


Abstract
Detailed target-selectivity information and experiment-based efficacy prediction tools are primarily available for Streptococcus pyogenes Cas9 (SpCas9). One obstacle to develop such tools is the rarity of accurate data. Here, we report a method termed ‘Self-targeting sgRNA Library Screen’ (SLS) for assaying the activity of Cas9 nucleases in bacteria using random target/sgRNA libraries of self-targeting sgRNAs. Exploiting more than a million different sequences, we demonstrate the use of the method with the SpCas9-HF1 variant to analyse its activity and reveal motifs that influence its target-selectivity. We have also developed an algorithm for predicting the activity of SpCas9-HF1 with an accuracy matching those of existing tools. SLS is a facile alternative to the much more expensive and laborious approaches used currently and has the capability of delivering sufficient amount of data for most of the orthologs and variants of SpCas9.
INTRODUCTION
Since the adaptation of SpCas9 for genome engineering, Cas9 nucleases have become versatile tools for a range of applications (1–13). These nucleases cleave target sequences that are complementary to the 5′ ‘spacer’ region of their single guide RNA (sgRNA), provided that they are adjacent to a short protospacer adjacent motif (PAM) sequence that is specific to each particular Cas9 nuclease (14). Most experimental data have been generated for the SpCas9 nuclease that is considered to be the most powerful among the orthologs. High resolution X-ray crystallographic data on the structure of SpCas9 nuclease has revealed that the protein interacts with the DNA–sgRNA hybrid helix via the sugar-phosphate backbone, without making any sequence-specific contact with the bases (15–17). Nonetheless, the nuclease exhibits considerable sequence-dependent variation in its DNA cleavage activity (18–22).
The underlying structural determinants of the sequence selectivity have primarily been characterized for the wild type (WT) SpCas9 (18–29), although several orthologs are known and several mutant variants with increased fidelity or altered PAM specificity have been generated with apparently differing sequence selectivity (30–35). A target cleavage efficacy prediction algorithm has also been generated for AsCas12a (36). An understanding of their target-selectivity and in silico target prediction tools such as those available for the wild type SpCas9 are much needed to facilitate their routine usage. However, the development of accurate prediction tools is a challenging, labour intensive and exceedingly costly task, primarily due to the need for reproducible cleavage activity information for a very large number of target sequences. This kind of information for the variants and orthologs of SpCas9 is rare. Even in the case of SpCas9, the on-target efficiency prediction tools developed on the basis of cleavage activity data derive from experiments that exploited only a relatively small number of targets, not exceeding a few tens of thousands of different sequences: this number is dwarfed by the number of sequences, exceeding a trillion (420 = ca. 1012), theoretically targetable by SpCas9 (18–22).
The limitation on generating cleavage information on a larger number of sequences derives from the need for the joint presence of the target sequence and its matching sgRNA in one cell in a manner that allows the identification of the outcome of the corresponding cleavage reaction. This criterion makes it difficult to use randomized libraries. The feasibility of studying systematic arrays of designed target sequences decreases sharply with the increasing number of target/sgRNA pairs involved. Here, we exploit the fact that SpCas9 requires only the presence of an intact stem structure of the sgRNA adjacent to its spacer sequence without being sensitive to the actual sequence of the stem (12,15,37–39). This allowed us to modify the stem sequences of the sgRNAs to contain an NGG sequence motif, the PAM of SpCas9, thereby, generating self-targeting sgRNAs (stgRNAs) that target their own coding sequences (Figure 1A, B) (12,38,39). Such self-targeting sgRNAs effectively solve the problem of presenting the matching sgRNA and target jointly in one cell when randomized sequence libraries are used.
Figure 1.

Principle of the Self-targeting sgRNA Library Screen (SLS) method. (A, B) Representation of sgRNAs with canonical and self-targeting scaffolds. Position 22–23 (TT dinucleotides, (green) (A) in the coding sequence of the sgRNA are altered to GG (blue) to generate a PAM motif, resulting in the construction of a self-targeting sgRNA (B). To maintain the stem structure, the complement strand is also modified. (C) Self-targeting sgRNAs, in contrast to canonical sgRNAs, target their own coding DNA sequence. The spacer sequence is shown in orange, the PAM in yellow, the sgRNA scaffold in blue, the plasmid backbone in gray. (D) Flowchart of the SLS method developed to identify SpCas9-HF1’s spacer preference. The plasmids harboring effectively-cleaved target sequences derived from the one million sequence library are depleted under antibiotic selection. Sequencing both the initial and the nuclease-processed libraries, cleaved and uncleaved spacer sequences can be identified.
Based on this rationale we generated randomized target/sgRNA libraries with more than a million different sequences for mapping the sequence specificities of SpCas9-HF1. For assaying its cleavage activity, a negative bacterial selection scheme was applied that made possible a high throughput assessment of the full library.
MATERIALS AND METHODS
Materials
Restriction enzymes, T4 ligase, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, Turbofect, Shrimp Alkaline Phosphatase (SAP), Qubit™ dsDNA HS Assay Kit and penicillin/streptomycin were purchased from Thermo Fischer Scientific. DNA oligonucleotides and the GenElute HP Plasmid Miniprep kit used in plasmid purifications were acquired from Sigma-Aldrich. Q5 High-Fidelity DNA Polymerase, NEB5-alpha competent cells, HiFi Assembly Master Mix were from New England Biolabs Inc. NucleoSpin Gel and the PCR Clean-up kit used to clean up DNA from agarose gels were purchased from Macherey-Nagel. ZymoPURE Plasmid Midiprep Kit was from Zymo Research.
Plasmid construction
Vectors were constructed using standard molecular biology techniques. For detailed cloning, primer and sequence information see Supplementary Information. The sequences of all plasmid constructs were confirmed by Sanger sequencing (Microsynth AG).
Plasmids acquired from the non-profit plasmid distribution service Addgene (http://www.addgene.org/) were the following: pdCas9-bacteria (#44249 (6)), pWN10042 (#89052 (40)), pmCherry-gRNA (#80457 (34)), pX330-Flag-dSpCas9 (#92113 (34)), pMJ806 (#39312 (1))
The following plasmids used in this study are available from Addgene: pAT-9208 (#124221), pAT9218 (#124225), pAT-9222 (#124222), pAT-9251 (#124226), pKH-1699 (#124227).
Library construction
The randomized oligonucleotide was annealed (4 μM) with a primer (4 μM) and filled by one-step PCR using Q5 polymerase and the resulting dsDNA was gel purified. The cloning vector (pAT-9208) was synthesized (Genscript Inc.), PCR amplified, digested with DpnI enzyme (3 h) and gel purified. The final product was assembled by HiFi Assembly using 25 ng PCR amplified vector and a molar 1:5 vector:insert ratio, incubating at 50°C for 1 h. The mix was then transformed into in-house made ‘ultra-competent’ Escherichia coli (NEB5-alpha) prepared by the Inoue method (41) and plated on Bioassay plates (Nunc). The transformation was continued until roughly 1 200 000 individual colonies (named ‘1M library’) were acquired (by combining two libraries containing about 500 000 and 700 000 sequences) the colonies were then washed off, and plasmid DNA was purified with ZymoPURE Plasmid Midiprep Kit. An independent, ∼100 000 colony library was also constructed (named ‘100K library’) using the method described above.
