

Abstract
RNA capping and decapping tightly coordinate with transcription, translation, and RNA decay to regulate gene expression. Proteins in the DXO/Rai1 family have been implicated in mRNA decapping and decay, and mammalian DXO was recently found to also function as a decapping enzyme for NAD+-capped RNAs (NAD-RNA). The Arabidopsis genome contains a single gene encoding a DXO/Rai1 protein, AtDXO1. Here we show that AtDXO1 possesses both NAD-RNA decapping activity and 5′-3′ exonuclease activity but does not hydrolyze the m7G cap. The atdxo1 mutation increased the stability of NAD-RNAs and led to pleiotropic phenotypes, including severe growth retardation, pale color, and multiple developmental defects. Transcriptome profiling analysis showed that the atdxo1 mutation resulted in upregulation of defense-related genes but downregulation of photosynthesis-related genes. The autoimmunity phenotype of the mutant could be suppressed by either eds1 or npr1 mutation. However, the various phenotypes associated with the atdxo1 mutant could be complemented by an enzymatically inactive AtDXO1. The atdxo1 mutation apparently enhances post-transcriptional gene silencing by elevating levels of siRNAs. Our study indicates that AtDXO1 regulates gene expression in various biological and physiological processes through its pleiotropic molecular functions in mediating RNA processing and decay.
INTRODUCTION
In eukaryotic cells, the 5′ end of an mRNA molecule typically consists of a 7-methylguanylate (m7G) cap. The capping process is co-transcriptionally catalyzed by capping enzymes soon after RNA synthesis is started (Mao et al. 1995). In addition to regulation of RNA decay, the m7G cap plays an essential role in almost all aspects of gene expression, including transcription, RNA processing and modification, nucleocytoplasmic transport, assembly of translation initiation complexes, and post-transcriptional gene silencing (Jiao et al. 2010; Trotman et al. 2017). Regulated decapping of mRNAs is a key step in controlling gene expression. In plants, the removal of the 5′ m7G cap is processed by a decapping complex comprising DECAPPING 1 (DCP1), DCP2, and VARICOSE (VCS) (Xu et al. 2006), followed by RNA degradation through 5′ to 3′ exoribonucleases (XRNs) (Chiba and Green 2009).
In addition to the canonical m7G cap, noncanonical 5′ caps such as the NAD cap have been found in RNAs of both prokaryotes and eukaryotes (Chen et al. 2009; Kowtoniuk et al. 2009; Cahová et al. 2015; Jiao et al. 2017), implying an additional layer to regulation of gene expression through capping or decapping. Recently, it was reported that NAD-capped RNAs (NAD-RNAs) are produced from at least several thousand Arabidopsis genes, most of which are protein-coding genes (Wang et al. 2019; Zhang et al. 2019). However, the molecular and biological functions of NAD-RNAs remain unclear. In prokaryotes, the NAD cap was found to protect RNA from degradation (Cahová et al. 2015). However, in mammalian cells synthetic NAD-mRNAs were not translated but instead underwent degradation, partly through the DXO/Rai1 decapping pathway (Jiao et al. 2017). In Arabidopsis, indirect evidence suggests that endogenous NAD-RNAs could be translated (Wang et al. 2019; Zhang et al. 2019).
DXO/Rai1 proteins belong to a small protein family in eukaryotes and have been shown to be involved in the quality control of the eukaryotic mRNA 5′ cap in yeast and mammals (Jiao et al. 2010; Jurado et al. 2014; Zhai and Xiang 2014). DXO/Rai1 proteins are distinct from the better-known decapping enzyme Dcp2, which removes the m7G cap from mRNA (m7GpppRNA) (Wang et al. 2002). In yeast, Rai1 has RNA 5′ pyrophosphatase (PPH) activity (Xiang et al. 2009) and can also decap an unmethylated GpppRNA molecule, but has much weaker decapping activity on m7GpppRNA (Jiao et al. 2010). Another homolog in yeast, Dxo1, can hydrolyze the cap of both GpppRNA and m7GpppRNA, but lacks the PPH activity (Chang et al. 2012). In S. cerevisiae, incompletely capped mRNAs accumulate in rai1 cells under nutritional stress (glucose or amino acid starvation) (Jiao et al. 2010) or in rai1dxo1 under normal, non-stress conditions (Chang et al. 2012). These data indicate that Rai1 and Dxo1 function with partial redundancy in maintaining the fidelity of the 5′ RNA cap. Mammalian DXO possesses multiple activities, namely the PPH activity, decapping activity of both unmethylated and methylated RNA caps, and 5′-3′ exoribonuclease activity (Jiao et al. 2013). Recently, mammalian DXO was found to decap NAD-RNAs (Jiao et al. 2017). The Drosophila DXO/Rai1 family protein, Cuttoff, lacks the m7GpppRNA decapping activity and is thought to bind to the 5′ end of an mRNA, preventing its degradation (Zhang et al. 2014; Munafò et al. 2018). Arabidopsis has a single DXO gene (AtDXO1), which was recently reported to be involved in RNA turnover and chloroplast function, but these functions were found to be independent of its enzymatic activity (Kwasnik et al. 2019).
Defects in the degradation of aberrant mRNAs, which can be caused by mutations in genes encoding 5′-3′ exoribonucleases (XRN2, XRN3, XRN4) or the RNA helicase AtSKI2, can trigger post-transcriptional gene silencing (PTGS) in plants (Liu and Chen 2016). A fraction of the aberrant transcripts may be converted to double-stranded RNAs (dsRNAs) by RNA-DE-PENDENT RNA POLYMERASEs (RDRs). The resultant dsRNAs would be processed by Dicer-like (DCL) proteins into small interfering (si)RNAs, which subsequently direct ARGONAUTE (AGO)-mediated cleavage of their homologous transcripts (Vazquez and Hohn 2013; Liu and Chen 2016). Loss-of-function of both AtXRN4 and AtSKI2 causes a severe growth defect or embryo-lethality, but these defects can be significantly suppressed by disruption of the core PTGS components, such as RDR6, Suppressor of Gene Silencing 3 (SGS3), DCL2 and DCL4, or AGO1 (Zhang et al. 2015).
Here, we report that Arabidopsis DXO1 possesses both 5′-3′ exonuclease and NAD-RNA decapping activities. Loss of function of DXO1 enhances the stability of NAD-RNAs. The dxo1 mutant exhibits pleiotropic growth and developmental defects and autoimmunity; however, these phenotypes are independent of its NAD-RNA decapping and exoribonuclease activity. The dxo1 mutation enhances accumulation of siRNAs. Our study shows that DXO1 has multiple functions as an essential component in RNA processing and decay.
RESULTS
Loss-of-function of DXO1 causes multiple growth and developmental defects and lowers chlorophyll content
Arabidopsis thaliana has one gene encoding a homolog of the DXO/RAI1 family, AT4G17620, which was designated DXO1 (Kwasnik et al. 2019). The predicted DXO1 protein has 544 amino acids (aa). Its 344-aa C-terminal region shares approximately 30% sequence identity and approximately 45% similarity to both Schizosaccharomyces pombe RAI1 (SpRAI1) and Mus musculus DXO (mDXO) (Figure S1A). The residues around the catalytic site in DXO1 are highly conserved with those of the fungal and mammalian DXO/Rai1 family proteins. The 200-aa N-terminus of DXO1 is apparently plant-specific (Figure S1B).
To reveal the subcellular localization of DXO1, the DXO1-GFP fusion protein was transiently expressed in protoplasts from Arabidopsis leaves. The GFP signal was found to be localized to both the cytosol and the nucleus (Figure S2A).
