

Abstract
The global emergence of drug-resistant bacteria leads to the loss of efficacy of our antibiotics arsenal and severely limits the success of currently available treatments. Here, we developed an innovative strategy based on targeted-antibacterial-plasmids (TAPs) that use bacterial conjugation to deliver CRISPR/Cas systems exerting a strain-specific antibacterial activity. TAPs are highly versatile as they can be directed against any specific genomic or plasmid DNA using the custom algorithm (CSTB) that identifies appropriate targeting spacer sequences. We demonstrate the ability of TAPs to induce strain-selective killing by introducing lethal double strand breaks (DSBs) into the targeted genomes. TAPs directed against a plasmid-born carbapenem resistance gene efficiently resensitise the strain to the drug. This work represents an essential step toward the development of an alternative to antibiotic treatments, which could be used for in situ microbiota modification to eradicate targeted resistant and/or pathogenic bacteria without affecting other non-targeted bacterial species.
INTRODUCTION
The worldwide proliferation of drug-resistant bacteria is predicted to cause a dramatic increase in human deaths due to therapeutic failures in the next decades (1). The constant emergence of bacterial resistances and the current low rate of antibiotic discovery emphasize the need to develop innovative antibacterial strategies that represent a real alternative to the use of antibiotics. Moreover, antibiotics generally lack specificity as they target processes that are essential to bacterial proliferation. Antibiotics consequently affect the whole treated bacterial community without discriminating between harmful and commensal strains, and lead to the population enrichment in drug-resistant strains.
Recent reports have demonstrated the possibility to achieve specific antimicrobial activity through the use of clustered regularly interspaced short palindromic repeats (CRISPR) and the associated Cas proteins. CRISPR/Cas systems can achieve bacterial killing through the induction of double-strand breaks (DSBs) to the chromosome by the Cas9 nuclease (2,3). The expression of specific genes can also be inhibited through CRISPR interference (CRISPRi), when using the dead catalytic Cas9 enzyme (dCas9) (4,5). CRISPR targeting relies on the ∼16–20 nucleotide (nt) target-specific guide RNA (gRNA) sequence, which allows the recruitment of the Cas nuclease to the complementary DNA sequence (2,6). Yet, to be used as practical antibacterial tools, CRISPR/Cas genes need to be delivered to the targeted bacterium. Bacterial DNA conjugation precisely offers the possibility to transfer long DNA segments to a range of bacterial species, with the transfer specificity depending on the considered conjugation system. Methodologies using conjugation to deliver CRISPR/Cas systems have been recently developed to target Escherichia coli (7–10) or Salmonella Typhimurium (11). These methods rely on the RK2 plasmid conjugation machinery that perform broad-host range transfer, but with the drawback of low efficiency. Besides, these studies report the targeting of a single bacterial strain within mono-species populations only. One major challenge is to develop an antibacterial strategy that selectively alter one or several targeted bacterial strains, without affecting the other species present in a multispecies population. This objective requires the development of bioinformatics tools to identify gRNA sequence able to achieve such strain-specific targeting.
In this work, we present an innovative antibacterial methodology based on mobilizable plasmids that carry CRISPR/Cas systems designed to induce antibacterial activity into specifically targeted recipient strains. These so-called targeted-antibacterial-plasmids (TAPs) use the conjugation machinery (tra genes) encoded by a F plasmid to be efficiently transferred to E. coli strains and to closely related Gram-negative Enterobacteriaceae. TAPs were designed to produce the CRISPR Cas system in a constitutive manner and can easily be redirected against any bacterial species of interest by changing gRNA sequence in one-step cloning. The gRNA sequence carried by the TAP determines the targeting of the antibacterial activity towards specific recipient strains only. To identify strain-specific gRNA, we have developed a bio-informatic program CSTB (CRISPR Search Tool for Bacteria) that allows the rapid and robust identification of ∼16–20 nt sequences on the basis of their presence or absence in the genome of bacterial strains selected on a phylogenetic tree. Consequently, the CRISPR/Cas system will only be active in recipients that contain the DNA sequence complementary to the chosen gRNA sequence, while being inactive in other strains of a multispecies population mix. Here we demonstrate TAPs ability to induce efficient and strain-specific antibacterial activity against a range of Gram-negative Enterobacteriaceae within multi-species population in vitro.
MATERIALS AND METHODS
Bacterial strains, plasmids, primer and growth culture conditions
Bacterial strains construction and growth procedures
Bacterial strains, plasmids and primers are listed in Supplementary Tables S1, S2 and S3, respectively. Plasmid cloning were done by Gibson Assembly (12) and verified by Sanger sequencing (Eurofins Genomics). Chromosome mutation were transferred by phage P1 transduction to generate the final strains. Strains were grown at 37°C in Luria-Bertani (LB) broth, M9 medium supplemented with glucose (0.2%) and casamino acid (0.4%) (M9-CASA) or M63 medium supplemented with glucose (0.2%) and casamino acid (0.4%) (M63). When appropriate, the media were supplemented with the following antibiotics: 50 μg/ml kanamycin (Kn), 20 μg/ml chloramphenicol (Cm), 10 μg/ml tetracycline (Tc), 20 μg/ml nalidixic acid (Nal), 20 μg/ml streptomycin (St), 100 μg/ml ampicillin (Ap), 10 μg/ml gentamycin (Gm), 50 μg/ml rifampicin (Rif). When appropriate 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and 40 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) were added for screening of LAC phenotype.
TAPs construction and one-step-cloning change of the spacer sequence on the TAPs
Plasmid construction was performed by IVA cloning (13), expect for changing the spacer sequence in the TAPs, which was performed by the replacement of the spacer in pEGL129 by a SapI-spacer-SapI DNA sequence. The nsp (non-specific) spacer sequence is flanked by two SapI restriction sites that allow for liberation of non-cohesive DNA ends upon SapI digestion. To replace the nsp spacer, a new spacer is constructed by annealing two oligonucleotides (listed in Supplementary Table S3) with oriented complementary sequences to the non-cohesive ends generated by SapI restriction of TAP-Cas9-nsp or TAP-dCas9-nsp plasmids. Ligation production between the new spacer fragment and the TAP backbone was transformed into DH5α or TB28 strains. Constructions were verified by PCR reaction and sequencing.
Congo red assay
Curli production colony assay
Escherichia coli strain OmpR234 with or without plasmids were plated on Congo Red medium (10 g bacto tryptone, 5 g yeast extract, 18 g bacto agar, 40 μg/ml Congo Red and 20 μg/ml Coomassie Brilliant blue G) and incubated 4 days at 30°C. Colonies were visualized at ×10 magnification with a M80 stereomicroscope (Leica). Digital images were captured with an IC80-HD integrated camera coupled to the stereomicroscope, operated via LASv4.8 software (Leica).
Liquid aggregation test
Overnight culture of E. coli strain OmpR234 with or without plasmids were diluted to an A600 of 0.05 in 1 ml M9-CASA medium supplemented with 25 μg/ml of Congo Red. Culture were grown without agitation at 30°C for 24 h and image captured.
Conjugation assay
Overnight cultures grown in LB of donor and recipient strains were diluted to an A600 of 0.05 and grown until an A600 comprised between 0.7 and 0.9 was reached. 50 μl of donor and 150 μl of recipient cultures were mixed into an Eppendorf tube to obtain a 1:3 donor to recipient ratio. At time 0 min, 100 μl of the mix were diluted into 1 ml LB, serial diluted and plated on LB agar supplemented with antibiotics selecting for donor, recipient and transconjugant cells. The remaining 100 μl were incubated for 1h30 at 37°C. 1 ml of LB was added gently and the tubes were incubated at 37°C for another 1h30, 4h30 or 22h30. Conjugation mix were then vortexed, serial diluted and plated as for time 0 min.
Long-term conjugation experiment
Conjugation mixes were prepared and incubated at 37°C without agitation. Every 24 h, 100 μl of the mix were diluted into 1 ml of LB and re-incubated at 37°C. The remaining of the mixes were vortexed, serial diluted and plated on LB agar supplemented with antibiotics selecting for donor, recipient and transconjugant cells. At day 1 and day 7, 100 clones of the resulting ampicillin resistant recipients mixed with the TAP-dCas9-OXA48 carrying donor were streaked on LB agar supplemented with Tc or Kn to evaluate the presence of the F-Tn10 or TAP-dCas9-OXA48 plasmids respectively.