Bacterial selection and NGS
The bacterial SpCas9-HF1 and WT-SpCas9 expressing plasmids (pAT-9251, pAT9218) were constructed from the pdCas9-bacteria plasmid. This plasmid was used to transform NEB5alpha cells from which ‘ultra-competent’ cells were made by the Inoue method (41). Next, the competent SpCas9-HF1 and WT-SpCas9 expressing cells were transformed with either the 1M or the 100K randomized plasmid libraries in three parallel experiments until at least 50× transformation coverage had been achieved (in each parallel experiment). These measures are also safeguards against the falsifying effects of artefacts, such as double transformants that are also minimized under the applied conditions (a concentration of 1 ng library plasmid per 200 μl competent cells (42,43)). After transformation, bacteria were grown in a 3D culture (in antibiotic containing semi-solid agarose-LB medium) to minimize overgrowth. For the 3D culture 0.3 m/V % of SeaPrep Agarose (Lonza) was mixed in LB medium, autoclaved and cooled to 37°C. Antibiotics and 100 μl of transformed bacteria were then mixed with the medium in 50 ml batches and the mixture was cooled in ice-cold water until a jelly-like consistency was achieved. The culture was incubated overnight at 37°C until visible colonies formed. The colony containing medium was homogenized at 37°C with a magnetic stirrer, bacteria were pelleted by centrifugation (3000 g, 10 min, RT), and DNA was isolated with ZymoPURE Plasmid Midiprep Kit.
The target sequence (promoter, spacer, sgRNA scaffold) was amplified by PCR from the original ‘uncut’ (1M, 100K), and the cut libraries, then DNA was gel purified. Sample concentrations were measured using the Qubit dsDNA HS Assay Kit (Invitrogen), then they were pooled and sequenced on the Illumina HiSeq4000 platform (BGI Genomics). At least 50× sequencing coverage was achieved per sample. The deep sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA643977.
Cell culture
N2a (neuro-2a mouse neuroblastoma cells, ATCC, CCL-131) cells were grown at 37°C in a humidified atmosphere of 5% CO2 in high glucose DMEM supplemented with 10% heat-inactivated fetal bovine serum, 4 mM l-glutamine (Gibco), 100 units/ml penicillin and 100 μg/ml streptomycin.
GFxFP assay
For experiments when the stgRNA and the canonical sgRNA scaffolds were compared all spacers were cloned into pmCherry-gRNA (canonical scaffold), and into an stgRNA cloning plasmid with the same backbone (pPIK8691) between BpiI sites. The GFxFP assay was used as described previously (40,44). Briefly, N2a cells cultured on 48-well plates were seeded a day before transfection at a density of 3 × 104 cells/well. 10 ng of GFxFP, 150 ng of Cas9 (SpCas9-HF1,WT-SpCas9 or dSpCas9) and 90 ng of sgRNA-mCherry coding plasmid was mixed with 1 μl Turbofect reagent in 50 μl serum free DMEM and incubated 30 min prior adding to the cells. Three parallel transfections were made from each sample. Cells were analysed by flow cytometry 2 days after transfection.
For the experiment on Figure 3A the GFxFP target plasmid was modified in such a manner that both the sgRNA coding and target site were on the same plasmid (self-targeting sgRNA). A spacer cloning plasmid (pAT-9222) was constructed by inserting a human U6 promoter and self-targeting sgRNA between the GFP halves of the GFxFP plasmid (pWN10042). All spacers were cloned into this plasmid between BpiI sites.
Figure 3.

Self-targeting sgRNA Library Screen (SLS) results provide information relevant to the activity data in mammalian cells. (A) Fifty-seven targets were picked from the bacterial selection library containing both efficiently (20) and weakly (37) cleaved targets and were tested in mammalian N2a cells in a plasmid context using the GFP recovery assay (GFxFP assay). Sequences that were more efficiently cleaved in the bacterial system (blue dots) were also more efficiently cleaved in the GFxFP assay, compared to the weakly cleaved ones (red dots). The difference between the cleaved and uncleaved target groups are statistically significant (P< 0.0001, independent samples t-test with Kolmogorov–Smirnov's normality test). Dots on the figure represent the means of three parallel transfections, for each of which the background GFP signal of the negative controls has been subtracted from that of the SpCas9-HF1 treated samples. (B, C) Validation of motifs that have an adverse effect on the activity of SpCas9-HF1 identified by SLS on a 50 000 target sequence library (59). Heatmaps show the deviations in the average indel-inducing ability of SpCas9 variants on target sequences containing the indicated motifs versus those that do not contain them. The motifs are the 15 most unfavourable sequences among the four (B) and five (C) nucleotide-long motifs identified by SLS for SpCas9-HF1 (see Supplementary Figure S4a and b). Data show that these motifs are unfavourable for the activity of SpCas9-HF1 in mammalian cells too. Some of the motifs affect the two increased-fidelity variants only (upper panel), while the others affect all variants. (D) Comparison of Hi-CRISPR predictions with DeepHF on five data sets tested in other studies (31,32,34,65) and on the DeepHF library (59) used for training the DeepHF tool as assessed by G-mean values. The predictors compared are Hi-CRISPR A (magenta), Hi-CRISPR B (blue), Hi-CRISPR C (yellow) and DeepHF (black).
N2a cells cultured on 48-well plates were seeded a day before transfection at a density of 3 × 104 cells/well. 70 ng GFxFP–sgRNA, 100 ng Cas9 (SpCas9-HF1 or the inactive dSpCas9) and 80 ng mCherry coding plasmid (to monitor transfection efficiency) was mixed with 1 μl Turbofect reagent in 50 μl serum free DMEM and incubated 30 min prior adding to the cells. Three parallel transfections were made from each sample. Cells were analysed by flow cytometry 2 days after transfection.
The SpCas9-HF1 coding plasmid was pKH-1699. As negative controls, the background GFP level was determined in the case of each GFxFP plasmid by co-transfecting a nuclease-inactive dSpCas9 coding plasmid (pX330-Flag-dSpCas9). The 57 spacers used in these experiments were chosen by BiSearch (45) from those sequences in the 1M library that are unique to either the human or mouse genomes. These sequences were removed from the training dataset.
Flow cytometry
Flow cytometry analysis was carried out using an Attune NxT Acoustic Focusing Cytometer (Applied Biosytems by Life Techologies). In all experiments, a minimum of 10 000 viable single cells were acquired by gating based on side and forward light-scatter parameters. The GFP signal was detected using the 488 nm diode laser for excitation and the 530/30 nm filter for emission. The mCherry signal was detected using the 561 nm diode laser for excitation and a 615/20 nm filter for emission. For data analysis Attune Cytometric Software v.2.1.0 was used.