To elucidate the biological function of DXO1 in Arabidopsis, we obtained two T-DNA insertion lines for the DXO1 gene, SALK_103157 and SALK_032903. Salk_103157 has a T-DNA insertion in the second exon and was previously named dxo1-1 (Kwasnik et al. 2019), and SALK_032903 (dxo1-2) has a T-DNA insertion in the fifth intron (Figure 1A, B). Both dxo1-1 and dxo1-2 were apparently null mutants, as normal DXO1 transcript could not be detected in these two mutants by semi-quantitative reverse transcription PCR (RT-PCR) (Figure S3A). Along with these two T-DNA insertion mutants, another knockout mutant (dxo1-3) was generated using the CRISPR-Cas9 system to introduce a 13-bp deletion in the fourth exon, causing a premature stop codon (Figure 1C, D). These three dxo1 mutant lines displayed similar morphological phenotypes, with profound growth and developmental defects. The fact that homozygous mutant progeny were obtained from heterozygous plants indicates that at least some dxo1 pollen and ovules are functional and that the DXO1 gene is not essential for embryogenesis and seed development. However, true leaves of the mutant seedlings were pale and morphologically abnormal compared to the wild type (WT) seedlings (Figure 1B). Mutant leaves were narrow, often pointed, curled, or needle-like. The pistils and siliques were often de-formed and curled as well (Figure S3B). The rosette leaves of the mutants were not positioned as in WT plants (Figure S3C). At the bolting stage, the mutant plants showed a loss of apical dominance, with thin, short stems (Figure S3C). Since the multiple mutant lines showed similar phenotypes, the mutant phenotypes were attributed to loss-of-function of DXO1. For further confirmation, we generated transgenic complementation lines. The DXO1 genomic DNA, including its native promoter, was introduced into the dxo1-2 mutant. From the ten lines we obtained, two individual complementation lines were further characterized and showed full complementation of the morphological defects of dxo1-2 (Figure 1E), further demonstrating that the phenotype of the mutants is caused by the loss-of-function mutation of DXO1.
Figure 1. Loss-of-function mutation of DXO1 causes growth and developmental defects in Arabidopsis.
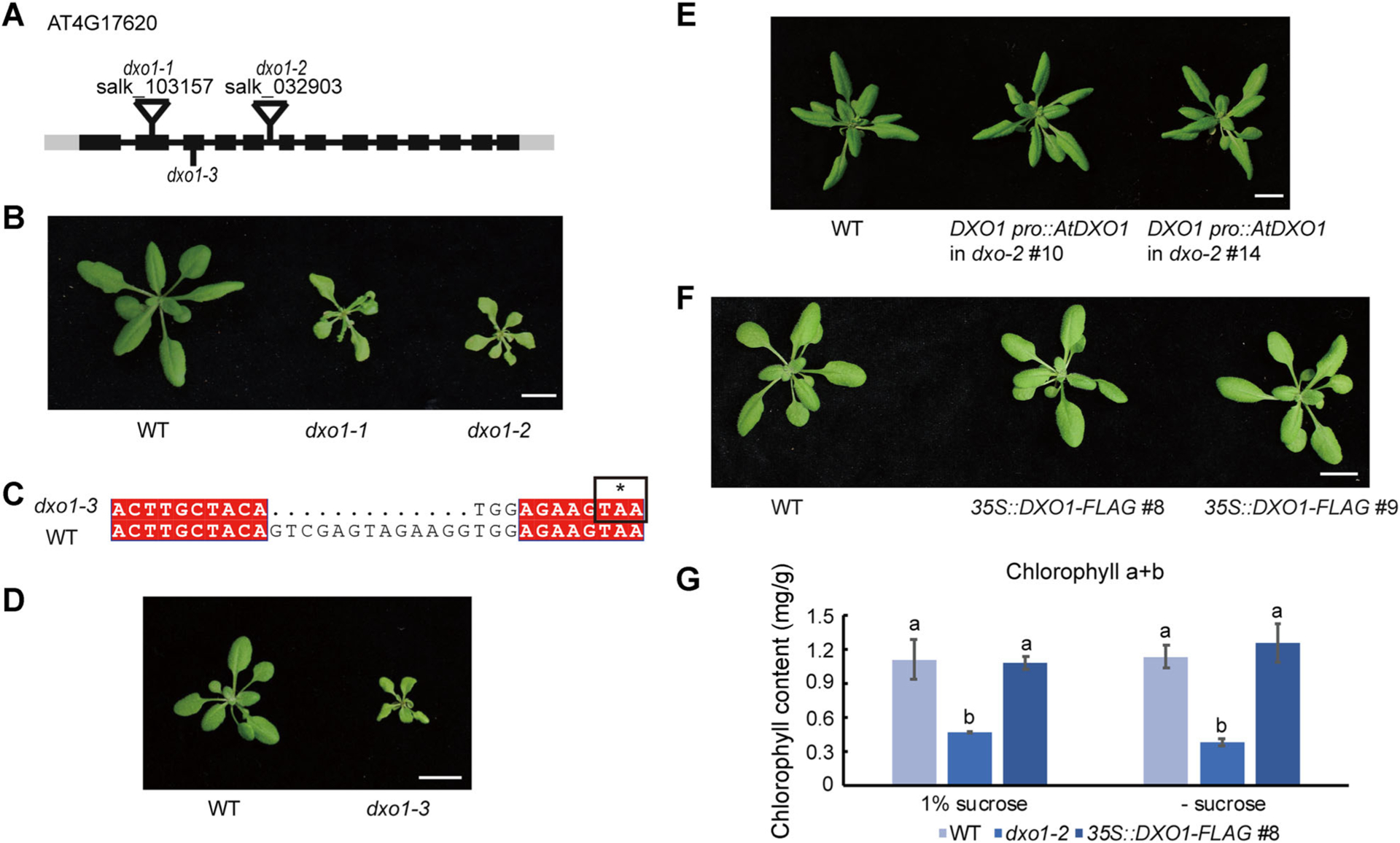
(A) Schematic diagram of the DXO1 gene structure, the T-DNA insertion sites and the deleted regions in three mutants. The boxes represent exons, the lines represent introns, and the grey boxes represent untranslated regions (UTR). (B) 28-day-old wild type (WT), dxo1-1, and dxo1-2 plants. Bar: 1 cm. (C) The sequence of the genomic region of DXO1 where a 13-bp deletion was introduced in Cas9 line #93 (dxo1-3). The * represents a stop codon caused by the deletion. (D) Twenty-one-day-old WT and dxo1-3 plants. Bar: 1 cm. (E) Twenty-eight-day-old plants of WT and dxo1-2 transgenic lines complemented by the genomic clone of AtDXO1. Bar: 1 cm. (F) 25-day-old plants of WT and DXO1-overexpressing transgenic lines (35S::DXO1-FLAG #8 and #9). Bar: 1 cm. (G) Chlorophyll content of WT, dxo1-2, and 35S::DXO1-FLAG #8 grown on 1/2 MS medium with and without sucrose. The error bars represent standard deviation with three biological replicates. A t-test was used to analyze statistical significance; different letters represent a significant difference between the samples.
We also generated transgenic lines overexpressing DXO1 fused to the FLAG tag under the control of the 35S CaMV promoter in the WT background (Figure 1F). The DXO1 mRNA levels in these two lines were 350 to 600 times higher than in WT, as determined by real-time RT-PCR (qPCR) (Figure S3D). However, there was no obvious morphological difference between the two overexpression lines and WT.
As the dxo1 mutants showed yellowish leaves and stems, we measured the chlorophyll content in seedlings grown on 1/2 MS medium with 1% sucrose. The chlorophyll content (Chlorophyll a + b) of dxo1-2 was approximately 50% of the WT level, whereas the overexpression plants showed no difference from WT in chlorophyll content (Figure 1G). Similar results were obtained when seedlings were grown on the same medium without exogenous sucrose, indicating that the low content of chlorophyll in the dxo1 mutant is not dependent on exogenous sucrose supply.
