

Abstract
Epstein-Barr virus (EBV), a herpes virus also termed HHV 4 and the first identified human tumor virus, establishes a stable, long-term latent infection in human B cells, its preferred host. Upon induction of EBV’s lytic phase, the latently infected cells turn into a virus factory, a process that is governed by EBV. In the lytic, productive phase, all herpes viruses ensure the efficient induction of all lytic viral genes to produce progeny, but certain of these genes also repress the ensuing antiviral responses of the virally infected host cells, regulate their apoptotic death or control the cellular transcriptome. We now find that EBV causes previously unknown massive and global alterations in the chromatin of its host cell upon induction of the viral lytic phase and prior to the onset of viral DNA replication. The viral initiator protein of the lytic cycle, BZLF1, binds to >105 binding sites with different sequence motifs in cellular chromatin in a concentration dependent manner implementing a binary molar switch probably to prevent noise-induced erroneous induction of EBV’s lytic phase. Concomitant with DNA binding of BZLF1, silent chromatin opens locally as shown by ATAC-seq experiments, while previously wide-open cellular chromatin becomes inaccessible on a global scale within hours. While viral transcripts increase drastically, the induction of the lytic phase results in a massive reduction of cellular transcripts and a loss of chromatin-chromatin interactions of cellular promoters with their distal regulatory elements as shown in Capture-C experiments. Our data document that EBV’s lytic cycle induces discrete early processes that disrupt the architecture of host cellular chromatin and repress the cellular epigenome and transcriptome likely supporting the efficient de novo synthesis of this herpes virus.
INTRODUCTION
Viruses exploit their hosts at the cellular or organismic level to support viral propagation and spread. Towards this end, they also manipulate the infected cellular host with their own toolkit to suppress various antiviral defense mechanisms. For example, viruses can inhibit several levels of interferon responses (1 and references therein), counteract cellular cytidine deaminases with potent antiviral activities (2), reduce cellular immunity with viral micro RNAs (3), or even mimic histone tails to interfere with antiviral responses of the infected cells (4).
The manipulation of the host's antiviral programs is especially important for herpes viruses. Commonly, they turn the infected cell into a virus factory, but they can also initiate their temporal coexistence in certain cells to establish long-lasting, non-productive, latent infections. During latency, the herpesviral, genomic DNA acquires a genuine cellular and highly repressive chromatin signature, which blocks transcription of most viral genes. Herpes viruses can escape from the latent phase and reactivate virus production. In this step, herpes viruses instruct their cellular host to remove the repressive epigenetic signature from viral chromatin to allow and activate massive viral transcription of all lytic viral genes within hours. During lytic reactivation, the host cell must also be manipulated to provide chemical energy, macro-molecules, and nuclear space for the so-called viral replication compartments (5,6) or amplification factories (7). Additionally, the cell's transcription machinery needs to be redirected to support an efficient virus de novo transcription within a few hours after reactivation. Many molecular details of these fundamental processes are unknown.
A ubiquitous human herpes virus is Epstein-Barr virus (EBV, HHV4), which infects ∼ 95 % of the human population. B lymphocytes are the preferred target cells in which EBV establishes a latent infection. EBV reactivates from this latent state with the help of the viral BZLF1 protein, which is expressed upon differentiation of EBV-infected memory B cells to plasma cells (8), and induces the switch from latency to EBV’s lytic phase (9,10). BZLF1, also called EB1, ZEBRA, zta or Z, is known to act as an essential viral transcription and replication factor (9–11). BZLF1 binds two classes of DNA binding motifs (ZREs) in the viral genome, one of which needs to be methylated to be bound efficiently (12,13). These sites are termed meZREs or CpG-ZREs and are mainly positioned in promoters of important lytic viral genes (12,14). Upon initial infection during EBV’s pre-latent phase (15), BZLF1 is transiently expressed (14,16), but it does not bind its many viral meZREs because the incoming viral DNA is free of methylated CpG dinucleotides (17). As a consequence, the virus cannot activate its lytic promoters in the pre-latent phase, but it rather initiates its latent program, which leads to the restricted expression of the few latent viral genes only. CpG methylation of viral DNA is a slow process in newly infected primary B cells that takes several weeks to completion (18), but CpG methylation further supports the epigenetic silencing of all viral lytic genes (19). It is likely that EBV uses this strategy to prevent the premature expression of its lytic genes in newly infected cells, which would induce a massive antiviral immune response of EBV-specific T cells and would readily eliminate the virus-infected cells (20).
BZLF1 is a homo-dimeric bZIP transcription factor. Its DNA binding domain shows a strong homology to the cellular AP-1 protein family including the Jun, Fos, Fra, and ATF subfamilies. AP-1 binding motifs and ZREs are related sequences of about seven nucleotides (21,22). Similar to BZLF1, c-Jun/c-Fos hetero-dimers can also bind to and regulate cellular genes via methylated DNA motifs, which are related to meZRE motifs of BZLF1 (23). Collectively, these findings suggest that BZLF1 may widely influence cellular transcription since several hundred thousand motifs of members of the AP-1 family are known in the genome of mammalian cells (24), some of which are located in enhancers (25–29). AP-1 family members participate in regulating genes involved in cellular proliferation, differentiation, and apoptosis (30–33) – pathways which EBV also manipulates in its pre-latent, latent, and lytic phases.
Much is known about the impact of BZLF1 on the viral genome after induction of EBV’s lytic phase and the ensuing alterations in viral chromatin (18,34), but if and how herpes viruses in general – and EBV in particular – manipulate the chromatin of the host cell during the early hours of lytic, productive infection is not known in detail. Therefore, we examined the global modification of the host genome in the lytic, pre-replicative phase of EBV. We chose the Raji cell line as our preferred model for one main reason. In Raji cells, EBV’s latent phase is very tightly controlled and, upon induction, the majority of the cells readily enter EBV’s lytic phase. The lytic phase, however, is incomplete and does not support the amplification the viral DNA because Raji cells carry a defective EBV genome (35–37). Thus, Raji cells allow studying the very early modifications in cellular chromatin in the pre-replicative phase, when the cells transit from viral latency to lytic reactivation, but prior to the formation of replication compartments or amplification factories. In EBV-infected cells, the chromatin modifications become microscopically visible as early as 16–24 h after lytic phase induction and are characterized by the local accumulation of EBV DNA and the occlusion of cellular chromatin constituents such as histones (7). This report by Chiu et al. also indicated that the nuclear architecture of the lytic cycle-induced cells undergoes substantial modifications prior to the formation of the amplification factories, with nuclei showing an initial ‘honeycombed’ structure, suggestive of an early reorganization of the cellular chromatin or nuclear 3D architecture.
BZLF1 and certain members of the cellular AP-1 transcription factor family are structurally and functionally similar. It seems therefore plausible that BZLF1 may take part in manipulating the chromatin and transcriptome of its cellular host during viral reactivation and the subsequent lytic phase. Our focus was to detect global changes within EBV’s host cell upon induction of the lytic cycle. Therefore, we considered the possible role of BZLF1 in inducing modifications with respect to the cell's transcriptome, chromatin accessibility, and chromatin-chromatin interactions. To investigate these cellular processes when BZLF1 is expressed in the absence of other viral proteins, we also included DG75 – an EBV-negative B cell line – in our studies.
We discovered that the induction of BZLF1 induces global alterations in the host cell transcriptome and chromatin within 6–15 h. We found (i) an up- and down-regulation of cellular RNA causing considerable transcriptional noise, (ii) a prevalent reduction or even collapse of chromatin-chromatin interactions, concomitant with a general reduction of open, accessible chromatin compared to non-induced cells and (iii) a localized opening of chromatin at the majority of about 105 cellular BZLF1 binding sites. These changes were generally more pronounced in Raji cells than in the EBV-negative DG75 cells. Only in EBV-positive Raji cells we detected a global and impressive reduction of cellular transcripts, indicative of the virus-induced host shut-off. Together, these findings document that the host cell is subject to global nuclear changes already in the pre-replicative phase of the virus’ lytic cycle.
MATERIALS AND METHODS
Eukaryotic cell lines
The cell lines B95-8 (EBV-positive) (38), DG75 (EBV-negative) (39) and Raji (EBV-positive) (40) and their derivatives were cultured in RPMI 1640 medium (Life Technologies) supplemented with 1 mM sodium pyruvate (Life Technologies), 100 μg/ml streptomycin, 100 units/ml penicillin (Life Technologies) and 8 % fetal bovine serum (FBS) (Bio&Sell GmbH) in an atmosphere with 5 % CO2, 95 % humidity at 37 °C. 1 (0,4) μg/ml puromycin (Invitrogen) was added to Raji (DG75) cells to select for the maintenance of oriP plasmids with two conditional doxycycline-inducible BZLF1 alleles: iBZLF1 full-length (p4816) and iBZLF1 AD-truncated (p5694) encode the wild-type BZLF1 and the activation domain (AD)-truncated alleles, respectively. On average, the cells were kept at a density of 5×105 cells/ml. HEK 293 cells (41) were kept at a confluency of ∼ 70 %. HEK 293 cells that produce the EBV strain wt/B95.8 (2089) upon induction were cultivated with 100 ng/ml hygromycin as described (42). The 9G10 HEK293 cell line carries a recombinant EBV genome, which encodes puromycin resistance, but lacks the gene encoding green fluorescent protein (GFP). The cells were stably transduced with a retroviral vector that encodes BZLF1 fused to the hormone binding domain variant ERT2 of the human estrogen receptor. The cells were cultivated in the presence of 1 μg/ml puromycin.
Plasmids
The doxycycline-inducible BZLF1-expressing plasmids p4816 and p5694, which encode full-length iBZLF1 and the activation domain (AD)-truncated iBZLF1 respectively, are described in Woellmer et al. (18). The two plasmids also express the green fluorescent protein (GFP) upon doxycycline addition (43). The BZLF1 plasmid p3928 expresses a truncated BZLF1 (aa 149–245) with an amino-terminal tandem step-tag as described (44). The retroviral vector plasmid termed p7087.1 encodes BZLF1:ERT2.
Quantitative BZLF1 expression analysis
BZLF1 protein was purified from HEK 293 cells transiently transfected with the BZLF1-strep-tag expressing plasmid p3928 for 48 h. Cells were lysed in RIPA buffer (1 % NP-40, 0.1 % SDS, 0.5 % sodium deoxycholate, 150 mM NaCl, 50 mM Tris–HCl pH 8.0, 1× proteinase inhibitor cocktail (Roche)), incubated (20 min) and sheared on ice with a Branson sonifier 250-D (10 s on/50 s off, 1 min, 20 % amplitude). The lysates were centrifuged (15 min, 16 000 g, 4 °C) and the supernatants were purified with IBA Strep-Tactin sepharose beads (IBA, 2-1201-002) on Poly-Prep Chromatography Columns (731-1550, BioRad) with IBA’s buffer's W (washing) and E (elution). The concentration of purified protein was determined by Coomassie staining of 14 % gels after SDS-PAGE with BSA standards obtained from the Pierce BCA protein assay kit (23209, Thermo Fisher Scientific). The purified BZLF1 protein was used as a quantitative standard and reference for subsequent Western blot analyses.
For BZLF1 expression level analysis 2.5 × 106 DG75, B95-8 cells, non-induced Raji iBZLF1 cells or Raji iBZLF1 cells induced for 6 h were lysed in RIPA buffer and sheared with the BioRuptor (Diagenode) four times (5 min, 30 on/off, high) in ice cold water. 5× Laemmli loading buffer containing 15 % DTT (1 M) and 0.75 % beta-mercaptoethanol were added to the lysates. Aliquots of these lysates corresponding to different cell numbers were loaded onto 14 % SDS-PAGE gels and analysed by Western blot immune detection. BZLF1 was detected with the BZ1 antibody (45), which was used at a 1:50 dilution in combination with a secondary anti-mouse HRP antibody (1:10 000, Cell Signaling #7076S). ECL Select (GE Healthcare) was used for signal detection and bands were quantified with a Fusion FX (VILBER) system. Calculation and visualisation of BZLF1 dimers was done with R (46).
Intracellular BZLF1 staining
For the intra-cellular staining of BZLF1 Fix & Perm permeabilisation Kit (Invitrogen) was used. 1×106 cells were centrifuged (300 g, 10 min) and washed with 100 μl PBS. 100 μl of Kit reagent A were added (15 min, in the dark). Samples were washed with 2 ml wash buffer (PBS, 50 % FBS, 0.1 % NaN3) before 100 μl Kit reagent B was added. 1 μl BZ1 antibody (45) coupled with Alexa647 was added before vortexing (2 s) and incubation (20 min, RT, in the dark). Samples were washed with 2 ml wash buffer and taken up in 0.5 ml wash buffer afterwards for flow cell analysis (BD Fortessa).
Virus titration
The 2089.2.22.7 version of HEK 293 cells stably transduced with the maxi-EBV plasmid p2089 releases the EBV strain wt/B95.8 (2089) upon BZLF1 expression (42). This cell line was transfected with the inducible iBZLF1 expression plasmid p4816 and selected with 500 ng/ml puromycin (34). 4×105 HEK 293 2089 cells were induced with 25, 100 or 200 ng doxycycline/ml for three days or the cells were left non-induced as a control. The supernatants were filtered through a filter with a pore size of 1.2 μm and 25, 50 or 100 μl were used to infect 105 parental Raji cells. GFP positive Raji cells (Green Raji units) were identified via flow cell analysis as described in detail (47). The virus concentrations were calculated and visualised with R.
Analysis of cytoplasmic and nuclear BZLF1 signals and plasma membrane-associated gp350 in the 9G10 HEK293 cell line upon tamoxifen induction
Sample preparation
Round 18 mm coverslips were transferred to individual wells of a 12-well cluster plate and incubated with 80 % EtOH (5 min) for sterilization. After drying, the coverslips were coated with 1 ml of a 0.01 % poly-l-lysine solution (diluted in H2O) for 5 min. Poly-l-lysine was removed and the coverslips were washed twice with sterile water and dried for 2 h.
2×105 9G10 cells were seeded onto a poly-l-lysine treated coverslip in a single well of a 12-well cluster plate and incubated for at least 8 h to allow cellular adherence. The cells were induced in 2 ml induction medium (D-MEM without supplements) for 12 and 72 h with tamoxifen in the following concentrations [nM]: 0, 5, 10, 20, 40, 80, 160, 320, 640, 1000.
Immunofluorescence analysis
The coverslips were washed with 1 ml PBS prior to adding 500 μl fixation buffer (4 % PFA in PBS; 20 min, RT). Cell were washed again with 1 ml PBS, permeabilised with 500 μl 0.3 % Triton in PBS (RT, 20 min) and blocked twice in 500 μl 3 % BSA with 0.1 % Triton in PBS (RT, 10 min). The coverslips were transferred onto parafilm in a humidified chamber. Subsequently, a single coverslip was covered with 150 μl staining buffer (PBS, 1 % BSA, 0.1 % Triton) containing 0.5 μl anti-BZ1-Alexa 647, anti-CD147-Alexa 488 (9G2), anti-gp350-Alexa 568 (6G4) and 1 μl DAPI (10 mg/ml) and incubated (RT, 2 h, in the dark). Coverslips were transferred to wells of 12-well cluster plates containing 1 ml 1 % BSA, 0.1 % Triton in PBS. Cells were washed 5 times with 1 ml 1 % BSA, 0.1 % Triton and 3 times with 1 ml PBS. The coverslips were embedded in Mowiol and sealed with nail polish after Mowiol hardened over night.