Multispecies conjugation
Overnight cultures grown in LB of donor and recipient strains were diluted to an A600 of 0.05 and grown until an A600 comprised between 0.7 and 0.9 was reached. A recipient mix is prepared by mixing C. rodentium, E. cloacae, E. coli EPEC and E. coli HS recipients strains in indicated proportions (Figure 5C). This mix is serial diluted and plated on LB agar supplemented with antibiotics to select for each recipient. 100 μl of donor and 100 μl of the recipient mix were added to an Eppendorf tube to perform mating. At time 0 min, 100 μl of the mix were diluted into 1 ml LB, serial diluted and plated on LB agar supplemented with antibiotics to select for donor, recipients and transconjugants. The remaining 100 μl were incubated for 1h30 at 37°C. 1 ml of LB was gently added and the tubes were incubated for an additional 1h30 at 37°C. Conjugation mix were then vortexed, serial diluted and plated on LB agar supplemented with antibiotics to select for donor, recipients and transconjugants. In the figures, the efficiencies of conjugation are represented either as the final concentration of transconjugant cell (CFU/ml) or as the percentage of transconjugant cells calculated from the ratio (T/R + T).
Figure 5.

Efficient and strain-specific killing of TAPs within a multispecies recipient population. (A) Efficiency of TAP-Cas9-nsp transfer from E. coli (LY1369) donor to C. rodentium, E. cloacae, E. coli EPEC or HS recipients. Histograms show the percentages of tranconjugants (T/R + T) after 24 h of conjugation for C. rodentium, E. cloacae, E. coli EPEC recipients and 3 h for E. coli HS recipient; mean and SD are calculated from at least three independent experiments. (B) TAPs carrying specific spacers identified with the CSTB algorithm were tested against each recipient cells. To account for the variability of TAP transfer in the different recipient strains, the histograms show the relative abundance of viable transconjugants normalized by viable transconjugants obtained for the TAPKn-Cas9-nsp. Numbers in brackets indicate replicates with detection limit of transconjugants below 10−8. Mean and SD are calculated from 3 independent experiments. One-way ANOVA with Sidak's multiple comparisons test were performed on log10-transformed values and p-value show comparison with the TAP-Cas9-nsp data. ns = non-significant P-value >0.05; **P-value <0.0021; ****P-value <0.0001. (C) Proportion of recipients estimated by plating assay before mating with donors. Mean and SD calculated from three independent experiments are indicated for each recipient strains. (D) Each TAP carrying specific spacers were tested through conjugation between E. coli donors and a recipient population containing all recipient species. Histograms show the proportion of viable transconjugants in the mixed population after 3 h of mating. Numbers in brackets indicate replicates with detection limit of transconjugants below 10−8. Mean and SD are calculated from three independent experiments. One-way ANOVA with uncorrected Fisher's LSD test were performed on the log10-transformed values and P-value show comparison with the TAP-Cas9-nsp data. ns = non-significant P-value >0.05; *P-value<0.05; **P-value<0.0021; ****P-value<0.0001. Donors TAP-Cas9-nsp (LY1369), TAP-Cas9-Cr1 (LY1597), TAP-Cas9-Ecl (LY1566), TAP-Cas9-EPEC (LY1618), TAP-Cas9-EEC (LY1665); recipients C. rodentium (LY720), E. cloacae (LY1410), E. coli EPEC (LY1615) or HS (LY1601).
Transformation assay
Overnight cultures grown in LB were 1/100 diluted and grown until an A600 comprised between 0.4 and 0.6. Cells were treated with Rubidium Chloride and 90 μl of the resulting competent cells transformed with 100 ng of plasmid and heat shock. Following the 1h incubation at 37°C for phenotypic expression, cells were centrifugated 5 min at 5000 rpm, resuspended in 100 μl of LB, and plated on LB-agar plates supplemented with the appropriate antibiotics.
Live-cell microscopy imaging and analysis
Time-lapse experiments
Overnight cultures in M9-CASA (between E. coli) or M63 (between E. coli and C. rodentium) of donor and recipient cells were diluted to an A600 of 0.05 and grown until an A600 comprised between 0.7 and 0.9. 25 μl of donor and 75 μl of recipient were mixed into an Eppendorf tube and 50 μl of the mix was loaded into a B04A microfluidic chamber (ONIX, CellASIC®). Nutrient supply was maintained at 1 psi and the temperature maintained at 37°C throughout the imaging process. Cells were imaged every 10 min for 3 h.
Image acquisition
Conventional wide-field fluorescence microscopy imaging was carried out on an Eclipse Ti-E microscope (Nikon), equipped with ×100/1.45 oil Plan Apo Lambda phase objective, FLash4 V2 CMOS camera (Hamamatsu), and using NIS software for image acquisition. Acquisition were performed using 50% power of a Fluo LED Spectra X light source at 488 and 560 nm excitation wavelengths. Exposure settings were 50 ms for sfGFP and 50 ms for mCherry produced from the TAPs; 100 ms for RecA-GFP, HU-mCherry and DnaN-mCherry.
Image analysis
Quantitative image analysis was done using Fiji software with MicrobeJ plugin (14). The Manual-editing interface of MicrobeJ was used to optimize cell detection and the Mean intensity fluorescence, skewness and cell length parameters were automatically extracted and plotted. We defined the timing of TAP acquisition (time t = 0) by analyzing the increase of the fluorescence signal conferred by the TAPs (sfGFP or mCh). Plasmid acquisition was validated when a 15% sfGFP or a 30% mCherry fluorescence increase was observed in the transconjugant cells. Fluorescence profiles of each cells were then aligned according the defined t = 0 to generate the graphs presented in Figures 2B, C, E, F, 3C and Supplementary Figure S5d.
Figure 2.

Real-time visualization of E. coli killing after acquisition of TAP. (A) Upper panel: diagram of the fluorescent reporter system allowing microcopy visualization of TAP transfer and subsequent nucleoid disorganization in transconjugants. Donor cells exhibit diffuse green fluorescence due to sfGFP production from TAP; HU-mCherry recipients exhibit red nucleoid associated fluorescence; transconjugants are identified by the production of both green and red fluorescence. Lower panel: time-lapse microscopy images performed in a microfluidic chamber. D (donor), recipient (R), and transconjugant (T) cells are indicated. Scale bar 1 μm. Donors TAP-Cas9-nsp (LY1371) or TAP-Cas9-lacZ2 (LY1380); recipient HU-mCherry lac+ (LY248). (B, C) Single-cells time-lapse quantification of transconjugants (B) bacterial and (C) nucleoid lengths. Average and SD are indicated (n cells analysed). The time of TAP acquisition (red dashed line at 0 min) corresponds to a 15% increase in the green fluorescence in the transconjugant cells. (D) Upper panel: diagram of the fluorescent reporter system. Donor cells exhibit diffuse red fluorescence from the mCherry produced by TAP; recipients exhibit diffuse RecA-GFP fluorescence; transconjugants are identified by the production of red fluorescence followed by RecA-GFP polymerization. Lower panel: time-lapse microscopy images performed in a microfluidic chamber. Donor (D), recipient (R), and transconjugant (T) cells are indicated. Scale bar 1 μm. Donors TAP-Cas9-nsp (LY1537) or TAP-Cas9-lacZ2 (LY1538), recipient RecA-GFP (LY844). (E, F) Single-cells time lapse quantification of transconjugants (E) cell length and (F) skewness of RecA-GFP fluorescence signal. Average and SD are indicated (n cells analysed). The time of TAP acquisition (red dashed line at 0 min) corresponds to a 30% increase in the green fluorescence in the transconjugant cells. (B–C and E–F) Multiple t-test were performed corrected with Holm–Sidak method. Stars indicate the time with significant difference (P-value < 0.05). Significant difference was observed from this point until the end of the analysis.
Figure 3.