Calculating cleavage efficiencies
Paired-end reads for all libraries were merged with BBMerge (v37.22) using default settings and the merged reads were aligned to the sgRNA reference sequence with BLASTn (v2.6.0). To ensure that efficiency only depended on the spacer sequence, reads that had any mutations or indels either in the promoter or sgRNA scaffold region and reads with truncated or extended spacers were eliminated (Supplementary Table S1). The reads for every individual spacer were then counted for each library.
We normalized the corresponding spacer counts between initial and cleaved libraries to calculate the cleavage efficiency for each spacer, which in turn was compared between the three replicates. Normalization was based on the hypothesis that low efficiency, uncut spacer sequences should have high counts in the cleaved library and the ratio of their cleaved and initial counts could be used as a normalization factor between the two libraries. For each of the cleaved libraries, we repeated the following process using its respective initial library. First, we sorted spacer counts in a cleaved library in descending order and determined the first 5% of this list. Then, the ratios between the raw cleaved and initial counts were calculated for every spacer in this set. To remove outliers that could introduce a bias in the process, we removed those ratio values that were not between the first and third quartile of the data. To calculate the normalized cleaved counts, the remaining ratio values were averaged and every spacer count in the cleaved library was divided by this value. The efficiency of a spacer (further referred to as the cleavage efficiency parameter) was calculated by dividing the normalized and the initial counts.
Raw datasets contained three cutting efficiency measurements (from three parallel experiments) and a read count for each spacer sequence. Spacers with less than 10 initial reads were eliminated. All subsequent analyses involving cutting efficiency data employed the mean of the three parallel measurements. The raw dataset originally consisted of 1 667 787 spacers of which 1 222 805 were left after the removals and were used as the final training set (Source Data Figure 2 – 1M-library). An independent dataset, that initially contained 281 834 spacers of which 136 556 remained after filtering, was used for testing prediction performance (Source Data Figure 2 – 100K-library). To test prediction methods with datasets containing either a balanced or unbalanced composition (see Results section) spacers were picked randomly from the 100K test dataset. In case of SpCas9-HF1 three unbalanced test sets, each containing 330 uncleaved and 4670 cleaved spacers, in case of WT-SpCas9 95 uncleaved and 4905 cleaved spacers were picked. In case of SpCas9-HF1 the balanced dataset consisted of 3 × 2360/2360 cleaved/uncleaved spacer sequences, because there were only 3 × 2360 uncleaved sequences available in the 100K library. In case of WT-SpCas9, the balanced dataset consisted of 3 × 695/695 cleaved/uncleaved spacer sequences for the same reason. (Source Data Figure 2 – balanced, unbalanced datasets).
We used a cutoff value of 0.3 to divide the datasets into positive and negative sets (i.e. spacers with high and low cutting efficiency, respectively).
Spacer feature calculations
Standard converted numerical constants for fifty physicochemical DNA properties (PCP) were taken from the literature (46,47). Spacer sequences were divided into overlapping di- or trinucleotides, and their corresponding PCP constants were added to obtain a total PCP value for the spacer. The free energy of the spacer and of the whole sgRNA was calculated using the ViennaRNA package 2.4.3. (48).
Calculation of nucleotide composition features (NucCom)
The nucleotide composition feature (NucCom) is an index number that was used to characterize the expected sequence-specific cutting ability of a spacer. To generate NucCom values, first a NucCom dictionary was generated from the 1M training dataset. The dictionary contained all possible sequence permutations of a given length along the span of the 20-nucleotide long spacer (e.g. four nucleotides in case of NucCom4). Motif values for these subsequences were calculated by averaging the cutting efficiencies of those spacers that contained the given motif at the given position.
The NucCom# value for any particular test spacer sequence was calculated by summing the differences of the motif values stored in the dictionary for all positional subsequences contained in the spacer and the mean cutting efficiency of the training set.
All data manipulation and analysis were performed with R scripts, using the standard R 3.4.4 distribution with no external code packages.
A Python script was developed to calculate NucCom4 values for any given spacer sequences and it is available on Github (https://github.com/welkergroup/HiCRISPR).
Development of Hi-CRISPR B and C
All 154 147 weakly-cutting spacers were collected from the 1M library and in a one-to-two ratio 308 294 well-cutting spacers were randomly selected to form a balanced SpCas9-HF1 training set.
For the WT-SpCas9, all 43 204 weakly-cutting spacers were collected from the 1M library and in a one-to-two ratio 86 408 well-cutting spacers were randomly selected to form a balanced WT SpCas9 training set.
For-Hi-CRISPR B, twenty-three nucleotide-long input sequences were built for each spacer sequence including 20-bp spacer and 3-bp PAM sequences. The fully convolutional neural network (CNN) developed by Chuai et al. (49) was used by employing the script at https://github.com/bm2-lab/DeepActiveCRISPR/blob/master/cnn/transfer/ontar_raw_cnn_pretrain.py. The training was carried out through a hundred epoch.
For-Hi-CRISPR C, the twenty nucleotide-long spacer sequences were used as input sequences. The training exploited the neural network built by H.K.Kim et al. (29) that can be found at https://github.com/MyungjaeSong/Paired-Library/blob/DeepCRISPR.info/DeepCas9/DeepCas9_TestCode.py using a batch size of 700 and an iteration of 2000. The script used for training can be found at https://github.com/welkergroup/HiCRISPR.
Statistics
Differences between samples were tested using Welch's one-way Anova with Games–Howell post hoc tests for samples with unequal variances and/or sample size and by one-way Anova with Tukey's post-hoc test for homoscedastic samples. Homogeneity of variances was tested by Levene's test. Statistical tests were performed using SPSS Statistics v.20 (IBM).
Performance metrics
To assess the performance of the different prediction methods we used the following metrics. In the equations, TP are true positive, FP are false positive, TN are true negative, FN are false negative predictions, MCC is Matthews correlation coefficient and G-mean is the geometric average of sensitivity and specificity.
 |
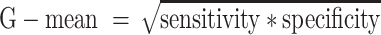 |
Thresholds for each NucCom values were determined as the value of that specific NucCom where the absolute difference between specificity and sensitivity was the lowest for the 100K test library. These thresholds were later applied to all datasets to calculate performance values.
To calculate sgRNA Designer scores and DeepCRISPR values, Azimuth (18), and DeepCRISPR v2.0 (49) was used, respectively. CRISRSPRScan (19), CRISPRater (21), SSC (22) and sgRNA scorer (20)scores were compiled by Haeussler et al. (50) for literature datasets.
RESULTS
First, we altered the coding sequence of the wild type sgRNA (Figure 1A), by replacing the 22nd and the 23rd T nucleotides downstream of the spacer sequence with Gs, thereby creating an NGG PAM motif to generate a sgRNA with a self-targeting scaffold (Figure 1B). To maintain the stem structure the complement strand was also modified. These sgRNAs cut the plasmid which they are expressed from when transfected into a cell (Figure 1C). We found that the self-targeting sgRNAs demonstrated high activity when expressed in E. coli (data not shown) in line with lineage-tracing experiments in different species reported earlier (12,38,39).
