

Abstract
Light absorption and scattering by metal nanoparticles can drive catalytic reactions at their surface via the generation of hot charge carriers, elevated temperatures, and focused electromagnetic fields. These photoinduced processes can substantially alter the shape, surface structure, and oxidation state of surface atoms of the nanoparticles and therefore significantly modify their catalytic properties. Information on such local structural and chemical change in plasmonic nanoparticles is however blurred in ensemble experiments, due to the typical large heterogeneity in sample size and shape distributions. Here, we use single-particle dark-field and Raman scattering spectroscopy to elucidate the reshaping and surface restructuring of individual silver nanodisks under plasmon excitation and during photocatalytic CO2 hydrogenation. We show that silver nanoparticles reshape significantly in inert N2 atmosphere, due to photothermal effects. Furthermore, by collecting the inelastic scattering during laser irradiation in a reducing gas environment, we observe intermittent light emission from silver clusters transiently formed at the nanoparticle surface. These clusters are likely to modify the photocatalytic activity of silver nanodisks and to enable detection of reaction products by enhancing their Raman signal. Our results highlight the dynamic nature of the catalytic surface of plasmonic silver nanoparticles and demonstrate the power of single-particle spectroscopic techniques to unveil their structure–activity relationship both in situ and in real time.
Keywords: surface restructuring, silver nanodisks, plasmon excitation, photocatalysis, single-particle spectroscopy
Introduction
Light is known to enhance the catalytic activity of metal nanoparticles via the excitation of localized surface plasmon resonances (LSPRs). These resonances, which are due to the collective oscillation of free electrons in metal nanostructures, can give rise to nonthermalized (hot) charge carriers, elevated temperatures, and enhanced electromagnetic fields.1,2 These plasmon decay mechanisms can be exploited to activate catalytic reactions such as ethylene epoxidation and organic coupling reactions on silver nanoparticles,3−5 carbon dioxide reduction on silver and gold,6−8 and ammonia decomposition on plasmonic alloys.9 However, to date, a complete understanding of these photocatalytic processes along with the relative contributions of these plasmon decay mechanisms remains contentious.9−14
The catalytic and photocatalytic properties of metal nanoparticles are highly dependent on their chemical composition and the morphology of their surface.15−17 However, during most heterogeneous catalytic reactions, atomic-scale structural and chemical modifications occur on the catalyst surface, leading to dramatic changes in their activity and selectivity.18−21 Such surface modifications are also known to markedly alter the catalytic properties of metal nanoparticles in plasmon-driven reactions.22,23 Despite abundant research on plasmon-assisted photocatalysis, most results currently focus on the molecular products, while the fate of the plasmonic nanoparticles remains poorly understood. To tackle this challenge, single-particle spectroscopic techniques are emerging as a powerful tool to characterize the structure–activity relationship of plasmonic catalysts under irradiation.6,24,25
In this study, we use a suite of spectroscopic techniques to follow light-driven structural changes of individual plasmonic nanoparticles under different reactive gas environments. We follow the structural evolution of individual silver nanodisks (Ag NDs) fabricated using electron-beam lithography technique, by measuring their dark-field (DF) scattering spectra under photocatalytic conditions. Furthermore, we elucidate their structure–activity relationship by correlating the LSPR of the Ag NDs measured in DF with their photocatalytic activity under laser illumination, using surface-enhanced Raman spectroscopy (SERS). We observe that Ag NDs reshape significantly under mild (photothermal) heating even under inert nitrogen atmosphere. Such nanocatalyst reshaping is accompanied by dynamic restructuring of their surface, leading to silver cluster formation (Agn) under plasmon excitation in a CO2 and H2 mixture, as confirmed by their intermittent Stokes emission. These silver clusters are likely to strongly affect the CO2 reduction reaction, as suggested by the correlated Stokes emissions from the nanoclusters and the SERS signals of the photocatalytic products.
Results and Discussion
Microscopy Setup and Sample Design
Silver nanodisks with an LSPR close to the 532 nm laser irradiation are fabricated on a glass substrate using electron-beam lithography (EBL). In a typical sample, the nanodisks are ∼95 nm in diameter and 40 nm in thickness and adhered to glass with a 2 nm Ge layer. The nanodisks are arranged in a square lattice and separated by 10 μm. Figure 1a shows the DF image of the fabricated Ag NDs along with a few representative scanning electron microscopy (SEM) images of silver disks fabricated on a Si substrate, showing particle-to-particle heterogeneity in the sample (also see the Supporting Information Section S1). Such heterogeneity arises from the polycrystalline nature of the EBL-deposited nanodisks.
Figure 1.

Sample design and microscopy setup. (a) (Top) Dark-field image of Ag NDs fabricated on a glass substrate (scale bar, 10 μm) and (bottom) SEM images of four individual Ag NDs on a silicon substrate (scale bar, 50 nm). (b) Experimental setup for single-particle DF scattering and SERS measurements. (c) Dark-field images and corresponding scattering spectra of four selected Ag NDs in air along with the FDTD simulated scattering spectrum of a single Ag ND with a diameter of 95 nm and a height of 40 nm on a glass substrate. The vertical dashed line represents the position of the 532 nm laser.
Single-particle scattering spectra of the NDs are recorded in an inverted microscope equipped with a monochromator and an electron-multiplying charge-coupled device (EMCCD) camera using a DF white light illumination geometry.26−30 The microscope setup (Figure 1b) can be switched from broadband to monochromatic illumination, allowing the acquisition of single-particle surface-enhanced Raman scattering spectra as well. The transparent glass substrate supporting the Ag NDs is used as the bottom window of a gas flow cell. Three mass flow controllers (N2, CO2, H2) allow us to regulate the gas composition during photocatalytic experiments. In Figure 1c, we show the measured dark-field scattering image and the corresponding scattering spectra of four Ag NDs in air on a glass substrate, along with the spectrum of an ideal Ag ND simulated using finite-difference time domain (FDTD). Most silver particles have LSPRs close to the ideal one, with differences that can be attributed to their deviation from a perfectly round disk.
(Photo)thermal Stability in Inert Atmosphere
To characterize the thermal stability of our nanoparticles, we first separately track the DF spectral evolution of 16 individual Ag NDs during conventional heating under nitrogen atmosphere. We find that the surface plasmon resonance of the NDs blue-shifts with increasing temperatures (Figure 2a). Such spectral shifts of the plasmon resonance are attributed to the reshaping of slightly anisotropic NDs into (more) round particles (Supporting Information Section S2).31 These shape transformations are due to the surface diffusion of silver atoms, which follows an Arrhenius-type relation with temperature.32,33 Since the thermal reshaping is characterized by a blueshift in LSPR, we quantify the degree of particle reshaping using the plasmon resonance blueshift, ΔλLSPR, and by following the relation
| 1 |
where Ea is the activation energy for the surface diffusion of silver atoms, kB is the Boltzmann constant, and T is the temperature. A linear fit of ln(ΔλLSPR) as a function of the inverse temperature (Figure 2b) yields an activation energy of 0.28 eV/atom, which is in excellent agreement with previously reported values for the surface diffusion of silver atoms.33,34
Figure 2.
