

Abstract
DNA repair pathways are essential to maintain the integrity of the genome and prevent cell death and tumourigenesis. Here, we show that the Barrier-to-Autointegration Factor (Banf1) protein has a role in the repair of DNA double-strand breaks. Banf1 is characterized as a nuclear envelope protein and mutations in Banf1 are associated with the severe premature aging syndrome, Néstor–Guillermo Progeria Syndrome. We have previously shown that Banf1 directly regulates the activity of PARP1 in the repair of oxidative DNA lesions. Here, we show that Banf1 also has a role in modulating DNA double-strand break repair through regulation of the DNA-dependent Protein Kinase catalytic subunit, DNA-PKcs. Specifically, we demonstrate that Banf1 relocalizes from the nuclear envelope to sites of DNA double-strand breaks. We also show that Banf1 can bind to and directly inhibit the activity of DNA-PKcs. Supporting this, cellular depletion of Banf1 leads to an increase in non-homologous end-joining and a decrease in homologous recombination, which our data suggest is likely due to unrestrained DNA-PKcs activity. Overall, this study identifies how Banf1 regulates double-strand break repair pathway choice by modulating DNA-PKcs activity to control genome stability within the cell.
INTRODUCTION
DNA double-strand breaks (DSBs) are viewed as one of the most cytotoxic lesions to damage the genome and as such the inability to repair these breaks may lead to genomic instability or cell death. Consequently, this may in turn lead to cellular transformation, promoting tumourigenesis (1). It is therefore essential that DSBs in human cells are detected, signalled and repaired efficiently. Cells contain a vast armoury of proteins that function in signalling networks to detect and signal DSBs, as well as proteins that function in a highly orchestrated manner to repair these breaks directly. There are two main pathways available for the repair of these cytotoxic DSBs. The homologous recombination (HR) pathway repairs DSBs that specifically occur in the S or G2 phases of the cell cycle when a sister chromatid is available (2). The second pathway, non-homologous end joining (NHEJ) is the predominant pathway and may be used to repair DSBs in any phase of the cell cycle. NHEJ essentially results in the re-ligation of the break site with no or limited DNA processing. If processing does occur during NHEJ, this can lead to loss of genetic information. The major effector of the NHEJ process is the DNA-PK complex comprising of the phosphatidylinositol-3-kinase like kinase (PIKK) family member, DNA-dependent Kinase catalytic subunit (DNA-PKcs), and the Ku70/80 heterodimer (3). Once a DSB is induced it is recognized by the Ku70/80 heterodimer and DNA-PKcs is also recruited and stabilized at the DSB. The DNA ends are then stabilized and DNA end processing occurs if required. The DNA ends are ligated back together by several proteins, including DNA Ligase IV and XRCC4. Multiple layers of regulation of DSB repair pathway choice have been proposed, including the DNA break complexity, the stage of the cell cycle, chromatin complexity, regulation of resection and modulation of ATM, DNA-PKcs, Ku70/80, 53BP1 and BRCA1 (reviewed in (4)).
Barrier-to-autointegration factor (Banf1/BAF) is a small non-specific DNA binding protein that is conserved amongst multicellular eukaryotes. Banf1, functions as a dimer, and binds to the phosphate backbone of the DNA, compacting the DNA in a looping process (5). The loss of Banf1 results in a loss of nuclear envelope integrity and aberrant chromatin organization. Our previous work characterized the role of Banf1 in regulating PARP1 activity following oxidative stress (6). Specifically, induction of oxidative lesions promotes the binding of Banf1 to PARP1, a critical NAD+-dependent DNA repair protein. In cells with increased Banf1 this leads to the inhibition of PARP1 auto-ADP-ribosylation and defective repair of oxidative lesions.
A single point-mutation in the N-terminal domain of Banf1 is associated with the severe premature aging syndrome, Néstor–Guillermo Progeria Syndrome (7–9). Premature aging is intrinsically linked with the genome stability pathways (10,11). Consistent with this, cells from NGPS patients exhibit defective PARP1 activity and impaired repair of oxidative lesions, supporting a model whereby Banf1 is crucial to reset oxidative-stress-induced PARP1 activity (6).
In this study, we demonstrate that Banf1 also functions in DNA DSB repair. Banf1 responds to the induction of DSBs by relocating from the nuclear envelope to the break sites. Banf1 binds to DNA-PK and inhibits its activity, and subsequently NHEJ, possibly to allow HR to occur at a subset of breaks. Here, we establish a new Banf1-dependent mechanism regulating the pathway choice between HR and NHEJ.
MATERIALS AND METHODS
Reagents
Chemical reagents
All chemical reagents were purchased from Sigma.
Antibodies
The antibodies used were as follows: anti-Banf1 N-terminus (SAB1409950, Sigma-Aldrich, 1:1000 for WB and 1:500 for IF), anti-Banf1 C-terminus (PRS40170604, Sigma-Aldrich, 1:1000 for WB and 1:500 for IF), anti-Emerin (5430, Cell Signalling Technology, 1:500 for IF), anti-Flag M2 Antibody (F3165, Sigma-Aldrich, 1:1000 for WB and 1:300 for IF), anti-γ-Tubulin (T6557, Sigma-Aldrich, 1:2000 for WB), anti-H3 (4499, Cell Signaling Technology, 1:2000 for WB), anti-β-actin (612656, BD Biosciences, 1:2000 for WB), anti-P-ATM S1981 (5883, Cell Signaling Technology 1:1000), anti-Mre11 (HPA002691, Sigma-Aldrich, 1:1000 for WB), anti-MDC1 (PLA0016, Sigma-Aldrich, 1:1000 for IF), anti-γ-H2AX (ab26350, abcam, 1:1000 for WB; 9718, Cell Signalling Technology, 1:1000 for IF, ab81299, abcam for ChIP), anti-p-DNA-PK S2056 (Abcam, ab124918, 1:1000 wb), anti-DNA-PKcs (12311, Cell Signaling Technology 1:1000 for western blot and ab70205 1:200 for IF), anti-Ligase IV (ab193353, Abcam, 1:1000), anti-Ku70 (ab92450, Abcam, 1:1000) and anti-nucleolin (14574, Abcam, 1:1000). Fluorescent secondary antibodies used were: Donkey anti-Mouse 800 nm (LiCor; IRDye 800CW 926–32212, 1:5000 for WB), Donkey anti-Rabbit (LiCor; IRDye 680LT 926–28023, 1:5000 for WB) and Alexa Fluor 488 (Cat# A32766, Molecular Probes, 1:200 for IF) and 594 (Cat# A32754, Molecular Probes, 1:200 for IF).
Biological resources
Cell lines
The U2OS and HEK293T cells were obtained from CellBank Australia (catalogue numbers 92022711 and 85120602, respectively). U2OS cells were grown in RPMI media, supplemented with 10% FCS, HEK293T cells were grown in DMEM media, supplemented with 10% FCS. Cell lines were grown at 37°C, 5% CO2 and at atmospheric O2, unless otherwise stated.
Constructs
The Flag-Banf1 construct was synthesized by Genscript in the pcDNA3.1+N-DYK vector in the BamHI-XhoI cloning sites. These constructs were sequenced using the CMV primer (5′-CGCAAATGGGCGGTAGGCGTG-3′). The His-Banf1 was synthesized by Genscript in the pET-28a(+) vector in the NdeI–XhoI cloning sites. These constructs were sequenced using the T7 primer (5′-TAATACGACTCACTATAGG-3′).
siRNA
Control and Banf1 pooled esiRNA were purchased from Sigma. Control and Banf1 siRNA were purchased from GenePharma (Banf1, GGGUUUUGACAAGGCCUAUdTdT). Control and Banf1 Silencer Select siRNA were purchased from ThermoFisher. Cells were typically assayed 72 h after transfection.