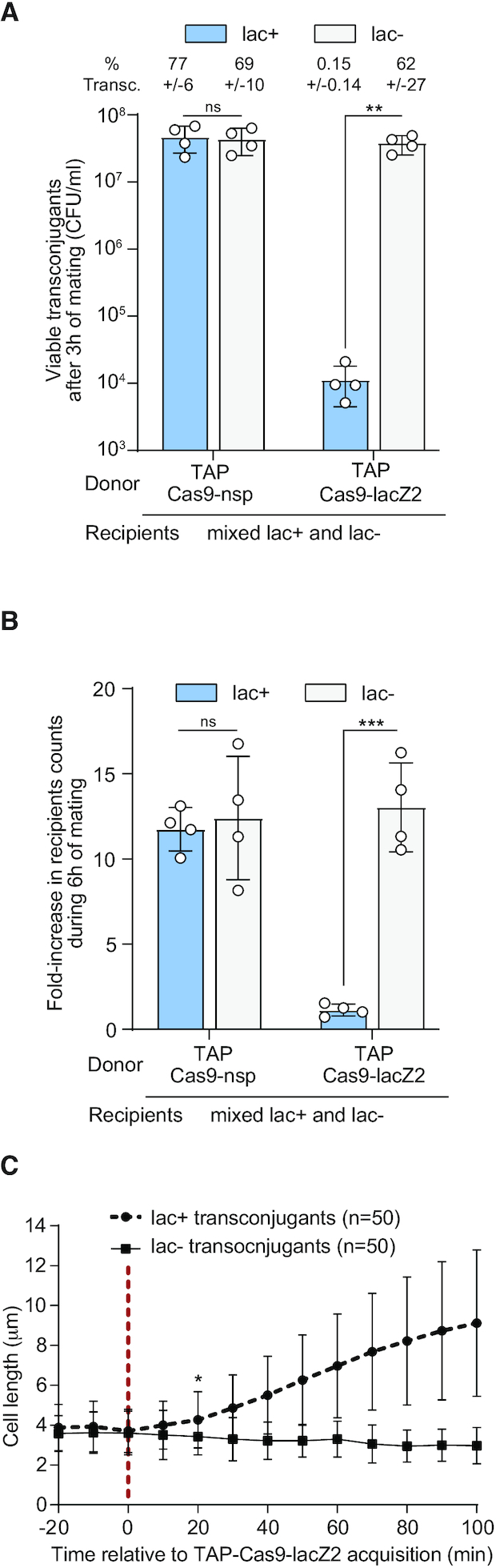
TAP system specifically kills targeted recipients in a mix of targeted and non-targeted E. coli recipient cells. (A) Viable transconjugant cells and percentage of transconjugants (ratio T/R + T) through TAPkn-Cas9-nsp or TAPkn-Cas9-lacZ2 transfer from donor to a mixed lac+ and lac– recipient population. Two-tails unpaired t-test was performed on log10-transformed values. ns = non-significant P-value >0.05; ***P-value <0.0002. (B) Quantification of fold-increase in lac+ and lac– recipient populations counts over the 6h of mating. Mean and SD are calculated from 4 independent experiments. Donors: TAP-Cas9-nsp (LY1369) or TAP-Cas9-lacZ2 (LY1370); recipients lac+ (LY827) and lac– (LY848). Two-tails unpaired t-test was performed. ns = non-significant P-value >0.05; **P-value <0.0021. (C) Single-cell quantification showing cell length increase in the targeted lac+ transconjugant cells but not non-targeted lac- transconjugants. The time of TAP acquisition (red dashed line at 0 min) corresponds to a 15% increase in the green fluorescence in the transconjugant cells. Cell length average is indicated with SD (n cells analysed). Donor TAP-Cas9-lacZ2 (LY1380); recipients HU-mCherry lac+ (LY248) and DnaN-mCherry lac– (LY1423). Multiple t-test were performed corrected with Holm–Sidak method. Stars indicate the time with significant difference (P-value < 0.05). Significant difference was observed from this point until the end of the analysis.
Flow cytometry
Conjugation was done as described in the conjugation assay section in 0.1 μm filtered LB. At time 90 min and 180 min, conjugation mix were diluted to an A600 of 0.03 in 0.1 μm filtered LB and analysed into an Attune N×T acoustic focusing cytometer at a 25 μl/min flow rate. Forward scattered (FSC), Side scattered (SSC) as well as fluorescence signal BL1 (sfGFP) and YL2 (mCherry) were acquired with the appropriate PMT setting and represented with the Attune™ N×T analysis software. To verify the absence of toxicity of the Cas9 or dCas9 constitutive expression from the TAPs, we compared the growth of E. coli MS388/TAP with the cas9 or dcas9 or without any cas9 gene. Those strains were grown overnight in 0.1 μm filtered LB and diluted to an A600 of 0.05 in 0.1 μm filtered LB. They were grown during 8 h and the A600 and CFU/ml were estimated by plating assays at 0, 2, 4, 6 and 8 h. In parallel, at 1 h, 2 h and 5h30 the strains were analysed into the Attune NxT acoustic Focusing cytometer at a 25 μl/min flow rate. Forward scattered (FSC) was acquired and represented with the Attune™ NxT software.
Analysis of TAP-escape mutants
In E. coli. The 31 TAP-escape mutants were streaked on medium supplemented with X-Gal and IPTG to determine their LAC phenotype. TAP-escape mutants exhibiting lac+ phenotype were classified as ‘Blue’ and the others as ‘White’ in Supplementary Figure S3. To determine the acquisition of point mutation or deletion that modify the targeted lacZ2 locus, a PCR was realized with OL240 and OL654 that amplify a fragment of 748 pb encompassing the lacZ2 locus in wt strain. For escape mutants that exhibited no deletion of the lacZ2 locus but still had an active TAP CRISPR system, the PCR product was sequenced and the mutations identified. A PCR was also done with OL655 and OL656 to amplify a larger fragment around lacZ2 and observe large deletion as previously described (15). To determine the activity of the TAPs extracted from escape mutants, conjugation was performed between the TAP-escape mutants and an E. coli MS388 lac+ strain as described in the conjugation assays section. In parallel, the activity of the TAPs extracted with the Machery Nagel NucleoSpin® Plasmid kit from escape mutants were verified by transformation into lac+ and lac− strains as described in the transformation assay section. Seven inactive TAPs were sequenced to identify mutations inactivating CRISPR system.
C. rodentium. For the 20 TAP-Cas9-Cr1-escape mutants, a PCR with OL686 and OL687 was performed to determine deletions in the chromosome locus. To verify the CRISPR activity of the TAPs from C. rodentium TAP-escape mutants, conjugation was performed during 5 h between the C. rodentium mutants and the E. coli MS388 strain to generate new E. coli TAPs donors. Then conjugation was performed during 24 h between those new donors and fresh C. rodentium recipients and plated to select for recipient and transconjugants. To confirm the activity of the TAPs isolated form C. rodentium escape mutants, TAPs were extracted with Machery Nagel NucleoSpin® Plasmid kit and transformed by electroporation (2.5 kV) into wt C. rodentium cells treated with 10% sucrose. Following 1 h of incubation at 37°C, cells were plated on LB-agar supplemented or not with Kn to evaluate the transformation efficiency. Two inactive TAP-Cas9-Cr1 and two inactive TAP-Cas9-Cr22 isolated from escaper clones were sequenced.
CSTB algorithm
The CSTB web site can be freely accessed at https://cstb.ibcp.fr. The CSTB web service enables the comparative analysis of CRISPR motifs across a wide range of bacterial genomes and plasmids. Currently considered motifs are NGG-anchored sequences of 18–23 bp long. The CSTB back-end database indexes all occurrences of these CRISPR motifs present in 2914 complete genomes labeled as representative or reference in the release 99 of RefSeq (03/12/20). In addition, seve bacterial genomes and five plasmids of interest were added. The mean number of distinct motifs among bacterial genomes is 55 923 (5719 and 2 729 570 as respective minimum and maximum). Genomes are classified according to the NCBI taxonomy (07/22/20). Each genome is inserted in the database of motifs by processing the corresponding complete fasta using the following procedure. Firstly, all words satisfying the CRISPR motif regular expression are detected and their chromosomal coordinates stored in a database of motifs. Secondly, all unique words are converted into an integer representation using a 2 bits per base encoding software we developed (https://github.com/glaunay/crispr-set). These integers are then sorted in a unique flat file per genome. The indexing of CRISPR motifs as integers enables computationally efficient comparison of the sets of motifs across several organisms. Finally, the original fasta file is added to a blast database. All related software can be freely accessed at https://github.com/MMSB-MOBI/CSTB_database_manager. The CSTB input interface displays the 2914 genomes available for searching as two taxonomic trees. The left-hand tree allows for the selection of species whose genomes have to feature identical/similar CRISPR motifs. This set of genomes defines the targeted CRISPR motifs. Meanwhile, the right-hand tree allows for the selection of ‘excluded’ organisms, which must have no motif in common with the targeted ones. The set of motifs that satisfies the user selections will effectively be equal to the union of the motifs found in the selected organisms subtracted from the intersection of the motifs found in the ‘excluded’ organisms. Computation time ranges from seconds to a few minutes according to the size of the selections and an email is sent upon completion.
All the solutions CRISPR motifs are presented in an interactive table of gRNA sequences and their occurrences in each selected organism. The table has sorting and filtering capabilities on motif counts and sequence composition. This allows for the easy selection of motifs of interest. Detailed information can be downloaded for the entire set of solutions or for the selected motifs only. This detailed information is provided in tabulated file with lines featuring the coordinates of each sgRNA motif in the targeted organisms. Alternatively, the user may explore the results using a genome-based approach. Hence, each targeted genome has its graphical view. The graphic is a circular histogram of the entire distribution of solution sgRNA motifs in a selected genome. The graphic is interactive to display the local breakdown of sgRNA distribution.
