

Abstract
The ability to perceive and interact with the world depends on a diverse array of neural circuits specialized for carrying out specific computations. Each circuit is assembled using a relatively limited number of molecules and common developmental steps, from cell fate specification to activity-dependent synaptic refinement. Given this shared toolkit, how do individual circuits acquire their unique properties? We explore this question by comparing development of the circuitry for seeing and hearing, highlighting a few examples where differences in each system’s sensory demands necessitate different developmental strategies.
One Sentence Summary:
During the assembly of neural circuits specialized for vision and audition, many familiar developmental events take place but with subtle differences, illustrating the value of comparing wiring strategies across systems.
An enduring mystery is how undifferentiated cells in the early embryo are transformed into intricately connected networks of neurons that allow us to engage with the surrounding environment. Early insights came from the keen observations of Santiago Ramón y Cajal, who inferred principles of axon guidance and neural circuit formation from his morphological study of individual neurons in their natural context (1). In seeking answers, Ramón y Cajal did not limit himself to one region of the nervous system or one model organism. Rather, by surveying widely, he was able to appreciate common features of developing neurons across systems and species, laying the foundation for our modern understanding of the general mechanisms governing neural circuit assembly. But how is the wide variety of specialized networks underlying animal perception and behavior created using a common toolkit? To explore this question, we contrast the development of two systems with strikingly different patterns of organization – vertebrate seeing and hearing. The details of auditory and visual circuit assembly have been reviewed comprehensively previously (2–6). Instead, we discuss ways in which common mechanisms and molecules function differently to meet the wiring demands of each system. In doing so, we highlight lessons learned and identify opportunities for more multimodal approaches and intellectual cross-pollination to unravel the subtleties of neural development. Although cross-species comparisons are also informative, we focus largely on mammalian systems to facilitate more direct comparisons. Throughout, we highlight opportunities for more detailed analyses of both systems in the future, so that we can begin to understand the flexibility and complexity of mechanisms used to build a functional nervous system.
Shared and specialized features of visual and auditory circuits
The visual and auditory systems face unique challenges in detecting and perceiving sensory stimuli. Dynamic three-dimensional scenes are captured by photoreceptors in the retina, creating an internal map of visual space that is transmitted to the brain by retinal ganglion cell (RGC) afferents (Fig. 1A). Local microcircuits in the vertebrate retina perform parallel computations to parse contrast, color, movement and direction, and changes in intensity (2, 3). The output of these computations is communicated by a variety of RGC subtypes, which project their axons to the brain via the optic nerve. Many RGC axons cross the optic chiasm, but a variable fraction stays ipsilateral, depending on the degree of stereopsis in a species (7). The axons project to a wide variety of targets, including the dorsal lateral geniculate nucleus of the thalamus, the superior colliculus, and numerous accessory and non-image-forming retinorecipient nuclei. Retinotopy is the prevailing organizational principle throughout this pathway, followed by binocularity and RGC subtype identity (3).
Fig. 1. Organization of visual and auditory pathways.
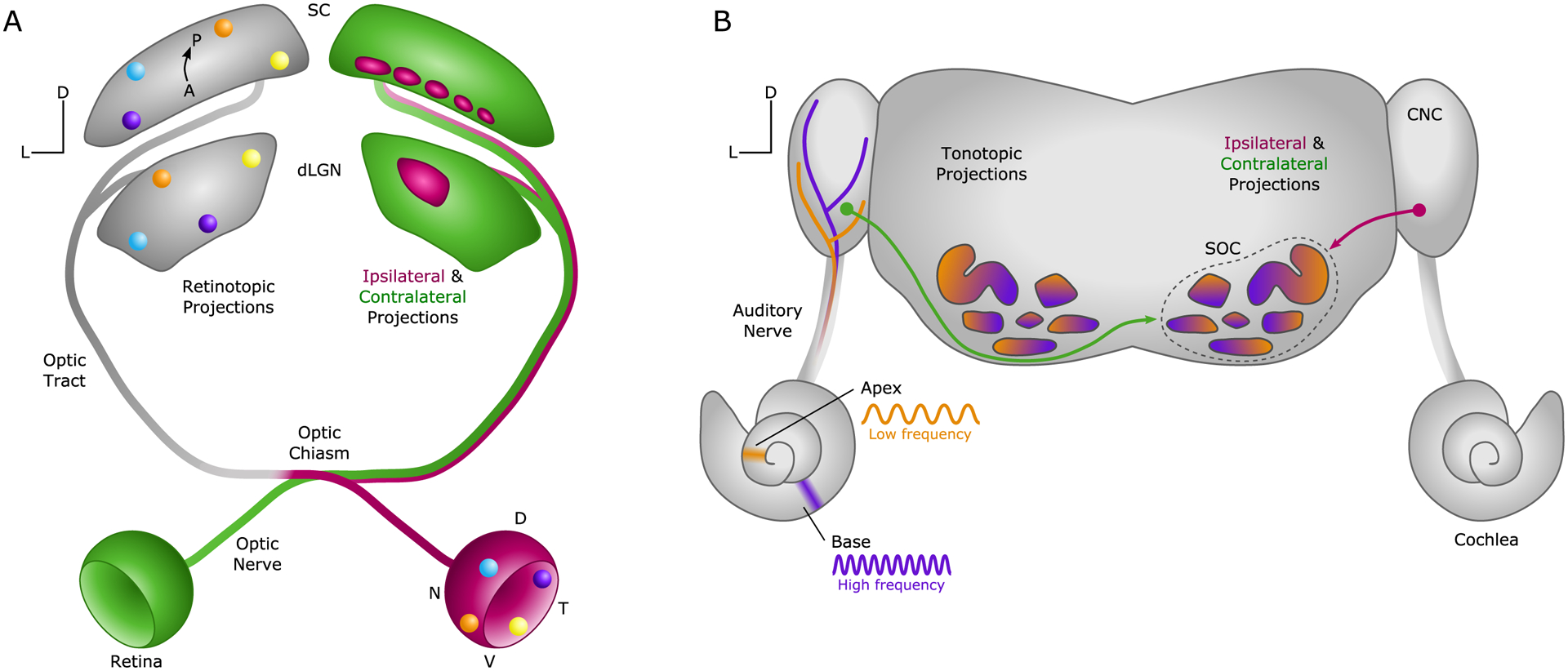
The circuitry connecting the eye and the ear to the brain use similar mapping strategies to convey sensory information centrally. (A) Retinal ganglion cell axons form the optic nerve and travel through the chiasm and tract to innervate the dorsal lateral geniculate nucleus (dLGN) and superior colliculus (SC). The system is organized by retinotopy (left, corresponding to colored circles on right retina) and binocularity (right). Retinal ganglion cell subtypes also target discrete regions of each target (not illustrated, see (3)). (B) Spiral ganglion neurons are arranged by frequency tuning along the cochlea (high frequency in purple, low in orange). Their axons project via the auditory nerve to the ipsilateral cochlear nucleus complex (CNC), where they form tonotopically organized bifurcations (left). Binaural comparisons occur in the superior olivary complex (SOC), which receives both ipsilateral and contralateral CNC inputs (right) and is also tonotopically ordered. D = dorsal, L = lateral, A = anterior, P = posterior.