Microscopy
Images were acquired on a Leica SP8 confocal microscope with a Plan Apochromat 40× NA 1.3 oil immersion objective. Laser power was adjusted to obtain signal intensities within the dynamic range of the detector. Typical settings were as follows: DAPI signal was acquired with a 405 nm diode laser at 0.8 % on a photo-multiplier with a detection window set to 410–475 nm and voltage gain at 768 V and 0.1 % offset. Alexa 488 signal was acquired on a Hybrid Detector with the white light laser set to 488 nm at 2.5 %, a detection window of 494–562 nm and a time gate of 0.3–6 ns. Alexa 568 signal was acquired on a Hybrid Detector with the white light laser set to 568 nm at 3.5 %, a detection window of 573–641 nm and a time gate of 0.4–6 ns. Alexa 647 signal was acquired on a Hybrid Detector with the white light laser set to 647 nm at 1.0 %, a detection window of 653–773 nm and a time gate of 0.5–6 ns. DAPI and Alexa 647 channels were acquired simultaneously, the other channels separately. We scanned images at 0.75 zoom factor, 2048 × 2048 pixels, 400 Hz, at 2× line averaging and in bi-directional scan mode, yielding an effective pixel size of 189 nm. Depending on the number of cells per field of view, 15–30 images were acquired per experimental condition, either in tile mode or individually.
Image processing and analysis
For downstream processing, we wrote a custom Python script based on the bioformats library to extract individual channel images from the Leica LIF image files to save them as individual TIFF files. To identify nuclei, we used the image segmentation tool StarDist (48) with the previously published neuronal network to detect fluorescent cells. Centroids of StarDist labels were found with the scikit-image (49) regionprops module and used as seeds for RACE (50) segmentation of the CD147 channel. We used a typical RACE pipeline for seed-assisted segmentation with some adjustments: Median filter was set to 2 pixels, Sigma of the Hessian filter to 2 pixels and the Morphological Operations filter maximum radius set to 8 pixels.
Subsequently, we combined the cell segmentation result and the output of the N-scale Morphological Watershed filter with the StarDist nuclear label. We only considered objects as nuclei if the area was larger than 500 px2 (corresponding to 17.8 micron2) and smaller than 15,000 px2 (corresponding to 535.8 micron2) to remove cell fragments, speckles and dirt and/or wrongly classified larger objects, respectively, from our analysis. Subsequently, we identified matching cell-nuclei pairs by a global distance map that excluded pairs of >100 px distance (>18.0 micron), followed by individually testing whether a nucleus lies within a cell region. Finally, from matching nuclei and cell objects, we constructed masks to measure BZLF1 and gp350 intensity parameters in the nucleus, cytoplasm or at the cell border, the latter representing a dilated outline around the cell object. Data were saved in csv format for further data analysis.
Data analysis
Data processing and visualisation was conducted in R. All samples were analysed separately regarding replicate, tamoxifen concentration and time point. These sets were size sampled to the smallest number of cells analysed from the four replicates to continue with an identical number of sampled cells per replicate. Subsequently, the mean signal intensities were calculated based on the intensity values of single cells regarding cytoplasm, nucleus and membrane within each replicate. All non-induced samples were set to 1 and all other signal intensities were normalised accordingly. The mean of four replicates was plotted together with the standard error for each concentration.
Next generation ChIP-sequencing
For immuno-precipitation, two samples with 1×108 cells each were adjusted to a concentration of 5×105 cells/ml in fresh medium and were left non-induced or were induced with a final concentration of 100 ng/ml doxycycline (Sigma-Aldrich) for 15 h. Nuclei were extracted with hypotonic buffer (10 mM KCl, 340 mM sucrose, 1.5 mM MgCl2, 10 mM HEPES pH 7.9) containing 10 % proteinase inhibitor cocktail (PIC, Roche) and lysed in RIPA buffer + 1× PIC. The chromatin was shared in a BioRuptor on wet ice (4 cycles, 5 min each, 30 on/off, high). The chromatin was immuno-precipitated with the BZ1 antibody (45), 10 % input was used as a control. The precipitates were washed with different salt concentrations and the proteins were digested with proteinase K. For Raji iBZLF1 cells, the library preparations and sequencing (Illumina, paired-end, 150 nt) were performed by Vertis Biotechnology AG (Freising, Germany), while DG75 iBZLF1 and DG75 cells were prepared with the NEBNext Ultra II DNA Library Prep Kit and the NEBNext Multiplex Oligos for Illumina kits. The sequences were mapped with bowtie2 2.2.6 (51), formats were transformed with samtools 1.0 (52) and bedtools v2.25.0 (53) and the peak calling of the samples versus input was done by MACS2 2.1.0 (54,55). The overlapping peaks of two replicates were merged with bedtools intersect and the DREME algorithm (56) of the MEME-Chip 4.10.1 suite (57) was used for motif identification. Raji 15 h DREME motifs shown in Figure 1C resulted from the merger of the two motifs TGAGYVA and TGTGYVA.
Figure 1.
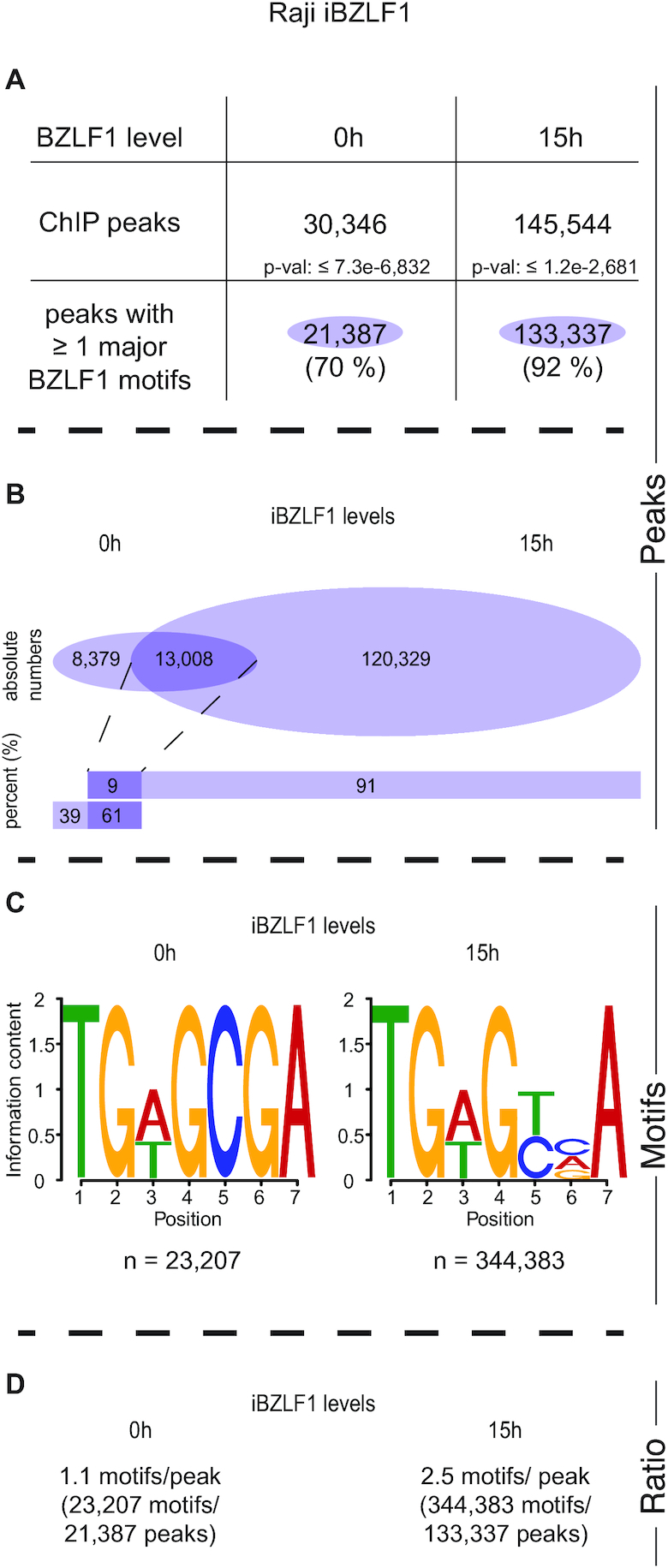
Identification of ChIP-seq peaks and BZLF1 binding motifs in chromatin of Raji iBZLF1 cells. (A) Numbers of peaks at 0 and 15 h levels of BZLF1 (i.e. in the non-induced and induced states, respectively) in EBV-positive Raji iBZLF1 cells after ChIP-seq with the BZLF1 specific BZ1 antibody. (B) The intersections of the 0 and 15 h peak sets with at least one identified motif indicate that the majority of 0 h-level peaks are maintained when BZLF1 is induced. The abundance of peaks increases more than 5-fold at 15 h BZLF1 levels compared with the peak number at 0 h BZLF1 levels. (C) At 0 h BZLF1 levels, the known BZLF1 binding motif TGWGCGA predominates in individual peaks. At 15 h BZLF1 levels, the less specific TGWGYVA motif was identified as the major motif, which encompasses the previously identified meZRE motif TGWGCGA (12). (D) The average frequencies of the number of BZLF1 motifs per ChIP-seq peak is provided.
ATAC-sequencing
106 Raji or DG75 cells, with either inducible BZLF1 plasmid (iBZLF1, p4816) or AD-truncated BZLF1 (p5694), were treated with 100 ng/ml doxycycline for 15 h or left untreated. The cells were FACS-sorted for living cells (untreated) and living, induced cells (15 h induced), controlled with trypan blue and prepared for NGS. Omni-ATAC was performed as previously described (58), with minor modifications. Briefly, 50,000 FACS-sorted cells were washed in PBS, re-suspended in 50 μl of ATAC-seq re-suspension buffer (RSB: 10 mM Tris–HCl, pH 7.4, 10 mM NaCl and 3 mM MgCl2) containing 0.1 % NP40, 0.1 % Tween-20 and 0.01 % digitonin (Promega) and were incubated on ice for 3 min. Following lysis, 1 ml of ATAC-seq RSB containing 0.1 % Tween-20 was added, and nuclei were collected at 500 g (4 °C, 10 min). Pelleted nuclei were re-suspended in 50 μl of transposition mix (25 μl 2 × TD buffer, 2.5 μl Tagment DNA enzyme) (Illumina Nextera DNA Library Preparation Kit, cat. FC-121–1030), 16.5 μl PBS, 0.5 μl 1 % digitonin, 0.5 μl 10 % Tween-20 and 5.25 μl water) and incubated (37 °C, 30 min) in a thermo-mixer shaking (1000 rpm). DNA was purified using Qiagen PCR clean-up MinElute kit (Qiagen). The transposed DNA was subsequently amplified in 50 μl reactions with custom primers, as described (59). After four cycles, libraries were monitored with qPCR: 5 μl PCR sample in a 15 μl reaction with the same primers. qPCR output was monitored for the delta reporter value (ΔRN); 0.25 ΔRN cycle number was used to estimate the number of additional cycles of the PCR reaction needed for the remaining PCR samples. Amplified libraries were purified with the Qiagen PCR clean-up MinElute kit (Qiagen) and size-selected for fragments < 600 bp using the Agencourt AMPure XP beads (Beckman Coulter). Libraries were quality controlled by Qubit and Agilent DNA Bioanalyzer analysis. High-throughput sequencing was performed by the Laboratory for Functional Genome Analysis (LAFUGA) of the Ludwig-Maximilian-University, Munich, on an Illumina Hi-Seq 1500, using 50 nt single-end reads.
The data were mapped on the hg19 genome with Bowtie 1.1.2. The HOMER 4.9 software was used to calculate tag directories. The data for the metaplots and heatmaps were calculated with HOMER’s annotatePeaks.pl tool on data from three independent experiments and visualised with R (3.5.1) (46). For heatmap visualisation, the data.table package (60) and the superheat package (61) were used.
Next generation RNA-sequencing
For RNA sequencing, parental Raji and DG75 cells and their derivatives carrying the conditional expression plasmids encoding full-length iBZLF1 or AD-truncated iBZLF1 were employed. At an initial cell concentration of 5×105 cells/ml, a total of 2×107 non-induced cells or 4×107 cells induced with 100 ng/ml doxycycline for 6 h were analysed. Doxycycline leads to the co-expression of BZLF1 and the truncated human NGF-receptor. To limit the analysis to BZLF1 expressing cells, only, we sorted NGF-R-positive cells with magnetic beads (MACS, Miltenyi Biotec) and the primary anti-NGF-R antibody (HB8737-1, IgG1) and a secondary anti-mouse IgG (1:10, Miltenyi Biotech) antibody. 7.5×105 cells were lysed in 1 ml Trizol (Thermo Fisher Scientific), snap frozen in liquid nitrogen and stored at −80 °C.
Prior to RNA isolation, identical molar amounts of ERCC spike-in control RNAs (Ambion) were added to 3×105 non-induced or doxycycline-induced Raji iBZLF1 (0 h/6 h) and DG75 iBZLF1 cells. The subsequent processes and steps were identical for all cells. RNA was extracted with the Direct-zol RNA MiniPrep Kit (Zymo Reseach). The RNA concentration and quality were controlled with Nanodrop (Thermo Fisher Scientific), Qubit (Thermo Fisher Scientific) and Bioanalyzer (Agilent), and the RNAs were treated with dsDNase (Thermo Fisher Scientific). The Encore Complete RNA-Seq Library System kit (NuGEN), which uses not-so-random hexamer primers for rRNA depletion, was used for library preparation. The samples were sequenced on an Illumina HiSeq 1500 instrument (100 nt, single-end reads).
For bioinformatic analysis, the samples were mapped with Tophat2 (62) on a Galaxy server (63) to the hg19 genome, locally with HiSat2 2.0.1 to EBV-Raji (KF717093), and reads were assigned to annotated genes with HTSeq-count 0.6.1p1 (64). The counts were compared between induced (6 h) and non-induced (0 h) cells with the R package DESeq2 1.12.3 (65). Samples with added ERCC spike-in RNAs were normalised to these. The visualisation was done with R including the packages ‘Extrafont’ 0.17 (66) and ‘RColorBrewer’ 1.1–2 (67).
Promoter analysis
Bedtools intersect was used to identify peaks which are located within the promoters [−1 kb/+5 kb relative to TSS (UCSC, RefSeq Genes, Hg19)] of regulated and non-regulated genes. R was used to remove duplicates, calculate and visualize the number of BZLF1 peaks or motifs within the promoters. Genes with fewer than 20 reads were excluded from the analysis.
Capture-C
For Capture-C analysis, 107 non-induced Raji iBZLF1 and DG75 iBZLF1 cells or cells induced for 6 and 15 h with 100 ng/ml doxycycline were used. The 6 h time period was analysed only in Raji iBZLF1 cells. Cells were washed (PBS), filtered for single cells, and fixed (1 % formaldehyde, 10 % fetal bovine serum in PBS). The fixation was quenched on ice with 1 M glycine and washed with PBS before cells were homogenized two times with 15 strokes each with a 20G syringe in 3C lysis buffer (10 mM Tris–HCl (pH 7.5), 10 mM NaCl, 0.2 % NP-40 in ddH2O + PIC). Cells were washed and taken up in DpnII restriction buffer (NEB). SDS was added to a concentration of 0.1 % and the cells were incubated (65 °C, 40 min) while shaking (1200 rpm) and 20 min at 37 °C shaking (1200 rpm). Triton X-100 was added to a final concentration of 4 % and the cells were incubated (37 °C, 1 h, 1200 rpm). The cell suspension was divided into four aliquots of equal volume and incubated each with 100 units DpnII at 37 °C while shaking (1200 rpm) overnight followed by another incubation with 100 units of DpnII for 2.5 h. Samples were joined and the reaction was stopped with SDS at a final concentration of 1.6 %. The samples were incubated (65 °C, 1200 rpm, for 30 min), diluted with 5760 μl 1.25× 3C ligation buffer (62.5 mM Tris–HCl (pH 7.5), 12.5 mM MgCl2, 1.25 mM ATP, 12.5 mM DTT in ddH2O) and 720 μl Triton X-100 (final concentration of 1 %) and incubated again (37 °C, 1 h, carefully inverting every 10 min). 100 units T4 DNA Ligase (Affymetrix or Thermo Scientific) was used for DNA fragment ligation (16 °C for 4.5 h and RT for 45 min) followed by proteinase K (20 μg/μl) treatment at 65 °C over-night to revert cross-linking. RNAs were degraded with RNase A (10 μg/μl), 37 °C, 1 h). DNA was extracted with organic solvents (2/3 v/v phenol–chloroform and 1/3 v/v butanol), precipitated (EtOH, 100 %), washed (EtOH 70 %) and re-hydrated (TE buffer).
