmid to clone pheS_A294G between T7 promoter and T7 terminator, and induced its expression with Isopropyl β-d-1-thiogalactopyranoside (IPTG).
eMutaT7 rapidly generates mutations in the target gene in vivo
To test the in vivo mutagenesis, we prepared the Δung strain carrying the mutator plasmid and the target plasmid, and started mutagenesis by adding arabinose and IPTG. We also assayed the expression of no mutator (empty plasmid), unfused PmCDA1 and T7RNAP, and T7RNAP only as negative controls. After three cycles of 4-h growth and 100-fold dilution to new media (Figure 2A), cells taken at each cycle were grown on agar plates either with or without p-Cl-Phe to determine the suppression level of the PheS_A294G toxicity (Supplementary Figure S1). Cells with an empty vector express neither T7RNAP nor PheS_A294G, and thus were not included in the suppression experiment. Indeed, cells expressing eMutaT7 showed a notable increase of suppressor frequency over three cycles (12 h), compared to those producing T7RNAP only or unfused proteins (Figure 2B). In particular, the suppressor frequency was almost saturated after three cycles, indicating that most cells have either the inactivated pheS_A294G gene or off-target mutations that suppress the PheS_A294G toxicity. Additional tests showed that the wild-type strain had lower suppressor frequency than the Δung strain, and that cell growth in different setups—Erlenmeyer flask, test tube, and 96-well plate—did not yield significant difference (Supplementary Figure S2).
Next, we determined if eMutaT7 indeed introduces mutations in the target gene. To accumulate mutations, we continued the mutagenesis up to 27 cycles (108 h or 4.5 days) in the absence of p-Cl-Phe. Then, we randomly picked up five clones at various cycles to sequence the target gene by Sanger method. Remarkably, mutations accumulated very quickly: the average mutation rate was ∼3.7 mutations day−1 kb−1 or ∼9.4 × 10−5 mutations base−1 generation−1 in ∼1.1 kb of the target gene region (Figure 2C and Supplementary Figure S3A). This mutation rate is 7–11-fold higher than that of MutaT7 (11), paralleling those of typical directed evolution experiments using in vitro mutagenesis (typically 2–5 mutations gene−1) (30). By contrast, we found no mutation in cells grown without eMutaT7 (Figure 2C). The number of mutations increased with the cycle number, suggesting that eMutaT7 has not been inactivated during mutagenesis cycles. As expected in the scheme (Figure 1), the C→T mutation (208 mutations, 96%) dominated G→A (eight mutations, 4%). These mutations were widely distributed throughout the target gene region (Supplementary Figure S3B).
Because the mutator expression is induced by arabinose, we reasoned that we could control the mutation rate by changing the amount of arabinose. Indeed, the lower amounts of arabinose reduced suppressor frequency, indicating that this in vivo mutagenesis is tunable (Figure 2D). High mutation frequency in the genome may result in lower cell viability, as shown by the 1000-fold reduction of cell number with MP6, which randomly generates mutations in the genome (31) (Figure 2E and Supplementary Figure S4). eMutaT7, however, did not show any notable reduction of cells, indicating that eMutaT7 has no significant toxicity (Figure 2E). To evaluate the level of off-target mutagenesis in the genome, we determined the rifampicin resistance frequency of cells at cycle 27 with or without mutator expression. We found that the rifampicin resistance frequencies were similar between cells grown with and without eMutaT7, suggesting that genome-wide off-target mutagenesis is not frequent with eMutaT7 (Figure 2F). We also tested fusion of PmCDA1 to the C-terminus of T7RNAP, various linker lengths between PmCDA1 and T7RNAP, and T7RNAP mutations that reduce transcription elongation speed (32,33) and possibly increase the time for deamination, but found the original form to be the best among all variants we tested (Supplementary Figure S5).
High-throughput sequencing confirms the gene-specific mutagenesis by eMutaT7
To comprehensively analyze the gene-specific mutagenesis, we sequenced ∼3.3 kb DNA around the target gene from cells taken at cycle 27 by Illumina sequencing. We found that, among all substitution types, only the C→T and G→A mutations were significantly accumulated in the presence of eMutaT7 (1.72% and 0.22%, respectively; Figure 3A). Among three regions—upstream, the target gene, and downstream—the target gene region had the highest average frequency of C→T mutation (3.6%) compared to the upstream (0.27%) and downstream (0.96%) regions (Figure 3B). This result indicates that eMutaT7 preferentially modifies the target gene. eMutaT7 generally raised the mutation frequency in all three regions, and, in particular, the downstream region showed a higher mutation frequency than the upstream region, suggesting leakage of gene targeting. However, we believe that the off-target mutations were not randomly introduced in the genome, as indicated in the low rifampicin resistance frequency (Figure 2F). Interestingly, we found that the G→A mutations were mainly enriched in the ∼280 bp region from the transcription start site (Figure 3C). One possibility is that the longer the newly synthesized RNA, the better it protects the template DNA strand (the non-coding strand) from PmCDA1-mediated deamination. We also found that pyrimidine-C-purine (YCR) is the preferred but not exclusive sequence for eMutaT7 (Supplementary Figure S6).