The auditory system detects and localizes sounds, often in the midst of competing background noises. Like the retina, cochlear circuitry is organized topographically, but according to sound frequency (i.e., tonotopy) rather than sound location. Auditory stimuli are detected by hair cells that are organized by frequency, spiraling from high frequencies in the base of the cochlea to low in the apex (Fig. 1B). Stimulus frequency and intensity are encoded by the activity of spiral ganglion neurons (SGNs), which are tonotopically ordered in parallel to the hair cells. SGNs receive direct input from hair cells on their peripheral processes and send their central axons from the cochlea to the brainstem via the auditory nerve. Upon entering the brainstem, the axons bifurcate to innervate the ventral and dorsal divisions of the cochlear nucleus complex, the sole cochleorecipient target (4). Tonotopy is preserved in the auditory nerve, cochlear nucleus complex, and in most higher order auditory nuclei (5). Within the auditory brainstem nuclei, multiple computations are performed in parallel to localize sound in space, coordinate binaural processing, separate salient sounds from background, and adapt to loud noises (8).
Thus, two different modalities – visual space and sound frequency – are represented in topographic maps from the sense organ to the brain, a principle shared across sensory systems more broadly. However, whereas numerous visual computations are initiated locally in the retina, analogous auditory computations mostly occur in the brainstem. As such, the burden on the afferent neurons in the two systems differs, perhaps reflected in the degree of heterogeneity in the sense organs. In the mouse retina, over 40 RGC subtypes receive and transmit highly parsed sensory information centrally (3, 9). In the cochlea, by contrast, there are only four molecularly distinct SGN subtypes, which rapidly transmit sensory information directly from hair cells to the brainstem (4). The afferent neurons also show marked differences in their central projections. RGC axons innervate nearly 50 central nuclei in rodents (10) and project either ipsilaterally or contralaterally at the midline to give rise to binocular vision. Mammalian SGN axons, on the other hand, project only to the cochlear nucleus complex and are entirely ipsilateral. Information from the two ears is first compared in the superior olivary complex, a hub of auditory nuclei that receives binaural input from the cochlear nucleus complex (11). In sum, the auditory and visual systems differ in the number and roles of afferent neuron subtypes, the balance between peripheral and central computations in the circuit, and the overall pattern of central wiring, including where information from both sides of the head is integrated. As a result, some common developmental events unfold slightly differently in each system.
Circuit specificity at the sensory source
Generating diversity: sensory receptor cell fate
The nature of the physical world constrains how the visual and auditory sense organs detect their respective stimuli, leading to some fundamental differences in the composition and organization of sensory receptor cells. Nonetheless, the initial patterning events are similar. The retina and cochlea both contain two discrete types of sensory receptor cells that transduce stimuli to primary sensory neurons. Rod and cone photoreceptors in the outer retina (Fig. 2A) detect dim and bright light, respectively, with cones providing additional information about color. In the cochlea, two types of mechanoreceptive hair cells reside in the auditory sensory epithelium, the organ of Corti, with one row of inner hair cells separated from three rows of outer hair cells (Fig. 2C). However, only inner hair cells directly capture sound information; the outer hair cells amplify this signal. In each system, these basic sensory receptor cell fates are established by early-acting transcriptional hierarchies. Otx2 and Prdm1 drive retinal progenitors towards photoreceptor fates that are further subdivided by the master regulator NRL, which coordinates the gene regulatory network for rods while inhibiting the cone fate (12). Similarly, Atoh1 and Gfi1 induce cochlear progenitors to assume hair cell fates (13, 14), while Insm1 and Helios promote the outer hair cell fate (15, 16). Thus, although different transcription factors are involved, the general principle is the same, as it is throughout the nervous system: define a generic sensory receptor fate and then execute additional fate decisions to progressively restrict cell identity.