For Raji and DG75 iBZLF1 cells, 1.5 μg and 3.0 μg DNA were sheared, respectively, using a Covaris M-series instrument (Covaris, peak incident power: 50 W; duty factor 20 %; cycles/burst: 200 counts, duration 200 s). Samples were cleaned with Agencourt AMPure beads (Beckman Coulter). The library preparation and sequence capture for Raji iBZLF1 cells were done as described in the SureSelectXT2 Target Enrichment System for Illumina Paired-End Multiplexed Sequencing manual (Agilent, Version: E1, June 2015). For DG75 iBZLF1 cells the SureSelectXT Target Enrichment System was used (Agilent, Version C2, December 2018). The desired fragments were captured with Dynabeads (Thermo Fisher Scientific). Finally, the samples were sequenced on a HiSeq 1500 (Illumina, paired-end reads, 100 nt read length).
Overlapping paired-end reads were joined using the flash tool (68). The sequences were DpnII-digested in silico with the dpnII2E.pl script (kindly provided by James Davies, Oxford, UK) and mapped with Bowtie 1.1.0 (69). The perl script dpngenome3_1.pl (kindly provided by James Davies, Oxford, UK) was used to digest the hg19 genome in silico with DpnII. Interactions between distant DNA fragments were identified with the CCAnalyser2.pl script (kindly provided by James Davies, Oxford, UK). The data were averaged between triplicates, normalised by the total number of interactions for all captured fragments per time point, and visualised with R. Additionally, ChIP-seq and RNA-seq results were added to the visualisation. The analysed genes can be found in Supplementary Table S2. The CHiCAGO package (70) was used to calculate and visualize the loss or gain of significant chromatin interaction upon induction of EBV’s lytic cycle.
RESULTS
The aim of this study was to investigate the alterations of the cellular chromatin structure and the transcriptome of latently-infected B cells when BZLF1 is expressed and EBV’s lytic cycle starts. First, we determined the physiological levels of BZLF1 protein in cells that naturally support virus de novo synthesis. Next, we stably introduced two conditional oriP plasmids into Raji and DG75 cells, which are EBV-positive and EBV-negative Burkitt lymphoma cell lines, respectively. Upon addition of doxycycline, the two plasmids express regulated levels of full-length BZLF1 or BZLF1 devoid of its trans-activation domain. The established human B cell line Raji is latently infected with EBV, does not support EBV’s lytic phase spontaneously, but enters it rapidly and synchronously upon the induced expression of full-length BZLF1. We used this cell line (and parental controls) throughout our studies and investigated the cells in their non-induced state and upon addition of doxycycline for up to 15 h covering the pre-replicative lytic phase of reactivated EBV. Indistinguishable from full-length BZLF1, BZLF1 without its trans-activation domain binds to its many cognate sequence motifs, but cells that express it do not enter EBV’s lytic phase (18,34). We therefore used Raji cells engineered to express the conditional AD-truncated BZLF1 for certain control experiments as well as DG75 cells, an EBV-negative human B cell line, in which we addressed the functions of BZLF1 in the absence of other viral genes. Last, we studied the function of the > 5×105 binding sites of BZLF1 in cellular chromatin, which we had identified in our initial experiments. In a third cellular model, we found that the abundant number of BZLF1 binding sites acts as a sink to prevent the induction of EBV’s lytic phase upon low level expression of BZLF1. In turn, the many BZLF1 binding sites implement a dichotomous function that can only become active when BZLF1 accumulates at high molar concentration in the nucleus of a cell latently infected with EBV.
Lytic viral gene expression requires high levels of BZLF1
When latent EBV-infected memory B cells come in contact with their cognate antigens, the viral BZLF1 gene is activated and virus synthesis is induced (8). In vivo, only single memory B cells respond to this trigger, which makes it technically impossible to determine the protein levels of BZLF1 required to induce and/or support the lytic phase of EBV’s life cycle in these cells. Therefore, we turned to the B95-8 cell line because a small fraction within its population spontaneously enters the lytic phase and releases infectious virions. We first determined this fraction of cells in the lytic cycle to be 2.8 % on average by intra-cellular staining with an Alexa 647 fluorophore-coupled monoclonal antibody directed against BZLF1 and flow cytometry.
We then obtained whole cell protein lysates from a known number of B95-8 cells and compared the BZLF1 signal intensities by western blot immuno-detection with known molar amounts of a truncated BZLF1 protein (Supplementary Figure S1A) that was recombinantly expressed in HEK 293 cells and purified to high homogeneity (Supplementary Figure S1B). We also engineered Raji cells to carry a conditional BZLF1 allele on the oriP-based plasmid p4816 (18), added doxycycline for 6 h (Raji 6 h iBZLF1), and loaded protein lysates of non-induced Raji cells (Raji 0 h iBZLF1) onto the same gel. Depending on the cell line and its state of induction, protein equivalents that corresponded to different cell numbers were also loaded and the signal intensities were quantified as shown in Supplementary Figure S1A. Based on this approach, we estimated that each B95-8 cell that was found to express BZLF1 by intra-cellular staining contained 1.3×106 ± 0.2×106 BZLF1 dimers on average. BZLF1 levels in Raji iBZLF1 cells induced for 6 h with doxycycline were ∼ 3.5-fold higher (Supplementary Figure S1C), while the population of non-induced Raji iBZLF1 cells still expressed detectable levels of BZLF1 that were lower by a factor of about 42 (Supplementary Figure S1C). Using flow cytometry, we reproducibly found that the population of non-induced Raji iBZLF1 cells contained a very small fraction with low to medium levels of BZLF1 protein (data not shown).
We were concerned that the conditional expression plasmid p4816 is leaky and might express steady state levels of BZLF1, even in the absence of doxycycline that are sufficient to induce EBV’s lytic phase in latently-infected cells. Since Raji cells are incapable of fully supporting de novo virus synthesis (35), we turned to a derivative of HEK 293 2089 cells that releases the EBV strain wt/B95.8 (2089) upon BZLF1 expression (42). We stably introduced the p4816 plasmid into these cells termed HEK293 2089 iBZLF1 and evaluated the concentration of EBV virions in the cells’ supernatant, both in the absence and presence of different concentrations of doxycycline (Supplementary Figure S1D). The supernatants were used to infect Raji cells that turn GFP-positive upon infection with the EBV strain wt/B95.8 (2089). Virus amounts were expressed as Green Raji units (GRU), as described (42,47). As expected, only HEK 293 2089 iBZLF1 cells released infectious wt/B95.8 (2089) EBV in a dose-dependent fashion after adding increasing concentrations of doxycycline, but not the parental HEK 293 2089 cells (Supplementary Figure S1D). Without doxycycline, the supernatants of HEK 293 2089 iBZLF1 cells contained very low numbers of virus, similar to supernatants obtained from the parental HEK 293 2089 cells (Supplementary Figure S1D). In all subsequent experiments, a doxycycline concentration of 100 ng/ml was used in cells with the conditional expression plasmid p4816 (iBZLF1) to reach BZLF1 levels comparable to lytically-induced B95-8 cells (Supplementary Figure S1C).
In RNA-seq experiments using our conditional Raji iBZLF1 cell model, we compared viral lytic genes prior to and after induction of BZLF1 to test how the expression of BZLF1 influences their expression. Before induction, lytic genes were expressed at very low levels only, while latent genes were strongly expressed (Supplementary Tab. S1). Upon adding doxycycline for 6 h, Raji iBZLF1 cells readily expressed many viral genes that clearly mark the onset of EBV’s lytic phase. Supplementary Figure S1E shows the log2 fold differential expression of viral genes comparing non-induced and induced Raji iBZLF1 cells after ERCC spike-in normalisation (see below for experimental details). Very few viral genes were down-regulated such as EBNA1 and EBNA2 among other latent genes (Supplementary Tab. S1). Five of the six early lytic genes known to be essential for lytic amplification of viral DNA (71) were up-regulated 60- to > 250-fold (highlighted genes: BBLF3-BBLF2, BBLF4, BALF5, BMLF1-BSLF1 and BMRF1; Supplementary Figure S1E), except BALF2, which is deleted in Raji EBV DNA (37). Additional genes that contribute to the lytic replication of viral DNA (BKRF3, BRLF1, BHLF1 and BMLF1) (72) were strongly induced as well as BZLF1. The most strongly up-regulated gene, BDLF3.5, was induced almost 300-fold. Supplementary Table S1 provides a list of viral genes and their regulation.
BZLF1 binds many DNA sites in cellular chromatin
The binding sites and sequence motifs of BZLF1 were already identified and further analysed in the viral genome (12,73,74). BZLF1 is a member of the cellular AP-1 family of transcription factors (75), which are estimated to bind ∼ 500,000 sites in human DNA. We therefore hypothesised that BZLF1 might also bind to many accessible sites in the chromatin of human B cells. To address this possibility, we used our conditional iBZLF1 Raji cell model in comparison with DG75 iBZLF1 cells to identify BZLF1 binding sites in a genome-wide ChIP-seq approach in EBV-positive and EBV-negative cells, respectively.
We analysed different experimental conditions using the BZ1 monoclonal antibody directed against the dimerisation domain of BZLF1 (45) in our ChIP-seq analysis: Raji iBZLF1 and DG75 iBZLF1 cells in their non-induced state and after induction of full-length BZLF1 with 100 ng/ml doxycycline for 15 h. Parental EBV-negative DG75 cells served as a negative control.
In the parental EBV-negative DG75 cells, we found few mapping reads after ChIP-seq that rarely accumulated to peaks. This finding suggested that our ChIP-seq protocol delivers data with a very low background, because DG75 cells do not express BZLF1.
When BZLF1 is not inducibly expressed (0 h level) in Raji iBZLF1 or DG75 iBZLF1 cells in the absence of doxycycline (Supplementary Figure S2A) we found about 30,000 or 11,000 ChIP peaks, respectively, of which 70 % (Figure 1A) and 82 % (Supplementary Figure S2E) contained the known BZLF1 binding motif TGWGCGA (Figure 1C; Supplementary Figure S2G) also termed meZRE (12,18). BZLF1 preferentially binds this motif when its CpG dinucleotide contains methylated 5′-cytosine residues (12). When the cells were induced with doxycycline for 15 h more than half of the peaks with this motif were preserved (Figure 1B; Supplementary Figure S2F), but the total number of peaks increased dramatically (Figure 1A, B; Supplementary Figure S2E, F) in both Raji and DG75 iBZLF1 cell lines. In the induced state, about 146,000 and 231,000 ChIP peaks were identified, respectively, of which 92 % (Figure 1A) and 88 % (Supplementary Figure S2E) contained one or more copies of the less precisely defined consensus motif TGWGYVA (Figure 1C; Supplementary Figure S2G). At 0 h BZLF1 levels, this motif was not among the highly ranked motifs.
In a minor fraction of called peaks the MEME-Chip suite (57) failed to identify a common sequence motif for unknown reasons. We therefore turned to a visual inspection of the DNA sequences within the peak regions of this ChIP fraction. After inspection of peaks, in which no other motif could be identified before, we found an additional and related sequence motif TGWGYVT. This motif is very similar to the meZRE motif TGWGYVA except its last nucleotide residue. We continued our analysis with a computational search for this possible motif in the sets of called ChIP peaks. Data shown in Supplementary Figure S2A and S2I indicate that 58 % and 54 % of all peaks in Raji and DG75 iBZLF1 cells, respectively, contained the motif TGWGYVT when BZLF1 was expressed for 15 h. Taking both motifs with terminal A or T residues into consideration, 96 % respectively 95 % of all peaks contained the consensus motif TGWGYVW at 15 h BZLF1 levels in Raji (Figure 2A) and DG75 iBZLF1 cells (Figure 2D).
Figure 2.

Motif abundance as a function of BZLF1 levels in Raji and DG75 iBZLF1 cells. (A, D) The Venn diagram shows the number of peaks that contain either the TGWGYVA (A) or the TGWGYVT (T) motifs, both (A/T) or no identifiable motif (none). In Raji and DG75 iBZLF1 cells at 15 h BZLF1 levels 54 % and 46 % of the peaks contain both motifs (A/T), respectively. Within 96 % and 95 % of all peaks in Raji and DG75 iBZLF1 cells, respectively, at least one motif could be identified. (B, E) The number of the four BZLF1 binding motifs found in ChIP-seq peaks is plotted at 0 h and 15 h expression levels of BZLF1. At both levels and in both cell lines, the number of motifs ending with the residue A exceeds the number of motifs ending with T. The finding suggests that BZLF1’s ranked motif preference is TGWGCGA > TGWGCGT > TGWGYVA > TGWGYVT. (C, F) Calculation of the number of motifs per peak.
In total, in induced Raji and DG75 iBZLF1 cells, 145,544 and 231,019 peaks contained 579,911 and 794,729 TGWGYVW motifs, respectively, of which 344,383 and 431,282, respectively, terminated with A (Figure 1C, Supplementary Figure S2G) and 235,528 and 363,447, respectively, terminated with T (Supplementary Figure S2C, K). The peak number in Raji iBZLF1 cells includes the peaks we found in the viral genome (34). Our analysis also revealed a clear hierarchy of the BZLF1 sequence motifs in Raji and DG75 iBZLF1 cells as shown in Figure 2B and E. The highest affinity at 0 h BZLF1 levels appears to correlate with the motif TGWGCGA, whereas the weakest binding is associated with the motif TGWGYVT, to which BZLF1 binds also less frequently at 15 h BZLF1 levels compared with the TGWGYVA motif.
At 15 h BZLF1 levels, on average, in Raji and DG75 iBZLF1 cells each peak was found to contain about 4.0 and 3.4 BZLF1 binding motifs, respectively (Figure 2C and F).
High BZLF1 levels induce open chromatin at cellular BZLF1 binding sites, but a genome-wide loss of open chromatin
Next, we investigated the chromatin of EBV’s host cells and studied the consequences of inducing EBV’s lytic cycle and the binding of BZLF1 to cellular DNA. BZLF1 is a viral pioneer factor, binds to nucleosomal DNA, recruits chromatin remodelers such as INO80, and induces the local opening of repressed EBV chromatin (34). To extend this insight, we analysed the chromatin accessibility of non-induced and induced Raji and DG75 iBZLF1 cells by ATAC-seq. In these experiments, both cell lines engineered to carry a conditionally expressed BZLF1 gene lacking its transactivation-domain (AD-truncated BZLF1) were used as negative controls.
We combined the ATAC-seq data with the BZLF1 peaks identified at 15 h levels in Raji and DG75 iBZLF1 cells (Figure 1A, Supplementary Figure S2E) to analyse the local chromatin accessibility as a function of BZLF1 binding. Supplementary Figure S3 illustrates the bioinformatic approach. Average chromatin accessibility along all BZLF1 peaks in cellular chromatin is shown as meta-plots for Raji and DG75 iBZLF1 cells in Figure 3A and C, and as heatmaps in Supplementary Figure S4A and D, respectively. BZLF1 peak coverage and input signals are shown in Supplementary Figure S5, panels A–C and D–F, respectively, as meta-plots and heatmaps.
Figure 3.