MATERIALS AND METHODS
Transfections
All DNA constructs were transfected using Fugene HD (Promega) as per manufacturer's instructions. RNAiMax (Invitrogen) was used to transfect esiRNA and siRNA as per manufacturer's instructions.
Immunoblotting
Cells were lysed [lysis buffer: 20 mM HEPES pH7.5, 250 mM KCl, 5% glycerol, 10 mM MgCl2, 0.5% Triton X-100, protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Cell Signaling)] and sonicated. Lysates were cleared by centrifugation. Typically, 30 μg of protein lysate was separated on a 4–12% SDS-PAGE gel (Invitrogen) blocked in Odyssey buffer (LiCor Biosciences) and immunoblotted with the indicated antibodies. Immunoblots were imaged using an Odyssey infrared imaging system (LiCor).
Immunoprecipitation
Cells were treated as stated then lysed using Pierce lysis buffer (25 mM Tris•HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) and sonicated as for immunoblotting. Lysates were transferred to a new tube and incubated with Pierce™ Universal Nuclease for Cell Lysis to digest DNA. Digested lysates were then incubated with the indicated antibodies or equivalent amount of IgG for the relevant species for 1 h on rotation at 4°C. Protein A or G magnetic Dynabeads (Thermofisher) were then added to the tubes and were incubated for 1 h on rotation at 4°C. Beads were then washed five times in lysis buffer and boiled in 2× SDS loading dye before immunoblotting.
Recombinant Banf1 purification and direct protein interactions
The method of Banf1 recombinant protein purification was as in (6). For Banf1:DNA-PK interactions, purified DNA-PK (Promega) was incubated with wild-type Banf1 for 30 min in lysis buffer (50 mM Tris–HCl, pH 7.8, 10 mM MgCl2 and 1 mM DTT) at room temperature. Banf1 was then immunoprecipitated and immunoblotted using Banf1 antibodies as above.
Mass spectrometry
U2OS cells were transfected with Flag vector and Flag-Banf1 and prepared as per the immunoprecipitation protocol. On-bead tryptic digest produced peptides that were subsequently cleaned, desalted and concentrated using SCX staged tips. Samples were then separated using LC/MS-MS on an Orbitrap Q Exactive plus HF.
The dried peptides were resuspended and analysed using ThermoFisher Scientific Ultimate 3000 RSLC nano ultra high pressure liquid chromatography system (nUHPLC) interfaced to hybrid quadrupole-Orbitrap Q Exactive Plus mass spectrometer. Acidified peptides were loaded onto Acclaim Pepmap trap (300 μm I.D. × 5 mm, C18, 5um particle size, 100 Å) at 10 μl/min and separated in-line using a pre-equilibrated analytical column (ThermoFisher Scientific, Easy-Spray C18, 75 μm I.D. × 500 mm, 2 μm particle size, 100 Å) at a flow-rate of 0.25 μl/min and temperature of 40°C. The Q Exactive Plus was operated in a data-dependent mode to automatically switch between Orbitrap-MS and Orbitrap-MS/MS acquisition. Survey full scan MS spectra (from m/z 350–1400) were acquired in the Orbitrap with resolution r 70 000 at m/z 400. Depending on the signal intensity, up to 10 most intense ions were sequentially isolated, fragmented in the HCD cell and recorded in the orbitrap at a resolution of 17 500. For accurate mass measurements the lock mass option was enabled in MS mode and the polydimethylcyclosiloxane ions (protonated Si(CH3)2O6; m/z 445.120025 from ambient air) were used for real time internal recalibration.
Protein identification and label-free quantification were performed using MaxQuant (version 1.6.8.0) (12). MaxQuant was used to extract peak lists from the Xcalibur .raw files (ThermoFisher Scientific, Germany) and the embedded database search engine Andromeda (13) was used to assign peptide sequences to the fragmentation spectra. The database searched consisted of the reference proteome for Homo Sapiens (20 369 canonical non-redundant sequences downloaded from www.uniprot.org on 5 September 2019), reversed sequences and the MaxQuant contaminant database. For protein identification, the PSM and protein FDRs were set to 0.01 and both unique and razor peptides were used for label-free quantification. For this dataset a minimum of two peptides were required for identification of a protein.
Quantitative interaction proteomic analysis was performed on MaxQuant-processed data by Perseus (version 1.6.8.0). Normalized protein group intensities were filtered for contaminants and reverse hits. Statistical significance by comparing the bait pull-down to its control replicates was determined on the basis of a hyperbolic curve threshold of s0 = 2.5 and FDR 0.001 (Class A interactors) and s0 = 2.5 and FDR 0.005 (Class B interactors).
DNA-PK activity assay
The activity of DNA-PK (DNA-PKcs and Ku70/80) was determined, according to manufacturer's instructions, using a DNA-PK kinase enzyme system (V4106, Promega) and ADP-Glo assay (V9101, Promega), using increasing doses of recombinant Banf1 (100, 200 and 500 ng), in triplicate. The Banf1-suspension buffer was also added to the DNA-PK alone sample to control for potential buffer-induced inhibition of DNA-PK. Luminescence was measured on an EnSpire 2300 Multi-label reader (Perkin Elmer).
Immunofluorescence
Cells were seeded the day before siRNA transfection. Following siRNA transfection cells were allowed to grow for 72 h before treatment or mock-treatment with the indicated DNA damaging agent. After treatment cells were treated with an extraction buffer (20 mM HEPES, 20 mM NaCl, 5 mM MgCl2, 0.5% IGEPAL), to remove soluble proteins to enable study of chromatin-bound proteins (14) for 5 min before fixation in 4% PFA. Cells were permeabilized with 0.2% Triton X-100 for 5 min and blocked in 3% BSA for 30 min. Cells were incubated with indicated primary antibodies and Alexa-conjugated secondary antibodies for 1 h each at room temperature. In order to visualize the colocalization between Banf1 and DNA-PK cells were treated as above but with the addition of 0.3 mg/ml RNase A to the extraction buffer (15). Cells were stained with DAPI, before imaging on a Delta Vision PDV microscope, 60×/1.42 or 100×/1.42 oil objective (Applied Precision, Inc). All immunofluorescence figures were assembled using ImageJ. High content imaging was performed using the InCell Analyzer 6500 Imaging System (GE Healthcare Life Sciences). Nuclear staining intensity was analysed using the InCell Investigator software (GE Healthcare Life Sciences) with a minimum of 500 nuclei quantified per each independent experiment and the results shown represent the mean and S.D. of three independent experiments. The average Pearson coefficient, r, was calculated from the analysis of 10 cells in each condition using the ImageJ Coloc 2 analysis software.
Nuclear envelope quantification
Immunofluorescence was performed as previously using anti-Banf1 antibodies and images were taken using a DeltaVision PDV microscope (6). To assess the proportion of cells with Banf1 at the nuclear envelope, cells were treated with 6 Gy ionizing radiation and 50 cells in each condition were assessed for the localization of Banf1 using a Delta Vision PDV microscope.