Fig. 2. Local circuitry for seeing and hearing.

In retinal (A, B) and cochlear (C, D) circuits, sensory stimuli are detected by primary sensory receptor cells (yellow and orange) and encoded by the activity of primary afferent neurons (blues and greens). In the retina, discrete types of photoreceptors are intermingled in a mosaic, and local interneuron microcircuits process sensory information before reaching a specific retinal ganglion cell (RGC) subtype (A, cross-section, blues and greens). RGCs extend fasciculated bundles of axons through the retina and into the optic nerve via the optic disc (B, wholemount). In the cochlea, each inner hair cell transmits information directly to multiple Type I spiral ganglion neuron (SGN) subtypes (C, cross-section), which extend their peripheral processes in tonotopically organized radial bundles (D, wholemount). Outer hair cells transmit information to Type II SGNs (C, green). Hair cell properties vary continuously along the tonotopic axis (D, inset).
The rules governing diversity within each sensory receptor population, however, differ. Cone photoreceptors are further subdivided based on the wavelength of light they best detect. By contrast, mammalian hair cells differ morphologically and functionally in a continuum along the tonotopic axis, with no discrete subtypes (6) (Fig. 2D, insets). Thus, whereas visual information is captured by photoreceptor subtypes arranged in a mosaic across a sensory epithelium sheet at the back of the retina, coding of auditory information depends on hair cell position along the length of a sensory epithelium strip. These different modes of sensory receptor organization make sense in the context of the sensory input: wavelengths of light hit the retina at discrete points, but wavelengths of sound travel along the length of the entire cochlea. Accordingly, different developmental mechanisms give rise to diversity in each system. The generation of discrete cone subtypes is achieved via thyroid hormone signaling and cis-regulatory interactions within the opsin loci (17). By contrast, gradients of signaling molecules appear to influence patterning of hair cells along the tonotopic axis (18), though the specific mechanisms remain unclear. More detailed analysis of both systems will continue to elucidate how different balances of intrinsic and extrinsic mechanisms create heterogeneity within pools of cells.
Finding local synaptic partners
Additional insights into neural development come from comparing how neurons find their synaptic partners in the retina and cochlea, which show significant differences in the logic and complexity of their local circuitry. In the rodent retina, there are 15 subtypes of bipolar cells (19), 63 amacrine cell subtypes (20), and 45 RGC subtypes (21), with many neuronal subtypes connecting to multiple preferred synaptic partners (2). Synapses among retinal neurons are organized within two discrete layers of neuropil, the inner and outer plexiform layers, and cell bodies are restricted to nuclear layers (Fig. 2A). The inner plexiform layer is further divided into sublaminae housing ON and OFF sub-circuitry (2). Here, cell identity features prominently in the logic of local connectivity. For example, in the direction-selective circuit, ON and OFF starburst amacrine cells extend their dendrites into two specific sublaminae of the inner plexiform layer (Fig. 2A) to make synapses with direction-selective RGC subtypes (2).
Two basic strategies help solve the daunting problem of creating appropriately localized, cell-type specific connections in the retina: lamination of processes followed by molecular pairing of partners within a layer. The first step is to project to the correct sublamina. Semaphorin ligands act through plexin receptors to confine early processes to the correct region of the inner plexiform layer (22). Additionally, adhesion molecules such as multiple epidermal growth factor-like domains protein 10 (MEGF10) (23) and Fat3 (24) help early-born amacrine cells form a proto-inner plexiform layer on which later-born amacrine cells, bipolar cells, and RGCs elaborate their processes (25, 26). Once localized to the appropriate sublamina, an immunoglobulin superfamily code matches pre- and post-synaptic partners. For instance, amacrine cells expressing Down syndrome cell adhesion molecule (Dscam) form synapses with Dscam-expressing RGCs (27), though a combinatorial code is likely needed for each subtype to recognize all of its partners (28). This system is intrinsically tied to cell identity through transcription factors such as Fezf2, which distinguishes ON starburst amacrine cells from OFF starburst amacrine cells (29). Thus, synaptic specificity arises at least in part from subtype specification, which endows a cell with the ability to project its dendrites to a specific layer and to subsequently pair with a specific collection of post-synaptic partners.
Although there are fewer discrete neuronal subtypes in the cochlea, synaptic pairing also starts with lamination, emphasizing the importance of this basic mechanism throughout the nervous system. The spiral ganglion of the cochlea contains two basic types of neurons that extend spatially segregated projections in the organ of Corti: Type I SGNs connect to inner hair cells and Type II SGNs connect to outer hair cells (Fig. 2C,D). Proper pairing is initiated by restricting Type I SGN projections to the one row of inner hair cells, while Type II SGN projections grow past the inner hair cells to spiral along the three rows of outer hair cells. Time-lapse imaging studies suggest that Type I fibers are segregated from Type II fibers by being kept out of the outer hair cell region and therefore laminating appropriately in the inner hair cell region (30). This is likely mediated by repulsion between the EphA4 guidance receptor in Type I SGNs and ephrin-A5 ligand on outer hair cells (31). Targeting of Type I and II processes is further shaped by semaphorin-plexin signaling (32), echoing the segregation of dendrites within the retinal inner plexiform layer. Indeed, both the general principles and molecular details governing where connections form are shared by both sensory systems, and the main difference is the greater degree of lamination in the retina.