RESULTS
Targeted-antibacterial-plasmids (TAPs) modular design
TAPs derivate from the synthetic pSEVA plasmid collection (16), and carry the pBBR1 origin of replication, a choice of resistant gene cassettes, and the oriTF origin of transfer of the F plasmid (Figure 1A). TAPs are consequently mobilizable by the conjugation machinery produced in trans from the conjugative F-Tn10 helper plasmid contained in the donor cells (Figure 1B and Supplementary Figure S1a) (17,18). We inserted the Streptococcus pyogenes wild-type cas9 (for CRISPR activity) or catalytically dead dcas9 gene (for CRISPRi activity) and the guide gRNA sequence composed of the constant tracrRNA scaffold and the target-specific crRNA spacer sequence (Figure 1A). Changing the crRNA spacer sequence in one-step-cloning allows reprogramming the targeting of the TAPs against any specific chromosome or plasmid DNA. Optionally, TAPs also carry either the superfolder green fluorescent protein (sfgfp) or the mcherry gene highly expressed from the broad-host range synthetic BioFab promoter (19) to serve as plasmid transfer fluorescent reporter in microscopy and flow cytometry assays (Figure 1A).
Figure 1.

Transfer of TAP by F plasmid machinery mediates killing of a targeted E. coli strain. (A) TAPs modules consist of CRISPR system composed of wild type (cas9) or catalytically dead cas9 (dcas9) genes expressed from the weak constitutive BBa_J23107 promoter and a gRNA module expressed from the strong constitutive BBa_J23119 promoter; the F plasmid origin of transfer (oriTF); the pBBR1 origin of replication (oriV), and a set of resistance cassettes (Ap, ampicillin; Kn, kanamycin; Cm, chloramphenicol; St, streptomycin; Gm, gentamycin), an optional cassette carrying the sfgfp or mcherry genes highly expressed from the broad-host range synthetic BioFab promoter. (B) Diagram of the TAP antibacterial strategy. A donor strain produces the F plasmid conjugation machinery to transfer the TAP into the recipient strain. Targeted recipient carries a sequence recognized by CRISPR(i) system that induces killing or gene expression inhibition. Non-targeted recipient lacking the spacer recognition sequence are insensitive to CRISPR(i) activity. (C) Histogram of TAPs transfer estimated by flow cytometry show that TAPkn-Cas9-nsp-GFP and TAPkn-Cas9-lacZ2-GFP are transferred with similar efficiency in recipient cells after 3 h of mating. Donors TAP-Cas9-nsp (LY1371) or TAP-Cas9-lacZ2 (LY1380), recipient HU-mCherry lac+ (LY248). Two-tailed unpaired t-test was performed. ns: non-significant P-value >0.05. (D) Histograms of the concentration of viable transconjugants estimated by plating assays show viability loss associated with the acquisition of TAP-Cas9-lacZ2. The corresponding percentage of viable transconjugants (ratio T/R+T) is shown above each bar. Two-tailed unpaired t-test was performed. **P-value <0.0021 (E) Fold-increase of the recipient population counts over the 3 h of mating. Donors TAP-Cas9-nsp (LY1369) or TAP-Cas9-lacZ2 (LY1370), recipient lac+ (LY827). Two-tailed unpaired t-test was performed. *P-value <0.05 (C–E) Mean and SD are calculated from three independent experiments.
Validation of TAPs CRISPR and CRISPRi activities
We addressed the ability of TAPs to induce efficient and specific Cas9-mediated killing (CRISPR) or dCas9-mediated gene expression inhibition (CRISPRi). First, TAPs ability to induce Cas9-mediated killing was confirmed using the previously described lacZ2 spacer that targets the lacZ gene of E. coli (15). Transformation of the TAP-Cas9-lacZ2 plasmid into the lac+ MG1655 wt strain was ∼1000-fold less efficient than in the isogenic lac– strain carrying a deletion of the targeted lacZ locus (Supplementary Figure S1b). By contrast, the TAP-Cas9-nsp plasmid, which contains a non-specific (nsp) crRNA spacer that does not target E. coli genome, was transformed with equal efficiency in both lac+ and lac– strains (Supplementary Figure S1b). Second, TAPs ability to induce dCas9-mediated CRISPRi activity was validated by using the csgB spacer that targets the csgB promoter driving the production of cell-surface curli fimbriae (20) in the MG1655 E. coli mutant strain OmpR234 (21). Congo Red (CR) staining on agar-plates and aggregation clumps formation in liquid medium were used as direct readouts for curli production (21,22). The TAP-dCas9-csgB efficiently inhibits curli production by the OmpR234 strain, as reflected by the formation of white colony in the presence of CR and the inability to form aggregation clumps (Supplementary Figure S1c). By contrast, the non-specific TAP-dCas9-nsp had no effect on curli formation or aggregation in the OmpR234 strain. Besides, we confirmed that the constitutive production of the Cas9 or dCas9 from the TAPs do not cause growth defects (Supplementary Figure S1d) or elongated cell morphology (Supplementary Figure S1e), contrasting with the toxic effects reported in some systems (23–26). These results demonstrate that TAPs ability to induce Cas9-mediated killing or dCas9-mediated gene expression inhibition is efficient and depends on the accurate targeting by the spacer sequence.
TAPs-mediated killing of targeted recipient cells
Next, we addressed the ability of the TAPs to be transferred by conjugation and induce antibacterial activity in E. coli recipient cells. Conjugation was performed using the E. coli MG1655 donor strain that contains the F-Tn10 helper plasmid and either the TAP-Cas9-nsp or TAP-Cas9-lacZ2 mobilizable plasmids. Using flow cytometry analysis, we quantified the transfer efficiency of these TAPs (carrying the sfGFP green fluorescent reporter) into a lac+ recipient strain that produces the red fluorescent histone-like protein HU-mCherry, encoded on the chromosome (Supplementary Figure S2a). Quantification of the transconjugants exhibiting combined red and green fluorescence show that TAP-Cas9-nsp and TAP-Cas9-lacZ2 are both transferred to ∼65% of the recipient cell population after 3h of mating (Figure 1C and Supplementary Figure S2b). As expected, TAPs transfer requires the presence of the F-Tn10 plasmid in the donor strain (Supplementary Figure S2c–e). Most importantly, the parallel plating of the conjugation mixes revealed a ∼3.5-log decrease in the viability of TAP-Cas9-lacZ2 transconjugants compared to TAP-Cas9-nsp transconjugants (Figure 1D). This killing activity is also reflected by the lack of increase in the total recipient cells count during the three hours of mating with the TAP-Cas9-lacZ2 donor strain, compared to a ∼20-fold increase with TAP-Cas9-nsp donors (Figure 1E). Importantly, no killing effect is observed for either TAPs when using the isogenic lac- recipient strain lacking the targeted lacZ locus (Supplementary Figure S2f). These results show that TAP-Cas9-nsp and TAP-Cas9-lacZ2 are transferred with equal efficiency through the F-Tn10 conjugation machinery. Yet, the acquisition of TAP-Cas9-lacZ2, but not TAP-Cas9-nsp, is associated with a loss of viability of the transconjugant cells.
Using live-cell microscopy, we characterized the cellular response of the recipient cells to the acquisition of TAPs (Figure 2). In these experiments, the TAPs carry the sfGFP reporter system that confers green fluorescence to the donor and transconjugant cells. The lac+ recipient cells produce the nucleoid-association protein HU-mCherry, which localization reveals the global organization of the chromosome. As expected, the acquisition of the TAP-Cas9-nsp reported by the production of sfGFP green fluorescence in red recipient cells has no impact on growth, cell morphology or nucleoid organization (Figure 2A–C and movie 1). By contrast, the acquisition of the TAP-Cas9-lacZ2 triggers the rapid disorganization of the nucleoid that grows into an unstructured DNA bulk, followed by cells filamentation and occasional cell lysis (Figure 2A–C and movie 2). Furthermore, we analyzed in recipient cells the localization pattern of a RecA-GFP fusion, which has been reported to polymerize into large intracellular structures in response to DNA-damage induction (27). In this experiment, TAPs carry the mCherry reporter system that confers red fluorescence to donors and transconjugant cells. Image analysis reveals that the acquisition of the TAP-Cas9-lacZ2 (Figure 2D and movie 4), but not the TAP-Cas9-nsp (Figure 2D and movie 3), is followed by cells filamentation (Figure 2E) as well as the RecA-GFP polymerization, which was quantified using fluorescence skewness analysis (Figure 2F, see Materials and Methods). Nucleoid disorganization, cell filamentation and RecA-GFP bundle formation confirm that TAP-Cas9-lacZ2 acquisition is followed by CRISPR-mediated induction of DSBs that result in the death of the transconjugants.