Homologous recombination and non-homologous end-joining assays
For the integrated homologous recombination and non-homologous end-joining assays; the pLCN DSB Repair Reporter (DRR) (Addgene plasmid #98895; http://n2t.net/addgene:98895; RRID:Addgene_98895) and pCAGGS DRR mCherry Donor EF1a BFP were a gift from Jan Karlseder (Addgene plasmid #98896; http://n2t.net/addgene:98896; RRID:Addgene_98896). Stable U2OS cells containing the pLCN DSB Repair Reporter were generated and the assay carried out as in (16). Briefly, the stable U2OS cells were transfected with the indicated siRNAs 48 h before transfection with Isce1 and an exogenous donor for HR (pCAGGS DRR mCherry Donor EF1a BFP). Forty-eight hours after transfection cells were processed for FACS to detect BFP (as a control for transfection efficiency) GFP or mCherry expression. Repair by NHEJ or HR leads to GFP or mCherry expression, respectively.
The homologous recombination DR-GFP assay was carried out as previously (17–19). Briefly, MCF7 stable cell lines were generated containing the DR-GFP reporter construct (19). Briefly, the stable MCF7 cells were transfected with the indicated siRNAs, 48 h before transfection with Isce1 (and a separate GFP construct to control for transfection efficiency). Forty-eight hours after transfection cells were processed for FACS to detect GFP positivity.
Subcellular fractionation
To detect binding of proteins to chromatin, subcellular fractionation was carried out using a Subcellular Protein Fractionation Kit for Cultured Cells, according to the manufacturer's instructions (Thermo Fisher Scientific).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (17,20). Briefly, MCF7 cells stably expressing DRGFP were transfected with Flag or Flag-Banf1. 24-post Flag transfection cells were transfected with Isce1 to induce a DSB. Immunoprecipitations with Flag and γH2AX antibodies were performed. Real-time PCR on ChIP samples used primers directed at 94–378 nucleotides from the DSB. The enrichment of Banf1 after induction of the DSB was compared with that of Flag control.
Comet assay
The neutral comet assay was employed to measure DSBs following IR treatment, as performed previously (21). Briefly, U2OS cells were treated or mock-treated with IR (6 Gy). Immediately after IR, or 0.5 or 4 h post-IR, cells were embedded in agarose, lysed and subjected to electrophoresis, according to the Trevigen neutral comet assay protocol, with a few variations. The lysis buffer used contained: 2.5 M NaCl, 100 mM EDTA, 10 mM Tris (pH 10), 1% Triton X-100. The electrophoresis was carried out with TBE buffer. Single cells were stained with Sybr Green I (Invitrogen) and at least 50 randomly selected cells per condition were analysed using imageJ software and the Olive tail moment calculated (22).
Colony-forming assays
U2OS cells were transfected with control or Banf1 siRNA. Forty-eight hours after transfection 500 cells were seeded into wells of a six-well plates. Cells were treated or mock-treated with the 0–6 Gy IR and incubated for 10 days. Colonies were stained with 4% methylene blue in methanol, manually counted and normalized back to the number of colonies in the untreated wells (23).
Statistical analyses
Histograms represent the average value ± standard error of the mean. Statistical analysis of the results was made using Prism software (GraphPad). T-test (two-tailed) were used for statistical analysis. Data are presented as means and standard deviation (SD) from ≥3 independent experiments (unless otherwise stated). Statistical significance is represented by *P-value < 0.05; **P-value < 0.01; ***P-value < 0.001; ****P-value < 0.0001.
Ethics
All experimental procedures were approved by the Queensland University of Technology; Human Research Ethics Committee (approval numbers 1700000940 and 1900000269).
RESULTS
Banf1 rapidly localizes to sites of DSBs
Banf1 mutation results in a severe premature aging syndrome, Néstor–Guillermo Progeria Syndrome (NGPS) (9). Premature aging syndromes are often associated with proteins that function directly or indirectly to control genome stability via DNA double-strand break repair, such as Werner Syndrome Helicase (WRN) and Lamin A (LMNA), which lead to Werner Syndrome and Hutchinson-Gilford Progeria, respectively (24–27). Given that we have previously shown that Banf1 responds to oxidative stress and single-strand breaks (6), we next examined whether Banf1 also responds to DNA double-strand breaks (DSBs).
Following DNA damage induction, the localization of repair proteins to sites of damage can be observed as nuclear foci. To determine if Banf1 may also have additional roles in maintenance of genome stability, we initially treated U2OS cells with ionizing radiation (IR) to induce DNA DSBs. In unperturbed cells we and others have previously shown that Banf1 is predominantly localized to the nuclear envelope (5–7). Using an extraction buffer to remove the soluble Banf1 protein from the cell before fixation, we visualized that within 1 h of IR treatment (6Gy), Banf1 relocalized from the nuclear envelope (as marked by Emerin antibody staining) in over 95% of cells, suggesting Banf1 mobilization was independent of cell cycle phase (Figure 1A, B, Supplementary Figure S1, for representative images of Banf1 and Emerin staining without pre-extraction. Supplementary Figure S2 shows Banf1 antibody specificity). This is consistent with the DSB-induced translocation of repair proteins, to the break site forming distinct chromatin bound foci that can be observed by immunofluorescence. Quantification of Banf1 foci showed an ∼3-fold increase within 15 min of IR suggesting the mobilization of Banf1 from the nuclear envelope was an early event in the response to IR-induced DNA damage (Figure 1C).
Figure 1.

Banf1 responds to ionizing radiation. (A) Banf1 relocalizes from the nuclear envelope following ionizing radiation in U2OS cells. Representative cells stained with the indicated antibodies at the indicated times post 6 Gy IR are shown. (B) Nuclear envelope localization of Banf1 (from a) was manually quantified using images taken on a Delta Vision PDV microscope. (C) Banf1 forms nuclear foci following IR. The number of Banf1 foci in U2OS cells treated as in a, were analysed via an InCell Analyser. (D) Banf1 protein levels stabilize following IR treatment. U2OS cells were transfected with control or Banf1 siRNA, treated with 6 Gy IR and whole cell lysates prepared at the indicated times post-IR. Lysates were immunoblotted with the indicated antibodies. P-ATM S1981 was used as a marker for DNA damage induction. (E) Banf1 is recruited to chromatin. U2OS cells were transfected with control or Banf1 siRNA, treated with 6 Gy IR and cells processed for cellular fractionation at the indicated times post-IR. The chromatin fraction was immunoblotted with the indicated antibodies. The accumulation of MRE11 was used as a marker for DNA damage and chromatin. Immunofluorescence scale bars represents 10 μm. Histogram data shown represent the mean and S.D. of three independent experiments and t-test was used for statistical analysis *P<0.05, ***P <0.001.
To further characterize Banf1 function we next examined Banf1 protein levels via immunoblot. Total Banf1 protein levels also showed a modest but significant increase from 0.5 h after IR treatment (Figure 1D, Supplementary Figure S3). In support of the immunofluorescence, cellular fractionations showed that prior to IR, Banf1 was localized predominantly to the chromatin fraction, but could also be detected at low levels in the cytoplasmic and cytoskeletal fractions (Supplementary Figure S2). However following IR, Banf1 was stabilized on the chromatin within 1 h of IR treatment (Figure 1E and Supplementary Figure S4).