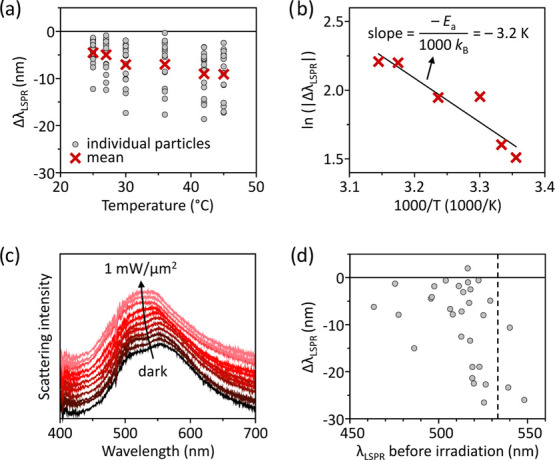
Thermal and photothermal stabilities of Ag NDs in inert atmosphere. (a) Temperature dependence of the LSPR shift (ΔλLSPR) of 16 individual Ag NDs after conventional heating in the dark in nitrogen atmosphere. (b) Arrhenius plot of the mean values from (a). The line is a linear fit using eq 1. (c) Time evolution of the dark-field scattering spectrum of an individual anisotropic Ag ND in nitrogen atmosphere illuminated by a 532 nm CW laser at increasing irradiance. The NDs were illuminated for 10 min before the accumulation of each DF spectrum. The spectra are vertically translated for clarity. (d) LSPR shift (ΔλLSPR) measured for 31 individual Ag NDs upon 532 nm CW illumination at 1 mW/μm2 in nitrogen atmosphere for 30 min, as a function of their initial LSPR. The dashed vertical line shows the position of the 532 nm laser. The horizontal line represents unaffected LSPR of irradiated Ag NDs.
At room temperature (∼18 °C) and in the absence of laser irradiation, the LSPR of individual Ag NDs remains constant over a 1 h period in N2, CO2, and H2 gas environments (Supporting Information Section S3). Under focused irradiation with a 532 nm continuous-wave (CW) laser, however, Ag NDs significantly reshape, as indicated by a characteristic blueshift of their plasmon resonance. In Figure 2c, we plot the LSPR evolution as a function of irradiation power in pure N2 atmosphere for an individual Ag ND showing two peaks in its initial scattering spectrum. Upon consecutive irradiation periods of 10 min at increasing laser powers, the LSPR blue-shifts and merges into a single peak, indicating a reshaping into a (more) symmetric or round disk.35 The LSPR blueshift converges to a constant value after about an hour of laser irradiation, suggesting that the particle has reconfigured into a stable shape.
Both continuous-wave and pulsed lasers have been known to cause dramatic shape change in metal nanoparticles.36−39 These transformations occur due to an increase in the nanoparticle temperature, as a result of the nonradiative decay of the LSPRs into heat. Under high illumination intensities by tightly focused lasers, plasmonic nanoparticles can in fact achieve temperatures as high as their melting points.39,40 However, significant reshaping can occur at milder temperatures well below the melting point of the bulk metal, thanks to the higher mobility of surface metal atoms with respect to bulk and to the low activation energy of diffusion in nanostructures with high curvatures.41−43 Hence, even under mild heating, surface atoms also change their equilibrium positions in order to minimize lattice stress and surface energy.44 As a result, reshaping and surface reconstruction can occur leading to the formation of pico- and nanocavities and surface protrusions.45
In Figure 2d, we plot the plasmon resonance shift measured for 31 individual Ag NDs after 30 min in 532 nm CW laser illumination at 1 mW/μm2, under a flow of N2 gas at atmospheric pressure, as a function of their initial LSPR. This laser irradiance is well below the typical threshold for ablation of plasmonic nanoparticles.46 Upon irradiation, most of the nanoparticles exhibit a blueshift in their LSPR. Interestingly, particles that had an original LSPR close to the 532 nm irradiation wavelength and were therefore resonantly excited show larger blueshifts due to plasmonic heating than particles with an initial plasmon resonance farther away from the laser wavelength. The scatter in the data can be attributed to the misalignment of the particles with respect to the Gaussian laser beam and to the varying degree of anisotropy of the individual nanodisks.
The temperature rise during plasmonic heating is dictated by the laser irradiance, the absorption cross section of the nanoparticle, and the thermal conductivity of the surrounding medium. For an irradiated Ag ND at the geometric center of the laser spot, the temperature rise, ΔT, can be estimated using47
| 2 |
where σabs is the absorption cross section of the nanodisk, I is the laser irradiance (power per unit area), κ is the average thermal conductivity of the surrounding medium, and r is the effective radius of the nanodisk. The latter is calculated by approximating a nanodisk of diameter d and height h, with a spherical particle of equal volume and therefore with a radius r given by47
| 3 |
For our Ag NDs, an absorption cross section of 1.13 × 10–14 m2 is calculated using FDTD simulations (see the Experimental Section). Assuming a thermal conductivity of 0.2 ± 0.1 W m–1 K–1 for a glass–air interface,48 and an irradiance of 1 mW/μm2, a temperature rise of the order of 100–200 K on a single Ag ND can be obtained from eq 2. Such temperature increases could lead to significant reshaping in the NDs. Equation 2 provides an upper bound of the nanoparticle temperature increase due to the potential misalignment of the nanoparticle with respect to the laser.
Photochemical Stability under CO2 and H2 Gas Atmosphere
Metal nanoparticles often undergo significant changes in surface structure and chemistry during (photo)catalytic processes.49−52 We therefore extend our single-particle approach to study the photochemical stability of Ag NDs under laser irradiation in different reactive gas environments. Under resonant excitation by a 532 nm laser at 1 mW/μm2, Ag NDs show changes in their scattering spectra that depend on the gas atmosphere around them. In addition to photothermal reshaping, the chemical environment and the thermodynamics of the processes occurring at the surface also affect their structure. Hence, a competition between photothermal reshaping and photochemical effects determines the overall spectral shifts observed. Depending on the gas environment, these two effects on the spectral position of the LSPR will either complement or cancel each other. In principle, the different thermal conductivities of the gasses used could also affect the photothermal reshaping. However, the thermal conductivities of N2 (0.026 W m–1 K–1), CO2 (0.017 W m–1 K–1), and H2 (0.187 W m–1 K–1) are all much lower than the one of the glass substrate (1 W m–1 K–1) and will therefore have a negligible effect on the photothermal heating under steady-state conditions.53
Similar to the case of N2, in an atmospheric flow of CO2, we observe a reshaping-induced blueshift of the plasmon resonance during the first 15 min of laser irradiation, after which the LSPR remains nearly constant (Figure 3a). In the CO2 environment, photocatalytic oxidation of silver catalysts has been previously observed.6,54 Such a surface oxidation would in principle lead to a redshift in the LSPR of the Ag NDs. In our experiments, however, the spectral signature of photothermal reshaping overcomes any potential contribution from surface oxidation and a net blueshift is thus observed.
Figure 3.