eMutaT7 introduces mutations faster than MutaT7
Although above results suggest that the mutation rate of eMutaT7 is faster than that of MutaT7, the apparent disparity may rise from different growth conditions, strains, and target genes. To directly compare the mutation rates of eMutaT7 and MutaT7, we constructed two plasmids expressing MutaT7 under control of either the PBAD promoter that is also used for eMutaT7, or the PA1lacO promoter that is adopted by the original MutaT7 system. To efficiently generate G→A mutations as well as C→T, we used the Shoulders group's dual promoter/terminator approach, in which a second pair of T7 promoter/terminator transcribes the target gene in the reverse direction (Figure 4A) (11). We found that this approach suppressed the pheS_A294G toxicity in the same level as the previous approach, but introduced many more G→A mutations (24%; Supplementary Figure S7). We prepared three strains expressing eMutaT7 or MutaT7 from either promoters, and subjected three independent cultures from each strain to nine cycles of 4-hour growth and 100-fold dilution (total 36 h). We randomly picked up two colonies from each culture, and determined the sequence of the target pheS_A294G gene (total six sequences from each strain). We found that PBAD_eMutaT7, PBAD_MutaT7 and PA1lacO_MutaT7 on average generated 6.8, 1.0 and 0.33 mutations, respectively (Figure 4B and Supplementary Figure S8). This result demonstrates that eMutaT7 indeed has the higher mutation rate than MutaT7.
Figure 4.
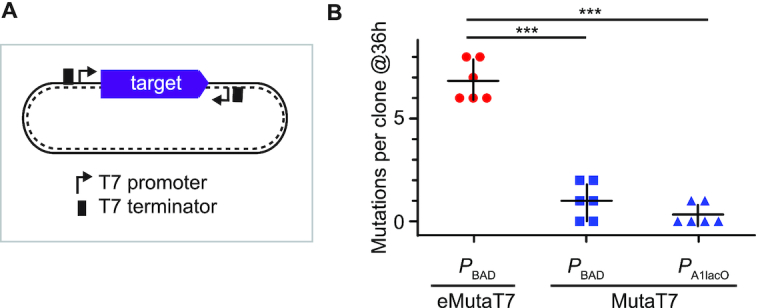
Comparison of eMutaT7 and MutaT7. (A) Dual promoter/terminator construct. (B) eMutaT7 generates more mutations than MutaT7. Cells containing PBAD_eMutaT7, PBAD_MutaT7 and PA1lacO_MutaT7 were subjected to evolution experiments for 36 hours. The number of mutations found in six clones are shown as dot plots with averages (short black lines) ±1 SD. ***P < 0.001 by Student's t-test.
eMutaT7 promotes rapid continuous directed evolution of model proteins
We next tested eMutaT7 for evolution of proteins using the dual promoter/terminator approach. We evolved a class A β-lactamase, TEM-1, for resistance against third-generation cephalosporin antibiotics, cefotaxime (CTX) and ceftazidime (CAZ) (Figure 5A). We tested multiple antibiotic concentrations at the same time, of which the cells grown at the highest concentration were subjected to the next round of growth in fresh media with higher amounts of antibiotics (Figure 5A). The minimum inhibitory concentrations (MICs) increased from 0.06 to 400–800 μg/ml in 32 h of in vivo CDE for CTX, and from 0.2 to 2000–4000 μg/ml in 24 h for CAZ (Figure 5B, C and Supplementary Figure S9). These results present a much faster evolution of antibiotic resistance than that of previous reports using traditional directed evolution methods: 3–5 rounds of in vitro mutagenesis, transformation, and selection for CTX (34,35), and 3–4 rounds for CAZ (36,37). Sanger sequencing of five colonies revealed that all the samples share two previously reported mutations (E102K and G236S for CTX; E102K and R162H for CAZ), which are located near the substrate binding site, and thus might alter the substrate specificity of the enzyme (Figure 5D and E). We also evolved AmpC, a class C β-lactamase, for resistance against carbenicillin (CB) and MICs increased from 16 μg/ml to 2000–4000 μg/ml in 28 h (Supplementary Figure S10). Although most clinical isolates that are resistant to ampicillin contain mutations on the promoter region of ampC (38), our experiment revealed some mutations near the substrate binding site (e.g. G214R/S, D217N and E196K) (Supplementary Figure S10).