Lamination works well to create parallel microcircuits among specific cell types in the retina, but in the cochlea, the primary challenge is instead preservation of the spatial relationships underlying tonotopy, rather than cell identity. SGN fibers accomplish this in part by forming radial bundles, groups of ~20–40 unbranched processes that grow together towards a few hair cells with similar characteristic frequencies (Fig. 2D). This directed growth depends on signals from surrounding non-neuronal cells: SGN peripheral processes extend from the ganglion to the nascent organ of Corti at the same time as neural crest-derived glia invade the ganglion (33). Glia may provide a scaffold for neurite outgrowth, as radial bundles fail to form in their absence (34, 35). The surrounding mesenchyme also influences radial bundle formation via repulsive Eph/ephrin signaling, such that SGN processes avoid mesenchymal cells and instead fasciculate with each other (36). Unlike the situation in the retina, though, these early events are common to all SGNs. Indeed, deletion of a pan-SGN transcription factor, Gata3, disrupts radial bundle formation (37), perhaps by preventing early peripheral neurites from reading glial and/or mesenchymal cues. The contribution of non-neuronal signals to peripheral process organization presents an interesting contrast to the retina, where local neuron-neuron interactions guide dendrite position and synaptic matching. Nonetheless, fasciculation also features in the retina, but in this case for directed growth of RGC axons towards the optic disc (Fig. 2B). Thus, lamination and fasciculation contribute to circuit formation to differing degrees in each system, with lamination dominating in the retina, where identity is paramount, and fasciculation dominating in the cochlea, where tonotopically appropriate partners are organized by position.
Once processes are properly localized by a combination of lamination and fasciculation, they must form subtype-appropriate synapses. Although there are four SGN subtypes, there is little evidence for the same kind of molecular matching system found in the retina. In contrast to the complex wiring among multiple classes of retinal neurons and their numerous subtypes, each inner hair cell in the cochlea signals to just three subtypes of Type I SGNs. These subtypes have distinct molecular and physiological signatures and their synapses are anatomically ordered along the base of the inner hair cell (38–42) (Fig. 2C). As such, cell identity is not a prominent driver in wiring the cochlear circuit. In fact, SGNs may be able to form synapses with hair cells rather promiscuously. In EphA4 mutants, for instance, some Type I SGNs project past the inner hair cells and form synapses with outer hair cells (31), suggesting that nothing in their Type I identity dictates their synaptic partner. In addition, fine-grain details of this connectivity are finalized by signals from the inner hair cells, which influence SGN subtype identity. Signature genes associated with each Type I SGN subtype show properly restricted expression only after synapses have begun to form. Further, the final stages of subtype diversification fail when signaling between hair cells and SGNs is altered, either by preventing glutamatergic transmission or blocking mechanotransduction (40, 42). Thus, proper connectivity between inner hair cells and Type I SGN subtypes depends on a combination of intrinsic molecular events and activity-dependent consolidation of subtype identity. In the retina, neuronal activity influences subcellular localization of synapses between amacrine cells and direction-selective RGCs after amacrine cell dendrites have already arrived at the appropriate laminar position (43). In this way, the order of developmental events in the retina and cochlea appears reversed: cell identity drives synapse specificity in the retina, which is refined by activity, whereas activity consolidates cell identity after synapses have formed in the cochlea. These two examples, along with the many mechanisms for establishing synaptic specificity found throughout the nervous system, further emphasize the value of studying how the interplay among fate specification, activity, and synapse formation creates circuits with distinct architectures.
Mechanistic versatility in wiring central connections
Organizing and guiding central inputs
By contrast with the obvious differences in the logic of connectivity in the sense organs, auditory and visual nerves share many features, and accordingly common mechanisms organize and guide SGN and RGC axons to their respective targets. The sequence of axon outgrowth from the sensory organ appears to influence axon order in both nerves, helping to maintain the hallmark topographic order en route (44). Because RGCs in the vertebrate retina develop from center to periphery (45) and SGNs in the mammalian cochlea similarly mature from base to apex (46), a neuron’s birthdate grossly corresponds to its position in the sense organ. This broad topography is maintained as development progresses, with earlier-born neurons extending axons while later-born neurons are still differentiating (46–48). Evidence from the visual system supports the hypothesis that neuronal birth order influences both molecular and cellular mechanisms of axon guidance and target innervation (49, 50). Retinal and cochlear afferents also follow similar general rules to reach their targets. First, long-range cues direct axons towards the target region, as demonstrated by transplantation (51–53) and ablation experiments (54, 55). Second, localized cues within the target provide appropriate “stop” and fine-grain organizational signals. The fact that transplanted axons intercalate with the native axons in both retino- and cochleorecipient targets (51, 56, 57) illustrates how local target-derived cues and axon-axon interactions cooperate to organize afferent inputs. These similarities therefore reinforce general principles of nerve development.
When establishing their central connections, however, RGC and SGN axons face different local guidance decisions. RGCs convey information about features of visual stimuli that have already been partially computed by complex microcircuits. Their axons traverse long distances, with highly organized midline crossing and abundant branching to innervate multiple central targets (3, 10). SGNs in the auditory system, on the other hand, directly link sensory receptor cells to their central target, with the majority of stimulus processing occurring in the auditory brainstem. Their axons extend a relatively short distance and only ipsilaterally to reach the sole cochleorecipient target, the cochlear nucleus, where they bifurcate to innervate three subdomains.