Changes in cellular chromatin accessibility after induction of BZLF1 in Raji and DG75 iBZLF1 cells. (A, C) The meta-plot summarizes the accessibility at the 145,477 and 231,019 BZLF1 binding sites (Figure 1A and Supplementary Figure S2E) in Raji and DG75 cell chromatin, respectively, prior to and after induction of full-length or AD-truncated BZLF1. The average ATAC-seq coverages in the four different Raji and DG75 cell samples are plotted according to the nucleotide coordinates of the centers of the 145,477 and 231,019 BZLF1 peaks. In non-induced Raji and DG75 iBZLF1 cells (BZLF1 full-length, 0 h) the average ATAC-seq coverage is congruent with the coverage found in induced and non-induced Raji and DG75 cells that carry the conditional AD-truncated BZLF1 allele. At induced BZLF1 levels (full-length, 15 h) the average ATAC-seq coverage is substantially increased indicating a gain in chromatin accessibility. The inset provides the ATAC-seq coverage of 145,477 and 231,019 randomly sampled sequences in the chromatin of Raji and DG75 iBZLF1 cells, respectively, expressing full-length BZLF1 at 15 h levels after doxycycline-mediated induction. (B, D) The meta-plot summarizes the ATAC-seq coverage at the about 81,000 and 105,000 called peaks of open host chromatin identified prior to the induction of BZLF1 in both Raji and DG75 iBZLF1 cell lines, respectively. After induced expression of full-length BZLF1 the chromatin accessibility is strongly reduced in Raji iBZLF1 cells indicating that previously open host chromatin becomes globally inaccessible upon induction of EBV’s lytic phase. In DG75 iBZLF1 cells the effect is apparent, but less pronounced. Compared with non-induced cells (0 h), the ATAC-seq coverage is barely affected when the AD-truncated BZLF1 protein is expressed in both cell lines. The data summarize three independent biological replicates.
We analysed the average coverage of ATAC-seq reads at these sites in the two pairs of Raji and DG75 cell lines that express full-length or the AD-truncated BZLF1 protein in their non-induced and induced states. The visualisation in Figure 3A and C documents the opening of silent, compact chromatin at cellular BZLF1 binding sites that occurs only at induced 15 h levels of BZLF1 in Raji iBZLF1 cells and also in the EBV-negative DG75 iBZLF1 cells, respectively. An example is shown in Supplementary Figure S6A and D. The average peak of chromatin opening co-locates in both cell lines exactly with the peak center of the > 105 cellular BZLF1 binding sites in induced cells that express full-length BZLF1. A truncated BZLF1 protein without its transcriptional activation domain does not induce chromatin remodeling (Figure 3A, C, Supplementary Figure S4A4, D4), although it binds inactive chromatin as efficiently as full-length BZLF1 (12). This finding clearly supports our view that BZLF1’s activation domain recruits chromatin remodelers such as INO80 to silent chromatin (34). As a consequence, the previously repressed chromatin becomes accessible (Figure 3A, C, Supplementary Figure S4A2,D2) and acquires activating histone marks (data not shown) or is kept open (Supplementary Figure S6B, E). Accessibility at random locations (n = 145,477 resp. n = 231,019 in Raji and DG75 iBZLF1 cells) was not affected (insets in Figure 3A, C, Supplementary Figure S4B, E).
Prior to BZLF1 induction, Raji and DG75 iBZLF1 cell chromatin demonstrates a high number of accessible, open regions that are clearly identifiable by ATAC-seq (Supplementary Figure S6C, F). In latent Raji and DG75 iBZLF1 cells, the MACS2 peak caller found about 81,000 and 105,000 peaks of accessible cellular chromatin, respectively, that allowed us to calculate the average ATAC-seq coverage of open chromatin prior to and after induction of EBV’s lytic cycle. As shown in Figure 3B and D and in the corresponding heatmaps (Supplementary Figure S4C, F), and in a representative IGV browser image shown in Supplementary Figure S6C and F, we found that open chromatin in non-induced Raji and DG75 iBZLF1 cell chromatin closes upon induction of EBV’s lytic phase. Reduction of chromatin accessibility was evident at the majority of ATAC-seq peaks identified to be accessible prior to induction of BZLF1 in both cell lines (Figure 3B, Supplementary Figure S6C, resp. Figure 3D, Supplementary Figure S6F), although the effect in Raji iBZLF1 cells was stronger compared to lytically-induced DG75 iBZLF1 cells.
The data suggest that the induced expression of BZLF1 alone reduces the accessibility of previously open cellular chromatin globally as shown in DG75 cells, but the ensuing activation of EBV’s lytic phase reduces it further as it is evident in Raji cells upon induction. On the contrary, chromatin loci bound by BZLF1 open up site-specifically upon its induced expression independent of other viral factors.
Induction of EBV’s lytic cycle drastically reduces cellular transcripts in Raji iBZLF1 cells
We investigated the consequences of the induction of EBV’s lytic phase and BZLF1’s binding to cellular chromatin with respect to transcriptional regulation. RNA-seq experiments were performed in three different experimental layouts:
Steady state RNA transcript levels of parental Raji or DG75 cells were analysed and compared with the same cells incubated with doxycycline for 6 h (Figure 4A, D). Doxycycline did not regulate any genes according to our threshold criteria, which are provided in the figure. The violin plots shown in Figure 4A and D indicate that the log2 fold change of 95 % of the expressed genes, subsequently termed ‘population spread’, was located in a very narrow range in parental Raji and DG75 cells (Figure 4A, D, bottom panels).
We repeated the experiment with Raji and DG75 cells engineered to express a truncated version of BZLF1 (AD-truncated) without its transcriptional activation domain (18). Upon adding doxycycline for 6 h, no gene was identified to be regulated (Figure 4B, E). The population spread in Raji iBZLF1 AD-truncated cells slightly exceeded the range found in parental Raji cells (0.28 versus 0.42 log2 values; Figure 4A,B), while DG75 iBZLF1 AD-truncated cells and parental DG75 cells were comparable (Figure 4D, E).
Raji iBZLF1 cells engineered to express the full-length BZLF1 protein were induced by adding doxycycline for 6 h and were analysed by RNA-seq (Figure 4C). We used the ERCC RNA spike-in mix to be prepared for global changes in the cellular transcriptomes, to analyse the basic performance metric of the RNA-seq libraries, and to normalize the data during the steps of subsequent bioinformatic analyses according to this standard. The quantitative detection of the external spike-in RNAs demonstrated the linearity, quality and reproducibility of the experimental approach in both cell lines (Supplementary Figure S7A–D).
Figure 4.

Gene regulation in Raji and DG75 cells upon doxycycline-induced expression of full-length BZLF1 compared with a BZLF1 variant lacking its transcriptional activation domain. Three pairs of cell lines, parental Raji (A) and DG75 (D) cells, Raji iBZLF1 AD-truncated (B) and DG75 iBZLF1 AD-truncated (E) cells, and Raji iBZLF1 (C) and DG75 iBZLF1 (F) were induced with doxycycline for 6 h and compared with their non-induced counterparts. The analyses are based on the hg19 reference genome and three replicates of each condition and cell line. Viral genes and NGFR (used as a doxycycline-regulated reporter gene) were excluded from the analyses. (A, D) The transcriptomes of parental Raji and DG75 cells were analysed by comparing their untreated versus doxycycline-treated (6 h) states. No gene with more than 20 sequencing reads (vertical purple line) was found to be up- (>2.5×) or down-regulated (<0.4×) in the MA plots (upper and lower green horizontal lines, respectively) in both parental cell lines. The median is centered at zero in the violin plot (lower panel). The distance between the quantiles that encompass 95 % of all data points describes the spread of the gene populations which was determined to be 20.28 and 20.5, in parental Raji and DG75 cells, respectively. (B, E) RNA expression in Raji and DG75 cells with a conditional activation-domain (AD)-truncated BZLF1 allele upon doxycycline induction for 6 h. No gene was considered up- or down-regulated when the previously introduced parameters were applied. The violin plots reveal the very narrow spread of the gene population of 20.42 and 20.41, respectively, comparable to the results shown in panel A and D. (C, F) RNA expression in Raji and DG75 cells upon doxycycline induction of full-length wild-type BZLF1 (iBZLF1) after data normalisation according to ERCC spike-in RNA reads. The MA plot shows a strong global reduction of cellular mRNA transcripts 6 h after induction of BZLF1 in the Raji iBZLF1 cells, while this effect was not observed in the DG75 iBZLF1 cells. 91 and 109 genes were upregulated (upper horizontal green line), while transcripts of 7174 and 93 genes were reduced by a factor of at least 0.4 (lower horizontal green line), in Raji and DG75 iBZLF1 cells, respectively. In the Raji iBZLF1 cells the violin plot shows that the median of the gene population is reduced by a factor of almost three (2–1.56) indicating a global reduction of mRNA steady state levels, while the median is around zero (2–0.03) and unchanged in DG75 iBZLF1 cells. The distance between the 2.5 and 97.5 quantiles shows a spread of the gene population of 23.00 for Raji iBZLF1 and 21.86 for DG75 iBZLF1 cells.
In Raji and DG75 iBZLF1 cells we found 91 and 109 cellular genes with increased transcript levels, while 7174 and 93 showed a decrease, respectively (Figure 4C, F). The population spreads were much higher in both cell lines when compared to their controls, but only in Raji iBZLF1 cells the median of the population was shifted to a strong negative value indicating that the majority of cellular transcripts was substantially reduced on average. The median of DG75 iBZLF1 cells was found unaltered indicating that full-length BZLF1 induces considerable transcriptional noise, but its expression does not repress cellular transcripts globally as in Raji iBZLF1 cells. Individual triplicates are shown as boxplots in Supplementary Figure S7E–H.
The results indicate that the induced expression of full-length BZLF1 for 6 h and subsequent induction of EBV’s lytic cycle led to a dramatic and global drop of transcript levels in Raji iBZLF1 cells, only. Fewer than hundred genes were found up-regulated upon BZLF1 expression, an unexpected finding, because BZLF1 is a known transcriptional activator in the viral genome, which also binds to the cellular genome frequently (Figure 1, Supplementary Figure S2).
BZLF1 binding sites in proximity to TSS do not correlate with the magnitude of gene regulation
Since BZLF1 binds more than 145,000 and 231,000 sites in the cellular chromatin of Raji and DG75 iBZLF1 cells, respectively (Figure 1, Supplementary Figure S2), it seemed plausible that it can regulate promoters of cellular genes similar to the many viral promoters of lytic EBV genes (12,14,18). To address this question, we looked for peaks identified by ChIP-seq within promoter regions of genes found to be regulated in our RNA-seq experiments (Figure 4C, F). We limited our search to a region 5 kb upstream to 1 kb downstream of the TSSs (Figure 5C). About half of the up-regulated genes comprised one or more peaks within the defined limits of their TSSs, while about two-thirds of the down-regulated genes did not contain recognizable peaks attributed to BZLF1 binding in both cell lines (Figure 5A, E).
Figure 5.

Regulated genes and their association with BZLF1 ChIP-seq peaks and binding motifs in promoter regions. (A, E) The numbers of ChIP-seq defined peaks are plotted on the x-axis versus the magnitude of gene regulation expressed as ‘log2 fold change’ on the y-axis after doxycycline induced expression of full-length BZLF1 in Raji iBZLF1 (A) and DG75 iBZLF1 cells (E) for 6 h. Among the regulated genes that did not encompass BZLF1 peaks within their defined promoter regions 41 and 52 genes were found up-regulated (45 resp. 48 % of all up-regulated genes) and about 4,500 and 63 genes were down-regulated (63 resp. 68 % of all down-regulated genes) in Raji and DG75 iBZLF1 cells, respectively, as indicated by the dashed horizontal green lines. Among the regulated genes with BZLF1 peaks in their promoter regions, 50 and 57 were found up-regulated (55 resp. 52 % of all up-regulated genes) and about 2.600 and 30 genes were down-regulated (37 resp. 32 % of all down-regulated genes) in Raji and DG75 iBZLF1 cells, respectively. (B, F) The number of BZLF1 motifs as defined in Figure 1C and Supplementary Figure S2 (panels C, G and K) are plotted on the x-axis versus the magnitude of gene regulation on the y-axis as in panel A. BZLF1 motifs downstream and upstream of TSSs entered the analysis. The distribution of regulated genes (y-axis) with or without BZLF1 binding sites follows the scheme in panel A. (C). The peak analysis includes the promoter proximal region −5 kb/+1 kb of the transcriptional start sites (TSS) as indicated. (D) The cartoon schematically depicts two single peaks upstream and downstream of a TSS with three and one BZLF1 motifs, respectively, illustrating the basics of this analysis and the principal location of the BZLF1 motifs.
We also searched for a possible correlation between the absolute numbers of BZLF1 motifs within the promoter regions and the magnitude of gene regulation. Similar to the results shown in Figure 5A and E, we did not find a correlation. Certain promoters with few BZLF1 binding motifs were more profoundly regulated than promoters with multiple binding motifs (Figure 5B, F). Few cellular genes seemed to be exceptions to this rule in both cell lines (Figure 5B, F). A very small number of genes contained > 50 and up to 142 and 111 BZLF1 binding motifs in Raji and DG75 cells, respectively, within their promoter regions but these genes were barely regulated upon BZLF1 expression.
Together, the many identified BZLF1 binding motifs in promoter regions of cellular genes did not provide a clear function that could be ascribed to BZLF1. BZLF1 was characterized as a transcription factor of viral promoters, initially, but BZLF1 has also been proposed to act as an enhancer factor (76) similar to its cellular homologous factor AP-1 (25,26).
The cellular genome is about eighteen thousand times larger than the EBV genome, contains up to 26,000 genes and a much higher number of regulatory regions (77,78). We asked if, upon induction of the lytic phase, EBV governs these regulatory regions at the level of chromatin interactions. We also asked if the frequent binding of BZLF1 to cellular DNA together with the remodeling of silent chromatin may have a discernable effect. Toward this end, we selected several up-, down- and non-regulated cellular genes in Raji iBZLF1 cells to look at their interacting chromatin regions, which BZLF1 might regulate to control the expression of cellular genes.
Induction of EBV’s lytic cycle reduces cellular chromatin interactions
The regulation of cellular transcription results from an interplay between transcription factors, histone and DNA marks and chromatin interactions. The latter govern the spatial proximity between enhancers (or repressors) and promoter regions and regulate the activity of genes. The likelihood of functional interactions increases when distant regions are organized into loops by chromatin organizing proteins such as CTCF and cohesin.
We applied the Capture-C technique (79) to study physical chromatin interactions of selected promoters as a function of BZLF1 levels. From our RNA-seq experiment with Raji iBZLF1 cells (Figure 4C) we chose promoters of genes that were up- or down-regulated, but we also included genes that were not affected when BZLF1 was induced. With synthetic single-stranded RNA probes that hybridize to regions of approximately 5 kb up- and downstream of the transcriptional start sites (TSS) we focused on 53 and 49 selected genes in Raji and DG75 iBZLF1 cells, respectively, and pulled-down their promoter regions.
After data normalisation, we identified about 1.4 and 1.6 million interactions in the two cell lines between the pulled regions and proximal or distal flanking DNA fragments that had been generated by DpnII cleavage following the Capture-C protocol. For about 17,000 and 33,000 DpnII fragments with more than four interactions on average, a fold change could be calculated in a pair-wise comparison of non-induced Raji and DG75 iBZLF1 cells with the two cell lines induced for 15 h (0 versus 15 h). After 15 h the total number of interactions decreased (Figure 6). About 23 % and 9 % of the DpnII fragment pairs had lost more than half of their interactions while only 0.4 % and 1.2 % pairs showed a > 2-fold gain of interactions in Raji and DG75 iBZLF1 cells, respectively.
Figure 6.