Since Banf1 relocalizes to chromatin after DNA damage we next sought to identify whether it is recruited to sites of DNA damage. One of the most robust responses to DNA DSBs is the phosphorylation of H2AX on serine 139 (known as γ-H2AX following its phosphorylation) in the chromatin surrounding the break. In cells treated with 2 Gy IR, 47% of γ-H2AX foci overlapped with Banf1 foci, suggesting that Banf1 is recruited to the sites of DSBs (Figure 2A). The Pearson coefficient r values were 0.02, prior to IR and 0.26 at 1 h post-IR. It should be noted that there were more Banf1 foci than γ-H2AX foci 1 h following IR and it is possible that these represent sites of IR-induced DNA single-strand breaks as we have characterized previously (6). This colocalization of Banf1 with γ-H2AX, and another DNA repair protein MDC1, could also be observed in cells treated with 6 Gy IR (Supplementary Figure S5a, b). Similarly to γ-H2AX, 42% of MDC1 foci overlap with Banf1 foci at 1 h post-IR. To further confirm the presence of Banf1 at DNA DSBs we also utilized a method using a MCF7 cell line with a stably integrated unique Isce1 endonuclease site. When the Isce1 endonuclease is expressed in the cells it induces a single DSB and chromatin immunoprecipitation (ChIP) can be used to study the recruitment of DNA repair proteins to the sites of damage (17). Using this method we found that similarly to γ-H2AX (28), Flag-Banf1 was bound close to the DNA DSB and could be detected 94–378 bp from the site of the ISCE-1-induced DSB, (Figure 2B), supporting that Banf1 localizes directly to the sites of DSBs. In contrast, Banf1 did not bind to DNA sequences 330–643 bp from the DSB site, supporting that it binds specifically to the region directly surrounding the break, within 94–378 bp.
Figure 2.

Banf1 localizes to DNA double-strand breaks. (A) Representative cells stained with the indicated antibodies at the indicated times post 2 Gy IR are shown. (B) ChIP analysis of Banf1 on a unique DSB induced by I-Sce1. Quantitative PCR was performed with primers at 94–378 bp from the break site in MCF7 DRGFP cells transfected with FLAG and Banf1-FLAG, ± I-Sce1. The enrichment of Banf1 after induction of a DSB was compared to the FLAG + I-Sce1 sample. γ-H2AX was also included as a control. (C) γ-H2AX foci in Banf1-depleted cells. U2OS cells were transfected with control or Banf1 siRNA. Seventy-two hours after transfection cells were treated with 2 Gy IR and fixed and stained with the indicated antibodies at the indicated time points. Representative cells are shown. (D) Cells from c were analysed for the number of γ-H2AX foci per cell using a Incell Analyser at the indicated times post-IR. Data points represent the γ-H2AX foci/nuclei from a minimum of 500 nuclei. Data shown show the mean and S.D. and are representative of three independent experiments. (E) Depletion of Banf1 promotes repair of IR-induced DNA damage. Neutral comet assay showing the relative Olive tail moment in U2OS control cells and cells depleted of Banf1 with siRNA at the indicated time points post-IR. +IR represents immediately following IR. Data points represent the Olive tail moment from a minimum of 100 cells. Data shown show the mean and S.D. of two independent experiments. Unless otherwise stated, data shown represent the mean and S.D. of three independent experiments and t-test was used for statistical analysis: ***P <0.001, ****P <0.0001. Immunofluorescence scale bars represent 10 μm.
We next examined the effect of Banf1 depletion on γ-H2AX formation to measure DSB repair kinetics (29). When Banf1 was depleted using siRNA we found that γ-H2AX foci were significantly decreased at 0.5 h and 1 h following 2 Gy IR (Figure 2C, D). The reduced numbers of γ-H2AX foci could represent down regulation of the regulatory pathways involved in its phosphorylation and dephosphorylation, including the catalytic activity of the ATM and DNA-PK kinases responsible for its phosphorylation. There is also the possibility that the DSBs are being repaired more quickly in Banf1-deficient cells. To address this possibility we next carried out neutral comet assays to measure the induction and repair of DSBs. Here, we found that Banf1-depleted cells had similar length comet tail moments to control cells prior to and directly after IR, representing a similar level of DNA damage induction to control cells (Figure 2E, Supplementary Figure S6). However, 0.5 and 4 h post-IR treatment Banf1-depleted cells had significantly shorter comet tails, compared to control cells. This is consistent with the fewer γ-H2AX foci observed in Banf1-deficient cells 0.5 h after IR in Figure 2C, D, suggesting that DNA DSBs are repaired more quickly in the absence of Banf1.
Depletion of Banf1 leads to decreased HR
Since we have shown that Banf1 responds to DNA DSBs and the absence of Banf1 leads to faster repair of DSBs, this raised the possibility that Banf1-deficient cells may utilize the faster DSB repair pathway of NHEJ, rather than HR to repair DSBs. To address this we next analysed the type of repair of DSBs in Banf1-depleted cells, using previously described cell reporter assays (Figure 3A) (16,20) to measure DSB repair via the two DSB repair pathways, NHEJ and HR. We found that in Banf1-depleted cells, HR was significantly reduced, whereas NHEJ was significantly increased (Figure 3B). BRCA1 depletion was also included as a positive control for disruption of HR. This reduction in HR was also observed using siRNA targeting a different region of Banf1 mRNA (Figure 3C) and this effect was rescued by expression of wild-type Banf1 (Figure 3D). Use of another HR assay using the DR-GFP system, also demonstrated a HR defect in Banf1-depleted cells (Supplementary Figure S7, (17–19)). Since HR usually only takes place in the S and G2 phases of the cell cycle, we next examined the effect of Banf1-depletion on the cell cycle. In Banf1-deficient cells, the cell cycle was confirmed to not be significantly altered in the absence of Banf1, suggesting that this was not the cause of the HR defect observed in these cells (Supplementary Figure S8).
Figure 3.

Banf1 depletion inhibits HR and promotes NHEJ. (A) Schematic showing the possible outcomes of the HR/NHEJ reporter assay. (B) Depletion of Banf1 results in decreased HR and increased NHEJ. U2OS cells stably expressing a HR/NHEJ reporter were depleted of Banf1 using siRNA. Cells were transfected with an Isce1-expressing plasmid and a donor plasmid and GFP and mCherry positive cells were detected via FACS. (C) Depletion of Banf1 using esiRNA results in decreased HR and increased NHEJ. U2OS cells stably expressing a HR/NHEJ reporter were depleted of the indicated proteins using esiRNA. Cells were then transfected with an Isce1-expressing plasmid and a donor plasmid and GFP and mCherry positive cells were detected via FACS. (D) Restoration of Banf1 restores HR activity. U2OS cells stably expressing a HR/NHEJ reporter were depleted of Banf1 using siRNA. Cells were then transfected with empty Flag or siRNA resistant Flag-Banf1, an Isce1-expressing plasmid and a donor plasmid and GFP and mCherry positive cells were detected via FACS. Unless otherwise stated, data shown represent the mean and S.D. of 3 independent experiments and t-test was used for statistical analysis: *P < 0.05 **P < 0.01, ***P < 0.001, ****P < 0.0001.
Since an increase in NHEJ activity can lead to changes in cellular sensitivity to IR, we next examined the colony forming ability of Banf1-depleted cells following IR. Consistent with an increase in NHEJ activity, Banf1 deficient cells displayed a modest increase in resistance to IR (Supplementary Figure S9).