Random sequence libraries of self-targeting sgRNAs were generated with a G nucleotide at position 1 of the spacer and randomized nucleotides at positions 2–20. We aimed to study about one-million sequences: this corresponds to the number of all theoretically possible variations of a 10-nucleotide-long nucleic acid segment (410), the length of a full turn of the double stranded DNA helix. Three libraries were generated, containing ∼500 000, 700 000 and 100 000 different spacer sequences respectively (Source Data Figure 2 – 1M-library, Source Data Figure 2 – 100K-library). The first two combined was used as the training dataset (1M library) while the third was used as the test dataset (100K library). Deep-sequencing of the libraries revealed a distribution slightly skewed to a lower T-content at all randomized positions, in contrast to a fully random distribution (Supplementary Figure S1). The skew decreases the presence of spacers in the library with long T nucleotide stretches that may become beneficial when the results are translated to a mammalian cell context. Such sequences act as transcription stop signals for the RNA polymerase III that is generally used for sgRNA transcription (51).
We set up a bacterial negative selection system (Figure 1D) that we named as a Self-targeting sgRNA Library Screen (or SLS) in which the SpCas9-HF1 expressing E. coli cells are transformed with plasmids containing a self-targeting sgRNA expression cassette. When the rate of plasmid cleavage outperforms the rate of replication and repair in the bacterial selection system, the bacteria cannot grow under antibiotic selection. By varying the expression level of Cas9 and the copy number of the target plasmid this threshold activity of Cas9 may be fine-tuned. For this study, we used a condition, in which the major fraction of the plasmids can be cleaved, or in other words, the condition under which a minor fraction of E. coli cells harbouring the uncleaved library is able to survive (see Methods). This facilitates the identification of factors and motifs that diminish the activity of SpCas9-HF1.
Assaying the activity of SpCas9-HF1 on a million target sequences
The libraries were transformed into SpCas9-HF1 expressing E. coli in three parallel experiments. To avoid the eventual overgrowth of some clones over others, instead of liquid medium we used 3D semi-solid agarose culture to grow the bacterial cells (see Materials and Methods). The sequences that are effectively cleaved by SpCas9-HF1 are depleted in the survival libraries, while those that are weakly cleaved are enriched in it. Plasmids were isolated from the survival cultures and sequenced by deep sequencing (Supplementary Figure S1). In order to characterize the cleavability of each sequence, following the normalization of the deep-sequencing data, we calculated a cutting efficiency parameter from the corresponding reads of initial and survival libraries, with 0 indicating no cleavage, and 1 indicating full cleavage of a sequence (described with more details in Methods). The parallel experiments showed excellent reproducibility with Pearson's correlation coefficients r = 0.92–0.93 (Supplementary Table S2). We also removed a few additional sequences from the training dataset to ensure that there was no overlap between the libraries used for training and testing. The final library comprised 1 223 805 spacers and was used as the training dataset, while 136 557 spacers comprised the test library and both were divided into efficiently- and weakly cleaved classes using a cutting efficiency value of 0.3 as the threshold (Source Data Figure 2 – 1M-library, Source Data Figure 2 – 100K-library). Due to the actual performance properties of SpCas9-HF1 in the chosen bacterial selection system, the two classes showed a very unbalanced distribution, and the weakly cleaved classes included only 6.6% of both libraries (Source Data Figure 2 – 1M-library, Source Data Figure 2 – 100K-library).
For comparison, we also tested the WT-SpCas9 on both the training and test libraries, which showed an even more unbalanced distribution, and the weakly cleaved classes included only 1.8% of both libraries (Source Data Figure 2 – 1M-library, Source Data Figure 2 – 100K-library).
We calculated over 60 physicochemical parameters for the sequences of the training 1M library several of them implicated in earlier studies (27,46,47) including position independent mono- and dinucleotide content, GC content, folding energy of the spacer and of the full guide RNA that are calculated using the ViennaRNA package 2.4.3. (48), or local GC content. Standard converted numerical constants for fifty physicochemical DNA properties (PCP) were adopted from the literature (46,47). Spacer sequences were divided into overlapping di- or trinucleotides, and their corresponding PCP constants were added to obtain a total PCP value for the spacer. A more detailed description of these parameters is available in (47). Several parameters were identified to correlate with the cutting efficiencies of SpCas9-HF1 (a few are shown in Supplementary Figure S2a), most of them have also been identified during the evaluation of WT-SpCas9 (Supplementary Figure S2b). Very high GC-content tends to decrease the activity of both WT- and SpCas9-HF1, whereas low GC-content decreases the activity of SpCas9-HF1 (Supplementary Figure S2a). Furthermore, we examined the correlation between the GC-content of different segments of the spacers and the mean cleavage efficiency of those spacers that contain the segments with identical GC-content. All sequences included in the segment between positions 6 and 13 with higher GC content tend to be present in spacers with higher mean cleavage efficiency, a property that neither has hitherto been revealed for SpCas9s, nor has been found here for WT-SpCas9 (Supplementary Figure S3).
Examining the sequence preferences of SpCas9-HF1 we found that motifs corresponding or overlapping to the GTNAC sequence between positions 10–14 have a large adverse impact on cleavage efficiency on SpCas9-HF1, but not on WT-SpCas9 (Supplementary Figure S4a, b). It is worthy of note that earlier studies, working with smaller target library sizes of Cas9 nucleases, have not captured such long motifs (18–20,22,52). A GCC triplet at the 3′ end of the spacer sequence, (i.e.: in PAM proximal position) has been identified as the strongest blocking motif for the WT-SpCas9 (52). Here we found that it also reduces the activity of both WT- and SpCas9-HF1 (Supplementary Figure S4). Interestingly, motifs containing a GCC triplet at positions shifted one or two nucleotides upstream are similarly detrimental to the activity of both nucleases that have not been reported for the WT protein (Supplementary Figure S4a, b). However, a 3′ TT dinucleotide that was also reported earlier (52) has no effect, consistent with the different termination requirements of the transcript in the bacterial cells and the improved scaffold of the self-targeting sgRNA having no consecutive T nucleotides downstream of the spacers (Supplementary Figure S4, Source Data Figure 2 – NucCom dictionaries). We found no preference for a cytosine nucleotide at position 18 at the DNA cleavage site that had also been reported for the WT-SpCas9 (22). This earlier finding might reflect the properties of the mammalian NHEJ DNA repair system (53–55).
Two parameters in particular seem to be most informative, the free energy of the full sgRNA and the free energy of the spacer sequence alone (Supplementary Figure S2). The more stable the structure (the minimum free energy conformation) of either the full sgRNA or the spacer sequence, the greater the likelihood that WT-SpCas9 and SpCas9-HF1 do not effectively cut the appropriate target sequence (Supplementary Figure S2). However, sequences with low enough values of any of these parameters to ensure at least a 50% probability of not being efficiently cleaved by SpCas9-HF1, amount to only about half of the weakly cleaved class (Supplementary Figure S2). Thus, they do not seem to provide sufficiently robust parameters on which to build a reliable prediction algorithm.