TAPs-mediated selective killing within a mixed E. coli recipient population
We verified the specificity of TAPs-mediated killing within a mixed recipient population composed of the targeted lac+ and the non-targeted lac- E. coli strains. We observed a ∼4 log-fold decrease in viable lac+ transconjugants compared to lac- transconjugants when using the TAP-Cas9-lacZ2, while no difference is observed with the TAP-Cas9-nsp (Figure 3A). TAP-Cas9-lacZ2 specific killing activity is also reflected by the stagnation of the targeted lac+ recipient total population, while the non-targeted lac- population is able to grow during the 6 h of mating (Figure 3B). We performed live-cell microscopy imaging where the lac+ and lac– recipients are distinguished by the typical localization pattern of nucleoid associated HU-mCherry and the replisome associated DNA clamp DnaN-mCherry, respectively. Time-lapse analysis shows that both strains receive the plasmids reported by the increase of green fluorescence, yet only the targeted lac+ transconjugants exhibits cell filamentation, symptomatic of Cas9-mediated DNA-damage induction (Figure 3C). These results recapitulate the effects obtained when using individual recipient strains, and demonstrate that the TAPs achieve selective killing of the targeted strain within a mixed population.
Analysis of TAP-escape mutants
Transfer of the TAP-Cas9-lacZ2 is associated with a ∼3.5-log viability loss of the lac+ transconjugant cells, yet we noticed a proportion of transconjugants that are able to survive despite the acquisition of the TAP (Figure 1D). Genotyping and sequence analysis of 31 clones escaping the TAP-Cas9-lacZ2 activity revealed two types of escape mutants (Supplementary Figure S3a and b). One third (12 out of 31) have acquired a transposase or IS insertion in the plasmid-born cas9 gene, thus inactivating the CRISPR system. Two-thirds have acquired mutations that modify the targeted lacZ chromosome locus, either by small or large deletions (12 out of 31) as already described (15), or by single point mutation in the seed region of the PAM (7 out of 31), which was shown to be key for recognition by the Cas9–gRNA complex (28) (Supplementary Figure S3c).
TAPs directed against carbapenem-resistant population
Conjugative plasmids are major contributors to the spread of multi-drug resistance in bacteria (29), those conferring carbapenem resistance being of severe clinical concern (30). The IncL/M pOXA-48a plasmid carries the blaOXA-48 gene that encodes the OXA-48 carbapenemase, which confer resistance to carbapenem and other beta lactams, such as imipenem and penicillin (31). We designed TAPs targeting the pOXA-48a and assessed their ability to sensitize the plasmid-carrying population to ampicillin. Using an OXA48 spacer that targets the 5′-end of the blaOXA-48 gene, we constructed the TAP-Cas9-OXA48 to induce Cas9-mediated DSBs on pOXA-48a, and the TAP-dCas9-OXA48 to inhibit blaOXA-48 gene transcription by CRISPRi (Figure 4A). Transfer of TAP-Cas9-OXA48 and TAP-dCas9-OXA48 plasmids into pOXA-48a-carrying E. coli recipients lead to a ∼4.5-log decrease in ampicillin resistance level, while the TAP-Cas9-nsp or the TAP-dCas9-nsp have no effect (Figure 4B). Unexpectedly, we observe that significantly less viable transconjugant are obtained without ampicillin selection when using the TAP-Cas9-OXA48 compared to TAP-dCas9-OXA48 (Figure 4B). We ruled out the possibility of a decrease in TAP-Cas9-OXA48 transfer ability as all four tested plasmids are acquired with similar frequency by pOXA-48a plasmid-free E. coli recipients (Supplementary Figure S4a). However, analysis of the pOXA-48a plasmid sequence revealed the presence of the pemIK toxin-antitoxin (TA) system, which is involved in plasmid stability by inhibiting the growth of daughter cells that do not inherit the plasmid (7,32,33). Indeed, the arrest of pemIK expression due to plasmid loss results in the rapid depletion of the labile PemI antitoxin, which can no longer repress the toxic activity of the more stable PemK toxin. This regulation was reported using CRISPR-associated phage therapy to cure antibiotic resistance carried by the pSHV-18 plasmid (7). We then hypothesized that the observed reduction of viable TAP-Cas9-OXA48 transconjugants could be due to PemK toxic activity in cells that have lost of the pOXA-48a targeted by the Cas9 cleavage. This possibility was confirmed by inserting a constitutively expressed antitoxin pemI gene into the TAP-Cas9-OXA48, which results in a ∼1.5 log increase in transconjugants viability, while retaining the inhibition of ampicillin resistance (Figure 4B).
Figure 4.

TAP re-sensitises pOXA48-carrying recipient cells and impedes resistance dissemination. (A) Diagram of the TAP-mediated anti-resistance strategy. TAP-Cas9-OXA48 targeting the blaOXA48 promoter is transferred from a donor to an ampicillin resistant recipient cells carrying the pOXA-48a plasmid. Acquisition of the TAP-Cas9-OXA48 induces double-strand-breaks (DSBs) into the plasmid, while the TAP-dCas9-OXA48 inhibits blaOXA48 gene expression. Both TAPs sensitize the transconjugant cells to ampicillin. (B) Histograms showing reduction of ampicillin resistance in transconjugants cells after acquisition of the TAPkn-Cas9-OXA48, TAPkn-dCas9-OXA48 and TAPkn-Cas9-OXA48-PemI. Percentages of transconjugants (ratio T/R + T) are indicated. (C) Histograms showing the frequency of donor cells acquiring ampicillin resistance through transfer of pOXA-48a from the recipients (as depicted in the above diagram). (D) Histograms show the frequency of ampicillin-resistance acquisition through pOXA-48a transfer into R#2 plasmid-free wt recipient that have received the TAPs (TAP-transc.#2) (as depicted in the above diagram). Mean and SD are calculated from at least three independent experiments. Donors TAP-dCas9-nsp (LY1524), TAP-Cas9-nsp (LY1369), TAP-dCas9-OXA48 (LY1523), TAP-Cas9-OXA48 (LY1522) or TAP-PemI-Cas9-OXA48 (LY1549); Recipients R#1 wt/pOXA-48a (LY1507) and R#2 wt (LY945). (B–D) Two-tailed unpaired t-tests were performed. ns = non-significant P-value >0.05; *P-value <0.05, **P-value <0.0021; ****P-value <0.0001.
We further investigated the long-term evolution of resistance of the strain carrying the pOXA-48a during conjugation with a TAP donor. We observed that while the TAP-dCas9-nsp had no effect, the transfer of the TAP-dCas9-OXA48 and the resulting re-sensitization of the recipient population to ampicillin reached equilibrium after 24 h (Supplementary Figure S4b). From this point on, a stable 90% of the recipients have received the TAP-dCas9-OXA48 and became sensitive to ampicillin. We hypothesized that the remaining 10% of ampicillin-resistant recipient cells could result from the acquisition of the F-Tn10 plasmid only, thus resulting in the establishment of the F-encoded exclusion systems in the recipient cells and the permanent inability to acquire the TAP through a subsequent conjugation event. This hypothesis was confirmed by showing that all ampicillin-resistant recipients present in the population after 1 and 7 days of co-culture do contain the F-Tn10 plasmid but not the TAP-dCas9-OXA48 (Supplementary Figure S4c). One way to modulate the transfer efficiency of the mobilizable TAPs would be to prevent the transfer of the F plasmid by deletion of its origin of transfer. First, this would prevent the acquisition of the F plasmid only and the consequent establishment of the exclusion mechanism in the recipient cells. Second, the recipient cells that receive the TAP only would be unable to transmit it to other recipient bacteria due to the absence of the F-encoded conjugation machinery. In this situation, TAPs are expected to disseminate more slowly, but potentially to all recipient cells in the population.