Finally, we attempted to find an allosteric mutation of DegP, the major heat-shock protease in bacterial periplasm. The proteolytic activity of DegP is carefully controlled to maintain cellular fitness under heat stress: the lower and higher activity reduce bacterial fitness by misfolded protein stress and hyper-proteolysis, respectively (16). Although many activity-lowering allosteric mutations have been found, only one activating mutation was reported (16). We started with a less active variant, DegP_A184S, which reduced the cell viability at high temperature, and evolved new variants that support better cellular growth at high temperature, by increasing temperature stepwise during growth cycles (Figure 6A). We expected that the new variants should have a compensating mutation that increases the proteolytic activity of DegP_A184S. Sanger sequencing of the degP gene from five colonies revealed P231L as the only common mutation (Figure 6A and Supplementary Figure S11). Indeed, the addition of P231L in DegP_A184S restored the cell viability at high temperature (43°C), and P231L alone reduced cell viability (Figure 6B). Also the in vitro enzymatic assay showed that P231L raises the basal activity, and the addition of P231L increased the basal activity of DegP_A184S to the level of wild-type DegP, indicating the rebalancing of proteolytic activity (Figure 6C). The P231 residue is located near the center of the trimeric DegP without direct contact with the substrate, suggesting that P231L allosterically increases activity (Figure 6D). Collectively, we demonstrated that our mutator allows rapid in vivo directed evolution of a target protein, given the continuous selection condition.
In conclusion, we demonstrate that the orthogonal RNA polymerase fused to a more efficient cytidine deaminase promotes rapid continuous directed evolution of proteins via gene-specific in vivo mutagenesis. To our knowledge, eMutaT7 has the highest mutation rate among the targeted in vivo mutagenesis methods for CDE of proteins. In theory, a 100 mL culture with a constant OD600 ∼0.1 (∼1010 cells) can generate 1010 independent variants of a 1-kb gene every 6 h with eMutaT7. Also, our method is (i) simple: two plasmids expressing an mutator and a target are sufficient for gene-specific CDE and any laboratory with a basic molecular biology setup can use this method; (ii) expandable: multiple genes under control of T7 promoter can be targeted and (iii) tunable: mutation rates can be controlled by the arabinose concentration. A major limitation of our method is the narrow mutational spectrum in which the C→T and G→A mutations dominate. However, we believe that fusion of different DNA-modifying enzymes to T7RNAP will help expand the substitution types, while maintaining specificity and speed of the in vivo mutagenesis. Alternatively, gene libraries constructed by in vitro mutagenesis can be a starting point of CDE, in which our in vivo mutagenesis method explores an additional layer of sequence space.
DATA AVAILABILITY
Illumina sequencing data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-9677.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Inseok Song for assistance in calculating statistics. We also thank Kyuhyun Kim, Sunhee Bae, Ga-eul Eom, Daege Seo, Younghyun Kim, Chanwoo Lee, Hyunjin Cho and Hyunbin Lee for helpful discussions. We thank Nam Ki Lee, Kayeong Lim and Sanggil Kim for providing purified DNAs. We thank Kayeong Lim, Euihwan Jeong and Sejong Choi for help in western blotting experiments.
Contributor Information
Hyojin Park, Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, South Korea.
Seokhee Kim, Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, South Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [2020R1F1A1054191, 2020R1A5A1019023]. Funding for open access charge: Seoul National University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Zeymer C., Hilvert D.. Directed evolution of protein catalysts. Annu. Rev. Biochem. 2018; 87:131–157. [DOI] [PubMed] [Google Scholar]
- 2. Morrison M.S., Podracky C.J., Liu D.R.. The developing toolkit of continuous directed evolution. Nat. Chem. Biol. 2020; 16:610–619. [DOI] [PubMed] [Google Scholar]
- 3. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hess G.T., Fresard L., Han K., Lee C.H., Li A., Cimprich K.A., Montgomery S.B., Bassik M.C.. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods. 2016; 13:1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Halperin S.O., Tou C.J., Wong E.B., Modavi C., Schaffer D.V., Dueber J.E.. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature. 2018; 560:248–252. [DOI] [PubMed] [Google Scholar]
- 6. Ravikumar A., Arzumanyan G.A., Obadi M.K.A., Javanpour A.A., Liu C.C.. Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell. 2018; 175:1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camps M., Loeb L.A.. Use of Pol I-deficient E. coli for functional complementation of DNA polymerase. Methods Mol. Biol. 2003; 230:11–18. [DOI] [PubMed] [Google Scholar]