Since SpCas9-HF1 is able to distinguish reproducibly between the targets of the two classes, we tried to use the sequences of the spacers themselves, which must code the features that discriminate the good and bad targets. We defined seven separate predictors named Nucleotide Composition 1–7 (NucCom1 to NucCom7), each based on position-dependent motifs with identical length from one up to seven nucleotides long, respectively. At first a value was calculated for each position-dependent motif (motif-values) from the cutting efficiencies of those spacers of the 1M library that contained the given motif at the appropriate position in the spacer (Source Data Figure 2 – NucCom dictionaries). Then, the NucCom values were generated for each spacer using the motif-values of those motifs that are present in the spacer sequence (as described in Materials and Methods). Using the experimental data of the 1M library we determined a threshold value for each of the seven NucCom parameters to divide the training library data into efficiently and weakly cleaved classes in such a way that sensitivity and specificity became equal. We used these threshold values to predict if a sequence in the 100K test library belongs to the efficiently or weakly cleaved classes by comparing its NucCom value to the corresponding threshold values (Source Data Figure 2).
Prediction of the cleavage activity of SpCas9-HF1 on bacterial data
To assess the quality of the classification on the 100K test library we used Matthews correlation coefficient (MCC, (56)) and G-mean (that is the geometric average of sensitivity and specificity, (57)) to provide a balanced assessment. Increasing the lengths of the motifs up to four nucleotides (NucCom4) improves the MCC (Figure 2A) and the G-mean (Figure 2B) values of the predictions for the 100K library reaching impressive G-mean values of 0.86 (Figure 2B). Similar results were achieved with WT-SpCas9 with G-mean values reaching 0.86 (Supplementary Figure S5a and b). Since the G-mean values decrease employing predictors that exploit longer motifs (>4) on the 100K library while they keep rising for the training set, we concluded that the size of the library is sufficiently large to allow four nucleotide motifs to generate reliable (stable) predictors (Figure 2). We therefore chose NucCom4, named Hi-CRISPR A hereafter, for subsequent studies.
Figure 2.

Prediction performance of Hi-CRISPR predictors on target-library data. (A, B) Binary classification results on the 1M training (plain bars) and on the 100K test libraries (chequered bars). NucCom 1–7 are position-dependent short (one to seven base long) motif-based predictors developed using the 1M library data. The quality of the classifications is assessed by the Matthews correlation coefficient (MCC) (A) and G-mean (i.e. the geometric average of sensitivity and specificity) (B). Increasing the lengths of the motifs up to four or seven nucleotides improves the G-mean and the MCC values of the predictions, respectively, on the 100K test libraries. (C, D) SpCas9-HF1 predictions developed in this study are compared on three, either balanced (50% efficiently cleaved, 50% weakly cleaved spacers – striped bars) or unbalanced (93.4% efficiently cleaved, 6.6% weakly cleaved – plain bars) target pools randomly selected from the 100K test dataset. Hi-CRISPR A (NucCom4 shown on A and B), Hi-CRISPR B (Deep-learning scheme based on (49)) and Hi-CRISPR C (Deep-learning scheme based on (29)). MCC (C), but not the G-mean (D) values are sensitive to whether balanced (striped bars) or unbalanced (plain bars) datasets are used. Columns represent means ± SD of the predictions on the three datasets.
To simplify testing, but also to see how different compositions of the two target classes in the test libraries influence prediction quality, we randomly picked a few thousand sequences from the 100K test dataset to obtain balanced (half of them effectively-cleaved and half weakly cleaved sequences) and unbalanced datasets (with 6.6% and 1.8% weakly cleaved sequences for SpCas9-HF1 and WT-SpCas9, respectively) and used these datasets for testing the algorithms. In addition to the Hi-CRISPR A algorithm, we applied a hybrid deep neural network built for developing DeepCRISPR (49) (Hi-CRISPR B) and a neural network used as the base model architecture to build DeepSpCas9 (29) (Hi-CRISPR C). The predictions reached G-mean values as high as 0.92 for SpCas9-HF1 on these data generated independently from the training data (Figure 2C, D). For WT-SpCas9 these values are lower presumably due to the highly unbalanced nature of the training WT data (Supplementary Figure S5c and d). The figures also show comparison with the best WT-SpCas9 prediction tools. Notably, in case of each prediction algorithm used, the MCC values differ greatly between the balanced and unbalanced sets (e g. 0.74 versus 0.28 for Hi-CRISPR A), while the G-mean values are similar. To decrease the dependence of the evaluation on the composition of the datasets we used G-mean in further experiments.
Comparing the cleavage activity of SpCas9-HF1 in mammalian and bacterial cells
Next, we were curious to know whether the cleavage activity results found in this bacterial system are relevant to the activity of SpCas9-HF1 in mammalian cells. We chose 57 sequences that had been tested in the bacterial system (Figure 3A, Source Data Figure 3), representing both efficiently cleaved and weakly cleaved sequences, and compared their activities on the same set of target sequences in mammalian N2a cells exploiting a plasmid-based GFxFP fluorescence-recovery assay we had used earlier (40,58). The assay exploits two GFP halves with overlapping sequences. The cleavage between the two halves induces DNA SSA repair that results in the formation of a functional GFP coding sequence (Supplementary Figure S6). The results of these experiments do not reveal much difference between the mammalian and bacterial activity of SpCas9-HF1 (Figure 3A): only three out of the 57 sequences fell into different classes when comparing the data derived from the two systems. This agreement between the bacterial and mammalian experimental data suggests that the different cellular milieu of each system does not, by itself, significantly influence the activity of SpCas9-HF1. The expression from a mammalian promoter (human U6) and the stability of these sgRNAs in mammalian cells are either not significantly different from the bacterial ones or these are not the major determinants for the mammalian SpCas9-HF1 activities in these experiments.
We also compared the impact of the difference between the self-targeting and canonical sgRNA scaffold on the cleavage activities of SpCas9-HF1 in the GFxFP recovery assay. The activities of SpCas9-HF1 on 20 targets showed excellent correlation (r = 0.98), indicating that the target sequence specificities of SpCas9-HF1 in mammalian cells are mainly determined by the characteristics of the mutant protein, rather than by the differences between the self-targeting and canonical sgRNA scaffold (Supplementary Figure S7).
During the preparation of this manuscript, a new study has provided SpCas9-HF1 cleavage activity data on ∼50 000 mammalian genomic target sequences and has reported on the development of a prediction algorithm, DeepHF, for the prediction of SpCas9-HF1 activity (59), which facilitates further assessment of our approach.