Following induction of DNA DSBs, multiple signalling events occur to initiate the repair processes, mediated primarily by phosphorylation of downstream substrates by the ATM and DNA-PK kinases. Depletion of Banf1 was not observed to significantly affect phosphorylation of DNA repair proteins, including DNA-PKcs, ATM, Chk2 or Chk1 (Supplementary Figure S10). Taken together, these data support a potential role for Banf1 in HR or in supressing NHEJ without affecting DNA damage signalling.
Banf1 interacts with DNA-PK
To identify the role of Banf1 in the cellular response to DSBs, we next performed immunoprecipitations on lysates from U2OS cells expressing Flag-Banf1. Immunoprecipitates were subjected to mass spectrometry to identify Banf1-interacting proteins. This led to the identification of several known Banf1-interacting proteins, including Emerin (EMD), Lamin A (LMNA) and Thymopoietin (TMPO) which are known to localize to the nuclear envelope and histone proteins such as Histone H1 (Figure 4A, B) (30–32). Significantly, one of the highest ranking Banf1-interacting proteins identified by mass spectrometry was the DNA damage responsive kinase, DNA-Dependent Protein Kinase catalytic subunit, DNA-PKcs (also known as PRKDC). To confirm this interaction between Banf1 and DNA-PK, Flag antibodies were used to immunoprecipitate Flag-Banf1. This showed that an interaction between DNA-PKcs and Banf1 could be detected pre- and post-IR treatment, however a moderate increase in this interaction was detected 0.5 h post-IR (Figure 4C–E). In addition, Ku70/80 could also be detected in immunoprecipitates suggesting that Banf1 binds to the DNA-PK complex. Immunoprecipitations using DNA-PKcs antibodies were also performed and Flag-Banf1 and Ku70/80 could be detected in the immunoprecipitates (Figure 4C, D). Using recombinant and purified proteins, Banf1 (Supplementary Figure S11) and DNA-PK were confirmed to interact directly (Figure 4F). It should be noted that the purified DNA-PK complex included DNA-PKcs, along with Ku70/80, supporting the evidence from immunoprecipitations that Banf1 can bind to the whole DNA-PK complex (Figure 4F).
Figure 4.

Banf1 interacts with DNA-Dependent Kinase (DNA-PK). (A) DNA-PK was identified as a Banf1 interacting protein via mass spectrometry. A list of significant Banf1-interacting proteins is shown, with a minimum cut-off of 12 peptides. (B) String interactions of significant proteins are shown. (C) Banf1 and DNA-PK are in a complex; Immunoprecipitations from HEK293T cells expressing Flag or Flag-Banf1 at the indicated times post-IR using Flag antibodies. Immunoprecipitates were immunoblotted with the indicated antibodies. (D) Immunoprecipitations from HEK293T cells expressing Flag-Banf1 using DNA-PK antibodies or IgG as a control. Immunoprecipitates were immunoblotted with the indicated antibodies. (E) The histogram represents the analysis of the relative Banf1 binding to DNA-PK at the indicated times post-IR via immunoprecipitation, from (C). (F) Banf1 and DNA-PK directly interact. The recombinant His-Banf1 protein was incubated with purified DNA-PK complex (comprising of DNA-PKcs and Ku70/80) before immunoprecipitation with His beads and immunoblotting with the indicated antibodies. Unless otherwise stated, data shown represent the mean and S.D. of three independent experiments and unpaired t-test was used for statistical analysis: *P < 0.05.
In order to observe whether Banf1 and DNA-PKcs colocalized after DNA damage, we treated cells with an extraction buffer plus RNase to digest RNA, allowing visualization of chromatin-associated DNA-PKcs following IR, as reported previously (15). Using this method we were able to detect that Banf1 foci overlapped with 27% and 31% of DNA-PKcs foci at 20 min and 1 h, respectively, following 6 Gy IR (Figure 5A).
Figure 5.
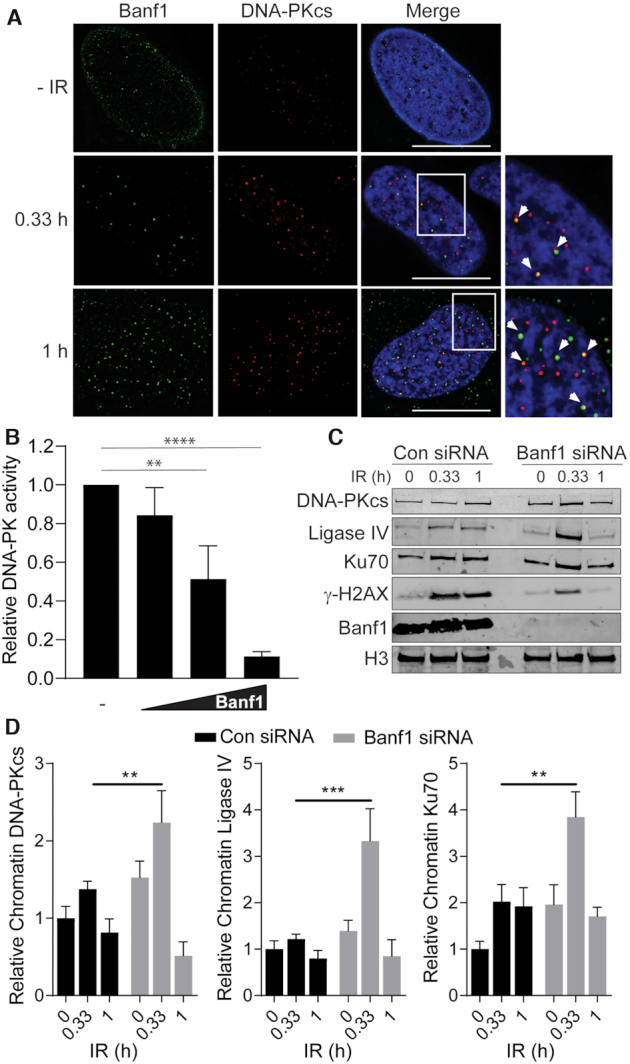
Banf1 inhibits the kinase activity of DNA-PKcs. (A) Banf1 and DNA-PKcs partially colocalize following ionizing radiation. U2OS cells were treated or mock-treated with 6 Gy IR and treated with extraction buffer (+ RNase) prior to fixation in PFA at the indicated timepoints. Cells were then stained with the indicated antibodies. (B) Recombinant WT Banf1-mediated inhibition of the kinase activity of purified DNA-PK complex (comprising of DNA-PKcs and Ku70/80) on an immobilized substrate peptide. (C) Depletion of Banf1 leads to increased recruitment of NHEJ proteins to chromatin. U2OS cells were transfected with control or Banf1 siRNA. Cells were treated or mocked treated with 6 Gy IR and cellular fractionations processed at the indicated time post-treatment. Cellular fractions were immunoblotted with the indicated antibodies. Data shown are representative of four independent experiments. (D) The bands of DNA-PKcs, Ligase IV and Ku70 were analysed via densitometry and normalized to Histone H3 bands. Immunofluorescence scale bars represent 10 μm. Histogram data shown represent the mean and S.D. of four independent experiments. Unpaired t-test was used for statistical analysis. Unless otherwise stated, histogram data shown represent the mean and S.D. of three independent experiments, t-test was used for statistical analysis: **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next investigated whether DNA-PK was required for Banf1 recruitment to DSBs. In cells depleted of DNA-PKcs, Banf1 still mobilized from the nuclear envelope to form a comparable number of nuclear foci to control cells in response to IR, (Supplementary Figure S12a–d). Similar results were also observed using a DNA-PK inhibitor (Supplementary Figure S13a–c).