To date, we know little about the mechanisms governing these key differences in SGN and RGC central axon navigation. As with other features of development, the simple presence or absence of a molecule can have profound effects. For instance, the stereotyped bifurcation of SGN axons within the cochlear nucleus complex depends on the Natriuretic peptide receptor 2 (Npr2) (58). The Npr2 ligand, C type natriuretic peptide, is expressed along the entire dorsal spinal cord and brainstem, and induces bifurcation of all entering somatosensory projections (59, 60). Thus, bifurcation of SGN axons is mediated by a broadly active system that can be accessed by any Npr2+ population. In contrast, the greater distances and complexities that RGC axons face in their central navigation likely require a wider array of molecules and mechanisms. Consistent with this idea, the development of topographically organized connections involves axon overshooting and refinement in the visual system (61). However, the same degree of overshooting is not evident in the auditory system, where the base-to-apex gradient of SGN development may be sufficient to establish a more precise topographic order in the cochlear nucleus complex. Additionally, SGN axons travel a short distance from the base of cochlea to reach the brainstem, and as such, the physical constraints of the cochlea may aid their guidance, whereas RGC axons likely require more active steering and a greater capacity for growth. Indeed, RGC axon growth capacity is transcriptionally regulated, with young axons extending more vigorously than old axons (62, 63). Finally, RGC axons, which innervate multiple central targets, branch more extensively than SGN axons. In the future, a focused comparison of molecules associated with SGN and RGC axon growth and guidance may point us towards the cellular and molecular mechanisms, like selective trafficking of molecules, that enable individual neurons to coordinate development of multiple branches.
Molecular flexibility: Ephs and ephrins
With a limited array of cues available for brain wiring, it is unsurprising that many molecules are shared across developing sensory systems. Examining how the same molecules function in different contexts highlights the importance of molecular versatility in creating circuit specializations. To illustrate this idea, we focus on the receptor tyrosine kinases Ephs and their ephrin ligands, a large family of contact-mediated guidance molecules. Ephs and ephrins interact promiscuously with each other, with binding partners capable of forward or reverse signaling and mediating repulsion or attraction, depending on the context (64). Such flexibility extends the range of effects that this family of molecules can have, allowing them to customize signaling output across developing systems.
Textbook views of topographic map formation largely stem from studies in the visual system. RGCs express Ephs and ephrins in topographically-defined gradients and their axons encounter corresponding gradients of ephrins and Ephs in their targets, which guide retinal axons to the appropriate area (61). Relative rather than absolute Eph and ephrin levels in the target and axon-axon interactions between retinal axons from neighboring retinal regions shape retinotopic map formation (61, 65, 66). Patterned neuronal activity acts synergistically with molecular cues by mediating RGC axon responsiveness to ephrin-A’s (67, 68). Eph and ephrin expression patterns in the auditory system suggest that they play similar roles in topographic mapping, but direct parallels with their functions in the visual system have been elusive. Consistent with a role in tonotopic map development, ephrinB2 heterozygous mice exhibit a broadening of tonotopy in the dorsal cochlear nucleus (69). Similarly, alterations of EphA4 levels perturb tonotopic connections within the chicken auditory brainstem (70). However, tonotopy appears unchanged in the cochlear nucleus complex of EphA4 mutant mice (69) and projections between auditory brainstem nuclei retain normal topography in ephrinA2/A5 double knock-out mice (71). Thus, the reliance on Eph/ephrin signaling for establishing tonotopic connections between the cochlea and brainstem remains unclear. Despite these differences, central tonotopy, like other topographic maps throughout the developing nervous system, is also refined by activity (5).
Perhaps the apparent discrepancy in Eph/ephrin contribution to topographic mapping in the two systems reflects differences in underlying cellular mechanisms. Ephs and ephrins might simply function differently in the brainstem than in the thalamus or forebrain, where they appear to play more familiar roles in topographic guidance (72, 73). They may influence tonotopic mapping by facilitating local axon-axon interactions within the nerve itself, much as semaphorin/neuropilin interactions mediate pre-targeting sorting in the olfactory nerve (74). Given the short distance SGN axons travel to the brainstem, pre-target sorting could partially obviate the need for more extensive molecular guidance within the target. In general, Ephs and ephrins appear to act more locally in the auditory system than in the visual system. For instance, Eph/ephrin signaling confines axons to appropriate target regions in the auditory brainstem, segregates ipsilateral and contralateral inputs onto shared target neurons (75), and bundles peripheral SGN processes within the cochlea, as discussed earlier (31, 36). However, it is also possible that similarities in molecular function are obscured by differences in circuit architecture. For example, in the cochlear nucleus complex, synapse elimination orchestrated by local Hox gene expression ensures the formation of 1:1 connections and in doing so, refines tonotopy (76). In this light, aberrant tonotopy in the cochlear nucleus complex of ephrinB2 mutants (69) could reflect a failure in refinement of SGN branches. This might mirror how the coordinate activity of Eph and ephrin forward and reverse signaling confines RGC axon interstitial branching to appropriate termination zones in visual targets (61).