The pOXA-48a is an autonomous conjugative plasmid that disseminates among Enterobacteriaceae, raising the possibility that the recipient containing the pOXA-48a could transfer ampicillin resistance to the TAPs-donors during mating. We observed that ampicillin resistance is indeed transmitted to ∼0.2% and 0.12% of donors carrying the TAP-dCas9-nsp or the TAP-Cas9-nsp (Figure 4C). However, donors carrying the TAP-dCas9-OXA48 or the TAP-Cas9-OXA48 do not acquire ampicillin resistance (Figure 4C). Assuming that the efficiency of pOXA48 transfer is insensitive to the presence of the TAPs in the cells, this result suggests that TAPs directed against OXA48 impedes the development of resistance, even if the pOXA48 plasmid is acquired. We tested this possibility by performing the same conjugation experiments with an additional plasmid-free recipient wt strain (R#2) in the conjugation mix (see diagram in Figure 4D). Among R#2 cells that have received the TAP-Cas9-nsp or the TAP-dCas9-nsp, ∼0.24% and 0.15% become ampicillin resistant, respectively. However, no ampicillin resistance is observed in R#2 cells that have received the TAP-Cas9-OXA48 or the TAP-dCas9-OXA48 (Figure 4D). Altogether, these results demonstrate that directing TAPs against the blaOXA-48 gene is an efficient strategy to sensitize the pOXA-48a-carrying strain to ampicillin. In addition, TAPs also appear to impede drug-resistance dissemination by protecting the donor and other plasmid-free recipients from developing the resistance.
CSTB software: targeting specific strains within multispecies bacterial population
Numerous bioinformatic tools have been developed to identify and design gRNA spacers specific of one species (34). However, designing TAPs that perform antibacterial activity against specific bacterial species, without affecting other bacterial strains, requires the robust identification of spacer sequences that are present in the genome of the targeted organism(s), but not in the genomes of other non-targeted strains. Since no such tools existed to achieve this task, we developed a ‘Crispr Search Tool for Bacteria’ CSTB algorithm (https://cstb.ibcp.fr.) that enables the comparative analysis of ∼18–23 nt long spacer sequences across a wide range of bacterial genomes and plasmids. The CSTB back-end database indexes all occurrences of these motifs present in 2919 complete genomes classified according to the NCBI taxonomy. CSTB allows identifying appropriate spacer sequences to reprogram TAPs against unique or multiple sites in the targeted chromosome or plasmid DNA.
We asked the CSTB algorithm to generate spacer sequences that target the attachment/effacement (A/E) pathogen Citrobacter rodentium strain ICC168 (Cr spacers), or the enteropathogenic E. coli EPEC strain E2348/69 (EPEC spacer), or the nosocomial pathogen Enterobacter cloacae (Ecl spacer), or the three of them (EEC spacer), without targeting other bacterial genome present in the database. TAPs directed against C. rodentium carry a Cr1 spacer that target a unique locus, or a Cr22 that targets 22 loci distributed throughout the genome (Supplementary Figure S5a). Transfer of TAP-Cas9-Cr1 from an E. coli donor reduces by 4-log the viability of C. rodentium transconjugant cells (Supplementary Figure S5b). Live-cell microscopy revealed that TAP-Cas9-Cr1 acquisition induces C. rodentium filamentation and lysis, while no growth defect was induced by the TAP-Cas9-nsp (Supplementary Figure S5c and d; movies 5 and 6). This indicates that, as observed in E. coli, the induction of a single DSB by the Cas9 is lethal to C. rodentium. Consistently, targeting 22 chromosome loci by the TAP-Cas9-Cr22 results in comparable transconjugant killing efficiency (Supplementary Figure S5b). However, multiple targeting unbalances the contribution of the mechanisms by which transconjugants escape to the TAPs activity. Analysis of twenty clones escaping Cr1 single targeting revealed either deletions of the targeted chromosomal locus or inactivation of the CRISPR system on the TAP, in equal proportion. By contrast, all 19 clones escaping the Cr22 multiple targeting carry mutations that inactivate the TAP CRISPR system (Supplementary Figure S6). This is consistent with the prediction that mutations of the 22 targeted chromosome sites within the same call is highly infrequent, if even possible.
TAP transfer through F conjugation machinery is highly efficient towards MG1655 E. coli laboratory strain reaching up to 90% efficiency in 3h of mating (Figures 1, 2 and Supplementary Figure S2). We quantified the efficiency of TAP-Cas9-nsp transfer in non-laboratory strain and observed a disparity between recipients with an overall ∼7- to 900-fold decrease in TAP acquisition frequency in comparison to MG1655 E. coli (Figure 5A). To account for this variability, we normalized the frequency of viable transconjugants obtained for TAP-Cas9-Cr1, -Ecl, -EPEC and -EEC to the frequency of TAP-Cas9-nsp transconjugants in the corresponding bacterial strain (Figure 5B). We quantified that the TAP-Cas9-Cr1 induces a transconjugant viability loss only in C. rodentium, TAP-Cas9-Ecl in E. cloacae, TAP-Cas9-EPEC in E. coli EPEC, while the TAP-Cas9-EEC targets the three pathogenic strains. As a control, we show that the commensal E. coli HS recipient, which genome is not targeted by any spacer, is affected by none of these antibacterial TAPs (Figure 5B). These results demonstrate that the spacer sequences generated by the CSTB algorithm allow the robust reprograming of the TAPs for efficient and strain-specific antibacterial activity on mono-species recipient populations. It also demonstrates that one given TAP can target several species at the time.
Next, we addressed TAPs ability to induce strain-specific antibacterial activity within a multi-species recipient population composed of the three pathogenic strains and the commensal E. coli HS (Figure 5C). The proportion of transconjugants obtained after 3 h of mating with TAP-Cas9-nsp varies (Figure 5D) and reflects the efficiency of TAP transfer among the different recipient strains (Figure 5A). We observed that within the multispecies recipient mix, C. rodentium transconjugant viability is dramatically reduced by TAP-Cas9-Cr1, that of E. cloacae by TAP-Cas9-Ecl and that of E. coli EPEC by TAP-Cas9-EPEC, while all three species are affected by the triple-targeting TAP-Cas9-EEC. The viability of transconjugants of the control commensal E. coli HS is not affected by any of the antibacterial TAPs (Figure 5D). These results validate that TAPs achieve selective killing within a multispecies mixed recipient population without affecting the non-targeted species. Although the antibacterial TAPs impact selectively the viability of the transconjugant populations, their activity is not significantly reflected by the total recipient counts of each species (Supplementary Figure S7), due to the limited efficiency of TAP transfer to the pathogenic recipient strains and the differential fitness of these strains in competition within the conjugation mix.
DISCUSSION
Tools for in situ microbiota manipulation are currently in their infancy. Here, we demonstrate the ability of the TAP antibacterial strategy to exert an efficient and strain-specific antibacterial activity within multi-species populations in vitro. TAPs selective-killing activity induces a ∼4-log viability loss of the tested species. TAPs targeting the pOXA-48a carbapenem resistance-plasmids results in a 4- to 5-logs increase of the strain susceptibility to the drug. Most CRISPR delivery methodology currently in development focus on the use of bacteriophages, which have intrinsically narrow host-range (35,36,7). Besides, several recent studies successfully use the broad host range RK2 conjugation systems to deliver CRISPR system that target E. coli (7–9,37) or S. enterica (11) in vitro. One key advantage of our strategy over these approaches is the versatility conferred by the CSTB algorithm that enables the robust identification of gRNA that should be used to specifically re-target the TAPs against one or several bacterial strains of interest, without targeting other species. Despite the availability of numerous programs dedicated to the identification of CRISPR motifs, the CSTB has no equivalent so far (34). Another advantage is the modular design and the relatively small size of the mobilizable TAPs (compared to autonomous conjugation plasmids) that are easily modifiable. The spacer sequence that directs the TAP against the targeted strain(s), as well as the other constituent biobricks (origin of replication, origin of transfer, Cas9, resistance gene) can be changed in one-step-cloning (see methods), thus enabling the rapid construction of a library of TAPs adapted to the considered applications. Finally, the constitutive expression of the CRISPR system (and the fluorescent reporters) from promoter that are active in a wide range of Enterobacteriaceae avoids the requirement for an external inductor, rendering the TAP approach more suitable for the modification of natural bacterial communities in vivo.