Photochemical stability under reactive gas atmosphere. Time evolution of the dark-field scattering spectra of a single Ag ND irradiated by a 532 nm laser at 1 mW/μm2 in (a) CO2 and (b) H2 environment. DF spectra are recorded every 15 min for ∼2 h by switching between laser and white light source. The spectra in (a) and (b) are vertically translated for clarity. The black arrows are a guide to the eye.
Photoirradiation of an Ag ND in 1 bar H2 results in a redshift of its LSPR over a period of 2 h (Figure 3b). Hydrogen is a strong reducing agent and plasmon excitation via laser irradiation could lead to the reduction of the 1–2 nm thick native silver oxide at the surface of the nanodisk.55 A conformal reduction of the native oxide shell into metallic silver, however, would lead to a blueshift of the LSPR (see Supporting Information Section S4). To shed further light on the origin of the redshift observed experimentally, we performed a laser irradiation experiment under a continuous hydrogen flow on colloidally synthesized Ag nanoparticles deposited on a carbon-coated Cu TEM grid. The laser power was significantly lower, to avoid photothermal damage of the ultrathin carbon membrane. TEM measurements of the same nanoparticles before and after laser irradiation in hydrogen atmosphere reveal the photochemical transformation of smooth surfaces of silver nanoparticles into rough ones (see Supporting Information Section S5). The roughness is likely induced by the formation of silver clusters (Agn) on the silver nanoparticle surface, resulting in a Ag–Agn system. Strong evidence for silver cluster formation under photochemical experiments in reducing gas environments is discussed in the next section, where we show the appearance of a photoluminescence signal, which cannot be attributed to carbon species.
The optical properties of silver nanoparticles are typically modeled assuming a thin shell of native Ag2O around them.54,56 The mechanism of formation of silver clusters can then be understood by considering that Ag2O has a band gap of 2.25 eV (λ ≈ 554 nm), which nearly coincides with the LSPR of the Ag NDs.56 Upon irradiation in a hydrogen environment, a plasmon-induced reduction of the native oxide can therefore occur. Although we cannot exclude any plasmonic activation mechanism, including photothermal heating, hot charge carrier, and near-field driven processes, a similar effect has been observed by Linic et al. when irradiating plasmonic copper nanoparticles during the epoxidation of propylene to propylene oxide.22 Furthermore, the native silver oxide shell is often described as discontinuous and inhomogeneous in nature.57 Reduction of surface silver oxide can therefore lead to the generation of silver islands, clusters, or cavities at the surface of the Ag NDs. Such nano- and sub-nanostructures can give rise to electromagnetic hot spots under laser irradiation. These in situ formed hot spots and their uneven distribution on the surface of the nanoparticles result in a redshift and damping of the LSPR, as observed experimentally (Figure 3b and Supporting Information Section S6).58
Photocatalytic Restructuring in the CO2 and H2 Mixture
CO2RR is generally carried out in a reaction environment that can cause oxidation or reduction of the catalyst, hence modifying their surface structure and chemistry, depending on the overall gas composition. We study the combined effect of the constituting gases on individual Ag ND catalysts during a photocatalytic CO2RR by varying the volume ratios of CO2 and H2 in the gas mixture. For all of the measurements, the Ag ND catalysts are pretreated in nitrogen while irradiating with a 532 nm laser for 1 h. As observed before, during the pretreatment, the Ag ND particles display a blueshift of their plasmon resonance due to photothermal reshaping (see Supporting Information Section S7). After pretreatment in N2, we continue the photocatalytic experiment by switching to a H2/CO2 gas mixture and irradiating the NDs for periods of 15 min, followed by acquisition of the dark-field scattering spectra. Furthermore, during laser irradiation, we continuously record the inelastic scattering spectra of the catalysts. In Figure 4a, we plot the LSPR shift during photochemical irradiation of three single NDs under CO2/H2 mixtures of composition 90:10, 80:20, and 60:40. Interestingly, the higher the concentration of hydrogen, the larger is the redshift of the LSPR, indicating the formation of silver clusters similar to the one observed in pure hydrogen studies (Figure 3b).
Figure 4.
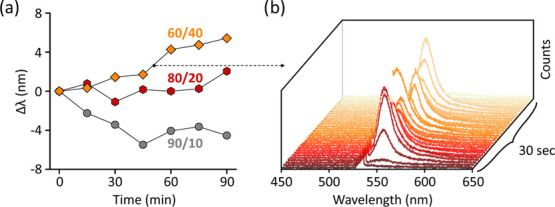
Catalyst restructuring during CO2RR under laser irradiation. (a) LSPR shift measured during 532 nm laser illumination at 1 mW/μm2 of single Ag NDs in gas mixtures with different CO2/H2 ratios: 90:10 (circles), 80:20 (hexagons), and 60:40 (rhombi). (b) Transient photoemission measured via inelastic scattering spectroscopy during the laser irradiation of the Ag ND in a CO2/H2 ratio of 60:40, corresponding to the time interval marked by the black dot in (a).
Interestingly, during one of the photocatalytic irradiation periods (marked by the black dot in Figure 4a), an intense green-red luminescence from the particles is observed (Figure 4b). Such a luminescence, which has been observed for multiple individual NDs studied under photocatalytic reduction conditions, is intermittent and typically centered between 540 and 650 nm (1.9–2.3 eV). Following the observation of Dickson et al.,59 we attribute this emission to the luminescence of the growing silver clusters resulting from their molecular-like electronic levels. The emission energy of the luminescence signal depends on the number of atoms of the cluster, N, according to the relation60
| 4 |
where Eg is the emission energy corresponding to the cluster’s band gap and EF is the Fermi energy of the metal (EF = 5.49 eV for silver61). Using eq 4, we find that all of the emission observed in our photocatalytic CO2RR experiments on various Ag NDs can be attributed to silver clusters ranging from 13 to 24 atoms (Supporting Information Section S8).62 No emission from clusters smaller than 13 atoms is observed in the blue part of the spectrum, as their band gap is larger than our 532 nm excitation energy (≈2.33 eV). As the clusters grow in size (N > 25), their emission red-shifts to wavelengths longer than 650 nm and therefore away from our spectral range. Interestingly, the silver cluster emission is only observed for a few seconds in our experiments, i.e., when the cluster is growing and their size lies between 13 and 24 atoms. Furthermore, as can be seen in Figure 4a, after measuring photoemission during laser irradiation, we observe a marked redshift of the plasmon resonance of the Ag ND. Both these optical effects are consistent with the nucleation and growth of nanoscale silver clusters at the disk surface under photocatalytic conditions. Finally, the intermittent nature of the measured luminescence signal also suggests the random nature of Agn cluster formation, growth, and dissolution, similar to what has been previously observed in nanoporous gold–silver catalysts.18
An alternative explanation for the observed luminescence signal could be the photocatalytic formation of amorphous carbon species or carbon dots on the silver surface. The luminescence signal from these species, however, is typically accompanied and often overshadowed by characteristic Raman signatures (broad D and G bands) that are not visible in our inelastic scattering spectra.63
It is well known that nanoscale metal clusters are highly active catalysts in a variety of chemical reactions.64−66 Their enhanced activity compared to bulk metals is due to the presence of corners and step sites with undercoordinated surface atoms and high surface energies. Furthermore, very small metal clusters possess molecular-like HOMO and LUMO electronic levels and could therefore efficiently mediate charge transfer reactions to and from adsorbed species.64
To examine the photocatalytic activity of the in situ formed Agn clusters, we perform correlated DF and SERS measurements on Ag NDs under a 60:40 volume ratio of CO2/H2. The particles are initially allowed to equilibrate under irradiation in pure N2 atmosphere for 1 h, during which their plasmon resonance blue-shifts (Figure 5a). For the particle shown in Figure 5a, upon switching the gas environment from N2 to the CO2RR mixture, no LSPR shift is observed during the first 45 min, implying the absence of any further structural change in the selected Ag ND. In addition, no SERS signal is detected during this initial period (Figure 5b, left). Such long induction periods have been ascribed to catalyst restructuring processes and are strongly dependent on the coverage density of reacting molecules on the catalyst surface.67,68 Driver et al. have also shown that surface reconstruction is a cooperative process requiring a critical number of adsorbates.69 In our studies, after 45 min of illumination in a photocatalytic gas environment, we observe a marked redshift in the LSPR (Figure 5b), indicating strong surface restructuring. The LSPR shift is accompanied by an increase in the Stokes background (photoluminescence) between 0 and 500 cm–1. Concomitantly, we also start observing SERS signal of molecules formed on the Ag ND surface during CO2RR between 800 and 1800 cm–1.