Parallels between the function of Ephs and ephrins in the auditory and visual systems are seemingly clearer in axon segregation at the midline. Classic work showed that EphB1 and ephrin-B2 mediate the divergence of ipsilateral RGC axons from midline radial glia at the mammalian optic chiasm (77). Though the auditory projection from the cochlea lacks an analogous structure to the optic chiasm, Eph signaling nonetheless guides development of contralateral projections within the auditory brainstem. When Ephs or ephrins are perturbed, contralaterally projecting axons from neurons in the ventral cochlear nucleus form aberrant ipsilateral branches to innervate both hemispheres of the superior olivary complex (71, 78, 79). Thus, despite acting at a midline choice point, the comparison is complicated by an additional role in branch elimination in the auditory brainstem, which is not found at the optic chiasm.
Identifying definitive similarities and differences between the two systems in the implementation of Ephs and ephrins is challenging, as we continue to discover additional complexity in their functional range. For example, they can influence topographic mapping and other modes of axon segregation independently in both systems. The failure to form a fully contralateral projection in the auditory brainstem in ephrinA2/A5 mutants occurs without any obvious effects on tonotopy (71). In the visual system of the same mutants, conversely, topographic mapping is aberrant, but RGC subtypes terminate in the correct laminar position (80). By studying the same molecules in circuits with fundamentally different patterns of organization, we can define the full range of their signaling repertoires and delineate the rules that determine what kind of signaling occurs and how cell behavior is affected. For example, broad examination of the nervous system has helped distinguish the relative contributions of forward and reverse Eph signaling to topographic mapping in the visual system and spinal cord (64), establishing bidirectional signaling as a key example of how the same molecules can elicit very different outcomes. Careful comparison across circuits will undoubtedly continue to reveal how differences in context can access and leverage this molecular versatility.
Neural development in the 21st century
Over the past hundred years, developmental neurobiologists have successfully defined the principles of nervous system wiring, from cell fate specification to axon guidance to topographic mapping, often by searching for commonalities across model systems and circuits. The next challenge is to learn how these individual events are customized and coordinated to create circuits with specialized features. At first glance, comparison of the auditory and visual systems highlights many familiar genetic, molecular, and activity-dependent mechanisms. A closer look, however, reveals points of divergence that bear further consideration. For instance, the progressive specification of two main sensory receptor cell fates follows generally similar rules in the retina and the cochlea, but acquisition of additional heterogeneity within a sensory receptor subtype differs. Both systems rely on lamination of cell bodies and synaptic zones to wire the local circuit, but to different degrees. Topographic mapping, a common organizational scheme for sensory projections, may arise via different mechanisms, and even shared molecules function in non-analogous ways in the two systems. Although these differences are sometimes subtle, they are consequential for circuit function and it is therefore important to understand how they arise.
Taking inspiration from Ramón y Cajal, if we look across systems to identify salient differences, we can better characterize the flexibility and dynamic range of developmental processes. For example, SGN axons employ multiple, likely redundant strategies to grow towards hair cells, and these strategies differ from those that guide RGC axons into the optic nerve or coordinate wiring among retinal neuronal subtypes. Likewise, SGN axons seem to be less reliant on Eph receptors than RGC axons for central targeting, highlighting the fact that there are multiple ways to create topographic maps. Building on what has been learned from dissecting the contributions of forward and reverse Eph/ephrin signaling, the availability of CRISPR/cas-9 approaches will facilitate efforts to study how versatility in other signaling systems extends the use of the same molecules to solve fundamentally different wiring problems. Cross-systems comparisons will also help us understand how networks of molecules orchestrate circuit assembly, beyond what can be learned from studying discrete events gene by gene. For instance, whole nervous system analysis in C. elegans led to the novel idea that circuits can be defined by shared transcription factor motifs (81). Similarly, examining how heterogeneity arises in SGNs and RGCs may provide clues as to whether populations with greater subtype diversity employ fundamentally different strategies than those with relatively less diversity. Perhaps local activity plays a more prominent role in defining the three Type I SGN subtypes than it does in shaping RGC heterogeneity, which may in turn require more complex transcriptional hierarchies. Such differences have important implications for our understanding of how malleable cell identity might be, both developmentally and in the mature brain. In a similar vein, the cochlea and retina each offer unique advantages and perspectives for studying how synapse heterogeneity and subcellular localization arise, features of circuit assembly for which the basic rules remain to be defined.
By studying many types of circuits with fundamentally different patterns, we can begin to appreciate the range of strategies used to wire the nervous system and uncover the rules that govern how and when specific strategies are employed. This information will not only enrich our understanding of the complexities of neural development, but also lay the foundation for designing ways to re-wire the nervous system in the future. Further, if we ever hope to understand the neural basis of individual differences among humans, it is essential to learn how to recognize and study the ways in which subtleties in circuit architecture emerge. With ever more powerful tools for imaging and genomic analysis, today’s neuroscientists are poised to extend our appreciation of shared developmental phenomena and explore the myriad ways in which a shared molecular genetic toolkit shapes specialized circuitry.
Acknowledgments:
We thank Dr. Corey Harwell and members of the laboratory, especially Dr. Evelyn Aviles and Dr. Lauren Kreeger, for helpful discussions and feedback on the manuscript, as well as Dr. Oliver Hobert for sharing related ideas. We are particularly grateful to the numerous outstanding researchers in auditory and visual neuroscience who have collectively provided so many important insights into neural development, but whose work we did not have room to discuss.
Funding:
The laboratory’s work on mechanisms of neural development is supported by NIH grants R01 EY024884, R01 DC015974, R01 DC009223, and R21 NS113562.
Footnotes
Competing interests: Authors declare no competing interests.