Gain and loss of chromatin interactions in Capture-C experiments 15 h after induction of BZLF1 in Raji iBZLF1 and DG75 iBZLF1 cells. The two MA plots summarize the dynamics of interactions between the promoter regions of 53 and 49 analysed genes and their captured distal DpnII fragments in Raji and DG75 iBZLF1 cells, respectively. The x-axis shows the number of identified interactions in log2 scale, the y-axis shows the log2 fold change between the paired time points (0 h versus 15 h). (A, C) After 15 h of doxycycline-induced BZLF1 expression, almost 23.4 % and 8.9 % fragments lost more than half of their interactions (lower green horizontal line) compared with the status prior to induction of the lytic phase in Raji and DG75 iBZLF1 cells, respectively. Conversely, 0.4 % and 1.2 % DpnII fragments showed a more than two-fold increase of interactions (upper green horizontal line) in Raji and DG75 BZLF1 cells, respectively. The two percentages correspond to about 17,000 and 33,000 DpnII fragments in total that reached or exceeded the threshold of four interactions (dashed horizontal orange line). (B, D) The violin plots summarize panels A and C and show a median of –0.61 and –0.28 and a population spread of 22.37 and 22.18 comparing induced versus non-induced Raji and DG75 iBZLF1 cells, respectively.
After bioinformatic analysis and data normalisation, we visualised the close, medium, and distant physical chromatin interactions of these promoter regions in a range of 400,000 nucleotides each on both flanks of the TSS (Figure 7, Supplementary Figures S8–S16). The y-axes of the graphs indicate the number of chromatin interactions prior to the induction of BZLF1 (0 h, gray bars, both cell lines), 6 h (orange bars, Raji iBZLF1 only) and 15 h (blue bars, both cell lines) after adding doxycycline. In addition, we plotted BZLF1 binding sites at 0 and 15 h levels in both cell lines.
Figure 7.

Loss of chromatin interactions between the MYC locus and the heavy chain enhancer in Raji iBZLF1 cells. On the x-axis, individual chromatin interactions are shown as thin vertical lines of different heights that indicate their interaction frequencies (plotted on the y-axis as ‘number of interactions’) with the MYC promoter region (approximate −5/ +5 kb of the TSS). The promoter region is depicted as a green bar in the center of the plot, a gray arrow head points in the direction of transcription, and the light blue line at position ‘1’ indicates the TSS. Gray vertical lines indicate the chromatin interactions prior to BZLF1 induction (0 h), orange and blue lines enumerate interactions 6 and 15 h after adding doxycycline, respectively. The bottom part of the graph shows the positions of BZLF1 binding sites prior to (big, pink dots) and 15 h after adding doxycycline (small, green dots). The x-axis indicates the relative nucleotide coordinates (hg19 genome reference) encompassing two flanking regions 400 kb up- and downstream of the TSS. Due to a chromosomal translocation (vertical dashed red line), the heavy chain enhancer (HCE) on chromosome 14 drives the expression of the MYC locus on chromosome 8 in Raji cells. Prior to induction of BZLF1, two regions of the heavy chain enhancer (HCE) indicated A and B make frequent contacts (gray vertical lines) with the captured MYC locus. Chromatin interactions decrease after 6 h (orange lines) and are further reduced to about 20 % (blue lines) 15 h after BZLF1 induction. Both interacted HCE regions A (chr14:106,031,165–106,031,487) and B (chr14:106,150,744–106,151,066) harbor the exact same DNA sequence of 324 bp in length and contain numerous BZLF1 ChIP-seq peaks with 18 BZLF1 motifs. In Raji cells, 6 h after BZLF1 induction the level of MYC transcripts is reduced to 2 % compared with the non-induced cells.
We visualised the actual situation for several genes, to inspect the individual gain and loss of chromatin interactions. Supplementary Figure S8 shows the CD68 gene, which is not regulated (0.99-fold) in Raji iBZLF1 cells, but transcriptionally up-regulated (2.53-fold) in DG75 iBZLF1 cells 6 h after induction of the lytic cycle. The number of locus interactions of the promoter region with flanking chromatin modestly increased by ∼ 7 % 6 h after induction of the lytic cycle relative to the interactions identified prior to its induction in Raji iBZLF1 cells. Fifteen hours after induction the interactions are reduced in both cell lines to about 63 % and 90 %, respectively, relative to their non-induced states. A very similar pattern of altered chromatin interactions was found in the locus of the strongly down-regulated BTG2 gene (Supplementary Figure S9) and the up-regulated COL2A1 gene (Supplementary Figure S10). In Raji cells, the number of interactions at the BTG2 and COL2A1 loci slightly increased by 12 % and 15 %, respectively, 6 h post-induction, but decreased by 30 % and 16 % in both Raji and DG75 iBZLF1 cell lines 15 h after induction following the general trend shown in Figure 6.
The altered numbers of interactions were particular impressive when the strong, long-range interactions of the CXCR4 (Supplementary Figure S11) and MIR155HG (Supplementary Figure S12) genes were considered. The CXCR4 gene is transcriptionally down-regulated in both cell lines (Supplementary Figure S11) while with the MIR155HG gene is considerably up-regulated in DG75 iBZLF1 cells (Supplementary Figure S12). Chromatin interactions of the CXCR4 and MIR155HG genes increased on average by 13 % and 12 % in Raji iBZLF1 cells 6 h after induction of BZLF1 (Supplementary Figures S11A and S12A, respectively). After 15 h, the long-range interactions of the CXCR4 and MIR155HG loci were reduced by 39 % and 34 % in Raji iBZLF1 cells and by 25 % and 19 % in DG75 iBZLF1 cells, respectively. The loss of interactions was particularly impressive in Raji cell chromatin (Supplementary Figures S11A and S12A), presumably because long-range interactions in Raji iBZLF1 cells were much more prominent prior to induction of BZLF1 when compared to DG75 iBZLF1 cells (Supplementary Figures S11 and S12).
All analysed genes but one followed the observed kinetics and showed a considerable loss of chromatin interactions over time. The exception is KCNQ5. Here, the interactions of the flanking region upstream but not downstream of the KCNQ5 gene remained stable or even slightly intensified 15 h after BZLF1’s induction (Supplementary Figure S13) in both cell lines.
Additional examples include E2F2 and ID3 as well as TLR6 and TLR10, which constitute two separate gene clusters as shown in Supplementary Figures S14 and S15. Again, these genes are in line with the global reduction of chromatin interactions as a late consequence of BZLF1 expression. Interestingly, the two neighboring genes in both examples share frequent interactions with distant DpnII fragments even in both cell lines. Still, each individual gene is also engaged in gene-specific chromatin-chromatin contacts.
The MYC gene, which showed a reduction of its steady state transcript levels by a factor of 50 in Raji iBZLF1 cells is an extreme example that demonstrates the reduction of chromatin interactions. In the Burkitt lymphoma cell line Raji, the MYC gene is not regulated by its genuine cis-acting elements, but the gene is under the control of the heavy-chain immunoglobulin enhancer due to a translocation between the chromosomes 8 and 14. The x-axis in Figure 7 is discontinuous and a vertical red dashed line upstream of the MYC promoter indicates the chromosomal break-point. The two DpnII fragments within the heavy chain enhancer, which make the most frequent contacts with the MYC promoter regions are indicated by A and B (Figure 7). Both interacting sites lose ∼ 20 % of their contacts with the MYC promoter locus already 6 h after induction of EBV’s lytic cycle. After 15 h, the number of chromatin contacts at the interacting sites A and B are reduced by 60 % and 80 %, respectively. The two distinct DpnII fragments, A and B, include a 323 nt long repeat that comprises 18 partially overlapping BZLF1 sequence motifs. In our ChIP experiments, both A and B fragments were found to be bound by BZLF1 in Raji and DG75 iBZLF1 cells.
In addition, we used the Chicago R package (70) to determine the statistical significance of interactions. The panels in Supplementary Figure S17 depict the Supplementary Figures S8–S15 in miniature, but it additionally highlights significant interactions and visualizes their enrichment or loss over time. This statistical analysis underscores our previous interpretations in both cell lines.
In summary, all analysed loci show reduced local and distal chromatin interactions 15 h after induction of EBV’s lytic cycle in Raji iBZLF1 cells. In DG75 iBZLF1 cells the effect is also visible, but its magnitude and significance (Supplementary Figure S17) are lower. The binding of BZLF1 to certain distant interacting sites is often apparent and occasionally impressive as in the example of the MYC gene in Raji iBZLF1 cells suggesting that its binding to these distal regions directly or indirectly reduces the frequency of chromatin interactions. However, this apparent linkage is not supported by our further analysis. In both cell lines, DpnII fragments bound by BZLF1 in our ChIP-seq experiments and enriched in the Capture-C experiments did not differ from DpnII fragments to which BZLF1 does not bind (Supplementary Figure S18). It thus appears as if BZLF1 alone (in DG75 cells) or in conjunction with other viral genes (in Raji cells) can initiate so far unknown mechanisms to reduce cellular chromatin interactions and alter the 3D structure of its host cell chromatin, presumably supporting EBV’s lytic phase.
Induction of EBV’s lytic cycle depends on a molar switch mechanism
Our results shown in Figure 2 and Supplementary Figure S2 indicate that > 5×105 BZLF1 binding motifs exist in cellular DNA. We also learnt that a B95–8 cell which expresses gp350 and thus supports EBV’s full lytic cycle contains 1.3×106 ± 0.2×106 BZLF1 dimers on average (Supplementary Figure S1C). Together, these observations suggest that the abundant BZLF1 binding motifs in cellular DNA together with the 85 binding motifs in EBV DNA (18) might cooperate to ensure that the viral lytic phase is only induced when a certain threshold concentration of BZLF1 dimers is reached in the nucleus. Thus, the many BZLF1 binding sites in cellular chromatin might act as a sink to prevent the induction of EBV’s lytic phase when BZLF1 is expressed at low and functionally insufficient levels. This hypothesis was further supported by the notion that EBV’s lytic cycle was not induced when BZLF1 was expressed at low levels in Raji cells (Supplementary Figure S1D). We tested this hypothesis in a third cellular model.
We engineered a cell line based on HEK293 cells that carries several copies of a recombinant EBV genome, which is stably maintained under continuous selection as described previously (42,80). Into this cell line, termed 9G10, we introduced a chimeric gene consisting of full-length BZLF1 fused to the ERT2 variant of the hormone binding domain of the human estrogen receptor (15). The BZLF1:ERT2 fusion protein is constitutively expressed at high level from the promoter of the chromosomally-integrated retroviral vector, but is localized in the cytoplasm, where the chimeric BZLF1:ERT2 transcription factor is tethered to heat-shock proteins. Only upon addition of tamoxifen, an estrogen derivative, to the cell culture medium, BZLF1:ERT2 is released and shuttles to the nucleus where BZLF1 binds to its cognate DNA binding motifs to activate downstream genes. Successful induction of EBV’s lytic phase can be easily monitored by the surface expression of the viral gp350 glycoprotein. The advantage of this model lies in its immediate, presumably synchronous and dose-depending loading of the cell's nucleus with BZLF1 as a function of tamoxifen added.
To evaluate the ensuing consequences, we analysed single cells using high resolution laser scanning microscopy coupled with an automated analysis pipeline to obtain statistically solid single cell data. We added different concentrations of tamoxifen to 9G10 cells cultured on cover slips to induce nuclear shuttling of BZLF1. Twelve and 72 h after addition of tamoxifen, cells were fixed and stained for the analysis of four markers: (i) BZLF1 to observe the dose-dependent transfer of BZLF1 from the cytoplasm to the nucleus; (ii) CD147, a universal surface marker to delineate the plasma membrane of cells together with (iii) the viral gp350 glycoprotein, which appears on the cell surface concomitant with the expression of late viral genes during EBV’s lytic phase and (iv) DAPI to assess the cellular nucleus.
Figure 8, panel A shows exemplary microscopic images of the location of BZLF1 in cells prior to (0 nM) and 12 h after having added increasing concentrations of tamoxifen. Without tamoxifen, the majority of BZLF1 molecules were clearly cytoplasmic. Already with 5 nM tamoxifen, BZLF1 visibly transferred to the cells’ nuclei (Supplementary Figure S19, upper panel). With increasing tamoxifen concentrations, the nuclear BZLF1 signal became stronger and appeared to be exclusively nuclear at only 20 nM tamoxifen (and beyond) upon microscopic inspection. Panel B of Figure 8 shows the level of gp350 expression as a function of increasing tamoxifen concentrations 72 h after induction. Starting at 80 nM tamoxifen, a clear signal of gp350 could be detected at the plasma membranes of many cells, but not at lower concentrations of tamoxifen. Supplementary Figure S19 shows the microscopy images in panel A and B of Figure 8 together with BZLF1, gp350, DAPI and CD147 signals 12 and 72 h after the addition of different concentrations of tamoxifen.
Figure 8.

Expression of the late lytic viral glycoprotein gp350 as a function of nuclear levels of BZLF1. The HEK293 cell line 9G10 was engineered to study the induction of EBV’s lytic phase with increasing doses of tamoxifen. The cell line carries several copies of a recombinant EBV genome and expresses a chimeric BZLF1:ERT2 protein, which is cytoplasmic. Upon addition of tamoxifen, BZLF1:ERT2 shuttles to the nucleus where BZLF1 binds to its cognate DNA binding motifs. Depending on a threshold concentration of nuclear BZLF1, tamoxifen can induce the lytic phase of EBV’s life cycle, which is monitored by the expression of viral gp350 glycoprotein at the plasma membrane. (A) Cytoplasmic and nuclear BZLF1 signals 12 h after the addition of different concentrations of tamoxifen as indicated. Scale bar indicates 10 micron. (B) Visualisation of the gp350 signal 72 h after the addition of different concentrations of tamoxifen. No gp350 signal was visible at the cell membrane in the absence and up to 20 nM tamoxifen. A clear gp350 signal becomes apparent at 160 nM tamoxifen in the series of selected microscopic images shown. Supplementary Figure S19 provides a complete set of images covering all four channels analysed (DAPI, BZLF1, CD147, gp350) and all incremental doses of tamoxifen and at both time points. The images are example derived from four biological and technical replicates. (C) Summary data from a total of about 18,000 cells per replicate (72,000 cells in total) analysed for cytoplasmic and nuclear BZLF1 signals 12 h after the addition of escalating doses of tamoxifen (left and middle plots; small numbers underneath the error bars indicate nM concentrations of tamoxifen). Expression of gp350 was determined 72 h after tamoxifen addition (right plot). Means and standard errors of fours experiments are shown for all tamoxifen concentrations tested (5, 10, 20, 40, 80, 160, 320, 640 and 1,000 nM) normalised to the situation without tamoxifen (0). BZLF1 signals are colored in red, gp350 signals are shown in orange. The y-axes show normalised intensity values. Supplementary Figure S20 describes the analysis pipeline of the microscopic images that led to the data shown in panel C.
For a statistically solid analysis of cytoplasmic versus nuclear BZLF1 signals as well as membranous gp350 signals in a population of cells, an automated analytic pipeline was set up. To identify single cells, their nuclei were defined with the help of the DAPI signal. This information was combined with the CD147 signal located at the cells’ plasma membranes to identify cell borders (see Supplementary Figure S20 for technical details). The resulting masks were used to define the cytoplasmic and nuclear compartments of the cells, followed by the unequivocal intra-cellular localization of BZLF1 signals and the calculation of their cytoplasmic or nuclear mean intensities together with signal intensities of gp350 at the cells’ plasma membranes as functions of tamoxifen concentrations and time.
Figure 8, panel C summarizes the results of this analysis, which is based on ∼ 72,000 cells in total. Levels of the cytoplasmic BZLF1 signal decreased by about half with only 5 nM and decreased further to reach the lowest value with 320 nM tamoxifen 12 h after tamoxifen induction. This trend broke with 640 and 1,000 nM tamoxifen for unknown reasons (Figure 8C, left plot). As a contra-movement, the nuclear BZLF1 signal increased continuously to reach very high levels at 40 nM and its peak at a concentration of 80 nM tamoxifen (Figure 8C, middle plot). This dynamic and steep increase saw some fluctuations and a slight decrease of signal intensities at higher tamoxifen concentrations.