Banf1 directly regulates DNA-PK kinase activity
Since we have shown that Banf1 binds to DNA-PK this raised the possibility that Banf1 could regulate DNA-PK kinase activity. To address this we used purified DNA-PK complex and assessed it's kinase activity against a substrate in the presence of increasing concentrations of purified recombinant wild-type Banf1 protein (Supplementary Figure S11). Banf1 inhibited DNA-PK activity in a dose-dependent manner (Figure 5B).
DNA-PK kinase activity is required for NHEJ and its overexpression has been previously shown to inhibit homologous recombination (33,34). In light of our data showing that Banf1 can inhibit the kinase activity of DNA-PKcs, we reasoned that the mechanism behind the increased NHEJ in Banf1-deficient cells might be an increase in DNA-PKcs activity. To further confirm that DNA-PKcs activity is altered in Banf1-deficient cells we next examined the recruitment of NHEJ factors to chromatin. Since we have shown that the majority of DSBs are repaired within 30 min post-IR in Banf1-deficient cells, we examined the recruitment of NHEJ factors to chromatin at 20 min and 1 h post-IR. In control cells a moderate increase of several NHEJ proteins on chromatin could be detected at 20 min and 1 h post-IR. However, a significant increase in NHEJ proteins, including DNA-PKcs, Ligase IV and Ku70, could be detected on chromatin in Banf1-deficient cells at 20 min post-IR, consistent with increased activity of DNA-PKcs and subsequent recruitment of other NHEJ proteins (Figure 5C, D). In contrast, the levels of these proteins were comparable in the cytoplasmic and nuclear fractions in control and Banf1-depleted cells (Supplementary Figure S14). Consistent with our previous observations, decreased γ-H2AX phosphorylation was also observed at 20 min and 1 h post-IR (Figures 2C, D and 5C).
To address whether the increased NHEJ observed in Banf1-deficient cells was due to increased DNA-PK activity, we next performed the NHEJ assay with the addition of a DNA-PK inhibitor. As expected, addition of DNA-PK inhibitor reduced the rate of NHEJ in both control and Banf1-depleted cells. The presence of the DNA-PK inhibitor also led to an increase of HR in control cells and also increased HR in the Banf1-deficient cells restoring HR to a similar level to wild type cells (Figure 6A). Similarly to Banf1, depletion of BRCA1 led to a defect in HR and an increase in NHEJ. However, in contrast to Banf1-depleted cells, treatment of BRCA1-depleted cells with DNA-PK inhibitor did not restore HR function, presumably since BRCA1 is an essential HR protein (Figure 6A). This suggests that the defective HR in the Banf1-deficient cells was likely to be due to unrestrained DNA-PKcs activity and not due to an additional role of Banf1 in HR.
Figure 6.
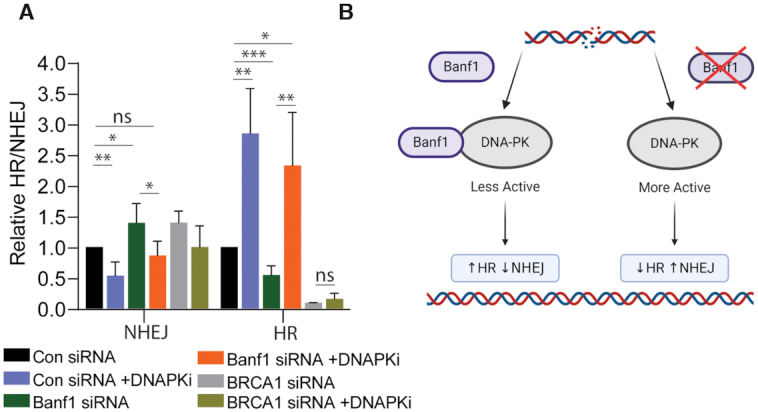
Inhibition of DNA-PKcs activity restores HR activity in Banf1-depleted cells. (A) Inhibition of DNA-PKcs can restore HR in Banf1-depleted cells. U2OS cells stably expressing a HR/NHEJ reporter were depleted of the indicated proteins using esiRNA. Cells were then treated or mocked treated with a DNA-PKi, transfected with an Isce1-expressing plasmid and a donor plasmid and GFP and mCherry positive cells were detected via FACS. (B) Model of Banf1-mediated control of DSB repair pathway choice. In the presence of Banf1, DNA-PKcs is less active, leading to HR. Conversely, when Banf1 is depleted DNA-PKcs is more active promoting NHEJ. Image was created using BioRender.com. Histogram data shown represent the mean and S.D. of three independent experiments, t-test was used for statistical analysis: *P < 0.05 **P < 0.01, ***P < 0.001.
These data support a model whereby the presence of Banf1 inhibits DNA-PK activity, potentially to promote homologous recombination repair and in the absence of Banf1 DNA-PKcs activity is increased, promoting NHEJ (Figure 6B).
DISCUSSION
Here, we define a role for Banf1 in the regulation of DNA-PKcs activity and subsequent DSB repair pathway choice. Following IR, Banf1 is rapidly stabilized and translocates from the nuclear envelope to the chromatin. Since this protein stabilization occurs within 30 min of IR treatment, it is likely due to stabilization of the protein, rather than a transcriptional response. However, further investigation is required to analyse the regulation of this increase of Banf1 protein. Banf1 was shown to be localized at sites of DSBs, following IR treatment, as marked by γ-H2AX foci. DNA-PK, along with ATM and ATR redundantly phosphorylates H2AX and paradoxically in the absence of Banf1 (where we propose there is increased DNA-PK activity) there is less γ-H2AX induction post-IR. We hypothesized that the reason for this could be that DSBs repaired by NHEJ are repaired significantly faster than breaks repaired by HR. Indeed, supporting this, one study estimated that the majority of NHEJ events are completed within 30 min whereas HR was observed to take up to 7 h (35). Since we have observed that the depletion of Banf1 leads to the increased recruitment of NHEJ proteins, upregulation of NHEJ and subsequent inhibition of HR, we suggest that a large proportion of DSBs and therefore γ-H2AX foci are resolved via NHEJ before the 30 min time point in Banf1-deficient cells, as observed in our comet assay data.
Mass spectrometry analysis of Banf1-interacting DNA repair proteins identified DNA-PK as one of the top ranked hits. DNA-PK was subsequently confirmed as a direct Banf1 interactor. Immunofluorescence also enabled the detection of colocalization of Banf1 and DNA-PKcs after IR. In addition to interacting with Banf1, DNA-PK has also been shown to interact with other nuclear envelope proteins, including Sun1, Sun2 and Emerin (36,37). DNA-PK also displayed colocalization with Sun1 and Sun2 along the inner side of the nuclear envelope (36).
Supporting a role for Banf1 in regulating DNA DSB repair, Banf1-deficient cells were more radioresistant, which is often observed in cells with upregulated DNA-PK and subsequently increased NHEJ (38). Consistent with this, Banf1-depleted cells were shown to undergo increased NHEJ events and decreased HR events compared to control cells. In support of this being due to upregulation of DNA-PK activity, inhibition of DNA-PK activity in Banf1-deficient cells, as expected reduced NHEJ, but also rescued the HR defect. In contrast, inhibition of DNA-PK catalytic activity was unable to rescue the HR defect in BRCA1-depleted cells, likely due to the essential role of BRCA1 in HR (39). Furthermore, DNA-PKcs activity has been shown to repress HR and overexpression of DNA-PKcs has been observed to inhibit HR to a comparable extent as Banf1-depletion in similar assays (34).