First, we assessed whether the effect of the sequence motifs that mostly affect the activity of SpCas9-HF1 revealed in the one-million bacterial data (as previously shown on Supplementary Figure S4a and b) are also discernible in the DeepHF mammalian data set. We selected those motifs, either four or five nucleotide-long, for which we found the lowest 15–15 motif-values and averaged the cutting efficiencies of those target sequences of the DeepHF data set, which contain the given motif. If the motif also has a strong adverse effect on the mammalian activity of SpCas9-HF1, the average cutting efficiencies of the selected, as well as of all targets should be clearly different. Indeed, they are (Figure 3). Both the four (Figure 3B) and five base-long motifs (Figure 3C) can be clustered into two groups on the mammalian data. One group involves the last four or five (PAM proximal) nucleotides of the target sequences and has a strong adverse effect on the activities of both the WT and the high-fidelity variant. Some of these motifs, that reduce the activities of the mammalian targets to 19%, contain the GCC triplet in positions 18–20, which had previously been identified as having a similar effect for the WT protein (52). Interestingly, most of the rest also contain a GCC triplet, but in positions 16–18 or 17–19, which are equally unfavourable, although these had not been identified for the WT protein in previous studies (Figure 3).
The other group of the motifs has an adverse impact only on the activities of SpCas9-HF1 without any discernible effect on the activity of the WT-SpCas9. Most of these belong or overlap with the GTNAC motif between positions 10–14 of the spacer (Figure 3B and C). To see if the effect of these motifs is specific to SpCas9-HF1 or may have a more general effect on other increased fidelity SpCas9 nucleases, we analysed the eSpCas9 cleavage data of these 50 000 sequences (59). The first group of the motifs decreases the activity of eSpCas9 similarly to that of WT and SpCas9-HF1. The effect of the second group is also discernible, but its extent is about half of that seen on the SpCas9-HF1 data (Figure 3B and C). Next, we examined whether the sequence motifs that proved to be the best in the one-million SpCas9-HF1 bacterial cleavage data are also discernible in the DeepHF mammalian data set. The effect of the best 15, either four (Supplementary Figure S8a) or five (Supplementary Figure S8c) base-long motifs is, on average, smaller than that of the unfavourable motifs for both increased fidelity variants and is comparable to that found in the bacterial library (Supplementary Figure S4c and d). However, exploiting randomly-selected motif-sets revealed that in contrast to the most unfavourable motifs (Supplementary Figure S8e and g), these effects, are not statistically significant (Supplementary Figure S8b, d, f, h). Thus, these analyses of the mammalian data show that the motifs identified in the bacterial cleavage data influence the activity of SpCas9-HF1 similarly, and partially affect the activity of eSpCas9 in mammalian cells (Supplementary Figures S4 and S8).
The accuracy of the mammalian on-target activity predictions of SpCas9-HF1 matches those of WT-SpCas9
We applied the Hi-CRISPR predictions trained on 1M bacterial sequences to test their performance on mammalian cleavage data in comparison with DeepHF (59). We compiled five smaller datasets generated in other studies, and the 50 000 target cleavage data used to develop the DeepHF prediction. The four prediction algorithms exhibit similar performances for these datasets, with Hi-CRISPRs reaching slightly higher average G-mean values on the five smaller datasets (0.86–0.89 versus 0.83 for all Hi-CRISPRs and DeepHF, respectively). As expected, DeepHF performed better on the large dataset on which it was trained (Figure 3D). The performance of Hi-CRISPRs and DeepHF on these datasets is well in the range of the performances of the best wild type SpCas9 prediction algorithms (Supplementary Figure S9). Indeed, when tested on the same five target sets but cleaved by the WT-SpCas9, DeepWT (59), which exhibited the best performance among the wild type prediction algorithms reached an average G-mean value of 0.81 (Supplementary Figure S9). Interestingly, the performance of these prediction algorithms exhibits considerable data set-dependent variability (Supplementary Figures S9 and S10) that makes it difficult to decide which prediction algorithm is the best in general.
Although a direct comparison on the same data is not possible, taken together, the results demonstrate that the performance of our SpCas9-HF1 prediction algorithms clearly reach a performance level generally achievable by the prediction algorithms for the WT-SpCas9 on mammalian genomic sequences.
Self-targeting sgRNAs can also be generated for the non-canonical PAM specificity of SpCas9 and for its orthologs
As a proof of principle, we tested whether SpCas9’s non-canonical PAM specificity (NAG PAM instead of NGG) would also be compatible with our approach (60,61). Mutation in the self-targeting sgRNA’s PAM to NAG was generated that showed wild type SpCas9-like activity in E. coli with a single spacer that had previously been tested in mammalian cells (Supplementary Figure S11). We generated a library of self-targeting sgRNAs with NAG PAM and found that WT-SpCas9 cleaved only 37% of the targets (Supplementary Figure S11). These experiments suggest that the sequence selectivity of other altered PAM specificity Cas9 variants could also be determined by the SLS method.
Several orthologs of SpCas9 have also been shown to be active in mammalian cells. However, from among them a target-selectivity prediction tool has only been developed for SaCas9 (28). We were intrigued to test if self-targeting sgRNAs can be generated for other orthologues as well. Type II-C Cas9 orthologues are particularly interesting because their small size makes them especially suitable for AAV delivery (62). We tested Neisseria meningitidis Cas9 (NmCas9), to see whether, despite being structurally and evolutionarily remote from SpCas9, it shares a similar tolerance toward mutations in its stem sequences immediately downstream from the spacer to that shown by SpCas9. We followed the same rationale as with SpCas9-HF1 previously (Figure 1): we altered the stem sequences of the crRNA and the corresponding nucleotides of the tracrRNA to contain a PAM sequence motif (NNNNGATT). We found that the self-targeting crRNA thus constructed, paired with the tracrRNA, enables the nuclease to demonstrate WT-like activity in E. coli, while a wt crRNA - self-targeting tracrRNA pair does not show activity, even though the corresponding protospacer-PAM target sequence is also placed on the plasmid (Supplementary Figure S12). These results suggest that many Cas9 orthologs may also be compatible with self-targeting sgRNAs and that their sequence specificity could be determined by our approach.
DISCUSSION
To reveal the full potential of RNA-guided nucleases for genome engineering it would be extremely beneficial to understand their sequence selectivity, as well as to have accurate in silico on-target prediction tools. Former mechanistic studies of SpCas9-HF1 exploited only a limited number of targets, altogether not exceeding a couple of hundred sequences, until most recently Wang et al. reported an impressive 50 000 sequences mammalian data set (59). Still, the advance that our study provides in generating cleavage activity data on more than one million sequences is apparent. Further advantages of the SLS approach include the much lower cost and effort necessary to deliver such enormous amounts of data. Since a major bottleneck in achieving prediction for Cas9 variants and orthologs is the paucity of accurate cleavage activity information, for which we have revealed a solution here, we expect that our approach will facilitate the understanding of the sequence specificity of a great number of orthologs and mutant variants of SpCas9, including other increased fidelity nucleases and/or PAM-altered alternatives, as we demonstrated on targets using both the NmCas9 nuclease (Supplementary Figure S12) and the alternative NAG PAM with SpCas9 (Supplementary Figure S11).