A gp350 signal at the plasma membrane was not detectable with concentrations of up to 20 nM tamoxifen 72 h after induction (Figure 8C, right plot). For comparison, at this concentration of tamoxifen, BZLF1 reached half maximal nuclear levels after 12 h (Figure 8C, middle plot). At a tamoxifen concentration of 40 nM, an initial gp350 signal became detectable, which increased in intensity to reach its maximum at a tamoxifen concentration of 160 nM, but decreased slightly at even higher tamoxifen concentrations. Supplementary Figure S19 provides a complete panel of micro-graphs for further inspection.
The careful analysis of this model supported our view that the many BZLF1 binding sites in cellular chromatin act as a sink to prevent an induction of EBV’s lytic phase when BZLF1 is expressed at insufficient levels. On the contrary, the observed regulation characteristics suggests that BZLF1 levels that reach a critical threshold lead to an immanent induction of EBV’s lytic phase in a dichotomous, i.e. binary manner.
DISCUSSION
After infection and a short period of pre-latency, EBV establishes its latent phase, which results in a very stable host–virus relationship. ln this phase, only very few proteins are expressed in vivo probably to avoid the recognition and elimination of virus-infected cells by cellular immunity. Upon escape from latency, the immediate early viral protein BZLF1 is the first protein of the lytic phase that is expressed. It induces the synthesis of additional viral proteins important for the complete transcriptional activation of the lytic phase, viral factors for the autonomous viral DNA replication, and viral genes that fend off the antiviral immunity of the host organism. Therefore, the host cell has to be manipulated and reorganized to free macro-molecules, energy, cellular machines, and transport capacity to support directly viral transcription, DNA replication, and viral morphogenesis, processes that also require space and occupy nuclear compartments to allow EBV’s production and assembly. Since BZLF1 shows a strong structural homology with members of the cellular AP-1 protein family (81) and binds their sequence motifs (82) genome wide, we not only examined global changes in the cellular genome and transcriptome, but also asked if BZLF1 is directly involved in these changes. We used two different cell lines, Raji (EBV-positive) and DG75 (EBV-negative) to be able to distinguish between effects directly induced by BZLF1 and effects induced by viral proteins induced by BZLF1.
BZLF1 reaches a high protein level in the lytic phase of B95-8 cells
In a first step, we investigated the BZLF1 protein levels in the fraction of B95–8 cells that are in the lytic phase of infection. We found very high molecule numbers per cell (Supplementary Figure S1C) that are also reached upon doxycycline-mediated induction in our Raji cell model. In this model, the conditional BZLF1 allele has a basal leaky expression, but at this level BZLF1 was incapable of inducing EBV’s lytic cycle in Raji iBZLF1 cells (18) and in HEK 293 2089 iBZLF1 cells (Supplementary Figure S1D, E). With this knowledge, we assumed that our model truly reflects the functions of BZLF1 in latently-infected and lytically-induced B cells.
BZLF1 binds two major motifs in the cellular genome with high frequency and specificity
At 15 h levels, BZLF1 binds to several hundred thousand sites in the Raji and DG75 cell genome, a finding which is in agreement with the diffuse nuclear distribution of BZLF1 (83). We identified two major sequence motifs in 96 % and 95 % of all called BZLF1 peaks in Raji and DG75 iBZLF1 cells, respectively. The high number of differently composed DNA sequence motifs led us to rank them according to their presumed relative affinities of BZLF1 binding (Figure 2B,E). Their composition partially confirms previous findings of Ramasubramanyan et al. (76), who, very much in contrast to our study, identified 5,000 BZLF1 binding sites in cellular genomic DNA, only. Our study now extends this set of BZLF1 binding motifs and reveals that BZLF1 also binds closely related motifs that terminate with a thymidine residue (Supplementary Figure S2C, K).
The many cellular BZLF1 binding motifs might have an obvious function supporting EBV’s life style. A cell that incompletely activates EBV’s lytic phase is likely prone to be eliminated by immediate antiviral T cell responses, because the many lytic immune evasive viral genes are not timely and properly expressed to fend off cytotoxic CD4+ and CD8+ effector T cells (84). It is therefore plausible that the transition from the latent to the lytic phase operates as a dichotomous switch and not as a rheostat. During a perhaps erratic expression of low BZLF1 levels the cellular genome with its abundant number of high-affinity meZRE binding motifs might act as a sink abrogating the activation of a subset of viral lytic promoters by chance.
In this context, it is also interesting to speculate that BZLF1’s numerous binding sites in cellular DNA have evolved to differ from the related DNA binding motifs of AP-1 sites because EBV does not intend to activate the discrete sets of cellular targets that AP-1 induces. Rather, the abundance of BZLF1 binding sites might buffer low level expression of BZLF1 to prevent an erroneous, incomplete and probably untimely induction of EBV’s lytic phase. Only a strong inducing signal leads to BZLF1 levels sufficient to bind both cellular and viral CpG containing high-affinity motifs (TGWGCGA, Figure 1C, Supplementary Figure S2G and TGWGCGT, Supplementary Figure S2C, K) and, subsequently, the two canonical non-CpG motifs (TGWGYVA, Figure 1C, Supplementary Figure S2G and TGWGYVT, Supplementary Figure S2C, K). We know that BZLF1 must bind certain low affinity binding sites in the viral lytic origin of DNA replication to support efficient viral DNA amplification (12,85). Thus, the diverse and abundant BZLF1 binding motifs in cellular DNA (>5 × 105) together with the 85 binding motifs within the 66 identified BZLF1 ChIP peaks in EBV DNA (18) likely cooperate to implement a molar switch that only flips when a certain threshold concentration of BZLF1 protein is reached. Since BZLF1 shows the same binding behavior in EBV positive and negative cells, it is very likely that it binds the cellular genome independently of other viral proteins.
The presumed functions of cellular BZLF1 binding sites
BZLF1's role in cellular chromatin is not obvious, but it might act at different levels. BZLF1 could induce or block the expression of a few cellular factors that serve important functions in EBV’s lytic phase similar to its role on viral genomic DNA. For example, BZLF1 has to induce the BMLF1-BSLF2 gene (86) such that they together can activate certain early EBV promoters (87). BZLF1 acts as a replication factor together with the viral factors BALF2, BALF5, BBLF2/3, BBLF4, BMRF1 and BSLF1 to initiate lytic viral DNA replication (88), but BZLF1 also induces the expression of these genes mainly via meZRE motifs (Figure 1C) with methylated CpG dinucleotides (12) and probably together with BRLF1. Similarly, BZLF1 has been reported to induce (or repress) numerous cellular genes (89–100), but a clear picture depicting how BZLF1 regulates cellular targets is lacking. BZLF1 is known to induce a DNA damage response (DDR), which seems to be needed for the full expression of BZLF1 and other downstream viral factors such as BMRF1. The induction of a DDR requires that BZLF1 binds DNA (101). Thus, cellular BZLF1 binding sites might contribute to DDR induction, but how a DDR could functionally support EBV’s lytic phase is uncertain. Lastly, the many cellular BZLF1 binding sites might be non-functional similar to 46–99 % of all binding sites of 59 different cellular transcription factors (102). The binding of transcription factors to these sites did not lead to a measurable change in transcription levels of putative target genes (102), but in the case of BZLF1 the many binding sites in cellular chromatin might act as a sink to prevent the erratic induction of EBV’s lytic phase and enable the activation of EBV's lytic phase via a dichotomous switch.
We challenged this hypothesis and designed an experimental approach shown in Figure 8 (and Supplementary Figures S19 and S20). We made use of a hormone-dependent nuclear shuttling of cytoplasmic BZLF1 protein to bypass the potential problem of a conditionally induced and thus heterogeneous de novo expression of the BZLF1 gene. We found that half maximal nuclear levels of BZLF1 (at 20 nM tamoxifen) were incapable of inducing the expression of gp350 at the cell surface (compare middle and right plots of Figure 8C), which serves as a proxy of EBV’s lytic phase. At a concentration of 40 nM tamoxifen nuclear BZLF1 levels were almost maximal when initial gp350 signals became detectable at this escalating dose 60 h later (ibid). This observation is in agreement with the idea that the vast number of meZREs in cellular chromatin are preferentially bound at low and medium levels of nuclear BZLF1 protein. For the activation of EBV’s full lytic phase BZLF1 also has to bind canonical ZREs with low binding affinities in the lytic origin of DNA replication, oriLyt, of EBV as mentioned above (12,85). The essential sites likely compete with several hundred thousand ZRE motifs in cellular DNA (we have no detailed information on HEK293 cells used in Figure 8, but information on Raji and DG75 cells in Figure 2 is concordant with this estimate). We conclude that a certain threshold level of BZLF1 dimers in cellular chromatin has to be reached to induce EBV’s lytic cycle. The conclusion supports our hypothesis of cellular BZLF1 binding sites to act as a sink that results in the implementation of a molar dichotomous switch. This switch might guarantee that molecular noise does not interfere with making a discrete biological decision that will generate virus progeny in the proper cells, eventually. Canceling of molecular noise and decision making are fundamental aspects in gene regulation in all cells (103).
It is pure speculation but certain cellular transcription factors and the regulation of their target genes could follow the viral principle and use an excess of binding sites as a functional sink. Many transcription factors including members of the AP-1 family have a plethora of binding sites in cellular chromatin. Their functionality has been mostly enigmatic (102), but they, too, could act as a sink for steady-state protein levels of transcription factors to allow an acute and powerful response to certain stimuli or stress but only when the level of these transcription factors exceeds a certain threshold upon induction. Similar to our viral model, this threshold could be set by the ratio of functional (i.e. gene-associated) versus non-functional (i.e. sink-associated) binding sites that a given cellular transcription factor can access and bind depending on epigenetic barriers.
BZLF1 induces open chromatin at its cellular binding sites, but open chromatin in non-induced cells closes upon expression of BZLF1
Regions of open chromatin allow the binding of cellular factors, protein complexes and large nuclear machines. Open chromatin is a prerequisite for chromatin-chromatin interactions that contribute to the formation of topologically associating domains (TADs), chromosome territories and, ultimately, the nuclear architecture. Our genome-wide ATAC-seq results indicate that the global accessibility of previously open cellular chromatin is considerably reduced upon induction of BZLF1 in Raji and DG75 iBZLF1 cells (Figure 3B and D). The effect is clearly stronger in Raji iBZLF1 cells and might be supported by additional viral proteins, which are induced by BZLF1. To our knowledge, only during embryonic development the loss of chromatin-chromatin interactions is a noted feature, which is seen in conjunction with the formation of new TADs (104). Alternatively, an early and general loss or even collapse of discrete chromatin-chromatin interactions might prepare for a subsequent spatial regulation of cellular chromatin including the increase in volume of the nucleus of herpes virus infected cells, when replication compartments or amplification factories form (6,7).
Very much in contrast to the global loss of previously open cellular chromatin, the binding of BZLF1 to silent chromatin increases its local accessibility in Raji iBZLF1 as well as DG75 iBZLF1 cells. BZLF1 is a pioneer factor (34) and shares this function with other pioneer factors that bind to repressed and generally inaccessible sequence motifs and recruit chromatin remodelers to mobilize or even evict nucleosomes at and in close proximity of their cognate binding sites. Since the effect is identical in both cell lines no additional viral protein is involved in the local opening of chromatin. It is currently unclear why BZLF1 opens chromatin at its many cellular binding sites but this phenomenon could contribute to the transcriptional noise seen in both cell lines upon BZLF1 expression (Figure 4C, F).
EBV’s lytic cycle induces a massive reduction of cellular transcripts in Raji cells
A recent report suggested that BZLF1 might directly or indirectly regulate ∼ 10 % of all protein-encoding genes in B cells (76). Our data from DG75 iBZLF1 cells are in strong contrast to this report and show that upon induction of BZLF1 only few genes are substantially up- or down-regulated in these cells. The expression of BZLF1 affects almost all cellular genes to a limited extent, but the regulation has no directionality, i.e. the median value in the transcriptome of DG75 cells does not change and thus gene regulation appears disordered (Figure 4F).
In total contrast, in Raji cells only few cellular genes are up-regulated, while the majority of transcripts (> 7,000) are strongly reduced (Figure 4C) together with a substantial spread of global transcriptional activity in the EBV-positive cells. Conversely, viral transcripts are massively up-regulated to accumulate to up to > 140,000 reads per viral gene (Supplementary Figure S1E).
The global reduction of cellular transcripts depends on BZLF1’s trans-activation domain (Figure 4B, C). BZLF1 can cause a restructuring of the cellular chromatin architecture as seen in Figures 6 and 7 and in numerous examples in Supplementary Data with a concomitant disturbance of cellular transcription in DG75 and Raji iBZLF1 cells. Since the global down-regulation of cellular genes only occurs in the EBV-positive Raji iBZLF1 cell line, it can be assumed that viral lytic genes other than BZLF1 cause this reduction. A likely viral candidate is the BGLF5 gene that is highly expressed (87,000 reads, 170-fold induced) already 6 h after the activation of EBV’s lytic phase. BGLF5 is an RNA exonuclease (105) that destroys viral as well as cellular mRNAs non-specifically (106) causing the so-called host-shutoff. Additionally, BGLF5 was described to potentially contribute to immune evasion (107,108) and to support the nuclear translocation of the PABPC protein together with BZLF1. A nuclear accumulation of PABPC leads to hyper-adenylated transcripts blocking their nuclear export and translation (109,110).
Ramasubramanyan et al. also analysed the transcriptome of Akata cells, an EBV-positive Burkitt lymphoma cell line (76), but did not see a global reduction of cellular transcripts in the lytic phase. The authors identified 2,300 genes of which ¾ were up- and ¼ down-regulated 24 h after lytic induction of Akata cells. In contrast, we found 99 % of cellular genes down- and 1 % up-regulated in Raji iBZLF1 cells 6 h post-induction (Figure 4C). The discrepancy between both studies can be explained by the chosen threshold levels of bioinformatic analysis and mainly by the method of normalisation. Based on the published literature (105,106,109,110) we expected the mRNA content between non-induced and induced cells to differ substantially. Therefore, we added known molar amounts of poly-adenylated ERCC spike-in RNAs to total RNA samples from identical numbers of viable cells. Conventional data processing and normalisation approaches mostly rely on average principles and center the mean of the fold change around zero independently of the RNA amount. We know from a conventional analysis of our data that we would have missed the profound global transcriptomic modifications in this model of viral activation had we chosen this bioinformatic approach.
Cellular gene regulation and promoter-binding of BZLF1 are not necessarily linked
BZLF1 is known to be a promoter factor in the viral genome and was also described to up- or down-regulate cellular genes in different cells (89–100). When we correlated cellular gene regulation and the occurrence of BZLF1 sites in promoter regions, many genes with BZLF1 binding sites were not regulated in both cell lines. This finding is in accordance with previous reports in which transcription factors were found to regulate only a subset of genes with which they are spatially associated (102,111,112). In Raji iBZLF1 cells, BZLF1 binding sites could be found in 37–55 % of apparently regulated genes, only (Figure 5), while these sites could be found in 32–52 % in DG75 iBZLF1 cells. This correlative finding contradicts the idea that BZLF1 binds to cellular promoters and induces cellular gene expression. No BZLF1 binding sites were identified in promoters of 45–63 % of genes in Raji iBZLF1 and 48–68 % in DG75 iBZLF1 cells, respectively, that are regulated after induced expression of BZLF1. Clearly, at the level of the lytically-induced cells, BZLF1 does not follow the rules found in the viral genome, where BZLF1 binds in close proximity to the transcriptional start sites of its regulated genes (12).
Upon induction of EBV’s lytic cycle, cellular chromatin connections detach
Using the Capture-C technique, we studied the chromatin structure of 53 and 49 selected cellular genes in Raji and DG75 iBZLF1 cells, respectively. We asked if BZLF1’s local binding contributed to their regulation. In latently-infected cells, we identified many distal regions that contact the promoters’ captured regions −5 kb/+5 kb of the transcriptional start sites. Uniformly, the number of chromatin interactions rises slightly 6 h post-induction, but is substantially reduced after 15 h in Raji iBZLF1 cells irrespective of whether the analysed genes were up-, down- or non-regulated. In DG75 iBZLF1 cells, the regulation of the chromatin-chromatin interactions shows a similar but reduced tendency as in Raji iBZLF1 cells.