TAPs designed to induce Cas9-mediated double-stranded-breaks (DSBs) to the chromosome of the targeted bacteria trigger significant viability loss. However, we observe the emergence of targeted bacteria that have evolved mutations allowing them to survive despite the acquisition of the TAP. The frequency of these TAP-escape mutants varies from ∼3 × 10−4 to 6 × 10−5 depending on the spacer and the recipient strains used (Figures 1D, 3A, 4B and 5B). The phenotypic and sequence analysis of E. coli and C. rodentium escape-mutants reveal two main mechanisms to escape TAP activity: (i) The first mechanism is the acquisition of insertions (transposase or Insertion Sequences, IS) or single nucleotide deletions that inactivate the cas9 gene carried by the TAP. These types of mutations allowing bacteria to escape CRISPR activity have been previously reported to occur with comparable frequency (7,38,39,11). It has also been reported that mutations or deletions within either tracrRNA or crRNA sequences is another way to inactivate CRISPR systems (7,40,39,38). Yet, no such mutations were found in the C. rodentium nor E. coli TAP-escape mutants analysed, potentially due to their lesser frequency of occurrence. (ii) The second mechanism we have identified is the acquisition of point mutations or deletion that modify the targeted locus, thus impeding the recognition by the gRNA. These were also previously described (15,28,11,38,7). When using TAPs that target one chromosome locus in E. coli and C. rodentium, escape-mutations occur by cas9-inactivation (first mechanism) in 38.7 and 42.8%, and by modification of the targeted locus (second mechanism) in 61.3% and 57.2%, correspondingly.
We show that the contribution of escape mutations by modification of the chromosome sequence can be dramatically, if not completely reduced by directing the TAPs against multiple chromosome loci. Indeed, the probability of mutating multiple chromosome sites within the same bacterial cell is expected to decrease as the number of targeted sites increases. When using the Cr22 spacer that targets 22 loci of C. rodentium chromosome, all the nineteen escape-mutants tested carry mutations that inactivate the TAP-born cas9 gene. Consequently, we observe a 2.9-fold decrease in the frequency of TAP-Cas9-Cr22 escapers (8.56 × 10−5) compare to TAP-Cas9-Cr1 (2.47 × 10−4) that targets one single chromosome locus. This decrease is consistent with our estimates of the relative contributions of each escape mechanisms.
The observed frequency of escape mutations by cas9 inactivation is likely related to the intrinsic rate of transposition estimated to oscillate between 10−5 to 10−6 in E. coli (41) and the rate of spontaneous mutations (10−8 and 10−10 per base pair and generation). In the case of TAPs, it is also possible that the induction of DSBs results in the increase of the mutation rates through the triggering of SOS-induced hypermutator phenotype. Cui and Bikard demonstrated that one way to improve CRISPR efficiency in E. coli host cell is to inhibit RecA activity, which is essential to DSB repair and to the induction of the SOS-response (15). Moreb et al. further proposed to inhibit RecA activity by co-expressing the CRISPR system with a dominant negative allele of the recA gene (42). These strategies are however less relevant in the case of the TAPs approach, as they would likely alter the recombination proficiency and thus the viability of the untargeted population.
We also addressed the possibility that part of the mutations in TAPs already emerge in the donor cells, thus resulting in the transfer of already inactive TAPs into the recipient target. This possibility is supported by the sequencing analysis of one C. rodentium escape mutant that has received TAP-Cas9-Cr1 from an E. coli donor. The sequencing revealed the insertion in cas9 of insAB genes coding for transposase elements present in E. coli but absent from C. rodentium genomes. This result indicates that mutation leading to TAPs inactivation can occur in the donor cells prior to transfer, without excluding that they might also emerge in the targeted recipient after plasmid acquisition.
Our work reveals that TAPs efficiency is primarily determined by two main limiting factors. The first limiting factor is the ∼10−4–10−5 frequency of escaper clones that acquire mutations inactivating the plasmid-born cas9 gene, or mutations that modify the targeted sequence. The occurrence of escaper clones due to the acquisition of TAPs that have been inactivated before transfer could be reduced by using a transposon-free E. coli donor strain (43). As shown in C. rodentium, the emergence of escaper clones by mutation of the targeted sequence can be avoided by targeting multiple sites on the genome. Alternatively, it has been shown that another way to avoid the emergence of escape mutants through the modification of the chromosome is to target essential genes, which mutation is often lethal for the cell (40). The second limiting parameter is the efficiency of TAPs transfer towards the targeted strain(s). This efficiency primarily depends on the conjugative system chosen to mobilize the TAPs. Here, we use the F plasmid as a helper plasmid that mediates relatively efficient TAPs transfer (10−1–10−2) to closely related Enterobacteriaceae. Therefore, TAPs appear appropriate to target a range of clinically relevant pathogenic or resistant Gram-negative bacteria (E. coli, Citrobacter, Enterobacter, Klebsiella, Salmonella, Yersinia, Shigella, Serratia,etc.). Recently, the narrow host range pPD1 plasmid has also been used to transfer CRISPR/Cas systems to the Gram-positive Enterococcus faecalis (44). Other reported antibacterial (7,11) or anti-drug (8–10) methodology using conjugation are mostly based on the incP RK2 conjugative system, which offer broad-host range, but low efficiency of transfer (10−4–10−5). Hamilton et al. have shown that transfer efficiency can be artificial increased using glass beads in vitro (11). It is also possible to isolate broad-host range conjugation systems with increased transferability. Such superspreader plasmid mutants have been successfully isolated through Tn-seq approach (45,46) and could represent an valuable option to widen the range of bacteria toward which TAPs could be efficiently transferred.
Translating the present in vitro proof of concept to in situ settings would represent an important step towards the development of a non-antibiotic strategy for the in situ manipulation of microbiota composition, in a directed manner. The efficiency of the TAPs methodology within host-associated bacterial communities would primarily depend on conjugation rates in situ, which often differs from rates achieved in vitro (47). For instance, it was recently reported that the IncI2 plasmid TP114 and IncX type plasmids are very actively transferred in the mouse intestinal tract (47) and in human fecal microbiomes (48), respectively, making them good candidate to mobilize TAPs in these given ecosystems. TAPs could be used for the inhibition of harmful pathogenic and resistance strains from an infected host or environments, or as anti-virulence strategy through inhibition of virulence effector genes or genes involved in biofilm formation. The future implementation of the TAPs approach in clinical or environmental settings would require the consideration of the rapidly evolving regulations on GMO, CRISPR and biocontainment (49–51).
DATA AVAILABILITY
Software and source codes: The CSTB web service can be freely accessed at https://cstb.ibcp.fr. The software we developed to convert all unique words (spacers) into an integer representation using a 2-bits per base encoding is also available at https://github.com/glaunay/crispr-set. All additional related software can be freely accessed at https://github.com/MMSB-MOBI/CSTB_database_manager.
Flow Cytometry: Data from flow cytometry experiments have been submitted to the FlowRepository (https://flowrepository.org/id/FR-FCM-Z35C and FR-FCM-Z35E).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Gregory Jubelin, François Cornet, Claire Prigent-Combaret, Pierre Bogaerts for the gift of strains and plasmids. Lisa Rubio, Brice Simon-Letcher, ShangNong Hu and Timothée Sluys, for earlier involvement in the project.
Author contributions: C.L. provided funding. C.L. and S.B. conceived the project, designed the study and wrote the paper. A.R., A.D. and S.B. performed the experiments and analyzed the data. The CSTB algorithm was conceived by G.L., C.H. and S.L. in discussion with E.G., S.B. and C.L. All the authors gave intellectual input throughout the project.
Contributor Information
Audrey Reuter, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Cécile Hilpert, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Annick Dedieu-Berne, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Sophie Lematre, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Erwan Gueguen, University of Lyon, Université Lyon 1, INSA de Lyon, CNRS UMR 5240 Microbiologie Adaptation et Pathogénie, 69622 Villeurbanne, France.
Guillaume Launay, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Sarah Bigot, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
Christian Lesterlin, Microbiologie Moléculaire et Biochimie Structurale (MMSB), Université Lyon 1, CNRS, Inserm, UMR5086, 69007 Lyon, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French National Research Agency [ANR-19-ARMB-0006-01]; Schlumberger Foundation for Education and Research [FSER2019]; Foundation for Innovation in Infectiology [FINOVI-2014]; Foundation pour la Recherche Médicale [FRM-ECO201806006855 to A.R.]. Funding for open access charge: French National Research Agency [ANR-19-ARMB-0006-01].
Conflict of interest statement. None declared.