The mechanism by which Banf1 may inhibit DNA-PKcs is currently unclear but we speculate there are several possibilities. Multiple mechanisms of DNA-PKcs regulation have been proposed previously, including its auto-phosphorylation and regulation of its interaction with DNA ends by Ku70/80, which is required for its activation (40). The precise mechanism of how the association of Ku70/80 regulates the catalytic activity of DNA-PKcs is not well characterized. However, it is considered likely that there are several layers of regulation and multiple regions of the DNA-PKcs protein are involved in this process. Therefore, it can be speculated that Banf1 may be involved in one or more of these complex regulatory processes. It has been proposed that the interaction of the Ku–DNA complex induces a conformational change in the DNA-PKcs FAT and FATC domains that surround the catalytic domain (41–43), facilitating alteration of the catalytic groups and/or the ATP binding pocket of DNA-PKcs, promoting its activity. In addition to the regulation at the C-terminus of DNA-PK, the N-terminus also has a regulatory impact on DNA-PK catalytic activity. This is highlighted by the deletion of the N-terminus leading to spontaneous activation of the enzymatic activity of DNA-PKcs (44,45). Therefore, it is tempting to speculate that binding of Banf1 to DNA-PK may inhibit the conformational changes in these domains in order to repress DNA-PKcs activity to promote HR at subsets of DSBs. DNA-PK is also heavily regulated by phosphorylation and to date over 40 phosphorylation sites have been identified, with many of these sites characterized as autophosphorylation sites (46–49) but other DNA-damage response kinases such as ATM and ATR have also been shown to phosphorylate DNA-PKcs (50,51). We have shown here that phosphorylation of DNA-PK on S2056 in Banf1-depleted cells is comparable to control cells. However, due to the unavailability of commercial antibodies towards these additional phosphorylation sites, we cannot exclude that Banf1 binding to DNA-PK can block the phosphorylation of one or more of these regulatory phosphorylation sites on DNA-PK and inhibit kinase activity or interaction with substrates or DNA. Given that Banf1 binding to DNA-PKcs also occurs in the absence of DNA damage, we also consider it likely that the conformation of Banf1 binding to DNA-PK is also tightly regulated.
The consequence of the downregulation of HR and reliance on NHEJ in Banf1-depleted cells on genome stability is unclear. Our comet assay data, here and previously, suggest there is no immediate increase in DNA damage in Banf1-depleted cells (6). We consider it is possible that over time or in the presence of, for example, replication stress, the reliance of Banf1-depleted cells on NHEJ for DSB repair may lead to an increase in genome instability, but further investigation is required to assess this.
We have previously shown that Banf1 also negatively regulates the activity of PARP1, leading to increased PARP1 activity in Banf1-depleted cells (6). PARP1 has been previously suggested to positively regulate DNA-PKcs activity (52,53), although another study suggested that PARP1 is not required for classical NHEJ (54). In light of the above, it may be predicted that the Banf1-mediated regulation of DNA-PKcs could also, at least in part, be through its regulation of PARP1 activity. However, although we can’t completely rule out a role for PARP1 in this process, we consider that at least part of the Banf1-mediated regulation of DNA-PKcs is likely to be direct and independent of PARP1, due to the ability of Banf1 to inhibit DNA-PKcs activity in the absence of PARP1 (Figure 5B).
Further study of the location of the interaction of Banf1 with DNA-PK and the interplay with PARP1 is required in order to shed light on the likely mechanism of Banf1-dependent regulation of DNA-PK activity and DSB repair pathway choice. In summary, these data support a model whereby Banf1 directly binds to and inhibits DNA-PK function in NHEJ.
Supplementary Material
Contributor Information
Joshua T Burgess, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Chee Man Cheong, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Amila Suraweera, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Thais Sobanski, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Sam Beard, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Keyur Dave, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Maddison Rose, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Didier Boucher, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Laura V Croft, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Mark N Adams, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Kenneth O’Byrne, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia; Princess Alexandra Hospital, Ipswich Road, Woolloongabba, Brisbane, Queensland 4102, Australia.
Derek J Richard, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
Emma Bolderson, Queensland University of Technology (QUT), Cancer & Ageing Research Program, Centre for Genomics and Personalised Health at the Translational Research Institute (TRI), Brisbane, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Cancer Council Queensland; J.T.B., L.V.C. and M.N.A. are supported by an Advance Queensland Early-career Research Fellowship; D.J.R. is supported by a Chenhall Research Foundation Fellowship. Funding for open access charge: QUT startup funds.
Conflict of interest statement. The authors declare competing financial interests; E.B, D.J.R. and K.J.O. are founders of Carpe Vitae Pharmaceuticals. E.B., K.J.O. and D.J.R. are inventors on provisional patent applications filed by Queensland University of Technology.
REFERENCES
- 1. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ceccaldi R., Rondinelli B., D’Andrea A.D.. Repair pathway choices and consequences at the Double-Strand break. Trends Cell Biol. 2016; 26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jette N., Lees-Miller S.P.. The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015; 117:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu Y., Xu D.. Repair pathway choice for double-strand breaks. Essays Biochem. 2020; 64:765–777. [DOI] [PubMed] [Google Scholar]
- 5. Segura-Totten M., Kowalski A.K., Craigie R., Wilson K.L.. Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 2002; 158:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolderson E., Burgess J.T., Li J., Gandhi N.S., Boucher D., Croft L.V., Beard S., Plowman J.J., Suraweera A., Adams M.N.et al.. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat. Commun. 2019; 10:5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paquet N., Box J.K., Ashton N.W., Suraweera A., Croft L.V., Urquhart A.J., Bolderson E., Zhang S.D., O’Byrne K.J., Richard D.J.. Nestor-Guillermo Progeria Syndrome: a biochemical insight into barrier-to-autointegration factor 1, alanine 12 threonine mutation. BMC Mol. Biol. 2014; 15:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puente X.S., Quesada V., Osorio F.G., Cabanillas R., Cadinanos J., Fraile J.M., Ordonez G.R., Puente D.A., Gutierrez-Fernandez A., Fanjul-Fernandez M.et al.. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 2011; 88:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cabanillas R., Cadinanos J., Villameytide J.A., Perez M., Longo J., Richard J.M., Alvarez R., Duran N.S., Illan R., Gonzalez D.J.et al.. Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am. J. Med. Genet. A. 2011; 155A:2617–2625. [DOI] [PubMed] [Google Scholar]
- 10. Gonzalo S., Kreienkamp R.. DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 2015; 34:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burla R., La Torre M., Merigliano C., Verni F., Saggio I.. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus. 2018; 9:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 13. Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M.. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011; 10:1794–1805. [DOI] [PubMed] [Google Scholar]