In both cell lines, DpnII fragments bound by BZLF1 do not show a stronger decrease of interactions, compared to DpnII fragments not bound by BZLF1 (Supplementary Figure S18). The regulation of the MYC locus could be an exception, because BZLF1 might be directly involved in the reduction of chromatin-chromatin contacts between the heavy chain enhancer (HCE) and the MYC locus (Figure 7 and Supplementary Figure S16). No BZLF1 binding sites are located in the promoter region of the MYC gene, but it forms strong contacts with HCE, which is rich in BZLF1 binding sites. Already 6 h after induction, HCE-MYC chromatin interactions decrease considerably in Raji iBZLF1 cells and the MYC mRNA level is reduced to two percent only (Figure 7). After 15 h, the contacts of the heavy chain enhancer with the MYC locus are mainly erased.
Clearly, studying 53 and 49 selected cellular loci in Raji and DG75 cells, respectively, considers only 0.1–0.5 % of about 10,000 to 50,000 3D chromatin interactions that exist in mammalian cells (113,114). The Capture-C technology requires a pre-selection of loci of interest, which clearly introduces a bias in the analysis and limits the significance of our findings. Thus, Hi-C data would uncover topologically associating domains within the entire 3D space and their presumed loss as a function of BZLF1 and at global resolution. It is interesting to note that preliminary 3C experiments revealed the loss a prominent chromatin interaction within the viral mini-chromosome within hours after lytic phase induction, suggesting that the loss of chromatin interactions is not restricted to about 50 selected cellular loci during the early lytic phase of EBV. This aspect needs more experimental work also in the light of a most recent paper that reports additional 3D modifications within genomic EBV DNA upon lytic phase induction in Akata cells (115).
For our aim, the Raji cell model is advantageous because induction of EBV’s lytic phase is a well-established and reliable function in this cell line. The viral BALF2 gene, which is deleted in Raji EBV DNA (37), blocks the progression of the lytic phase prior to viral DNA amplification and prevents the downstream formation of viral replication compartments. Clearly, their formation would introduce yet another layer of complexity in studying the architecture of chromatin in lytically-induced cells, but this block in Raji cells also limits our experimental analyses, which only encompass the very early events in EBV’s lytic phase. On the contrary, analyzing host chromatin composition in cells such as the EBV-positive Akata cell line that supports the entire lytic cycle including replication compartments (5,6) and amplification factories (7) would add to this study. Given the microscopically impressive alterations in the architecture of the nucleus (116) that occur in the more advanced phase of lytic viral DNA replication, it is fair to speculate that the entire chromatin composition including topologically associating domains will be disrupted but presumably remodeled to become compacted at the periphery of the nucleus (117).
Reduction in chromatin-chromatin interactions upon BZLF1 expression is certainly an indirect effect, but the molecular mechanisms need to be identified. The closing of previously open chromatin might contribute to this phenomenon and both effects combined could also cause the massive increase in transcriptional noise observed in Raji and DG75 iBZLF1 cells (Figure 4C and F). In Raji cells, BZLF1 regulated lytic viral genes such as BGLF5 are possible candidates that might further contribute to the reduced chromatin-chromatin interactions. This is because BGLF5 is not only an RNAse but also a DNAse that can affect genomic stability (118). BGLF5 acts as an endonuclease (119–121) and exonuclease (121) and was found to induce DNA damage in the cellular genome and instability of micro-satellites in epithelial cells (122). A second viral candidate that might contribute to the loss of chromatin interactions is BALF3, which induces DNA strand breaks, meditates genome instability (123) and supports efficient virion synthesis (124).
In conclusion, upon induction of EBV’s lytic phase, we found that cellular chromatin-chromatin interactions are lost and the transcriptome of the cell is profoundly reduced in Raji cells. Whereas BZLF1 seems to affect chromatin-chromatin interactions, it does not cause the global reduction of cellular transcripts. Regions of accessible, hence open chromatin in non-induced cells lose their accessibility upon expression of BZLF1, but, conversely, BZLF1 binding sites in repressed cellular chromatin gain accessibility, which is consistent with BZLF1’s role as a pioneer transcription factor (34). These alterations in cellular chromatin are massive and global, suggesting that they contribute to the success of lytic induction and viral de novo synthesis. We speculate that the alterations in the nucleus of the lytically-induced cell help to make space for the asynchronous and rapid viral DNA amplification (6,7) and/or for capsid morphogenesis and mobility (125). They redirect basic cellular functions including transcription, mRNA transport and export such that these resources and functions become available for the synthesis of viral transcripts and DNA and additional virus-related downstream processes. Since some of the molecular mechanisms of the underlying effects are unknown, the race is now open for the search for the responsible players.
DATA AVAILABILITY
NGS files have been deposited on ArrayExpress (126) using the web-based submission tool Annotare 2.0 (https://github.com/arrayexpress/annotare2). The data files of interest can be downloaded via https://www.ebi.ac.uk/arrayexpress/ using accession information listed in Supplementary Tables S3 and S4.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Alexander Graf (Laboratory for Functional Genome Analysis, Gene Center, Ludwig Maximilians University Munich, Munich, Germany) for the introduction to the Galaxy suit and the initial analysis of RNA-seq data, Sylvia Mallok (Laboratory for Functional Genome Analysis, Gene Center, Ludwig Maximilians University Munich, Munich, Germany) for the help with the preparation of the RNA-seq and Capture-C libraries, Tamas Schauer (Molecular Biology Division, Biomedical Center, Faculty of Medicine, Ludwig-Maximilians University Munich, Planegg-Martinsried, Germany) for the code for ERCC spike-in normalisation, Thomas Schwarzmayr (Institute of Human Genetics, Helmholtz Zentrum München, Germany) for the code for the visualisation of ERCC spike-in NGS sequencing reads, and James Davies (Trinity College, Bristol, UK) for supporting our understanding of the Capture-C analysis. We also thank our colleagues Reinhard Zeidler and Regina Feederle (Monoclonal Antibody Core Facility) for their generous gifts of monoclonal antibodies directed against gp350, CD147 and BZLF1 and their coupling to different fluorochromes. We would especially thank Hannah Busen and Elmar Spiegel (Institute of Computational Biology, Helmholtz Zentrum München, Germany) for their intellectual and practical support to understand the statistics of BZLF1′s binding to the pool of different binding motifs in cellular DNA.
Authors contributions: A.B., P.M.-G., S.K., H.B., F.M.C., A.E., D.P., B.M., T.S. and W.H. designed and performed experiments and analysed data. A.B., G.S., T.S. and W.H. designed and conceived the project. A.B. A.E. and W.H. wrote the manuscript.
Contributor Information
Alexander Buschle, Research Unit Gene Vectors, Helmholtz Zentrum München, German Research Center for Environmental Health and German Center for Infection Research (DZIF), Partner site Munich, Germany, Feodor-Lynen-Str. 21, D-81377 Munich, Germany.
Paulina Mrozek-Gorska, Research Unit Gene Vectors, Helmholtz Zentrum München, German Research Center for Environmental Health and German Center for Infection Research (DZIF), Partner site Munich, Germany, Feodor-Lynen-Str. 21, D-81377 Munich, Germany.
Filippo M Cernilogar, Division of Molecular Biology, Biomedical Center, Faculty of Medicine, Ludwig-Maximilians-Universität (LMU) München, 82152 Planegg-Martinsried, Germany.
Andreas Ettinger, Institute of Epigenetics and Stem Cells, Helmholtz Zentrum München, German Research Center for Environmental Health, Feodor-Lynen-Str. 21 D-81377 Munich, Germany.
Dagmar Pich, Research Unit Gene Vectors, Helmholtz Zentrum München, German Research Center for Environmental Health and German Center for Infection Research (DZIF), Partner site Munich, Germany, Feodor-Lynen-Str. 21, D-81377 Munich, Germany.
Stefan Krebs, Laboratory for Functional Genome Analysis (LAFUGA), Gene Center of the Ludwig-Maximilians-Universität (LMU) München, 81377 Munich, Germany.
Bianca Mocanu, Research Unit Gene Vectors, Helmholtz Zentrum München, German Research Center for Environmental Health and German Center for Infection Research (DZIF), Partner site Munich, Germany, Feodor-Lynen-Str. 21, D-81377 Munich, Germany.
Helmut Blum, Laboratory for Functional Genome Analysis (LAFUGA), Gene Center of the Ludwig-Maximilians-Universität (LMU) München, 81377 Munich, Germany.
Gunnar Schotta, Division of Molecular Biology, Biomedical Center, Faculty of Medicine, Ludwig-Maximilians-Universität (LMU) München, 82152 Planegg-Martinsried, Germany.
Tobias Straub, Bioinformatics Unit, Biomedical Center, Ludwig-Maximilians-Universität (LMU) München, 82152 Planegg-Martinsried, Germany.
Wolfgang Hammerschmidt, Research Unit Gene Vectors, Helmholtz Zentrum München, German Research Center for Environmental Health and German Center for Infection Research (DZIF), Partner site Munich, Germany, Feodor-Lynen-Str. 21, D-81377 Munich, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [SFB1064/TP A13, SFB-TR36/TP A04]; Deutsche Krebshilfe [70112875]; National Cancer Institute [CA70723 to W.H.]; Deutsche Forschungsgemeinschaft [SFB1064/TP A11, SFB1321/TP P13 to G.S.]; Deutsche Forschungsgemeinschaft [ET124/2-1 to A.E.]. Funding for open access charge: Institutional funds, extramural funding via Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1. Qian S., Fan W., Liu T., Wu M., Zhang H., Cui X., Zhou Y., Hu J., Wei S., Chen H.et al.. Seneca Valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J. Virol. 2017; 91:e00823-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng Y.H., Jeang K.T., Tokunaga K.. Host restriction factors in retroviral infection: promises in virus-host interaction. Retrovirology. 2012; 9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albanese M., Tagawa T., Buschle A., Hammerschmidt W.. microRNAs of Epstein-Barr virus control innate and adaptive anti-viral immunity. J. Virol. 2017; 91:e01667-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marazzi I., Ho J.S., Kim J., Manicassamy B., Dewell S., Albrecht R.A., Seibert C.W., Schaefer U., Jeffrey K.L., Prinjha R.K.et al.. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012; 483:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quinlan M.P., Chen L.B., Knipe D.M.. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell. 1984; 36:857–868. [DOI] [PubMed] [Google Scholar]
- 6. Monier K., Armas J.C., Etteldorf S., Ghazal P., Sullivan K.F.. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2000; 2:661–665. [DOI] [PubMed] [Google Scholar]
- 7. Chiu Y.F., Sugden A.U., Sugden B.. Epstein-Barr viral productive amplification reprograms nuclear architecture, DNA replication, and histone deposition. Cell Host Microbe. 2013; 14:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laichalk L.L., Thorley-Lawson D.A.. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005; 79:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Countryman J., Miller G.. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:4085–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takada K., Shimizu N., Sakuma S., Ono Y.. Transactivation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J. Virol. 1986; 57:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schepers A., Pich D., Hammerschmidt W.. A transcription factor with homology to the AP-1 family links RNA transcription and DNA replication in the lytic cycle of Epstein-Barr virus. EMBO J. 1993; 12:3921–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergbauer M., Kalla M., Schmeinck A., Gobel C., Rothbauer U., Eck S., Benet-Pages A., Strom T.M., Hammerschmidt W.. CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010; 6:e1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhende P.M., Seaman W.T., Delecluse H.J., Kenney S.C.. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 2004; 36:1099–1104. [DOI] [PubMed] [Google Scholar]
- 14. Kalla M., Schmeinck A., Bergbauer M., Pich D., Hammerschmidt W.. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jochum S., Ruiss R., Moosmann A., Hammerschmidt W., Zeidler R.. RNAs in Epstein-Barr virions control early steps of infection. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E1396–E1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wen W., Iwakiri D., Yamamoto K., Maruo S., Kanda T., Takada K.. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 2007; 81:1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalla M., Gobel C., Hammerschmidt W.. The lytic phase of Epstein-Barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J. Virol. 2012; 86:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woellmer A., Arteaga-Salas J.M., Hammerschmidt W.. BZLF1 governs CpG-methylated chromatin of Epstein-Barr virus reversing epigenetic repression. PLoS Pathog. 2012; 8:e1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kenney S.C., Mertz J.E.. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin. Cancer Biol. 2014; 26:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalla M., Hammerschmidt W.. Human B cells on their route to latent infection–early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell. Biol. 2012; 91:65–69. [DOI] [PubMed] [Google Scholar]
- 21. Lieberman P.M., Hardwick J.M., Sample J., Hayward G.S., Hayward S.D.. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 1990; 64:1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang Y.N., Dong D.L., Hayward G.S., Hayward S.D.. The Epstein-Barr virus Zta transactivator: a member of the bZIP family with unique DNA-binding specificity and a dimerization domain that lacks the characteristic heptad leucine zipper motif. J. Virol. 1990; 64:3358–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gustems M., Woellmer A., Rothbauer U., Eck S.H., Wieland T., Lutter D., Hammerschmidt W.. c-Jun/c-Fos heterodimers regulate cellular genes via a newly identified class of methylated DNA sequence motifs. Nucleic Acids Res. 2014; 42:3059–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou H., Zarubin T., Ji Z., Min Z., Zhu W., Downey J.S., Lin S., Han J.. Frequency and distribution of AP-1 sites in the human genome. DNA Res. 2005; 12:139–150. [DOI] [PubMed] [Google Scholar]
- 25. Lee W., Mitchell P., Tjian R.. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987; 49:741–752. [DOI] [PubMed] [Google Scholar]
- 26. Chavanas S., Adoue V., Méchin M.C., Ying S., Dong S., Duplan H., Charveron M., Takahara H., Serre G., Simon M.. Long-range enhancer associated with chromatin looping allows AP-1 regulation of the peptidylarginine deiminase 3 gene in differentiated keratinocyte. PLoS One. 2008; 3:e3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S.. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015; 17:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phanstiel D.H., Van Bortle K., Spacek D., Hess G.T., Shamim M.S., Machol I., Love M.I., Aiden E.L., Bassik M.C., Snyder M.P.. Static and dynamic DNA Loops form AP-1-Bound activation hubs during macrophage development. Mol. Cell. 2017; 67:1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vierbuchen T., Ling E., Cowley C.J., Couch C.H., Wang X., Harmin D.A., Roberts C.W.M., Greenberg M.E.. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell. 2017; 68:1067–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaulian E., Karin M.. AP-1 in cell proliferation and survival. Oncogene. 2001; 20:2390–2400. [DOI] [PubMed] [Google Scholar]
- 31. Shaulian E., Karin M.. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002; 4:E131–E136. [DOI] [PubMed] [Google Scholar]
- 32. Ameyar M., Wisniewska M., Weitzman J.B.. A role for AP-1 in apoptosis: the case for and against. Biochimie. 2003; 85:747–752. [DOI] [PubMed] [Google Scholar]
- 33. Eferl R., Wagner E.F.. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003; 3:859–868. [DOI] [PubMed] [Google Scholar]
- 34. Schaeffner M., Mrozek-Gorska P., Buschle A., Woellmer A., Tagawa T., Cernilogar F.M., Schotta G., Krietenstein N., Lieleg C., Korber P.et al.. BZLF1 interacts with chromatin remodelers promoting escape from latent infections with EBV. Life Sci. Alliance. 2019; 2:e201800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polack A., Delius H., Zimber U., Bornkamm G.W.. Two deletions in the Epstein-Barr virus genome of the Burkitt lymphoma nonproducer line Raji. Virology. 1984; 133:146–157. [DOI] [PubMed] [Google Scholar]
- 36. Hatfull G., Bankier A.T., Barrell B.G., Farrell P.J.. Sequence analysis of Raji Epstein-Barr virus DNA. Virology. 1988; 164:334–340. [DOI] [PubMed] [Google Scholar]
- 37. Decaussin G., Leclerc V., Ooka T.. The lytic cycle of Epstein-Barr virus in the nonproducer Raji line can be rescued by the expression of a 135-kilodalton protein encoded by the BALF2 open reading frame. J. Virol. 1995; 69:7309–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller G., Lipman M.. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U.S.A. 1973; 70:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ben-Bassat H., Goldblum N., Mitrani S., Goldblum T., Yoffey J.M., Cohen M.M., Bentwich Z., Ramot B., Klein E., Klein G.. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int. J. Cancer. 1977; 19:27–33. [DOI] [PubMed] [Google Scholar]
- 40. Pulvertaft J.V. Cytology of Burkitt's Tumour (African Lymphoma). Lancet. 1964; 1:238–240. [DOI] [PubMed] [Google Scholar]
- 41. Graham F.L., Smiley J., Russell W.C., Nairn R.. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977; 36:59–74. [DOI] [PubMed] [Google Scholar]
- 42. Delecluse H.J., Hilsendegen T., Pich D., Zeidler R., Hammerschmidt W.. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:8245–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bornkamm G.W., Berens C., Kuklik-Roos C., Bechet J.M., Laux G., Bachl J., Korndoerfer M., Schlee M., Holzel M., Malamoussi A.et al.. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 2005; 33:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schäffner M. Chromatin remodeling in Epstein-Barr virus after induction of the lytic phase: molecular characterization of the role of BZLF1 and its interactions. Fac. Biol. 2015; 1–142.PhD. [Google Scholar]
- 45. Young L.S., Lau R., Rowe M., Niedobitek G., Packham G., Shanahan F., Rowe D.T., Greenspan D., Greenspan J.S., Rickinson A.B.et al.. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J. Virol. 1991; 65:2868–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. R Core Team 2018; R: A language and environment for statistical computing.