References
- 1.Ramón y Cajal S, Histology of the nervous system of man and vertebrates. N Swanson & LW Swanson, Trans. 1995; New York, NY: Oxford University Press, Vols. 1, 2. (1911). [Google Scholar]
- 2.Hoon M, Okawa H, Della Santina L, Wong ROL, Prog. Retin. Eye Res 42, 44–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seabrook TA, Burbridge TJ, Crair MC, Huberman AD, Annu. Rev. Neurosci 40, 499–538 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Shrestha BR, Goodrich LV, in The Oxford Handbook of the Auditory Brainstem, Kandler K, Ed. (Oxford University Press, 2019). [Google Scholar]
- 5.Kandler K, Clause A, Noh J, Nat. Neurosci 12, 711–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann ZF, Kelley MW, Hear. Res 276, 2–15 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Petros TJ, Rebsam A, Mason CA, Annu. Rev. Neurosci 31, 295–315 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Young ED, Oertel D, in The Synaptic Organization of the Brain (Oxford University Press, 2004), pp. 125–164. [Google Scholar]
- 9.Sanes JR, Masland RH, Annu. Rev. Neurosci 38, 221–246 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Morin LP, Studholme KM, J. Comp. Neurol 522, 3733–3753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schofield BR, in The Inferior Colliculus, Winer JA, Schreiner CE, Eds. (Springer New York, New York, NY, 2005), pp. 132–154. [Google Scholar]
- 12.Brzezinski JA, Reh TA, Dev. 142, 3263–3273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY, Science. 284, 1837–41 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Wallis D, Hamblen M, Zhou Y, Venken KJT, Schumacher A, Leighton Grimes H, Zoghbi HY, Orkin SH, Bellen HJ, Development. 130, 221–232 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Wiwatpanit T, Lorenzen SM, Cantú JA, Foo CZ, Hogan AK, Márquez F, Clancy JC, Schipma MJ, Cheatham MA, Duggan A, García-añoveros J, Nature. 563, 691–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chessum L, Matern MS, Kelly MC, Johnson SL, Ogawa Y, Milon B, McMurray M, Driver EC, Parker A, Song Y, Codner G, Esapa CT, Prescott J, Trent G, Wells S, Dragich AK, Frolenkov GI, Kelley MW, Marcotti W, Brown SDM, Elkon R, Bowl MR, Hertzano R, Nature. 563, 696–724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viets K, Eldred KC, Johnston RJ, Trends Genet. 32, 638–659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Son EJ, Wu L, Yoon H, Kim S, Choi JY, Bok J, PLoS One. 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR, Cell. 166, 1308–1323.e30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan W, Laboulaye MA, Tran NM, Whitney IE, Benhar I, Sanes JR, J. Neurosci 40, 5177–5195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran NM, Shekhar K, Whitney IE, Jacobi A, Benhar I, Hong G, Yan W, Adiconis X, Arnold ME, Lee JM, Levin JZ, Lin D, Wang C, Lieber CM, Regev A, He Z, Sanes JR, Neuron. 104, 1039–1055.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuoka RL, Nguyen-Ba-Charvet KT, Parray A, Badea TC, Chédotal A, Kolodkin AL, Nature. 470, 259–264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray TA, Roy S, Kozlowski C, Wang J, Cafaro J, Hulbert SW, Wright CV, Field GD, Kay JN, Elife. 7, 1–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deans MR, Krol A, Abraira VE, Copley CO, Tucker AF, Goodrich LV, Neuron. 71, 820–32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chow RW, Almeida AD, Randlett O, Norden C, Harris WA, Development. 142, 2665–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godinho L, Mumm JS, Williams PR, Schroeter EH, Koerber A, Park SW, Leach SD, Wong ROL, Development. 132, 5069–79 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Yamagata M, Sanes JR, Nature. 451, 465–469 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Sanes JR, Zipursky SL, Cell. 181, 536–556 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Peng YR, James RE, Yan W, Kay JN, Kolodkin AL, Sanes JR, Neuron. 105, 464–474.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Druckenbrod NR, V Goodrich L, J. Neurosci 35, 16221–16235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Defourny J, Poirrier A-L, Lallemend F, Mateo Sánchez S, Neef J, Vanderhaeghen P, Soriano E, Peuckert C, Kullander K, Fritzsch B, Nguyen L, Moonen G, Moser T, Malgrange B, Nat. Commun 4, 1438 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Coate TM, Spita NA, Zhang KD, Isgrig KT, Kelley MW, Elife. 4, 1–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandell LL, Butler Tjaden NE, Barlow AJ, Trainor PA, Dev. Biol 385, 200–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris JK, Maklad A, Hansen LA, Feng F, Sorensen C, Lee KF, Macklin WB, Fritzsch B, Brain Res. 1091, 186–199 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao YY, Reiprich S, Wegner M, Fritzsch B, PLoS One. 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coate TM, Raft S, Zhao X, Ryan A, Crenshaw EB, Kelley MW, Neuron. 73, 49–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appler JM, Lu CC, Druckenbrod NR, Yu W-M, Koundakjian EJ, Goodrich LV, J. Neurosci 33, 3679–3691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberman MC, J. Acoust. Soc. Am 63, 442–455 (1978). [DOI] [PubMed] [Google Scholar]
- 39.Liberman MC, Science (80-. ). 216, 1239–1241 (1982). [DOI] [PubMed] [Google Scholar]