It is somewhat surprising that a prediction tool developed on bacterial data performs so well on mammalian cleavage activities, providing predictions comparable in quality to those developed using mammalian data (Figure 3D). We think this is attributable to a balance between the favourable effect of the much higher number of targets used in our approach and the unfavourable effect of the differences between the bacterial system used for training and the (mammalian) systems used for testing. Our data rather suggest that the difference between Cas9 activities in the bacterial and the mammalian cells, is not as large as the difference between the specific conditions of the actual experiments. In mammalian cells the readout is not the cleavage activity of SpCas9 per se, but rather the outcome of erroneous DNA repair (i.e., when the original sequence is altered). The error-prone repair outcome is strongly affected by the actual sequences around the double stranded breaks inflicted by Cas9 nucleases when indels are monitored (53–55). In contrast, local sequence features may influence the repair outcome less when the breaks are repaired by homology-directed repair. At present an efficacy prediction algorithm for SpCas9 is expected to forecast the cleavability of sequences regardless of species and cell types, DNA repairs, readout or transfection efficiencies involved in the actual experiments. These factors may affect the performance of the prediction algorithms as much as the bacterial environment.
Our data suggest that SpCas9-HF1’s altered sequence preference is localized primarily in the middle region of the spacer (Figure 3B, C; Supplementary Figures S3 and S4). It is interesting that eSpCas9 seems to share this feature (Figure 3B, C). The finding that the protein prefers a slightly higher GC-content in the middle region (positions 6–13) of the spacer compared to other regions (Supplementary Figure S3) seems plausible in light of its design (to destabilise the RNA–DNA hybrid helices during target recognition and cleavage), since favouring slightly higher GC-content in this region might counteract these effects. In general, the target binding of SpCas9-HF1 is not altered (32,34), rather the mutations regulate its target cleavage activity and restrict the transition of the nuclease from the conformational checkpoint during the target recognition/cleavage process (63). Our data suggest that the middle region of the spacer sequence may be a critical region affecting the ability of the high-fidelity nuclease to acquire its cleavage competent conformation.
Our method is suited to identify the sequence features that strongly inhibit SpCas9-HF1 cleavage. Thus, it can better identify sequences that should be avoided, rather than distinguishing between sequences that give average or higher than average activity. It does not fit very well to develop wild type SpCas9 cleavage activity prediction due to the very unbalanced nature of the generated data, however, it seems to be suitable for developing target cleavage efficacy prediction for orthologs and variants of SpCas9, since they are active on less targets (34,64).
DATA AVAILABILITY
The following plasmids used in this study are available from Addgene: pAT-9208 (#124221), pAT-9218 (#124225), pAT-9222 (#124222), pAT-9218 (#124225), pAT-9251 (#124226), pKH-1699 (#124227).
The deep sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA643977.
The Hi-CRISPR A, B and C prediction algorithms are available from https://hicrispr.welkergroup.hu and the codes are deposited at Github: https://github.com/welkergroup/HiCRISPR.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Zoltán Ligeti, Lőrinc Sándor Pongor, Sarah Krausz, Zsuzsa Bartos and Gábor Glatz for their valuable help. We are also grateful to Elfrieda Fodor for critical reading of the manuscript and helpful comments. We thank Bernadett Czene, Ildikó Szűcsné Pulinka, Judit Szűcs, Fanni Mráz, Orsolya Oravetz, Gábor Erdős, Balázs Bohár and Dávid Fetter for their excellent laboratory assistance.
Authors’ contributions: A.T. and E.W. conceived and designed the experiments, interpreted the results and wrote the manuscript with input from all authors. A.T. performed the bacterial library screen experiments, experiments with mammalian cells and NGS. K.H. fine-tuned the bacterial selection and contributed to NGS preparation. P.I.K. contributed to the mammalian cell experiments and to NGS preparation and calculated RNA folding energies. E.V. and E.T. contributed to the bacterial and mammalian cell experiments and to NGS preparation. J.K.V., G.E.T. processed the NGS data, calculated cleavage efficiency values, Z.W., P.F.P. and G.E. analysed the data, A.W. developed Hi-CRISPR B and C and Z.G. calculated the feature values. All authors read and approved the final manuscript.
Contributor Information
András Tálas, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary; School of Ph.D. Studies, Semmelweis University, Budapest, Hungary.
Krisztina Huszár, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary; Gene Design Ltd, Szeged, Hungary.
Péter István Kulcsár, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary; Biospiral-2006 Ltd, Szeged, Hungary; School of Ph.D. Studies, University of Szeged, Hungary.
Julia K Varga, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary.
Éva Varga, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary; School of Ph.D. Studies, University of Szeged, Hungary; Institute of Biochemistry, Biological Research Centre, Szeged, Hungary.
Eszter Tóth, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary.
Zsombor Welker, Biospiral-2006 Ltd, Szeged, Hungary.
Gergely Erdős, Institute of Advanced Studies, Kőszeg, Hungary.
Péter Ferenc Pach, Institute of Advanced Studies, Kőszeg, Hungary.
Ágnes Welker, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary.
Zoltán Györgypál, Institute of Biophysics, Biological Research Centre, Szeged, Hungary.
Gábor E Tusnády, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary.