Figure 5.

Correlated SERS and LSPR redshifts during photocatalytic CO2RR. (a) LSPR peak shift measured with DF spectroscopy of a single Ag ND during pretreatment in N2 and during photocatalytic CO2RR in a 60:40 CO2/H2 mixture under 532 nm laser irradiation at 1 mW/μm2. (b) SERS signal measured during the laser irradiation (left) before the onset of surface restructuring and (right) during surface restructuring showing strong intermittent Stokes emission.
The simultaneous occurrence of luminescence and SERS strongly suggests a correlation between the cluster formation and the photocatalytic activity of the Ag ND. Such correlation is indicative of a single source for the two processes of luminescence and catalysis, implying that the in situ formed cluster Agn is the active site for photocatalytic CO2RR. It is noteworthy that despite identical experimental conditions, the magnitude of the LSPR redshifts observed during CO2RR varies from particle to particle (Supporting Information Figure S9). We attribute this heterogeneity to particle-to-particle structural differences and to a varying degree of surface roughness induced in individual Ag NDs during photocatalysis.
Surface restructuring is crucial to the formation of cavities with electromagnetic hot spots,70 which are ideal for SERS detection and can significantly alter the photochemical activity of our catalysts toward CO2 hydrogenation reactions. Supporting Information Figure S10 shows a waterfall plot of the SERS signal observed during CO2RR on a different single Ag ND under identical photocatalytic reaction conditions, where blinking SERS signal is also observed. Dynamic restructuring of the Ag surface leads to the continuous formation and dissolution of active catalytic clusters. The diffusion of the reactants, intermediates, and products in and out of these active sites contributes to the stochasticity of the SERS signal. Our observation is consistent with a recent report attributing blinking SERS signature to in situ formed hot spots and cavities.70 Furthermore, compared to previous single-particle studies under 100% CO2 atmosphere, our use of a 60:40 CO2/H2 mixture leads to significant restructuring of the NDs surface and longer retention of their catalytic activity.6
Upon analyzing the spectral frames of CO2RR on individual Ag NDs, we identify some of the frequently observed CO2 reduction products (Figure 6).6,71−74 One of the most common products of reduction of CO2 on silver is formic acid, whose SERS signature was also observed in our measurement (Figure 6a). Owing to the high hydrogen concentration in the reaction mixture, we also observe the four-electron reduction product formaldehyde (Figure 6b). Figure 6c shows an intense signal from a molecule consisting of C–H stretching vibrations of methylene and methyl moiety as well as ring vibrations of aromatic hydrocarbon.71 Finally, we also observe the formation of amorphous carbon on silver surface (Figure 6d). In this study, we rarely observe any SERS signals from carbon monoxide, which can be attributed to high concentrations of hydrogen in our gas mixture, which prevents the formation of carbon monoxide, a commonly observed CO2RR product on silver.6
Figure 6.
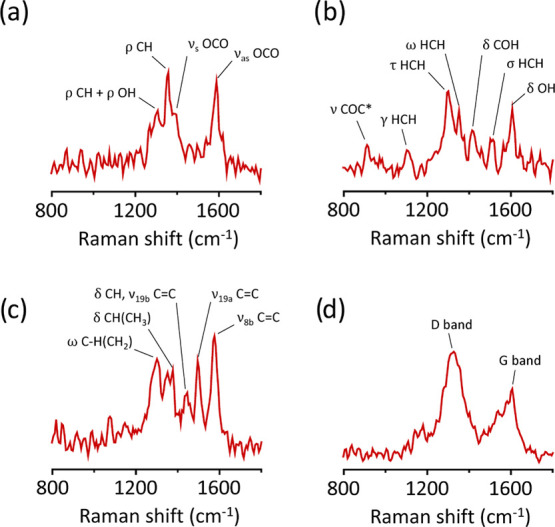
Photocatalytic CO2 reduction products. Spectral snapshots showing the SERS signature of (a) formic acid, (b) formaldehyde, (c) saturated and unsaturated hydrocarbons, and (d) amorphous carbon, observed on a single Ag ND under 532 nm excitation at 1 mW/μm2 in a 60:40 CO2/H2 reaction mixture. The SERS spectral acquisition time is 1 s. The Greek symbols represent symmetric stretch (νs), antisymmetric stretch (νas), rocking (ρ), scissoring (σ), wagging (ω), twisting (τ), out-of-plane deformation (γ), and deformation (δ). ν19a, ν19b, and ν8b are the ring vibrations of benzene-like molecules.
Conclusions
In conclusion, using single-particle spectroscopy, we demonstrate that plasmon excitation of metal nanoparticles can lead to significant thermal reshaping and photocatalytic restructuring of their surface, both in inert and reactive gas environments. Intermittent Stokes emission from Ag nanoparticles suggests a dynamic surface structure of silver photocatalysts under reducing gas environments, leading to the formation of nanoscopic and highly active silver clusters. Such photoinduced Ag–Agn nanoparticle-cluster formation and the presence of a high density of undercoordinated atoms/sites can be vital to the photocatalytic activity of silver nanoparticles. Our single-particle approach, while overcoming typical limitations of ensemble measurements, provides a clear understanding of the light-driven catalyst restructuring and its role in activating chemical reactions on their surface. Our results suggest that light, in conjunction with the selective reactive environment, offers a unique way to engineer highly active nanoparticle catalysts. Such photocatalytic surface modification and cluster formation are likely to influence the behavior of plasmonic nanoparticles used in a wide range of applications, from heterogeneous catalysis to single-molecule detection, and plasmonic sensing.