DXO1 possesses 5′-3′ exoribonuclease and NAD-RNA decapping activities
Proteins in the DXO/Rai1 family have been reported to have intrinsic 5′-3′ exoribonuclease activity (Jurado et al. 2014; Grudzien-Nogalska and Kiledjian 2017). Recombinant DXO1 and mouse DXO (mDXO) proteins were expressed in E. coli, purified (Figure S2B), and tested in an in vitro enzymatic assay using a 35-nt RNA with a 5′-end monophosphate and a 3′-end florescent label, 6-carboxyfluorescein (6-FAM). This uncapped RNA was degraded by DXO1 and mDXO (Figure 2A, B). While nuclease P1 completely degraded the substrates to single nucleotides, DXO1 and mDXO shortened the substrate but could not completely digest it, leaving several nucleotides at the 3′ region intact (Figure 2A). DXO1 was not able to degrade single-stranded DNA (ssDNA) (Figure 2C), indicating that the exonuclease activity of DXO1 is RNA-specific. We also generated two DXO1 mutations, E394A and K412Q (Figure S2B). E394 and K412 are conserved residues at the active site in DXO/RAI1 proteins. These DXO1 mutant variants displayed reduced 5′-3′ exoribonuclease activity (Figure 2B).
Figure 2. DXO1 has 5′-3′ exoribonuclease activity but no m7G-RNA decapping activit.
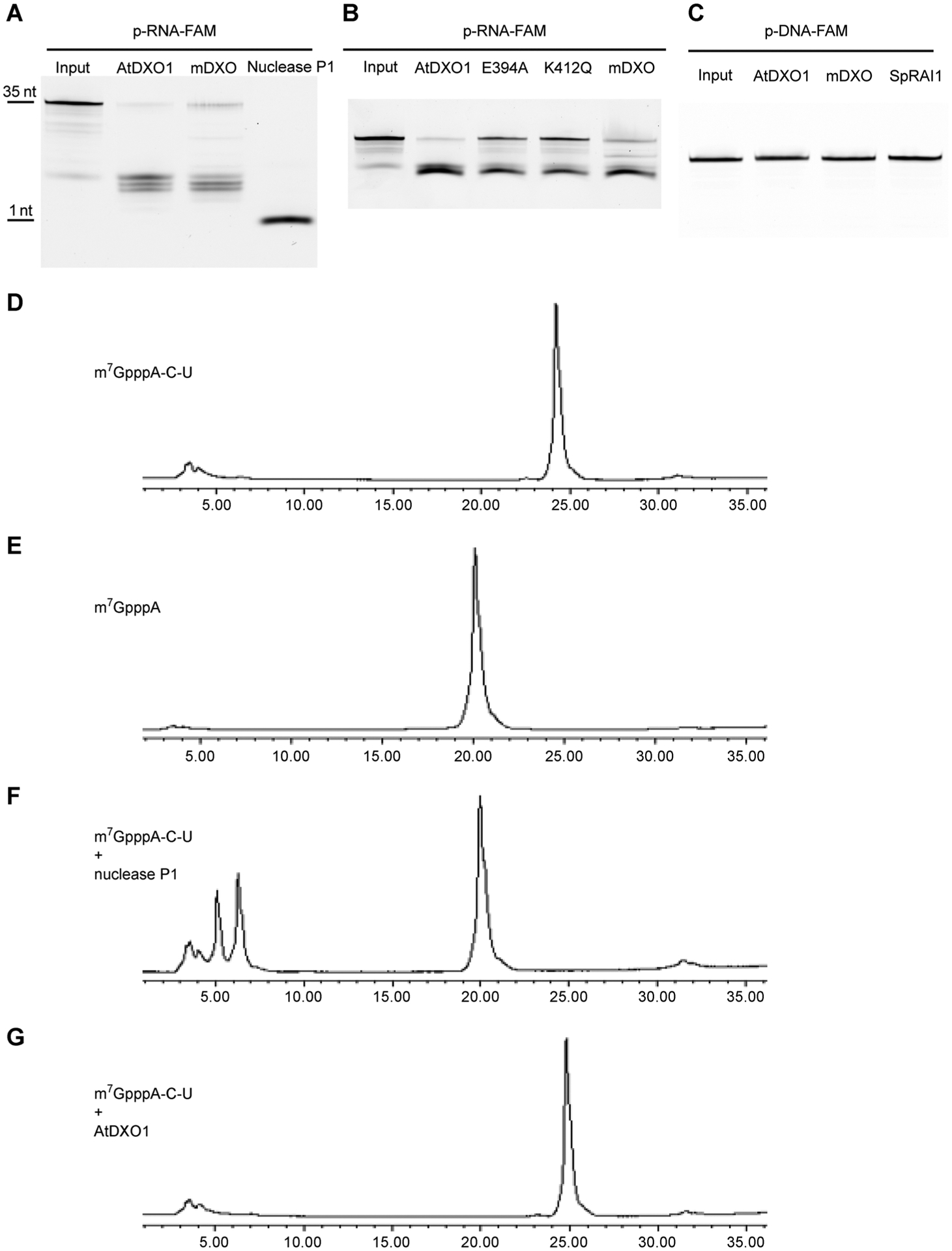
(A) and (B) 35-nt single-stranded 5′-monophosphate RNA with 3′ FAM was treated with 1 μM recombinant DXO1, mammalian (mDXO), or nuclease P1 at 37°C for 1 h (A) and with 1 μM recombinant DXO1, E394A, K412Q or mDXO at 37°C for 30 min (B). (C) Single-stranded, 35-nt monophosphate DNA with 3′ FAM was treated with 1 μM recombinant DXO1, mDXO, or SpRAI1 at 37°C for 1 h. (D) and (E) HPLC peaks of m7GpppA-C-U (D) and m7GpppA (E). (F) and (G) HPLC peaks of m7GpppA-C-U after digestion by 1 μM nuclease P1 (F) and 2 μM AtDXO1 (G) at 37°C for 1 h.
Mammalian DXO and yeast Dxo1 can remove the m7G cap of mRNA (Chang et al. 2012; Jiao et al. 2013). However, DXO1 did not display such an activity in the in vitro enzymatic assay (Figure 2D–G). Recently, mDXO was shown to hydrolyze the phosphodiester linkage between the NAD moiety and the following nucleotide in an NAD-RNA (Jiao et al. 2017). To determine whether DXO1 can also act as an NAD-RNA decapping enzyme, we synthesized a short NAD-capped RNA, NAD-C-U, by in vitro transcription using T7 RNA polymerase. DXO1 or its mutant variants were incubated with the NAD-RNA molecules. The reaction products were analyzed by high performance liquid chromatography (HPLC). DXO1 hydrolyzed NAD-C-U into NAD and p-C-U (Figure 3A–G). DXO1 also had NAD-decapping activity on a longer NAD-RNA template (38-nt) (Figure S4A). This decapping activity was impaired by the E394A mutation and abolished by the K412Q mutation on both the 38-nt NAD-RNA (Figure S4A) and the NAD-C-U substrate (Figure S4B–4F)
Figure 3. DXO1 hydrolyzes the NAD moiety from NAD-RNA in vitro and affects in vivo NAD-RNA stability.