- 40.Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV, Cell. 174, 1229–1246.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petitpré C, Wu H, Sharma A, Tokarska A, Fontanet P, Wang Y, Helmbacher F, Yackle K, Silberberg G, Hadjab S, Lallemend F, Nat. Commun 9, 3691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun S, Babola T, Pregernig G, So KS, Nguyen M, Su SM, Palermo AT, Bergles DE, Burns JC, Müller U, Cell. 174, 1247–1263.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bleckert A, Zhang C, Turner MH, Koren D, Berson DM, Park SJH, Demb JB, Rieke F, Wei W, Wong RO, Proc. Natl. Acad. Sci. U. S. A 115, E12083–E12090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sitko AA, Mason CA, in Axons and Brain Architecture, Rockland K, Ed. (Elsevier, 2016), pp. 267–288. [Google Scholar]
- 45.Nguyen-Ba-Charvet KT, Rebsam A, Int. J. Mol. Sci 21, 1–24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koundakjian EJ, Appler JL, Goodrich LV, J. Neurosci 27, 14078–14088 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsh C, Guillery RW, J. Neurosci 5, 3061–3069 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molea D, Rubel EW, J. Comp. Neurol 466, 577–591 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Bhansali P, Rayport I, Rebsam A, Mason C, Neural Dev. 9, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osterhout JA, El-Danaf RN, Nguyen PL, Huberman AD, Cell Rep. 8, 1006–1017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Constantine-Paton M, Law MI, Science (80-. ). 202, 639–641 (1978). [DOI] [PubMed] [Google Scholar]
- 52.Hankin MH, Lund RD, J. Comp. Neurol 263, 455–466 (1987). [DOI] [PubMed] [Google Scholar]
- 53.Elliott KL, Fritzsch B, Sci. Rep 8, 13819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maricich SM, Xia A, Mathes EL, Wang VY, Oghalai JS, Fritzsch B, Zoghbi HY, J. Neurosci 29, 11123–11133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reh TA, Pitts E, Constantine-Paton M, J. Comp. Neurol 218, 282–296 (1983). [DOI] [PubMed] [Google Scholar]
- 56.Gordy C, Straka H, Houston DW, Fritzsch B, Elliott KL, Dev. Neurobiol 78, 1064–1080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elliott KL, Houston DW, Fritzsch B, Sci. Rep 5, 8338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu CC, Cao XJ, Wright S, Ma L, Oertel D, Goodrich LV, PLoS Genet. 10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmidt H, Stonkute A, Jüttner R, Koesling D, Friebe A, Rathjen FG, Proc. Natl. Acad. Sci. U. S. A 106, 16847–16852 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ter-Avetisyan G, Rathjen FG, Schmidt H, J. Neurosci 34, 737–747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feldheim DA, O’Leary DDM, Cold Spring Harb. Perspect. Biol 2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goldberg JL, Klassen MP, Hua Y, Barres BA, Science. 296, 1860–4 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Steketee MB, Oboudiyat C, Daneman R, Trakhtenberg E, Lamoureux P, Weinstein JE, Heidemann S, Barres BA, Goldberg JL, Investig. Ophthalmol. Vis. Sci 55, 4369–4377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kania A, Klein R, Nat. Rev. Mol. Cell Biol 17, 240–256 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Suetterlin P, Drescher U, Neuron. 84, 740–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fiederling F, Weschenfelder M, Fritz M, Von Philipsborn A, Bastmeyer M, Weth F, Elife. 6, 1–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nicol X, Voyatzis S, Muzerelle A, Narboux-Nême N, Südhof TC, Miles R, Gaspar P, Nat. Neurosci 10, 340–347 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Pfeiffenberger C, Yamada J, Feldheim DA, J. Neurosci 26, 12873–12884 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miko IJ, Nakamura PA, Henkemeyer M, Cramer KS, J. Comp. Neurol 505, 669–81 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Huffman KJ, Cramer KS, Dev. Neurobiol 67, 1655–68 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Abdul-latif ML, Salazar JAA, Marshak S, Dinh ML, Cramer KS, Neural Dev. 10, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Intskirveli I, Metherate R, Cramer KS, PLoS One. 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellsworth CA, Lyckman AW, Feldheim DA, Flanagan JG, Sur M, J. Comp. Neurol 488, 140–151 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Imai T, Yamazaki T, Kobayakawa R, Kobayakawa K, Abe T, Suzuki M, Sakano H, Science. 325, 585–90 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Cramer KS, Bermingham-McDonogh O, Krull CE, Rubel EW, Dev. Biol 269, 26–35 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Karmakar K, Narita Y, Fadok J, Ducret S, Loche A, Kitazawa T, Genoud C, Di Meglio T, Thierry R, Bacelo J, Lüthi A, Rijli FM, Cell Rep. 18, 185–197 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Williams SE, Mann F, Erskine L, Sakurai T, Wei S, Rossi DJ, Gale NW, Holt CE, Mason CA, Henkemeyer M, Neuron. 39, 919–35 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Hsieh CY, Nakamura PA, Luk SO, Miko IJ, Henkemeyer M, Cramer KS, J. Neurosci 30, 9840–9849 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cramer KS, Cerretti DP, Siddiqui SA, Dev. Biol 295, 76–89 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Sweeney NT, James KN, Sales EC, Feldheim DA, Dev. Neurobiol 75, 584–593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pereira L, Kratsios P, Serrano-Saiz E, Sheftel H, Mayo AE, Hall DH, White JG, LeBoeuf B, Garcia LR, Alon U, Hobert O, Elife. 4, 1–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]