Experimental Section
EBL Sample Preparation
Square arrays of Ag ND with a diameter of 95 nm, a height of 40 nm, and a pitch of 10 μm in both x and y directions are fabricated by conventional electron-beam lithography (Raith EBPG 5250), followed by metal deposition and lift-off. A 22 × 40 mm2 glass substrate (Menzel-Gläser coverslip) or a superpolished silicon wafer was coated with 450 nm thick electron-beam resist (ZEP520A). Further, the glass substrate is coated with a conductive polymer (Electra 92). After electron-beam exposure and development, ∼2 nm of Ge and ∼40 nm of silver (Kurt J. Lesker, 99.999%) are deposited at a rate of 10 Å s–1 at a base pressure of ∼5 × 10–8 mbar. The deposition is followed by lift-off in acetone.
Synthesis of Colloidal Ag Nanoparticles
Citrate-capped silver nanoparticles are synthesized using the Lee Meisel method.75 Briefly, 18 mg of silver nitrate is dissolved in 100 mL of water and the solution is heated to boiling. Once the solution starts boiling, 2 mL of a 1 wt % aqueous solution of sodium citrate is added and the resulting mixture is kept under stirring for 1 h. The solution is then allowed to cool to room temperature and centrifuged at 8000 rpm (13 000g) for 10 min and redispersed in Milli-Q water.
Flow Cell Experiments
A glass flow cell is built using the methods described previously.6 To create a flow cell, two 1.6 mm diameter holes are drilled into a 1 mm thick glass slide (VWR) at a distance of 3 cm from each other. The glass slide is cleaned by etching away the top surface in a 2 M KOH solution at around 80 °C for 30 min. The slides are then thoroughly washed and sonicated for 30 min in Milli-Q water, and the process is repeated twice. The slides are blow-dried with a nitrogen jet. PTFE tubes (VWR) of diameter 1.6 mm are glued to the holes using two-part epoxy glue. Thereafter, coverslips with EBL-fabricated Ag nanodisks are attached to the glass slide using epoxy glue. For the gas flow experiments, H2, CO2, and N2 gases are purchased from Linde. The flow rates are digitally controlled using Brooks mass flow controllers of GF040 series calibrated for N2, CO2, and H2 gas. For all of the experiments, the net gas flow rate is 5 mL/min. For gas mixtures, the ratio is controlled by controlling the flow rate of the gases to be mixed. For example, a 60:40 volume mixture of CO2 and H2 is obtained by flowing the gases at rates of 3 and 2 mL/min, respectively.
Temperature-Dependent Measurements
For conventional heating experiments in the dark, the flow cell is mounted on a temperature-controlled stage (RC-30 confocal imaging chamber, Warner Instruments). N2 gas is flown at 5 mL/min. The temperature of the stage is varied stepwise from room temperature (∼18 °C) to 48 °C. For each set temperature, the sample is allowed to equilibrate for 10–15 min before recording single-particle dark-field scattering spectra. The drift in the stage due to temperature rise is corrected by readjusting its position using selected Ag NDs as markers in the dark-field imaging mode.
Single-Particle Spectroscopy
Single-particle dark-field imaging and scattering experiments are performed on an inverted Zeiss Axio Observer 7 microscope coupled to an Andor Shamrock SR-500i imaging spectrometer and an Andor Newton 970 EMCCD camera. The silver disks are illuminated with a halogen lamp focused through an EC Epiplan 50× objective (0.75 NA). The back-scattered light is collected with the same objective and sent to the spectrometer connected to the EMCCD. A slit size of 200 μm is chosen to select a single column of particles. Scattering spectra of individual particles are acquired with a grating of 150 lines per mm and centered at 550 nm. Only particles with resonances within 532 ± 10 nm are selected for the CO2RR experiments. Photocatalysis experiments are carried out under 532 nm laser excitation at a power of 3 mW distributed over a spot size of 2 μm in diameter, corresponding to a power density of ∼1 mW/μm2, unless otherwise specified. Typical acquisition times are 30 s for dark-field spectra and 1 s for photoluminescence and SERS spectra.
For SERS analysis, a selection criterion based on signal intensity with respect to the noise variation was set. If a peak intensity was 3 times or more than the noise level, it was identified as a Raman peak, otherwise it was ignored. A comparison of Raman peak positions and intensity was made to identify the molecules. Vibrational modes were assigned based on the Raman spectrum of known molecules such as formic acid, formaldehyde, and amorphous carbon, all of which are well established in the literature.6,71−74
Numerical Simulations
Optical modeling of plasmonic nanoparticles is performed using Lumerical FDTD. For the simulations, we consider a silver nanodisk (Ag ND) of 95 nm in diameter and 40 nm in height on a glass substrate. A total field scattered field source (TFSF) is incident from the glass side from a distance of 100 nm. Absorption and scattering cross section are calculated by placing the respective monitors in the simulation regions. Perfectly matched layer (PML) boundaries surround the simulation region. A simulation mesh of size 0.5 nm is used, which covers the nanoparticles and their immediate vicinity. Yang dielectric functions for silver and in-built dielectric function for glass is used. Dielectric function parameters are optimized by varying the fit tolerance and maximum coefficient in Materials Explorer, to obtain a good material data fit before simulating the scattering and absorption spectra.
Acknowledgments
The authors are grateful to all members of the Nanomaterials for Energy Applications group at DIFFER for fruitful discussions and valuable feedback. This work was supported by the Dutch Research Council (NWO) also through the Vidi award 680-47-550.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00478.
The authors declare no competing financial interest.