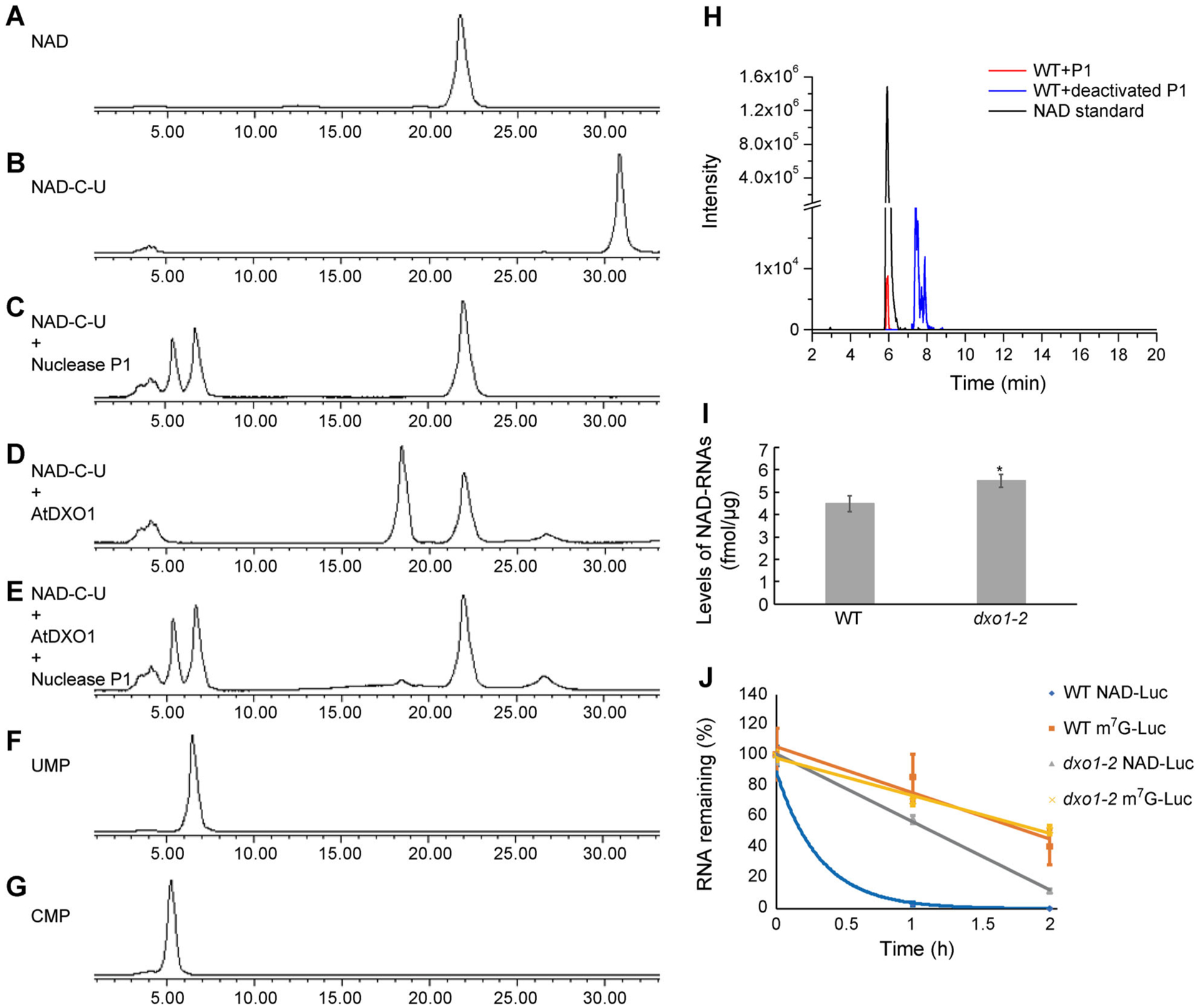
(A) and (B) HPLC peaks of NAD (A) and NAD-C-U (B). (C)–(E) HPLC peaks of NAD-C-U digested with 0.5 μM nuclease P1 at 37°C for 30 min (C), with 0.5 μM DXO1 at 37°C for 30 min (D), or with 0.5 μM DXO1 (30 min) followed by 0.5 μM nuclease P1 (30 min) at 37°C (E). (F) and (G) HPLC peaks of UMP (F) and CMP (G). (H) LC-MS detection of NAD from total RNAs of Arabidopsis after digestion by P1 nuclease. (I) Quantification of cellular NAD-capped RNA levels in WT and dxo1-2. Error bars represent standard deviation of three replicates; a t-test was used for statistical significance analysis; *Indicates statistically significant (P < 0.05). (J) Firefly luciferase mRNAs containing a 5′ NAD cap or an m7G cap and 3′ poly(A)60 were transfected into WT and dxo1-2 protoplasts. Protoplasts were harvested, and the remaining firefly luciferase mRNAs were detected by reverse transcription real-time PCR at the indicated time points. Error bars represent standard deviation of three replicates.
The above results suggested that DXO1 could be an NAD-RNA decapping enzyme in vivo. To determine whether NAD-RNAs are produced in Arabidopsis, total RNA was isolated from WT plants and hydrolyzed with active or heat-inactivated nuclease P1. The digested products were analyzed by liquid chromatography-mass spectrometry (LC-MS). The sample treated with P1 contained a fraction with the same retention time and the same NAD parent ion ([M-H]− m/z = 662.1018) as the NAD standard (Figures 3H, S5). No such product was found in the negative control in which the RNA sample was treated with heat-inactivated P1 (Figures 3H, S5). We then used NAD-capQ (Grudzien-Nogalska et al. 2018) to quantify and compare levels of NAD in total RNAs of the dxo1-2 mutant and WT. The dxo1-2 mutant was found to contain approximately 5.5 fmol NAD-RNA per μg of total RNAs, which was approximately 1.2-fold higher than the WT, which contained approximately 4.4 fmol NAD-RNA per μg of total RNAs (Figure 3I).
To test whether the dxo1 mutation affects the stability of NAD-RNAs in vivo, NAD-capped or m7G-capped luciferase RNAs with a poly(A60) tail were generated through in vitro transcription as previously described (Jiao et al. 2017) and transfected into WT and dxo1-2 protoplasts. Their levels were monitored by qPCR assay each hour after transfection. The results showed that the NAD-capped luciferase RNAs were less stable than the m7G-capped luciferase RNAs in both WT and dxo1-2 protoplasts. The NAD-capped luciferase RNAs were much more stable in the dxo1-2 protoplasts than in the WT protoplasts (Figure 3J), while the degradation rates of m7G-capped luciferase RNAs were not significantly different between the WT and dxo1-2 protoplasts. The in vivo result and our previous in vitro result indicate that DXO1 functions as an NAD-RNA decapping enzyme.
To test the link between the DXO1 enzymatic activity and the morphological phenotypes associated with the dxo1 mutant, the transgenes encoding the K412Q or E394A mutant forms under the control of the DXO1 promoter were introduced into the dxo1-2 mutant. Surprisingly, both the E394A and K412Q forms were able to rescue the phenotypes of dxo1-2 (Figure S6), indicating that restoring the developmental defects of the mutants does not require an enzymatically active DXO1.
The loss-of-function mutation of DXO1 elevates defense gene expression but lowers expression of photosynthesis-related gene
RNA-sequencing (RNA-Seq) was used to compare global gene expression patterns in rosette leaves of 16-d-old WT, dxo1-1 and dxo1-2 grown in soil. The transcriptome profiles in dxo1-1 and dxo1-2 were very similar but showed profound differences from that of WT, as shown in the heat map and hierarchical clustering generated based on quantile normalization of the Fragment Per Kilobase Million (Figure S7A). The transcriptome data are included in Table S1. We further selected differentially expressed genes (DEGs) based on the following criteria: at least a two-fold difference in transcript abundance between the different genotypes with an FDR-adjusted P-value of 0.01 or lower. Compared to WT, 2,959 genes were upregulated and 2,342 genes were downregulated in dxo1-1. In dxo1-2, 2,340 were upregulated and 1,689 genes were downregulated compared to WT. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the DEGs showed that among the upregulated genes in the dxo1 mutants, the enriched KEGG pathways include stress responses such as plant-pathogen interaction, protein processing in endoplasmic reticulum, and glutathione metabolism (Figure 4A). Among the highly upregulated genes were genes known to be induced by pathogen infection, such as PR1, PR2, and PAD4 (Table S1). The downregulated genes in the mutant were mostly in the pathways of photosynthesis, including porphyrin and chlorophyll metabolism, and antenna proteins. The reduced expression of genes involved in porphyrin and chlorophyll metabolism pathways, such as ALB1, HEMA1 and PPOX, could explain the yellowish phenotype of dxo1.