- 47. Steinbrück L., Gustems M., Medele S., Schulz T.F., Lutter D., Hammerschmidt W.. K1 and K15 of Kaposi's sarcoma-associated herpesvirus are partial functional homologues of latent membrane protein 2A of Epstein-Barr virus. J. Virol. 2015; 89:7248–7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt U., Weigert M., Broaddus C., Myers G.. Cell detection with star-convex polygons. International Conference on Medical Image Computing and Computer-Assisted Intervention. 2018; 265–273. [Google Scholar]
- 49. van der Walt S., Schönberger J.L., Nunez-Iglesias J., Boulogne F., Warner J.D., Yager N., Gouillart E., Yu T., scikit-image C.. scikit-image: image processing in Python. PeerJ. 2014; 2:e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stegmaier J., Amat F., Lemon W.C., McDole K., Wan Y., Teodoro G., Mikut R., Keller P.J.. Real-Time three-dimensional cell segmentation in large-scale microscopy data of developing embryos. Dev. Cell. 2016; 36:225–240. [DOI] [PubMed] [Google Scholar]
- 51. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000,G.P.D.P.S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng J., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011; 27:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME Suite. Nucleic Acids Res. 2015; 43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grothendieck G., Petzoldt T.. R help desk: date and time classes in R. R News. 2004; 4:29–32. [Google Scholar]
- 61. Barter R.L., Yu B.. Superheat: an R package for creating beautiful and extendable heatmaps for visualizing complex data. J. Comput. Graph. Stat. 2017; 27:910–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C.et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chang W. 2014; Extrafont: Tools for using fonts.
- 67. Neuwirth E. 2014; RColorBrewer: ColorBrewer palettes.
- 68. Magoc T., Salzberg S.L.. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cairns J., Freire-Pritchett P., Wingett S.W., Várnai C., Dimond A., Plagnol V., Zerbino D., Schoenfelder S., Javierre B.M., Osborne C.et al.. CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 2016; 17:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fixman E.D., Hayward G.S., Hayward S.D.. trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 1992; 66:5030–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fixman E.D., Hayward G.S., Hayward S.D.. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 1995; 69:2998–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dickerson S.J., Xing Y., Robinson A.R., Seaman W.T., Gruffat H., Kenney S.C.. Methylation-dependent binding of the Epstein-Barr virus BZLF1 protein to viral promoters. PLoS Pathog. 2009; 5:e1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Flower K., Thomas D., Heather J., Ramasubramanyan S., Jones S., Sinclair A.J.. Epigenetic control of viral life-cycle by a DNA-methylation dependent transcription factor. PLoS One. 2011; 6:e25922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Farrell P.J., Rowe D.T., Rooney C.M., Kouzarides T.. Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. EMBO J. 1989; 8:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ramasubramanyan S., Osborn K., Al-Mohammad R., Naranjo Perez-Fernandez I.B., Zuo J., Balan N., Godfrey A., Patel H., Peters G., Rowe M.et al.. Epstein-Barr virus transcription factor Zta acts through distal regulatory elements to directly control cellular gene expression. Nucleic Acids Res. 2015; 43:3563–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. International, H.G.S.C. Finishing the euchromatic sequence of the human genome. Nature. 2004; 431:931–945. [DOI] [PubMed] [Google Scholar]
- 78. Nguyen Q.H., Tellam R.L., Naval-Sanchez M., Porto-Neto L.R., Barendse W., Reverter A., Hayes B., Kijas J., Dalrymple B.P.. Mammalian genomic regulatory regions predicted by utilizing human genomics, transcriptomics, and epigenetics data. Gigascience. 2018; 7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hughes J.R., Roberts N., McGowan S., Hay D., Giannoulatou E., Lynch M., De Gobbi M., Taylor S., Gibbons R., Higgs D.R.. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014; 46:205–212. [DOI] [PubMed] [Google Scholar]
- 80. Pich D., Mrozek-Gorska P., Bouvet M., Sugimoto A., Akidil E., Grundhoff A., Hamperl S., Ling P.D., Hammerschmidt W.. First days in the life of naive human B lymphocytes infected with Epstein-Barr virus. mBio. 2019; 10:e01723-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Petosa C., Morand P., Baudin F., Moulin M., Artero J.B., Muller C.W.. Structural basis of lytic cycle activation by the Epstein-Barr virus ZEBRA protein. Mol. Cell. 2006; 21:565–572. [DOI] [PubMed] [Google Scholar]
- 82. Urier G., Buisson M., Chambard P., Sergeant A.. The Epstein-Barr virus early protein EB1 activates transcription from different responsive elements including AP-1 binding sites. EMBO J. 1989; 8:1447–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Park R., Heston L., Shedd D., Delecluse H.J., Miller G.. Mutations of amino acids in the DNA-recognition domain of Epstein-Barr virus ZEBRA protein alter its sub-nuclear localization and affect formation of replication compartments. Virology. 2008; 382:145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ressing M.E., van Gent M., Gram A.M., Hooykaas M.J., Piersma S.J., Wiertz E.J.. Immune Evasion by Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015; 391:355–381. [DOI] [PubMed] [Google Scholar]
- 85. Schepers A., Pich D., Hammerschmidt W.. Activation of oriLyt, the lytic origin of DNA replication of Epstein-Barr virus, by BZLF1. Virology. 1996; 220:367–376. [DOI] [PubMed] [Google Scholar]
- 86. Kenney S., Holley-Guthrie E., Mar E.C., Smith M.. The Epstein-Barr virus BMLF1 promoter contains an enhancer element that is responsive to the BZLF1 and BRLF1 transactivators. J. Virol. 1989; 63:3878–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chevallier-Greco A., Manet E., Chavrier P., Mosnier C., Daillie J., Sergeant A.. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 1986; 5:3243–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hammerschmidt W., Sugden B.. Replication of Epstein-Barr viral DNA. Cold Spring Harb. Perspect. Biol. 2013; 5:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Flemington E., Speck S.H.. Epstein-Barr virus BZLF1 trans activator induces the promoter of a cellular cognate gene, c-fos. J. Virol. 1990; 64:4549–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cayrol C., Flemington E.K.. Identification of cellular target genes of the Epstein-Barr virus transactivator Zta: activation of transforming growth factor beta igh3 (TGF-beta igh3) and TGF-beta 1. J. Virol. 1995; 69:4206–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lu J., Chen S.Y., Chua H.H., Liu Y.S., Huang Y.T., Chang Y., Chen J.Y., Sheen T.S., Tsai C.H.. Upregulation of tyrosine kinase TKT by the Epstein-Barr virus transactivator Zta. J. Virol. 2000; 74:7391–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Morrison T.E., Mauser A., Wong A., Ting J.P., Kenney S.C.. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity. 2001; 15:787–799. [DOI] [PubMed] [Google Scholar]
- 93. Morrison T.E., Mauser A., Klingelhutz A., Kenney S.C.. Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J. Virol. 2004; 78:544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chang Y., Lee H.H., Chen Y.T., Lu J., Wu S.Y., Chen C.W., Takada K., Tsai C.H.. Induction of the early growth response 1 gene by Epstein-Barr virus lytic transactivator Zta. J. Virol. 2006; 80:7748–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jones R.J., Seaman W.T., Feng W.H., Barlow E., Dickerson S., Delecluse H.J., Kenney S.C.. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer. 2007; 121:1274–1281. [DOI] [PubMed] [Google Scholar]
- 96. Hsu M., Wu S.Y., Chang S.S., Su I.J., Tsai C.H., Lai S.J., Shiau A.L., Takada K., Chang Y.. Epstein-Barr virus lytic transactivator Zta enhances chemotactic activity through induction of interleukin-8 in nasopharyngeal carcinoma cells. J. Virol. 2008; 82:3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tsai S.C., Lin S.J., Chen P.W., Luo W.Y., Yeh T.H., Wang H.W., Chen C.J., Tsai C.H.. EBV Zta protein induces the expression of interleukin-13, promoting the proliferation of EBV-infected B cells and lymphoblastoid cell lines. Blood. 2009; 114:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Heather J., Flower K., Isaac S., Sinclair A.J.. The Epstein-Barr virus lytic cycle activator Zta interacts with methylated ZRE in the promoter of host target gene egr1. J. Gen. Virol. 2009; 90:1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bristol J.A., Robinson A.R., Barlow E.A., Kenney S.C.. The Epstein-Barr virus BZLF1 protein inhibits tumor necrosis factor receptor 1 expression through effects on cellular C/EBP proteins. J. Virol. 2010; 84:12362–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zuo J., Thomas W.A., Haigh T.A., Fitzsimmons L., Long H.M., Hislop A.D., Taylor G.S., Rowe M.. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 2011; 7:e1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang’ondu R., Teal S., Park R., Heston L., Delecluse H., Miller G.. DNA damage signaling is induced in the absence of Epstein-Barr Virus (EBV) lytic DNA replication and in response to expression of ZEBRA. PLoS One. 2015; 10:e0126088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cusanovich D.A., Pavlovic B., Pritchard J.K., Gilad Y.. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014; 10:e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bressloff P.C. Stochastic switching in biology: from genotype to phenotype. J. Phys. A Math. Theor. 2017; 50:133001. [Google Scholar]
- 104. Kaaij L.J.T., van der Weide R.H., Ketting R.F., de Wit E. Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. 2018; 24:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Buisson M., Géoui T., Flot D., Tarbouriech N., Ressing M.E., Wiertz E.J., Burmeister W.P.. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 2009; 391:717–728. [DOI] [PubMed] [Google Scholar]
- 106. Horst D., Burmeister W.P., Boer I.G., van Leeuwen D., Buisson M., Gorbalenya A.E., Wiertz E.J., Ressing M.E.. The “Bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J. Virol. 2012; 86:9175–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rowe M., Glaunsinger B., van Leeuwen D., Zuo J., Sweetman D., Ganem D., Middeldorp J., Wiertz E.J., Ressing M.E.. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:3366–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zuo J., Thomas W., van Leeuwen D., Middeldorp J.M., Wiertz E.J., Ressing M.E., Rowe M.. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008; 82:2385–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kumar G.R., Glaunsinger B.A.. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell. Biol. 2010; 30:4996–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Park R., El-Guindy A., Heston L., Lin S.F., Yu K.P., Nagy M., Borah S., Delecluse H.J., Steitz J., Miller G.. Nuclear translocation and regulation of intranuclear distribution of cytoplasmic poly(A)-binding protein are distinct processes mediated by two Epstein Barr virus proteins. PLoS One. 2014; 9:e92593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gao X., Wang H., Sairenji T.. Inhibition of Epstein-Barr virus (EBV) reactivation by short interfering RNAs targeting p38 mitogen-activated protein kinase or c-myc in EBV-positive epithelial cells. J. Virol. 2004; 78:11798–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hu Z., Killion P.J., Iyer V.R.. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007; 39:683–687. [DOI] [PubMed] [Google Scholar]
- 113. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y.et al.. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015; 162:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Guo R., Jiang C., Zhang Y., Govande A., Trudeau S.J., Chen F., Fry C.J., Puri R., Wolinsky E., Schineller M.et al.. MYC controls the Epstein-Barr virus lytic switch. Mol. Cell. 2020; 78:653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nagaraju T., Sugden A.U., Sugden B.. Four-dimensional analyses show that replication compartments are clonal factories in which Epstein-Barr viral DNA amplification is coordinated. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:24630–24638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Chakravorty A., Sugden B., Johannsen E.C.. An epigenetic journey: Epstein-Barr virus transcribes chromatinized and subsequently unchromatinized templates during its lytic cycle. J. Virol. 2019; 93:e02247-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Baylis S.A., Purifoy D.J., Littler E.. The characterization of the EBV alkaline deoxyribonuclease cloned and expressed in E. coli. Nucleic Acids Res. 1989; 17:7609–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhang C.X., Decaussin G., de Turenne Tessier M., Daillie J., Ooka T.. Identification of an Epstein-Barr virus-specific desoxyribonuclease gene using complementary DNA. Nucleic Acids Res. 1987; 15:2707–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Stolzenberg M.C., Ooka T.. Purification and properties of Epstein-Barr virus DNase expressed in Escherichia coli. J. Virol. 1990; 64:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lin S.F., Hsu T.Y., Liu M.Y., Lin L.S., Yang H.L., Chen J.Y., Yang C.S.. Characterization of Epstein-Barr virus DNase and its interaction with the major DNA binding protein. Virology. 1995; 208:712–722. [DOI] [PubMed] [Google Scholar]
- 122. Wu C.C., Liu M.T., Chang Y.T., Fang C.Y., Chou S.P., Liao H.W., Kuo K.L., Hsu S.L., Chen Y.R., Wang P.W.et al.. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010; 38:1932–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chiu S.H., Wu C.C., Fang C.Y., Yu S.L., Hsu H.Y., Chow Y.H., Chen J.Y.. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget. 2014; 5:8583–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chiu S.H., Wu M.C., Wu C.C., Chen Y.C., Lin S.F., Hsu J.T., Yang C.S., Tsai C.H., Takada K., Chen M.R.et al.. Epstein-Barr virus BALF3 has nuclease activity and mediates mature virion production during the lytic cycle. J. Virol. 2014; 88:4962–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bosse J.B., Hogue I.B., Feric M., Thiberge S.Y., Sodeik B., Brangwynne C.P., Enquist L.W.. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5725–E5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kolesnikov N., Hastings E., Keays M., Melnichuk O., Tang Y.A., Williams E., Dylag M., Kurbatova N., Brandizi M., Burdett T.et al.. ArrayExpress update–simplifying data submissions. Nucleic Acids Res. 2015; 43:D1113–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS files have been deposited on ArrayExpress (126) using the web-based submission tool Annotare 2.0 (https://github.com/arrayexpress/annotare2). The data files of interest can be downloaded via https://www.ebi.ac.uk/arrayexpress/ using accession information listed in Supplementary Tables S3 and S4.