Supplementary Material
References
- Maier S. A.Plasmonics: Fundamentals and Applications; Springer Science & Business Media, 2007. [Google Scholar]
- Brown A. M.; Sundararaman R.; Narang P.; Goddard W. A.; Atwater H. A. Nonradiative Plasmon Decay and Hot Carrier Dynamics: Effects of Phonons, Surfaces, and Geometry. ACS Nano 2016, 10, 957–966. 10.1021/acsnano.5b06199. [DOI] [PubMed] [Google Scholar]
- Christopher P.; Xin H.; Linic S. Visible-Light-Enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures. Nat. Chem. 2011, 3, 467–472. 10.1038/nchem.1032. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Kumari G.; Heo J.; Jain P. K. In Situ Formation of Catalytically Active Graphene in Ethylene Photo-Epoxidation. Nat. Commun. 2018, 9, 3056 10.1038/s41467-018-05352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Zhao L. B.; Luo W. L.; Pang R.; Zong C.; Zhou J. Z.; Ren B.; Tian Z. Q.; Wu D. Y. Experimental and Theoretical Study on Isotopic Surface-Enhanced Raman Spectroscopy for the Surface Catalytic Coupling Reaction on Silver Electrodes. J. Phys. Chem. C 2016, 120, 11956–11965. 10.1021/acs.jpcc.6b02252. [DOI] [Google Scholar]
- Kumari G.; Zhang X.; Devasia D.; Heo J.; Jain P. K. Watching Visible Light-Driven CO2 Reduction on a Plasmonic Nanoparticle Catalyst. ACS Nano 2018, 12, 8330–8340. 10.1021/acsnano.8b03617. [DOI] [PubMed] [Google Scholar]
- Yu S.; Wilson A. J.; Heo J.; Jain P. K. Plasmonic Control of Multi-Electron Transfer and C-C Coupling in Visible-Light-Driven CO2 Reduction on Au Nanoparticles. Nano Lett. 2018, 18, 2189–2194. 10.1021/acs.nanolett.7b05410. [DOI] [PubMed] [Google Scholar]
- Yu S.; Wilson A. J.; Kumari G.; Zhang X.; Jain P. K. Opportunities and Challenges of Solar-Energy-Driven Carbon Dioxide to Fuel Conversion with Plasmonic Catalysts. ACS Energy Lett. 2017, 2, 2058–2070. 10.1021/acsenergylett.7b00640. [DOI] [Google Scholar]
- Zhou L.; Swearer D. F.; Zhang C.; Robatjazi H.; Zhao H.; Henderson L.; Dong L.; Christopher P.; Carter E. A.; Nordlander P.; Halas N. J. Quantifying Hot Carrier and Thermal Contributions in Plasmonic Photocatalysis. Science 2018, 362, 69–72. 10.1126/science.aat6967. [DOI] [PubMed] [Google Scholar]
- Kim N. H.; Meinhart C. D.; Moskovits M. Plasmon-Mediated Reduction of Aqueous Platinum Ions: The Competing Roles of Field Enhancement and Hot Charge Carriers. J. Phys. Chem. C 2016, 120, 6750–6755. 10.1021/acs.jpcc.6b00530. [DOI] [Google Scholar]
- Kamarudheen R.; Castellanos G. W.; Kamp L. P. J.; Clercx H. J. H.; Baldi A. Quantifying Photothermal and Hot Charge Carrier Effects in Plasmon-Driven Nanoparticle Syntheses. ACS Nano 2018, 12, 8447–8455. 10.1021/acsnano.8b03929. [DOI] [PubMed] [Google Scholar]
- Sivan Y.; Baraban J.; Un I. W.; Dubi Y. Comment on ‘Quantifying Hot Carrier and Thermal Contributions in Plasmonic Photocatalysis’. Science 2019, 364, eaaw9367 10.1126/science.aaw9367. [DOI] [PubMed] [Google Scholar]
- Baffou G.; Bordacchini I.; Baldi A.; Quidant R. Simple Experimental Procedures to Distinguish Photothermal from Hot-Carrier Processes in Plasmonics. Light: Sci. Appl. 2020, 9, 108. 10.1038/s41377-020-00345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarudheen R.; Aalbers G. J. W.; Hamans R. F.; Kamp L. P. J.; Baldi A. Distinguishing among All Possible Activation Mechanisms of a Plasmon-Driven Chemical Reaction. ACS Energy Lett. 2020, 5, 2605–2613. 10.1021/acsenergylett.0c00989. [DOI] [Google Scholar]
- Xu D.; Bliznakov S.; Liu Z.; Fang J.; Dimitrov N. Composition-Dependent Electrocatalytic Activity of Pt-Cu Nanocube Catalysts for Formic Acid Oxidation. Angew. Chem., Int. Ed. 2010, 49, 1282–1285. 10.1002/anie.200905248. [DOI] [PubMed] [Google Scholar]
- Narayanan R.; El-Sayed M. A. Shape-Dependent Catalytic Activity of Platinum Nanoparticles in Colloidal Solution. Nano Lett. 2004, 4, 1343–1348. 10.1021/nl0495256. [DOI] [Google Scholar]
- Chen M. S.; Goodman D. W. Structure–Activity Relationships in Supported Au Catalysts. Catal. Today 2006, 111, 22–33. 10.1016/j.cattod.2005.10.007. [DOI] [Google Scholar]
- Zugic B.; Wang L.; Heine C.; Zakharov D. N.; Lechner B. A. J.; Stach E. A.; Biener J.; Salmeron M.; Madix R. J.; Friend C. M. Dynamic Restructuring Drives Catalytic Activity on Nanoporous Gold-Silver Alloy Catalysts. Nat. Mater. 2017, 16, 558–564. 10.1038/nmat4824. [DOI] [PubMed] [Google Scholar]
- Xin H. L.; Alayoglu S.; Tao R.; Genc A.; Wang C. M.; Kovarik L.; Stach E. A.; Wang L. W.; Salmeron M.; Somorjai G. A.; Zheng H. Revealing the Atomic Restructuring of Pt-Co Nanoparticles. Nano Lett. 2014, 14, 3203–3207. 10.1021/nl500553a. [DOI] [PubMed] [Google Scholar]
- Tao F.; Grass M. E.; Zhang Y.; Butcher D. R.; Renzas J. R.; Liu Z.; Chung J. Y.; Mun B. S.; Salmeron M.; Somorjai G. A. Reaction-Driven Restructuring of Rh-Pd and Pt-Pd Core-Shell Nanoparticles. Science 2008, 322, 932–934. 10.1126/science.1164170. [DOI] [PubMed] [Google Scholar]
- Fujita T.; Guan P.; McKenna K.; Lang X.; Hirata A.; Zhang L.; Tokunaga T.; Arai S.; Yamamoto Y.; Tanaka N.; Ishikawa Y.; Asao N.; Yamamoto Y.; Erlebacher J.; Chen M. Atomic Origins of the High Catalytic Activity of Nanoporous Gold. Nat. Mater. 2012, 11, 775–780. 10.1038/nmat3391. [DOI] [PubMed] [Google Scholar]
- Marimuthu A.; Zhang J.; Linic S. Tuning Selectivity in Propylene Epoxidation by Plasmon Mediated Photo-Switching of Cu Oxidation State. Science 2013, 339, 1590–1593. 10.1126/science.1231631. [DOI] [PubMed] [Google Scholar]
- Linic S.; Christopher P.; Xin H.; Marimuthu A. Catalytic and Photocatalytic Transformations on Metal Nanoparticles with Targeted Geometric and Plasmonic Properties. Acc. Chem. Res. 2013, 46, 1890–1899. 10.1021/ar3002393. [DOI] [PubMed] [Google Scholar]