Figure 4. Mutation of dxo1 causes upregulation of defense-related genes and downregulation of photosynthesis-related genes.
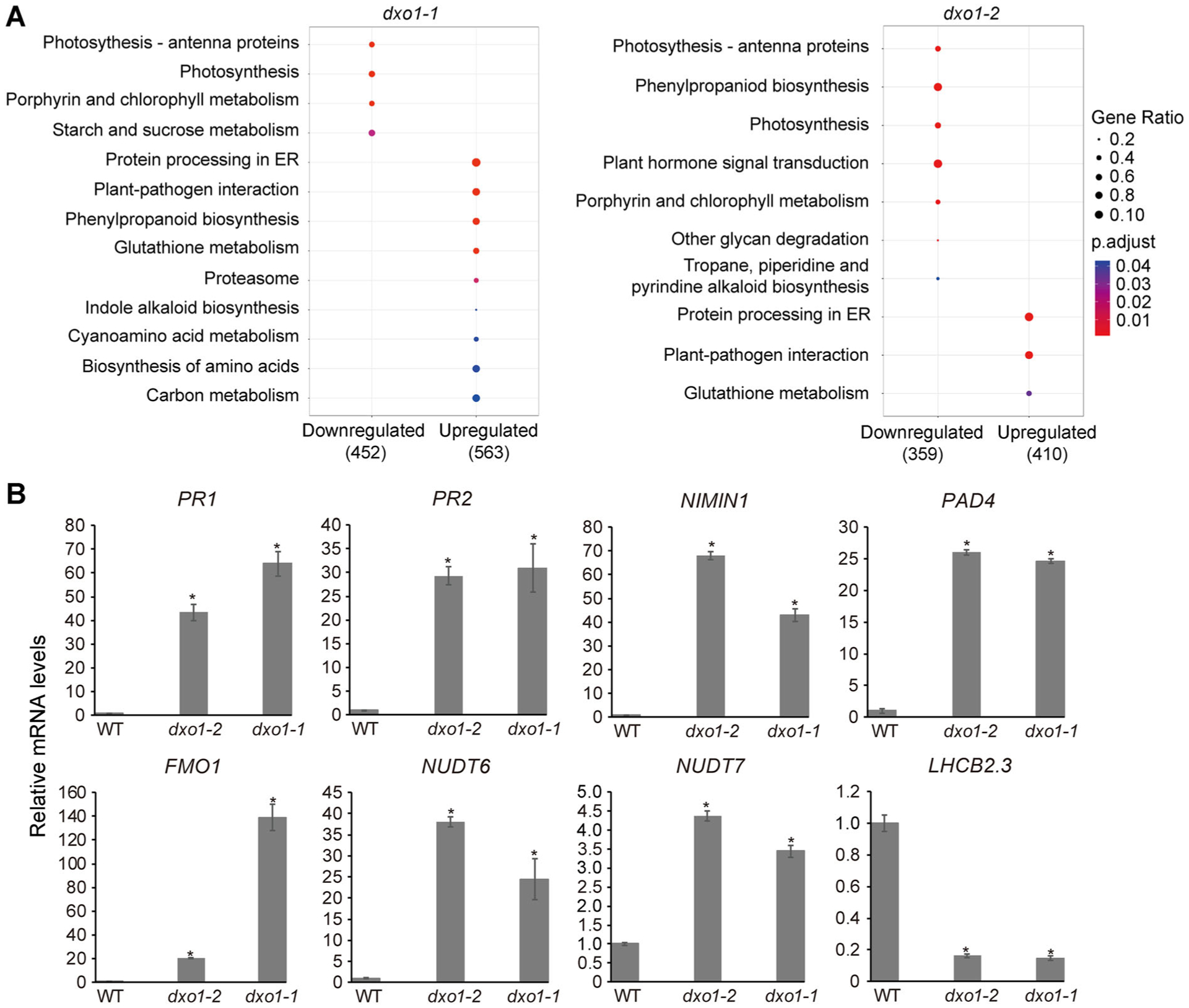
(A) KEGG pathway enrichment analysis of DEGs between the dxo1 mutants and WT. The dot size represents gene ratio (number of enriched genes/number of DEGs), and the color of the dots represents the P-value from high (blue) to low (red). (B) Expression levels of 10 DEGs in WT, dxo1-1 and dxo1-2 determined by qPCR. RNA was extracted from 16-d-old rosette leaves. Error bars represent standard deviation; a t-test was used for statistical significance analysis. *Indicates statistically significant (P < 0.05).
To further verify the RNA-Seq results, 30 DEGs identified from the RNA-seq experiment were selected for qRT-PCR analysis. The qRT-PCR results were highly consistent with those of RNA-seq (Figures 4B, S7B). Notably, the mRNA levels of defense marker genes (such as PR1 and PR2) were increased in both dxo1-1 and dxo1-2 by more than 30-fold compared to WT. These results indicated that the dxo1 mutation causes constitutive activation of the defense pathway.
The constitutive defense response phenotype of the dxo1 mutants was suppressed by elevated temperature or the eds1 or npr1 mutations
To examine whether the dxo1 mutation leads to an elevated level of reactive oxygen species (ROS), the hallmark of the defense response (Torres 2010), ROS accumulation was observed by staining leaves with DAB (3′,3′-diaminobenzidine), a reagent that displays a brownish color when it reacts with H2O2. The first pair of true leaves from 16-d-old dxo1-2 plants showed significantly more intense H2O2 accumulation compared to WT (Figure 5A).
Figure 5. Enhanced disease resistance by the dxo1 mutation was suppressed by high temperature, npr1, or eds1.