- 14. Bolderson E., Tomimatsu N., Richard D.J., Boucher D., Kumar R., Pandita T.K., Burma S., Khanna K.K.. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010; 38:1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Britton S., Coates J., Jackson S.P.. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013; 202:579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arnoult N., Correia A., Ma J., Merlo A., Garcia-Gomez S., Maric M., Tognetti M., Benner C.W., Boulton S.J., Saghatelian A.et al.. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017; 549:548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richard D.J., Bolderson E., Cubeddu L., Wadsworth R.I., Savage K., Sharma G.G., Nicolette M.L., Tsvetanov S., McIlwraith M.J., Pandita R.K.et al.. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008; 453:677–681. [DOI] [PubMed] [Google Scholar]
- 18. Bolderson E., Tomimatsu N., Richard D.J., Boucher D., Kumar R., Pandita T.K., Burma S., Khanna K.K.. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010; 38:1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pierce A.J., Johnson R.D., Thompson L.H., Jasin M.. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999; 13:2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodrigue A., Lafrance M., Gauthier M.C., McDonald D., Hendzel M., West S.C., Jasin M., Masson J.Y.. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 2006; 25:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolderson E., Petermann E., Croft L., Suraweera A., Pandita R.K., Pandita T.K., Helleday T., Khanna K.K., Richard D.J.. Human single-stranded DNA binding protein 1 (hSSB1/NABP2) is required for the stability and repair of stalled replication forks. Nucleic Acids Res. 2014; 42:6326–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olive P.L., Banath J.P., Durand R.E.. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 1990; 122:86–94. [PubMed] [Google Scholar]
- 23. Paquet N., Adams M.N., Ashton N.W., Touma C., Gamsjaeger R., Cubeddu L., Leong V., Beard S., Bolderson E., Botting C.H.et al.. hSSB1 (NABP2/OBFC2B) is regulated by oxidative stress. Sci. Rep. 2016; 6:27446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moser M.J., Kamath-Loeb A.S., Jacob J.E., Bennett S.E., Oshima J., Monnat R.J. Jr. WRN helicase expression in Werner syndrome cell lines. Nucleic Acids Res. 2000; 28:648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Le Merrer M.et al.. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003; 300:2055. [DOI] [PubMed] [Google Scholar]
- 26. Redwood A.B., Perkins S.M., Vanderwaal R.P., Feng Z., Biehl K.J., Gonzalez-Suarez I., Morgado-Palacin L., Shi W., Sage J., Roti-Roti J.L.et al.. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle. 2011; 10:2549–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saintigny Y., Makienko K., Swanson C., Emond M.J., Monnat R.J. Jr.. Homologous recombination resolution defect in werner syndrome. Mol. Cell. Biol. 2002; 22:6971–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caron P., Aymard F., Iacovoni J.S., Briois S., Canitrot Y., Bugler B., Massip L., Losada A., Legube G.. Cohesin protects genes against gammaH2AX Induced by DNA double-strand breaks. PLos Genet. 2012; 8:e1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stiff T., O’Driscoll M., Rief N., Iwabuchi K., Lobrich M., Jeggo P.A.. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004; 64:2390–2396. [DOI] [PubMed] [Google Scholar]
- 30. Lee K.K., Haraguchi T., Lee R.S., Koujin T., Hiraoka Y., Wilson K.L.. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001; 114:4567–4573. [DOI] [PubMed] [Google Scholar]
- 31. Shumaker D.K., Lee K.K., Tanhehco Y.C., Craigie R., Wilson K.L.. LAP2 binds to BAF.DNA complexes: requirement for the LEM domain and modulation by variable regions. EMBO J. 2001; 20:1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montes de Oca R., Shoemaker C.J., Gucek M., Cole R.N., Wilson K.L.. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS One. 2009; 4:e7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taccioli G.E., Gottlieb T.M., Blunt T., Priestley A., Demengeot J., Mizuta R., Lehmann A.R., Alt F.W., Jackson S.P., Jeggo P.A.. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 1994; 265:1442–1445. [DOI] [PubMed] [Google Scholar]
- 34. Neal J.A., Dang V., Douglas P., Wold M.S., Lees-Miller S.P., Meek K.. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol. Cell. Biol. 2011; 31:1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mao Z., Bozzella M., Seluanov A., Gorbunova V.. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.). 2008; 7:1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lei K., Zhu X., Xu R., Shao C., Xu T., Zhuang Y., Han M.. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr. Biol. 2012; 22:1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moser B., Basilio J., Gotzmann J., Brachner A., Foisner R.. Comparative interactome analysis of emerin, MAN1 and LEM2 reveals a unique role for LEM2 in nucleotide excision repair. Cells. 2020; 9:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sishc B.J., Davis A.J.. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancers (Basel). 2017; 9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moynahan M.E., Chiu J.W., Koller B.H., Jasin M.. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999; 4:511–518. [DOI] [PubMed] [Google Scholar]
- 40. Hammel M., Yu Y., Mahaney B.L., Cai B., Ye R., Phipps B.M., Rambo R.P., Hura G.L., Pelikan M., So S.et al.. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem. 2010; 285:1414–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rivera-Calzada A., Maman J.D., Spagnolo L., Pearl L.H., Llorca O.. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure. 2005; 13:243–255. [DOI] [PubMed] [Google Scholar]
- 42. Rivera-Calzada A., Spagnolo L., Pearl L.H., Llorca O.. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 2007; 8:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spagnolo L., Rivera-Calzada A., Pearl L.H., Llorca O.. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell. 2006; 22:511–519. [DOI] [PubMed] [Google Scholar]
- 44. Davis A.J., Lee K.J., Chen D.J.. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J. Biol. Chem. 2013; 288:7037–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meek K., Lees-Miller S.P., Modesti M.. N-terminal constraint activates the catalytic subunit of the DNA-dependent protein kinase in the absence of DNA or Ku. Nucleic Acids Res. 2012; 40:2964–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uematsu N., Weterings E., Yano K., Morotomi-Yano K., Jakob B., Taucher-Scholz G., Mari P.O., van Gent D.C., Chen B.P., Chen D.J.. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 2007; 177:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Douglas P., Cui X., Block W.D., Yu Y., Gupta S., Ding Q., Ye R., Morrice N., Lees-Miller S.P., Meek K.. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell. Biol. 2007; 27:1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Meek K., Douglas P., Cui X., Ding Q., Lees-Miller S.P.. trans Autophosphorylation at DNA-dependent protein kinase's two major autophosphorylation site clusters facilitates end processing but not end joining. Mol. Cell. Biol. 2007; 27:3881–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Povirk L.F., Zhou R.Z., Ramsden D.A., Lees-Miller S.P., Valerie K.. Phosphorylation in the serine/threonine 2609–2647 cluster promotes but is not essential for DNA-dependent protein kinase-mediated nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2007; 35:3869–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen B.P., Uematsu N., Kobayashi J., Lerenthal Y., Krempler A., Yajima H., Lobrich M., Shiloh Y., Chen D.J.. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007; 282:6582–6587. [DOI] [PubMed] [Google Scholar]
- 51. Yajima H., Lee K.J., Zhang S., Kobayashi J., Chen B.P.. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J. Mol. Biol. 2009; 385:800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han Y., Jin F., Xie Y., Liu Y., Hu S., Liu X.D., Guan H., Gu Y., Ma T., Zhou P.K.. DNAPKcs PARylation regulates DNAPK kinase activity in the DNA damage response. Mol. Med. Rep. 2019; 20:3609–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spagnolo L., Barbeau J., Curtin N.J., Morris E.P., Pearl L.H.. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012; 40:4168–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fouquin A., Guirouilh-Barbat J., Lopez B., Hall J., Amor-Gueret M., Pennaneach V.. PARP2 controls double-strand break repair pathway choice by limiting 53BP1 accumulation at DNA damage sites and promoting end-resection. Nucleic Acids Res. 2017; 45:12325–12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.