Ervin Welker, Institute of Enzymology, Research Centre for Natural Sciences, Budapest, Hungary; Institute of Biochemistry, Biological Research Centre, Szeged, Hungary.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of National Economy [GINOP-2.1.7-15-2016-00584]; National Research, Development and Innovation Office [K128188 to E.W., PD_125331 to E.T., 2018-1.1.1-MKI-2018-00167 to Z.W., EFOP 3.6.3-VEKOP-16-2017-00009 to E.V., 2018-1.1.1-MKI-2018-00081 to K.H.]; Hungarian Scientific Research Fund (OTKA) [K119287, K125607 to G.E.T.]; ‘Momentum’ Program of the Hungarian Academy of Sciences [LP2012/35]. Funding for open access charge: National Research, Development and Innovation Office [K128188].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gasiunas G., Barrangou R., Horvath P., Siksnys V.. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pineda M., Moghadam F., Ebrahimkhani M.R., Kiani S.. Engineered CRISPR systems for next generation gene therapies. ACS Synth. Biol. 2017; 6:1614–1626. [DOI] [PubMed] [Google Scholar]
- 6. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dominguez A.A., Lim W.A., Qi L.S.. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016; 17:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E.H., Weissman J.S., Qi L.S.. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013; 155:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S., Pederson T.. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qin P., Parlak M., Kuscu C., Bandaria J., Mir M., Szlachta K., Singh R., Darzacq X., Yildiz A., Adli M.. Live cell imaging of low-and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017; 8:14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalhor R., Mali P., Church G.M.. Rapidly evolving homing CRISPR barcodes. Nat. Methods. 2017; 14:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D.. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005; 151:2551–2561. [DOI] [PubMed] [Google Scholar]
- 15. Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O.. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014; 156:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S.. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014; 343:1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014; 507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.-D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J.. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods. 2015; 12:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chari R., Yeo N.C., Chavez A., Church G.M.. sgRNA Scorer 2.0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth. Biol. 2017; 6:902–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labuhn M., Adams F.F., Ng M., Knoess S., Schambach A., Charpentier E.M., Schwarzer A., Mateo J.L., Klusmann J.-H., Heckl D.. Refined sgRNA efficacy prediction improves large-and small-scale CRISPR–Cas9 applications. Nucleic Acids Res. 2018; 46:1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu H., Xiao T., Chen C.-H., Li W., Meyer C.A., Wu Q., Wu D., Cong L., Zhang F., Liu J.S.. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015; 25:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heigwer F., Kerr G., Boutros M.. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014; 11:122–123. [DOI] [PubMed] [Google Scholar]
- 24. Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E.. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016; 44:W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prykhozhij S.V., Rajan V., Gaston D., Berman J.N.. CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS One. 2015; 10:e0119372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park J., Bae S., Kim J.-S.. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015; 31:4014–4016. [DOI] [PubMed] [Google Scholar]
- 27. Wong N., Liu W., Wang X.. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015; 16:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Najm F.J., Strand C., Donovan K.F., Hegde M., Sanson K.R., Vaimberg E.W., Sullender M.E., Hartenian E., Kalani Z., Fusi N.. Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 2018; 36:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H.K., Kim Y., Lee S., Min S., Bae J.Y., Choi J.W., Park J., Jung D., Yoon S., Kim H.H.. SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Sci. Adv. 2019; 5:eaax9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F.. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016; 351:84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K.. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A.. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature. 2017; 550:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Casini A., Olivieri M., Petris G., Montagna C., Reginato G., Maule G., Lorenzin F., Prandi D., Romanel A., Demichelis F.. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018; 36:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kulcsár P.I., Tálas A., Huszár K., Ligeti Z., Tóth E., Weinhardt N., Fodor E., Welker E.. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017; 18:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hou Z., Zhang Y., Propson N.E., Howden S.E., Chu L.-F., Sontheimer E.J., Thomson J.A.. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim H.K., Min S., Song M., Jung S., Choi J.W., Kim Y., Lee S., Yoon S., Kim H.H.. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 2018; 36:239. [DOI] [PubMed] [Google Scholar]
- 37. Briner A.E., Donohoue P.D., Gomaa A.A., Selle K., Slorach E.M., Nye C.H., Haurwitz R.E., Beisel C.L., May A.P., Barrangou R.. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol. Cell. 2014; 56:333–339. [DOI] [PubMed] [Google Scholar]
- 38. Kalhor R., Kalhor K., Mejia L., Leeper K., Graveline A., Mali P., Church G.M.. Developmental barcoding of whole mouse via homing CRISPR. Science. 2018; 361:eaat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perli S.D., Cui C.H., Lu T.K.. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science. 2016; 353(6304):aag0511. [DOI] [PubMed] [Google Scholar]
- 40. Tóth E., Weinhardt N., Bencsura P., Huszár K., Kulcsár P.I., Tálas A., Fodor E., Welker E.. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol. Direct. 2016; 11:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Inoue H., Nojima H., Okayama H.. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990; 96:23–28. [DOI] [PubMed] [Google Scholar]
- 42. Weston A., Humphreys G., Brown M.G., Saunders J.. Simultaneous transformation of Escherichia coli by pairs of compatible and incompatible plasmid DNA molecules. Mol. Gen. Genet. MGG. 1979; 172:113–118. [DOI] [PubMed] [Google Scholar]
- 43. Goldsmith M., Kiss C., Bradbury A.R., Tawfik D.S.. Avoiding and controlling double transformation artifacts. Protein Eng. Des. Sel. 2007; 20:315–318. [DOI] [PubMed] [Google Scholar]
- 44. Tóth E., Czene B.C., Kulcsár P.I., Krausz S.L., Tálas A., Nyeste A., Varga É., Huszár K., Weinhardt N., Ligeti Z.. Mb-and FnCpf1 nucleases are active in mammalian cells: activities and PAM preferences of four wild-type Cpf1 nucleases and of their altered PAM specificity variants. Nucleic Acids Res. 2018; 46:10272–10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tusnady G.E., Simon I., Varadi A., Aranyi T.. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005; 33:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen W., Feng P.-M., Lin H., Chou K.-C.. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013; 41:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen W., Lei T.-Y., Jin D.-C., Lin H., Chou K.-C.. PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014; 456:53–60. [DOI] [PubMed] [Google Scholar]
- 48. Lorenz R., Bernhart S.H., Zu Siederdissen C.H., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorith. Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chuai G., Ma H., Yan J., Chen M., Hong N., Xue D., Zhou C., Zhu C., Chen K., Duan B.. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018; 19:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arimbasseri A.G., Rijal K., Maraia R.J.. Transcription termination by the eukaryotic RNA polymerase III. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013; 1829:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Graf R., Li X., Rajewsky K.. sgRNA sequence motifs blocking efficient CRISPR/Cas9-mediated gene editing. Cell Rep. 2019; 26:1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chakrabarti A.M., Henser-Brownhill T., Monserrat J., Poetsch A.R., Luscombe N.M., Scaffidi P.. Target-specific precision of CRISPR-mediated genome editing. Mol. Cell. 2019; 73:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M.. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019; 37:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen M.W., Arbab M., Hsu J.Y., Worstell D., Culbertson S.J., Krabbe O., Cassa C.A., Liu D.R., Gifford D.K., Sherwood R.I.. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018; 563:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta (BBA)-Protein Struct. 1975; 405:442–451. [DOI] [PubMed] [Google Scholar]
- 57. Kubat M., Holte R.C., Matwin S.. Machine learning for the detection of oil spills in satellite radar images. Mach. Learn. 1998; 30:195–215. [Google Scholar]
- 58. Mashiko D., Fujihara Y., Satouh Y., Miyata H., Isotani A., Ikawa M.. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013; 3:3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang D., Zhang C., Wang B., Li B., Wang Q., Liu D., Wang H., Zhou Y., Shi L., Lan F.. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019; 10:4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A.. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013; 31:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O.. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ibraheim R., Song C.-Q., Mir A., Amrani N., Xue W., Sontheimer E.J.. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 2018; 19:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dagdas Y.S., Chen J.S., Sternberg S.H., Doudna J.A., Yildiz A.. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Science advances. 2017; 3:eaao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schmid-Burgk J.L., Gao L., Li D., Gardner Z., Strecker J., Lash B., Zhang F.. Highly parallel profiling of Cas9 variant specificity. Mol. Cell. 2020; 78:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kulcsár P.I., Tálas A., Tóth E., Nyeste A., Ligeti Z., Welker Z., Welker E.. Blackjack mutations improve the on-target activities of increased fidelity variants of SpCas9 with 5′ G-extended sgRNAs. Nat. Commun. 2020; 11:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following plasmids used in this study are available from Addgene: pAT-9208 (#124221), pAT-9218 (#124225), pAT-9222 (#124222), pAT-9218 (#124225), pAT-9251 (#124226), pKH-1699 (#124227).
The deep sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA643977.
The Hi-CRISPR A, B and C prediction algorithms are available from https://hicrispr.welkergroup.hu and the codes are deposited at Github: https://github.com/welkergroup/HiCRISPR.