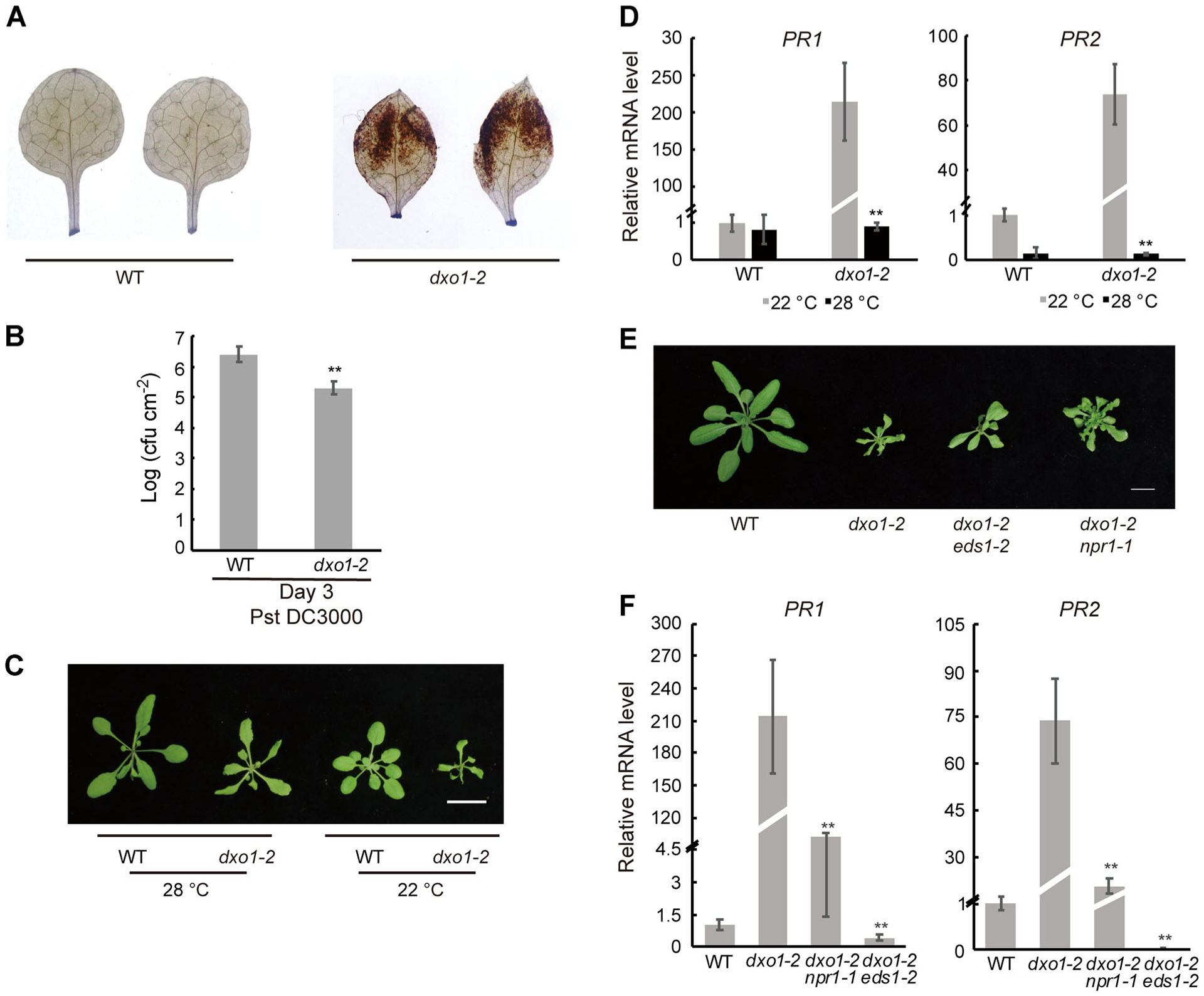
(A) DAB staining of WT and dxo1-2. The first pair of true leaves were detached from 16-d-old seedlings and stained with DAB for H2O2 detection. (B) Bacterial growth in WT and dxo1-2 plants inoculated with Pst DC3000. Bacterial numbers were counted 3 d post-inoculation. Error bars represent standard deviation; t-test was used for statistical significance; ** indicates statistically significant (P < 0.01). (C) 21-day-old WT and dxo1-2 plants grown at 28°C or 22°C. Bar: 1 cm.(D) Expression levels of PR1 and PR2 of 21-d-old plants of WT and dxo1-2 grown at 28°C or 22°C. Error bars represent standard deviation; t-test was used for statistical significance analysis comparing dxo1-2 at the different temperatures; **Indicates statistically significant (P < 0.01). (E) Twenty-eight-day-old WT, dxo1-2, eds1-2 dxo1-2, and npr1-1 dxo1-2 plants grown at 22°C. Bar: 1 cm. (F) Expression levels of PR1 and PR2 in 28-d-old plants of WT, dxo1-2, eds1-2 dxo1-2, and npr1-1 dxo1-2 grown at 22°C. Error bars represent standard deviation; t-test was used for statistical significance analysis in comparison to dxo1-2; **Indicates statistically significant (P < 0.01).
The transcriptome profiling data and elevated ROS accumulation in the mutant indicated that the dxo1 mutant might be more resistant to pathogen infection. To test that, the dxo1-2 mutant and WT were infected with the virulent Pseudomonas syringae pv. tomato (Pst) strain DC3000. The dxo1-2 mutant showed significantly increased resistance to Pst based on quantification of pathogen growth in the leaves 3 d post-inoculation (Figure 5B).
Many constitutive disease resistance mutants in Arabidopsis display growth retardation. In these mutants, a moderately high temperature, such as 28°C, is known to suppress the constitutive defense response and completely or partially restore normal growth (Alcázar and Parker 2011). We grew the dxo1 mutants at 28°C to observe changes in their growth phenotype. At 28°C, the mutants still grew slower than WT, but the difference was less striking than that between the mutant and WT under 22°C (Figures 5C, S2E, 2F). Moreover, the strong constitutive expression of PR1 and PR2 seen in dxo1-2 grown at 22°C was suppressed when grown at 28°C (Figure 5D). While these results indicated that high temperature largely suppressed the autoimmunity phenotype of the dxo1 mutant, the mutant plants remained pale compared to WT.
Many of the defense genes that were constitutively activated in the dxo1 mutants are known to be activated by the regulators of immunity in Arabidopsis, NPR1 and EDS1. Therefore, dxo1-2 was crossed with npr1-1 (Dong 2004) and eds1-2 (Falk et al. 1999). Compared to the single dxo1-2 mutant, the dxo1-2 npr1-1 and dxo1-2 eds1-2 double mutants were only slightly bigger (Figure 5E). However, the elevated expression of PR1 and PR2 seen in dxo1-2 was significantly repressed in both the dxo1-2 npr1-1 and dxo1-2 eds1-2 mutants (Figure 5F).
The dxo1 mutation leads to accumulation of ct-siRNAs (coding transcript-derived siRNAs) which is aggravated by the dcl4 mutation
Mutations in many components of the RNA decay pathway, which remove aberrant transcripts, are known to act as repressors of PTGS (Li et al. 2018). The DXO1-mediated RNA decay pathway might also interact with the PTGS pathway. To test that, dxo1-2 was crossed with loss-of-function mutants of several important PTGS components, including RNA-dependent RNA Polymerase 6 (RDR6), RNA-dependent RNA Polymerase 1 (RDR1), Dicer-like 2 (DCL2), and DCL4, to generate the various double mutants. The mutation of rdr1, rdr6, or dcl2 did not significantly alter the phenotype of dxo1-2 (Figure 6A). However, the loss-of-function mutation of DCL4, which is involved in biogenesis of 21-nt siRNA, aggravated the growth defect phenotype of dxo1-2. The dxo1-2 dcl4-2 double mutants could not survive after the second true leaves had emerged (Figure 6A).
Figure 6. Interactions between dxo1-2 and the PTGS pathway and ct-siRNA profiles of various mutants.