- Sambur J. B.; Chen P. Approaches to Single-Nanoparticle Catalysis. Annu. Rev. Phys. Chem. 2014, 65, 395–422. 10.1146/annurev-physchem-040513-103729. [DOI] [PubMed] [Google Scholar]
- Kamarudheen R.; Kumari G.; Baldi A. Plasmon-Driven Synthesis of Individual Metal@semiconductor Core@shell Nanoparticles. Nat. Commun. 2020, 11, 3957 10.1038/s41467-020-17789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringe E.; Sharma B.; Henry A. I.; Marks L. D.; Van Duyne R. P. Single Nanoparticle Plasmonics. Phys. Chem. Chem. Phys. 2013, 15, 4110–4129. 10.1039/c3cp44574g. [DOI] [PubMed] [Google Scholar]
- Zhi Y.; Yu X. C.; Gong Q.; Yang L.; Xiao Y. F. Single Nanoparticle Detection Using Optical Microcavities. Adv. Mater. 2017, 29, 1604920 10.1002/adma.201604920. [DOI] [PubMed] [Google Scholar]
- Cai Y. Y.; Liu J. G.; Tauzin L. J.; Huang D.; Sung E.; Zhang H.; Joplin A.; Chang W. S.; Nordlander P.; Link S. Photoluminescence of Gold Nanorods: Purcell Effect Enhanced Emission from Hot Carriers. ACS Nano 2018, 12, 976–985. 10.1021/acsnano.7b07402. [DOI] [PubMed] [Google Scholar]
- Foerster B.; Joplin A.; Kaefer K.; Celiksoy S.; Link S.; Sönnichsen C. Chemical Interface Damping Depends on Electrons Reaching the Surface. ACS Nano 2017, 11, 2886–2893. 10.1021/acsnano.6b08010. [DOI] [PubMed] [Google Scholar]
- Knight M. W.; Wu Y.; Lassiter J. B.; Nordlander P.; Halas N. J. Substrates Matter: Influence of an Adjacent Dielectric on an Individual Plasmonic Nanoparticle. Nano Lett. 2009, 9, 2188–2192. 10.1021/nl900945q. [DOI] [PubMed] [Google Scholar]
- Mahoney C.; Park K.; Jawaid A.; Kowalski B.; Gillman A.; Tondiglia V.; Treml B.; White T.; Vaia R. A. Low-Energy, Nanoparticle Reshaping for Large-Area, Patterned, Plasmonic Nanocomposites. J. Mater. Chem. C 2018, 6, 7157–7169. 10.1039/C8TC00780B. [DOI] [Google Scholar]
- Sonder E.; Tomizuka C. T. Self-Diffusion in Silver. Phys. Rev. 1956, 103, 1182–1184. 10.1103/PhysRev.103.1182. [DOI] [Google Scholar]
- Stark D. Diffusion Processes on Stepped Surfaces of Thin Metal Films: Migration of Silver Adatoms on Silver (111) Terraces. Surf. Sci. 1987, 189–190, 1111–1116. 10.1016/S0039-6028(87)80557-5. [DOI] [Google Scholar]
- Antczak G.; Ehrlich G.. Diffusion on Two-Dimensional Surfaces. In Surface Diffusion; Cambridge University Press: Cambridge, 2010; pp 261–422. [Google Scholar]
- Wang J.; Chen Y.; Chen X.; Hao J.; Yan M.; Qiu M. Photothermal Reshaping of Gold Nanoparticles in a Plasmonic Absorber. Opt. Express 2011, 19, 14726. 10.1364/oe.19.014726. [DOI] [PubMed] [Google Scholar]
- Inasawa S.; Sugiyama M.; Yamaguchi Y. Laser-Induced Shape Transformation of Gold Nanoparticles Below the Melting Point: The Effect of Surface Melting. J. Phys. Chem. B 2005, 109, 3104–3111. 10.1021/jp045167j. [DOI] [PubMed] [Google Scholar]
- Zijlstra P.; James W. M.; Chon M. G. White Light Scattering Spectroscopy and Electron Microscopy of Laser Induced Melting in Single Gold Nanorods. Phys. Chem. Chem. Phys. 2009, 11, 5866. 10.1039/b905203h. [DOI] [PubMed] [Google Scholar]
- Albrecht W.; Deng T. S.; Goris B.; Van Huis M. A.; Bals S.; Van Blaaderen A. Single Particle Deformation and Analysis of Silica-Coated Gold Nanorods before and after Femtosecond Laser Pulse Excitation. Nano Lett. 2016, 16, 1818–1825. 10.1021/acs.nanolett.5b04851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Ding T. Photothermal-Assisted Optical Stretching of Gold Nanoparticles. ACS Nano 2019, 13, 32–37. 10.1021/acsnano.8b06087. [DOI] [PubMed] [Google Scholar]
- Baffou G.Thermoplasmonics; Cambridge University Press, 2017. [Google Scholar]
- Petrova H.; Juste J. P.; Pastoriza-Santos I.; Hartland G. V.; Liz-Marzán L. M.; Mulvaney P. On the Temperature Stability of Gold Nanorods: Comparison Between Thermal and Ultrafast Laser-Induced Heating. Phys. Chem. Chem. Phys. 2006, 8, 814–821. 10.1039/b514644e. [DOI] [PubMed] [Google Scholar]
- Taylor A. B.; Siddiquee A. M.; Chon J. W. M. Below Melting Point Photothermal Reshaping of Single Gold Nanorods Driven by Surface Diffusion. ACS Nano 2014, 8, 12071–12079. 10.1021/nn5055283. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li W.; Wu D.; Deng Y.; Shao J.; Chen L.; Fang D. Size and Shape Dependent Melting Temperature of Metallic Nanomaterials. J. Phys.: Condens. Matter 2019, 31, 075701 10.1088/1361-648X/aaf54b. [DOI] [PubMed] [Google Scholar]
- Somorjai G. A. The Structure Sensitivity and Insensitivity of Catalytic Reactions in Light of the Adsorbate Induced Dynamic Restructuring of Surfaces. Catal. Lett. 1991, 7, 169–182. 10.1007/BF00764500. [DOI] [Google Scholar]
- Baumberg J. J. Hot Electron Science in Plasmonics and Catalysis: What We Argue About. Faraday Discuss. 2019, 214, 501–511. 10.1039/c9fd00027e. [DOI] [PubMed] [Google Scholar]
- Setoura K.; Okada Y.; Hashimoto S. CW-Laser-Induced Morphological Changes of a Single Gold Nanoparticle on Glass: Observation of Surface Evaporation. Phys. Chem. Chem. Phys. 2014, 16, 26938–26945. 10.1039/c4cp03733b. [DOI] [PubMed] [Google Scholar]
- Baffou G.; Quidant R.; García De Abajo F. J. Nanoscale Control of Optical Heating in Complex Plasmonic Systems. ACS Nano 2010, 4, 709–716. 10.1021/nn901144d. [DOI] [PubMed] [Google Scholar]
- Setoura K.; Okada Y.; Werner D.; Hashimoto S. Observation of Nanoscale Cooling Effects by Substrates and the Surrounding Media for Single Gold Nanoparticles Under CW-Laser Illumination. ACS Nano 2013, 7, 7874–7885. 10.1021/nn402863s. [DOI] [PubMed] [Google Scholar]
- Tao F. F.; Salmeron M. In Situ Studies of Chemistry and Structure of Materials in Reactive Environments. Science 2011, 331, 171–174. 10.1126/science.1197461. [DOI] [PubMed] [Google Scholar]
- Yoshida H.; Kuwauchi Y.; Jinschek J. R.; Sun K.; Tanaka S.; Kohyama M.; Shimada S.; Haruta M.; Takeda S. Visualizing Gas Molecules Interacting with Supported Nanoparticulate Catalysts at Reaction Conditions. Science 2012, 335, 317–319. 10.1126/science.1213194. [DOI] [PubMed] [Google Scholar]