REFERENCES
- 1. WHO Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 2015; ISBN: 9789241509763. [Google Scholar]
- 2. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gasiunas G., Barrangou R., Horvath P., Siksnys V.. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A.. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013; 41:7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anders C., Niewoehner O., Duerst A., Jinek M.. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 513:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Citorik R.J., Mimee M., Lu T.K.. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014; 32:1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dong H., Xiang H., Mu D., Wang D., Wang T.. Exploiting a conjugative CRISPR/Cas9 system to eliminate plasmid harbouring the mcr-1 gene from Escherichia coli. Int. J. Antimicrob. Agents. 2019; 53:1–8. [DOI] [PubMed] [Google Scholar]
- 9. Ruotsalainen P., Penttinen R., Mattila S., Jalasvuori M.. Midbiotics: conjugative plasmids for genetic engineering of natural gut flora. Gut Microbes. 2019; 10:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang P., He D., Li B., Guo Y., Wang W., Luo X., Zhao X., Wang X.. Eliminating mcr-1-harbouring plasmids in clinical isolates using the CRISPR/Cas9 system. J. Antimicrob. Chemother. 2019; 74:2559–2565. [DOI] [PubMed] [Google Scholar]
- 11. Hamilton T.A., Pellegrino G.M., Therrien J.A., Ham D.T., Bartlett P.C., Karas B.J., Gloor G.B., Edgell D.R.. Efficient inter-species conjugative transfer of a CRISPR nuclease for targeted bacterial killing. Nat. Commun. 2019; 10:4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 13. García-Nafría J., Watson J.F., Greger I.H.. IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Sci. Rep. 2016; 6:27459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ducret A., Quardokus E.M., Brun Y.V.. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 2016; 1:16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui L., Bikard D.. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016; 44:4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martínez-García E., Aparicio T., Goñi-Moreno A., Fraile S., de Lorenzo V.. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 2015; 43:D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nolivos S., Cayron J., Dedieu A., Page A., Delolme F., Lesterlin C.. Role of AcrAB-TolC multidrug efflux pump in drug-resistance acquisition by plasmid transfer. Science. 2019; 364:778–782. [DOI] [PubMed] [Google Scholar]
- 18. Crisona N.J., Clark A.J.. Increase in conjugational transmission frequency of nonconjugative plasmids. Science. 1977; 196:186–187. [DOI] [PubMed] [Google Scholar]
- 19. Mavridou D.A.I., Gonzalez D., Clements A., Foster K.R. The pUltra plasmid series: a robust and flexible tool for fluorescent labeling of Enterobacteria. Plasmid. 2016; 87–88:65–71. [DOI] [PubMed] [Google Scholar]
- 20. Bhoite S., van Gerven N., Chapman M.R., Remaut H.. Curli biogenesis: bacterial amyloid assembly by the Type VIII secretion pathway. EcoSal Plus. 2019; 8:doi:10.1128/ecosalplus.ESP-0037-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vidal O., Longin R., Prigent-Combaret C., Dorel C., Hooreman M., Lejeune P.. Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J. Bacteriol. 1998; 180:2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serra D.O., Hengge R.. Sauer K. Experimental Detection and Visualization of the Extracellular Matrix in Macrocolony Biofilms. c-di-GMP Signaling, Methods in Molecular Biology. 2017; 1657:NY: Springer; 133–145. [DOI] [PubMed] [Google Scholar]
- 23. Rock J.M., Hopkins F.F., Chavez A., Diallo M., Chase M.R., Gerrick E.R., Pritchard J.R., Church G.M., Rubin E.J., Sassetti C.M.et al.. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2017; 2:16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho S., Choe D., Lee E., Kim S.C., Palsson B., Cho B.-K.. High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synthetic Biology. 2018; 7:1085–1094. [DOI] [PubMed] [Google Scholar]
- 25. Zhang S., Voigt C.A.. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018; 46:11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Misra C.S., Bindal G., Sodani M., Wadhawan S., Kulkarni S., Gautam S., Mukhopadhyaya R., Rath D.. Determination of Cas9/dCas9 associated toxicity in microbes. Microbios. 2019; doi:10.1101/848135. [Google Scholar]
- 27. Lesterlin C., Ball G., Schermelleh L., Sherratt D.J.. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature. 2014; 506:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J.J., Severinov K.. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barlow M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol. Biol. 2009; 532:397–411. [DOI] [PubMed] [Google Scholar]
- 30. Codjoe F., Donkor E.. Carbapenem resistance: a review. Medical Sciences. 2017; 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poirel L., Bonnin R.A., Nordmann P.. Genetic features of the widespread plasmid coding for the carbapenemase OXA-48. Antimicrob. Agents Chemother. 2012; 56:559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003; 301:1496–1499. [DOI] [PubMed] [Google Scholar]
- 33. Mnif B., Vimont S., Boyd A., Bourit E., Picard B., Branger C., Denamur E., Arlet G.. Molecular characterization of addiction systems of plasmids encoding extended-spectrum beta-lactamases in Escherichia coli. J. Antimicrob. Chemother. 2010; 65:1599–1603. [DOI] [PubMed] [Google Scholar]
- 34. Alkhnbashi O.S., Meier T., Mitrofanov A., Backofen R., Voß B.. CRISPR-Cas bioinformatics. Methods. 2020; 172:3–11. [DOI] [PubMed] [Google Scholar]
- 35. Bikard D., Barrangou R.. Using CRISPR-Cas systems as antimicrobials. Curr. Opin. Microbiol. 2017; 37:155–160. [DOI] [PubMed] [Google Scholar]
- 36. Bikard D., Euler C.W., Jiang W., Nussenzweig P.M., Goldberg G.W., Duportet X., Fischetti V.A., Marraffini L.A.. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014; 32:1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ji W., Lee D., Wong E., Dadlani P., Dinh D., Huang V., Kearns K., Teng S., Chen S., Haliburton J.et al.. Specific gene repression by CRISPRi system transferred through bacterial conjugation. ACS Synth. Biol. 2014; 3:929–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fischer S., Maier L.-K., Stoll B., Brendel J., Fischer E., Pfeiffer F., Dyall-Smith M., Marchfelder A.. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J. Biol. Chem. 2012; 287:33351–33363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A.. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013; 31:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gomaa A.A., Klumpe H.E., Luo M.L., Selle K., Barrangou R., Beisel C.L.. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. mBio. 2014; 5:doi:10.1128/mBio.00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sousa A., Bourgard C., Wahl L.M., Gordo I.. Rates of transposition in Escherichia coli. Biol. Lett. 2013; 9:20130838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moreb E.A., Hoover B., Yaseen A., Valyasevi N., Roecker Z., Menacho-Melgar R., Lynch M.D.. Managing the SOS response for enhanced CRISPR-Cas-based recombineering in E. coli through transient inhibition of host RecA activity. ACS Synth. Biol. 2017; 6:2209–2218. [DOI] [PubMed] [Google Scholar]
- 43. Pósfai G., Plunkett G., Fehér T., Frisch D., Keil G.M., Umenhoffer K., Kolisnychenko V., Stahl B., Sharma S.S., de Arruda M.et al.. Emergent properties of reduced-genome Escherichia coli. Science. 2006; 312:1044–1046. [DOI] [PubMed] [Google Scholar]
- 44. Rodrigues M., McBride S.W., Hullahalli K., Palmer K.L., Duerkop B.A.. Conjugative delivery of CRISPR-Cas9 for the selective depletion of antibiotic-resistant Enterococci. Antimicrob. Agents Chemother. 2019; 63:e01454-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamaichi Y., Chao M.C., Sasabe J., Clark L., Davis B.M., Yamamoto N., Mori H., Kurokawa K., Waldor M.K.. High-resolution genetic analysis of the requirements for horizontal transmission of the ESBL plasmid from Escherichia coli O104:H4. Nucleic Acids Res. 2015; 43:348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poidevin M., Sato M., Altinoglu I., Delaplace M., Sato C., Yamaichi Y.. Mutation in ESBL plasmid from Escherichia coli O104:H4 leads autoagglutination and enhanced plasmid dissemination. Front Microbiol. 2018; 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neil K., Allard N., Grenier F., Burrus V., Rodrigue S.. Highly efficient gene transfer in the mouse gut microbiota is enabled by the Incl2 conjugative plasmid TP114. Commun. Biol. 2020; 3:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Munck C., Sheth R.U., Freedberg D.E., Wang H.H.. Recording mobile DNA in the gut microbiota using an Escherichia coli CRISPR-Cas spacer acquisition platform. Nat. Commun. 2020; 11:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fellmann C., Gowen B.G., Lin P.-C., Doudna J.A., Corn J.E.. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017; 16:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davison J., Ammann K.. New GMO regulations for old: determining a new future for EU crop biotechnology. GM Crops Food. 2017; 8:13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brokowski C., Adli M.. CRISPR ethics: moral considerations for applications of a powerful tool. J. Mol. Biol. 2019; 431:88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software and source codes: The CSTB web service can be freely accessed at https://cstb.ibcp.fr. The software we developed to convert all unique words (spacers) into an integer representation using a 2-bits per base encoding is also available at https://github.com/glaunay/crispr-set. All additional related software can be freely accessed at https://github.com/MMSB-MOBI/CSTB_database_manager.
Flow Cytometry: Data from flow cytometry experiments have been submitted to the FlowRepository (https://flowrepository.org/id/FR-FCM-Z35C and FR-FCM-Z35E).