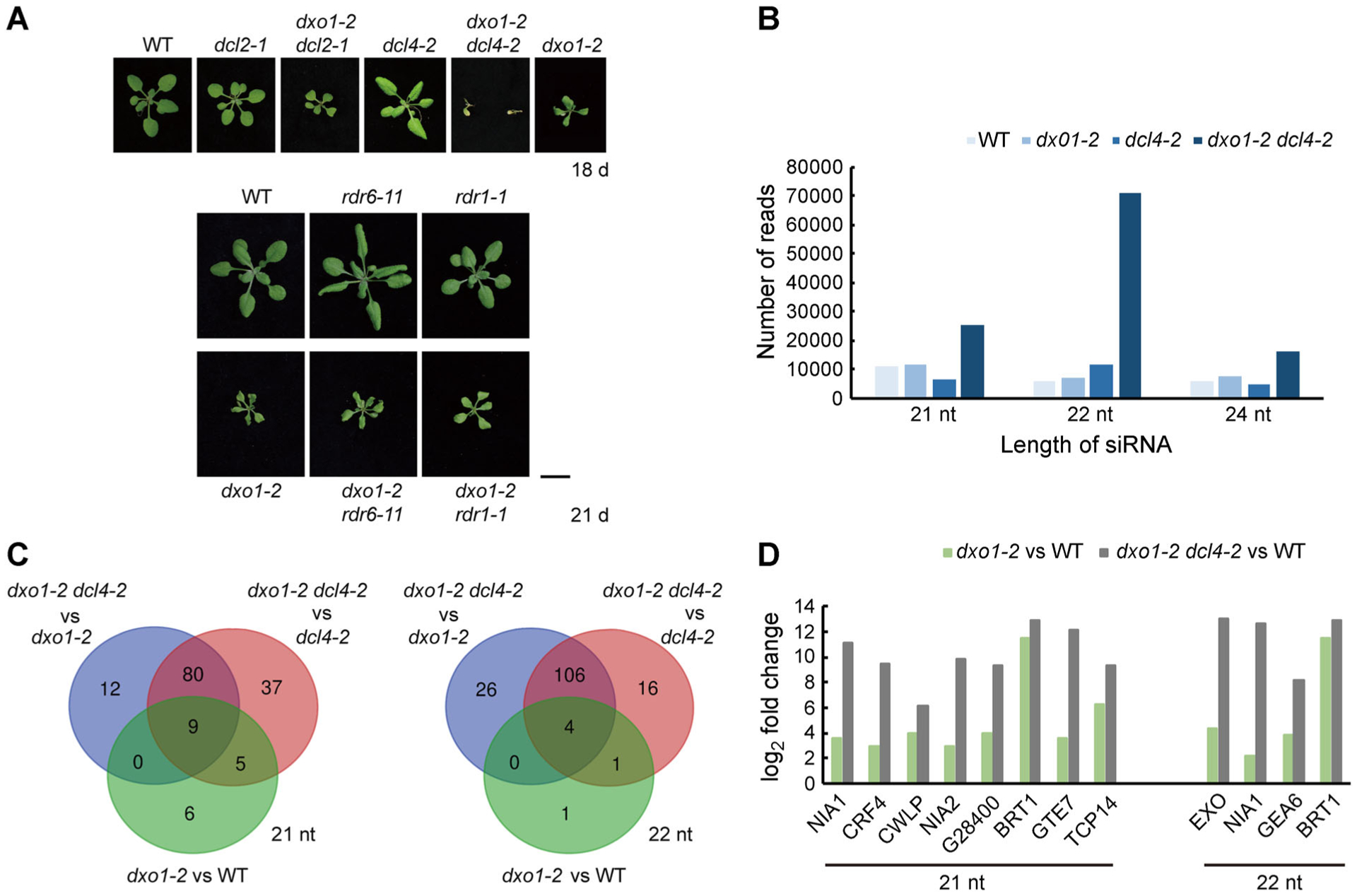
(A) Phenotypes of WT, dxo1-2, and the various double mutants between dxo1-2 and four mutants in the PTGS pathway. Bar: 1 cm. (B) Abundance of different sizes of ct-siRNAs in the different genotypes. Read counts were obtained from sRNA-seq of 10-d-old WT, dxo1-2, and dxo1-2 dcl4-2 plants and were normalized to reads per million. (C) Venn diagram of the number of genes with significant increases in the abundance of 21-nt or 22-nt ct-siRNAs in comparisons between dxo1-2 dcl4-2 and dxo1-2, between dxo1-2 dcl4-2 and dcl4-2, and between dxo1-2 and WT. Genes were selected if the ct-siRNA abundance differed by 2-fold or more with an FDR-adjusted P-value ≤ 0.01. (D) Log2 fold changes of 21- or 22-nt ct-siRNAs of the genes between dxo1-2and WTand dxo1-2 dcl4-2and WT.
sRNA-Seq (small RNA sequencing) was used to profile the sRNA production in 10-d-old WT, dxo1-2, dcl4-2, and dxo1-2 dcl4-2 seedlings grown on 1/2 MS medium. Following the method previously established for analyzing ct-siRNAs (Zhang et al. 2015), it was found that dxo1-2 had slightly higher levels of all 21-, 22- and 24-nt ct-siRNAs (Figure 6B; Table S2) compared to WT. The dxo1-2 dcl4-2 double mutation showed tremendously increased accumulation of these three types of ct-siRNAs compared to dxo1-2 or dcl4-2 (Figure 6B). In the dxo1-2 mutant, 21-nt ct-siRNAs from 20 genes and 22-nt ct-siRNAs from six genes were accumulated to higher levels than in WT. In the dxo1-2 dcl4-2 double mutant, 21-nt ct-siRNAs from 101 genes and 22-nt ct-siRNAs from 136 genes accumulated to higher levels than in dxo1-2 (Figure 6C). We identified nine genes with increased 21-nt ct-siRNAs and four genes with increased 22-nt ct-siRNAs in the dcl4 dxo1-2 double mutant compared to dxo1-2 (Figure 6C, D). The above results indicate that DXO1 might act as an RNA surveillance factor and that loss of its function enhances siRNA accumulation and the PTGS pathway.
DISCUSSION
DXO1 shares some functions with its homologs in fungi and mammals. DXO1 has 5′ to 3′ exoribonuclease activity and NAD-RNA decapping activity. While some DXO/Rai1 proteins hydrolyze methylated or unmethylated guanosine caps and act as an mRNA quality control monitor (Grudzien-Nogalska and Kiledjian 2017), our in vitro enzymatic assay showed that DXO1 can remove the NAD cap from NAD-RNAs but not the m7G cap. Our findings on the enzymatic activities are consistent with recently reported results (Kwasnik et al. 2019). We also found that NAD-capped luciferase RNA, but not m7G-capped RNA, was more stable in the dxo1-2 protoplasts than in the WT protoplasts, further supporting the idea that DXO1 is an NAD-RNA decapping enzyme.
DXO1 is apparently involved in multiple biological pathways. Its loss of function leads to pleiotropic phenotypic defects, including severe growth retardation, pale plants, low sterility, and autoimmunity. Comparison of transcriptomes between WT and the dxo1 mutant showed that genes involved in photosynthesis are generally downregulated. Similar morphological phenotypes associated with the dxo1 mutation were also observed in the previous report (Kwasnik et al. 2019).
We found that the dxo1 mutation also leads to an autoimmunity phenotype, including elevated activation of defense-related genes. The autoimmunity phenotype of dxo1 could be suppressed by npr1 and eds1. Interestingly, the dxo1 npr1 and dxo1 eds1 double mutants were still pale and grew much slower than WT, indicating that the defects of the mutant in growth and chlorophyll accumulation are not due to autoimmunity but occur through the loss of a different function of AtDXO1. These results indicated that DXO1 functions as a suppressor of plant immunity and separately plays an additional role(s) in various developmental processes. DXO1 could be directly involved in degeneration of transcripts from the defense-related genes or could affect expression of defense genes through its modulation of genes involved in defense signaling or another pathway.
E394 and K412 of DXO1 were predicted to be the conserved active site residues. In particular, the K412Q mutation completely abolished its NAD-RNA decapping activity and severely reduced its exoribonuclease activity. However, both the E394A and K412Q forms were able to rescue the morphological phenotypes of the dxo1 mutant, suggesting that DXO1’s function in the growth and developmental processes is independent of its enzymatic activities. Kwasnik et al. (2019) generated enzymatically inactive DXO1 by mutating other residues and also found that the inactive variant could complement the dxo1 mutant morphological phenotypes. Therefore, there is a possibility that DXO1 functions in the NAD-RNA decapping and decay process without acting as an enzyme. It has been shown for some proteins that their enzymatic activities are unnecessary for their biological function. For instance, GUARD CELL HYDROGEN PEROXIDE-RESISTANT1 (GHR1) was found to regulate stomatal closure and function as a kinase (Hua et al. 2012). However, further research found that the kinase activity of GHR1 is not required for stomatal closure and that GHR1 acts as a scaffolding component in stomatal closure (Sierla et al. 2018).
If DXO1 mediates RNA processing, decay, and quality control, cells without functional DXO1 should accumulate more aberrant RNAs that could enhance PTGS. Indeed, the dxo1 mutant accumulated higher levels of ct-siRNAs. The dcl4 mutation is known to exacerbate the phenotypes of mutants in the RNA decay pathway, such as ski2-2 or ein5-1, by generating additional molecules of destructive DCL2-mediated 22-nt ct-siRNA (Zhang et al. 2015). Stacking of the dcl4 mutation aggravated accumulation of 21-, 22-, and 24-nt ct-siRNAs in dxo1-2 dcl4. DCL4 is known to be required for generation of 21-nt siRNAs, so the increased level of 21-nt siRNAs in dxo1-2 dcl4 compared to dxo1-2 was unexpected. One possible explanation is that increased levels of siRNA precursors in the dxo1-2 dcl4-2mutant might lead other Dicer proteins to promiscuously cut some precursors into 21-nt siRNAs.