- Shen X.; Dai S.; Zhang S.; Lu Z.; Zhang C.; Graham G. W.; Lei Y.; Pan X.; Peng Z. Oxidation-Induced Atom Diffusion and Surface Restructuring in Faceted Ternary Pt-Cu-Ni Nanoparticles. Chem. Mater. 2019, 31, 1720–1728. 10.1021/acs.chemmater.8b05199. [DOI] [Google Scholar]
- Beaumont S. K.; Alayoglu S.; Pushkarev V. V.; Liu Z.; Kruse N.; Somorjai G. A. Exploring Surface Science and Restructuring in Reactive Atmospheres of Colloidally Prepared Bimetallic CuNi and CuCo Nanoparticles on SiO2 In Situ Using Ambient Pressure X-Ray Photoelectron Spectroscopy. Faraday Discuss. 2013, 162, 31–44. 10.1039/C2FD20145C. [DOI] [PubMed] [Google Scholar]
- Huber M. L.; Harvey A. H.. Thermal Conductivity of Gases. In CRC Handbook of Chemistry and Physics; CRC Press, 2011; Vol. 92. [Google Scholar]
- Kuzma A.; Weis M.; Flickyngerova S.; Jakabovic J.; Satka A.; Dobrocka E.; Chlpik J.; Cirak J.; Donoval M.; Telek P.; Uherek F.; Donoval D. Influence of Surface Oxidation on Plasmon Resonance in Monolayer of Gold and Silver Nanoparticles. J. Appl. Phys. 2012, 112, 103531 10.1063/1.4767688. [DOI] [Google Scholar]
- de Rooij A. The Oxidation of Silver by Atomic Oxygen. ESA J. 1989, 13, 363–382. [Google Scholar]
- Yin Y.; Li Z. Y.; Zhong Z.; Gates B.; Xia Y.; Venkateswaran S. Synthesis and Characterization of Stable Aqueous Dispersions of Silver Nanoparticles Through the Tollens Process. J. Mater. Chem. 2002, 12, 522–527. 10.1039/b107469e. [DOI] [Google Scholar]
- Sundaresan V.; Monaghan J. W.; Willets K. A. Visualizing the Effect of Partial Oxide Formation on Single Silver Nanoparticle Electrodissolution. J. Phys. Chem. C 2018, 122, 3138–3145. 10.1021/acs.jpcc.7b11824. [DOI] [Google Scholar]
- Rodríguez-Fernández J.; Funston A. M.; Perez-Juste J.; Alvarez-Puebla R. A.; Liz-Marzan L. M.; Mulvaney P. The Effect of Surface Roughness on the Plasmonic Response of Individual Sub-Micron Gold Spheres. Phys. Chem. Chem. Phys. 2009, 11, 5909–5914. 10.1039/b905200n. [DOI] [PubMed] [Google Scholar]
- Peyser L. A.; Vinson A. E.; Bartko A. P.; Dickson R. M. Photoactivated Fluorescence from Individual Silver Nanoclusters. Science 2001, 291, 103–106. 10.1126/science.291.5501.103. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Nicovich P. R.; Dickson R. M. Highly Fluorescent Noble-Metal Quantum Dots. Annu. Rev. Phys. Chem. 2007, 58, 409–431. 10.1146/annurev.physchem.58.032806.104546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasap S. O.Principles of Electronic Materials and Devices; Tata McGraw-Hill, 2006. [Google Scholar]
- McKee M. L.; Samokhvalov A. Density Functional Study of Neutral and Charged Silver Clusters Agn with N = 2-22. Evolution of Properties and Structure. J. Phys. Chem. A 2017, 121, 5018–5028. 10.1021/acs.jpca.7b03905. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Guo Y.; Zhu S.; Song Y.; Jin J.; Ji W.; Song W.; Zhao B.; Yang B.; Ozaki Y. Facile Synthesis of Silver Nanoparticles/carbon Dots for a Charge Transfer Study and Peroxidase-like Catalytic Monitoring by Surface-Enhanced Raman Scattering. Appl. Surf. Sci. 2017, 410, 42–50. 10.1016/j.apsusc.2017.03.049. [DOI] [Google Scholar]
- Luo Z.; Castleman A. W.; Khanna S. N. Reactivity of Metal Clusters. Chem. Rev. 2016, 116, 14456–14492. 10.1021/acs.chemrev.6b00230. [DOI] [PubMed] [Google Scholar]
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J.; Teo B. K.; Zheng N. Surface Chemistry of Atomically Precise Coinage–Metal Nanoclusters: From Structural Control to Surface Reactivity and Catalysis. Acc. Chem. Res. 2018, 51, 3084–3093. 10.1021/acs.accounts.8b00371. [DOI] [PubMed] [Google Scholar]
- Hervés P.; Pérez-Lorenzo M.; Liz-Marzán L. M.; Dzubiella J.; Lub Y.; Ballauff M. Catalysis by Metallic Nanoparticles in Aqueous Solution: Model Reactions. Chem. Soc. Rev. 2012, 41, 5577–5587. 10.1039/c2cs35029g. [DOI] [PubMed] [Google Scholar]
- Hrbek J.; Hoffmann F. M.; Park J. B.; Liu P.; Stacchiola D.; Hoo Y. S.; Ma S.; Nambu A.; Rodriguez J. A.; White M. G. Adsorbate-Driven Morphological Changes of a Gold Surface at Low Temperatures. J. Am. Chem. Soc. 2008, 130, 17272–17273. 10.1021/ja8081268. [DOI] [PubMed] [Google Scholar]
- Driver S. M.; Zhang T.; King D. A. Massively Cooperative Adsorbate-Induced Surface Restructuring and Nanocluster Formation. Angew. Chem., Int. Ed. 2007, 46, 700–703. 10.1002/anie.200603325. [DOI] [PubMed] [Google Scholar]
- Benz F.; Schmidt M. K.; Dreismann A.; Chikkaraddy R.; Zhang Y.; Demetriadou A.; Carnegie C.; Ohadi H.; De Nijs B.; Esteban R.; Aizpurua J.; Baumberg J. J. Single-Molecule Optomechanics in ‘Picocavities’. Science 2016, 354, 726–729. 10.1126/science.aah5243. [DOI] [PubMed] [Google Scholar]
- Mayo D. W.; Miller F. A.; Hannah R. W.. Course Notes on the Interpretation of Infrared and Raman Spectra; John Wiley & Sons, 2004. [Google Scholar]
- Castro J. L.; Otero J. C.; Marcos J. I. Anomalous SERS of Monocarboxylic Acids on Silver Sols. J. Raman Spectrosc. 1997, 28, 765–769. 10.1002/(sici)1097-4555(199710)28:10<765::aid-jrs145>3.3.co;2-f. [DOI] [Google Scholar]
- Möhlmann G. R. Raman Spectra of Aqueous Solutions of Formaldehyde and Its Oligomers. J. Raman Spectrosc. 1987, 18, 199–203. 10.1002/jrs.1250180310. [DOI] [Google Scholar]
- Ramsteiner M.; Wagner J. Resonant Raman Scattering of Hydrogenated Amorphous Carbon: Evidence for π-Bonded Carbon Clusters. Appl. Phys. Lett. 1987, 51, 1355–1357. 10.1063/1.98677. [DOI] [Google Scholar]
- Lee P. C.; MeiseL D. Adsorption and Surface-Enhanced Raman of Dyes on Silver and Gold Sols. J. Phys. Chem. A 1982, 86, 3391–3395. 10.1021/j100214a025. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.