- 8. Esvelt K.M., Carlson J.C., Liu D.R.. A system for the continuous directed evolution of biomolecules. Nature. 2011; 472:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berman C.M., Papa L.J. 3rd, Hendel S.J., Moore C.L., Suen P.H., Weickhardt A.F., Doan N.D., Kumar C.M., Uil T.G., Butty V.L.et al.. An adaptable platform for directed evolution in human cells. J. Am. Chem. Soc. 2018; 140:18093–18103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. English J.G., Olsen R.H.J., Lansu K., Patel M., White K., Cockrell A.S., Singh D., Strachan R.T., Wacker D., Roth B.L.. VEGAS as a platform for facile directed evolution in mammalian cells. Cell. 2019; 178:748–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moore C.L., Papa L.J. 3rd, Shoulders M.D.. A processive protein chimera introduces mutations across defined DNA regions in vivo. J. Am. Chem. Soc. 2018; 140:11560–11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen H., Liu S., Padula S., Lesman D., Griswold K., Lin A., Zhao T., Marshall J.L., Chen F.. Efficient, continuous mutagenesis in human cells using a pseudo-random DNA editor. Nat. Biotechnol. 2019; 38:165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y.et al.. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016; 353:aaf8729. [DOI] [PubMed] [Google Scholar]
- 14. Ochman H., Gerber A.S., Hartl D.L.. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988; 120:621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang S., Kim S., Rim Lim Y., Kim C., An H.J., Kim J.H., Sung J., Lee N.K.. Contribution of RNA polymerase concentration variation to protein expression noise. Nat. Commun. 2014; 5:4761. [DOI] [PubMed] [Google Scholar]
- 16. Kim S., Sauer R.T.. Distinct regulatory mechanisms balance DegP proteolysis to maintain cellular fitness during heat stress. Genes Dev. 2014; 28:902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim S., Sauer R.T.. Cage assembly of DegP protease is not required for substrate-dependent regulation of proteolytic activity or high-temperature cell survival. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:7263–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S., Grant R.A., Sauer R.T.. Covalent linkage of distinct substrate degrons controls assembly and disassembly of DegP proteolytic cages. Cell. 2011; 145:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Melancon C.E. 3rd, Schultz P.G.. One plasmid selection system for the rapid evolution of aminoacyl-tRNA synthetases. Bioorg. Med. Chem. Lett. 2009; 19:3845–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis J.H., Baker T.A., Sauer R.T.. Small-molecule control of protein degradation using split adaptors. ACS Chem. Biol. 2011; 6:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park H., Kim Y.T., Choi C., Kim S.. Tripodal lipoprotein variants with C-terminal hydrophobic residues allosterically modulate activity of the DegP protease. J. Mol. Biol. 2017; 429:3090–3101. [DOI] [PubMed] [Google Scholar]
- 25. Lada A.G., Krick C.F., Kozmin S.G., Mayorov V.I., Karpova T.S., Rogozin I.B., Pavlov Y.I.. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry (Mosc). 2011; 76:131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durniak K.J., Bailey S., Steitz T.A.. The structure of a transcribing T7 RNA polymerase in transition from initiation to elongation. Science. 2008; 322:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salter J.D., Bennett R.P., Smith H.C.. The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci. 2016; 41:578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mol C.D., Arvai A.S., Sanderson R.J., Slupphaug G., Kavli B., Krokan H.E., Mosbaugh D.W., Tainer J.A.. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995; 82:701–708. [DOI] [PubMed] [Google Scholar]
- 29. Kast P., Hennecke H.. Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J. Mol. Biol. 1991; 222:99–124. [DOI] [PubMed] [Google Scholar]
- 30. Cirino P.C., Mayer K.M., Umeno D.. Generating mutant libraries using error-prone PCR. Methods Mol. Biol. 2003; 231:3–9. [DOI] [PubMed] [Google Scholar]
- 31. Badran A.H., Liu D.R.. Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun. 2015; 6:8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonner G., Lafer E.M., Sousa R.. Characterization of a set of T7 RNA polymerase active site mutants. J. Biol. Chem. 1994; 269:25120–25128. [PubMed] [Google Scholar]
- 33. Radzicka A., Wolfenden R.. A proficient enzyme. Science. 1995; 267:90–93. [DOI] [PubMed] [Google Scholar]
- 34. Stemmer W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994; 370:389–391. [DOI] [PubMed] [Google Scholar]
- 35. Zaccolo M., Gherardi E.. The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 beta-lactamase. J. Mol. Biol. 1999; 285:775–783. [DOI] [PubMed] [Google Scholar]
- 36. Barlow M., Hall B.G.. Experimental prediction of the natural evolution of antibiotic resistance. Genetics. 2003; 163:1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujii R., Kitaoka M., Hayashi K.. RAISE: a simple and novel method of generating random insertion and deletion mutations. Nucleic Acids Res. 2006; 34:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Olsson O., Bergstrom S., Normark S.. Identification of a novel ampC beta-lactamase promoter in a clinical isolate of Escherichia coli. EMBO J. 1982; 1:1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina sequencing data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-9677.