Regulated decapping of eukaryotic mRNAs plays an essential role in controlling gene expression. Dcp2, a Nudix family protein, was once thought to be the only decapping enzyme; however, several more Nudix family proteins in mammals have been found to also function as m7G-RNA decapping enzymes (Song et al. 2010, 2013; Grudzien-Nogalska and Kiledjian 2017). In addition, a Nudix protein in E. coli, NudC, was recently found to hydrolyze the NAD cap of NAD-RNAs (Cahová et al. 2015; Zhang et al. 2016). The existence of different members of the DXO/Rai1 family proteins in fungi, mammals, and plants and their range of functions as decapping enzymes for the m7G cap, defective 5′ caps, and the non-canonical NAD cap further implies the complexity of gene regulation through RNA capping and decapping processes.
Collectively, we found that the loss-of-function mutation of DXO1 leads to pleiotropic phenotypes, indicating that DXO1 functions in multiple biological processes. DXO1 possesses NAD-RNA decapping and exonuclease activities but might also have other molecular functions. While recent reports of NAD as a noncanonical cap indicate another layer of gene regulation through decapping, the role of DXO1 in NAD-RNA decapping remains elusive. Further elucidation of the physiological significance of the NAD-RNA decapping activity of DXO1 could shed light on biological functions of NAD-RNAs in gene regulation.
MATERIALS AND METHODS
Plant materials, growth conditions, and inoculation of pathogens
Arabidopsis thaliana ecotype Col-0 was used as the WT in this study. The T-DNA insertion lines dxo1-1 (salk_103157) and dxo1-2 (salk_032903) were obtained from the Nottingham Arabidopsis Stock Centre (NASC) (http://arabidopsis.info/). dcl2-1 (salk_064627), dcl4-2 (GABI_160G05), rdr6-11, and rdr1-1 (SAIL_672F11) were provided by the laboratory of Hongwei Guo (Zhang et al. 2015). The double mutants between dxo1-2 and the other mutants were generated by hybridizing the corresponding mutants and then identifying homozygous mutants by PCR analysis of genomic DNA from the F2 plants. The primers used in the genotyping analysis are listed in Table S3.
Arabidopsis plants grown in soil were placed in a walk-in growth room at 22°C or 28°C (±2°C) with 50% humidity and a light intensity of 125 mol m−2 s−1 provided by cold, white fluorescent lamps under a 16-h light/8-h dark photoperiod. To grow seedlings on solid media, surface-sterilized seeds were placed on half-strength Murashige and Skoog (MS) Basal Salts (M5524; Sigma-Aldrich, St. Louis, MO, USA) containing 1% (w/v) sucrose (15503-022; Invitrogen, Carlsbad, CA, USA) and either 0.6% agar (A1296; Sigma-Aldrich), for horizontally placed plates, or 0.9% agar for vertically placed plates, with a pH of 5.8 adjusted by 1 M KOH. Modified media were prepared in the same way except with addition or elimination of some components. The plates with seeds were stratified in the dark at 4°C for 3 d and then placed in a growth chamber (Percival Scientific, Perry, IA, USA) at 22°C with 50% humidity and a light intensity of 125 mol m−2 s−1 under a 16-h light/8-h dark photoperiod.
For infection of plants with Pseudomonas syringae DC3000, 4-week-old plants grown in soil were sprayed with 5 × 105 colony-forming units (c.f.u.) mL−1 bacterial suspensions with 10 mM MgCl2 and bacterial growth was calculated 3 d after inoculation.
Generation of transgenic plants
The transgenic plants generated in this study included DXO1 pro::AtDXO1, DXO1 pro::E394A, DXO1 pro::K412Q, 35S::DXO1-FLAG and Cas9-DXO1. The DXO1 open reading frame was amplified from WT cDNA obtained from reverse transcription PCR using the primer pair DXO1-F and DXO1-R. The E394A and K412Q were generated using site-directed mutagenesis PCR from the DXO1 open reading frame. The cDNA fragments were cloned into 2,004 bp of the DXO1 promoter in a modified pCambia1305 to generate pCambia1305-DXO1 pro::AtDXO1, pCambia1305-DXO1 pro::E394A and pCambia1305-DXO1 pro::K412Q plasmids. DXO1 pro::AtDXO1, DXO1 pro::E394A and DXO1 pro::K412Q transgenic lines expressing DXO1 variants in dxo1-2 were generated by transforming dxo1-2 heterozygous plants with Agrobacterium tumefaciens strain GV3101 carrying the corresponding plasmids using the floral-dip method (Clough and Bent 1998). Transgenic plants were selected based on the resistance of the antibiotic resistance gene in the construct. The 35S::DXO1-FLAG construct was made by inserting full-length DXO1 cloned from WT genomic DNA in pCambia1305. The CRISPR-Cas9 knockout mutant lines were generated by transforming Col-0 with the modified pCambia1300 plasmid with the psgR-Cas9-At backbone (Mao et al. 2013) containing a pAtU9_sgRNA to target coding regions CTTGCTACAGTCGAGTAGA of AtDXO1 in exon 4 (refer to primer list). T1 plants were analyzed by PCR amplification of the genomic region using the primer pair M13F and dxo1-gRNA-R. The PCR products were sequenced to identify mutations. Presence or absence of the T-DNA in the F2 progeny was determined by PCR analysis of genomic DNA using the primer pair M13F and dxo1-gRNA-R. The homozygous state was confirmed by PCR amplification of genomic DNA using the dxo1-Cas9 seq-F and dxo1-Cas9 seq-R and sequencing of the PCR products. The homozygous knockout plants without T-DNA were used for the experiments.
Recombinant protein expression and purification
E394A and K412Q were cloned as mentioned. The mDXO and SpRAI1 DNA sequences were chemically synthesized (Thermo Fisher Scientific, Waltham, MA, USA). AtDXO1, E394A, K412Q, mDXO, and SpRAI1 were inserted into the expression vector pET28a, and the recombinant proteins with 6x His-tag were expressed by E. coli BL21 Rosetta cells with induction by IPTG. The transfected cells were grown at 20°C for 14–16 h and purified by Ni-NTA (Thermo Fisher Scientific) and gel filtration using an Amicon Ultra-0.5 Centrifugal Filter Unit (Millipore, Darmstadt, Germany). The proteins were stored in a buffer containing 20 mM Tris (pH 7.5), 100 mM NaCl and 5% (v/v) glycerol.
In vitro transcription of m7G- or NAD-capped RNAs
The short m7GpppA-RNA and NAD-RNA molecules were synthesized in vitro by T7 polymerase following the published method (Zhang et al. 2016). The double-stranded DNA templates were modified as follows. Our DNA template was formed by annealing the two single-stranded DNA oligonucleotides (5′-GATCACTA ATACGACTCACTATTACTGAAGCGGGC-3′ and 5′ GCCCGC TTCAGTAATAGTGAGTCGTATTAGTGATC-3′, the transcription start site is underlined). The transcription reactions were performed in 1 mL buffer containing 1 μM DNA template, 4 mM NAD or 4 mM m7GpppA, 4 mM rCTP, 4 mM rUTP, 50 mM Tris-HCl, pH 8.0, 20 mM MgCl2, 2 mM spermidine, 0.01% Triton X-100, 40 mM DTT and 0.1 mg/mL T7 RNA polymerase at 37°C for 4 h. The short NAD-RNA was isolated using a 1-mL Mono Q column (GE Healthcare, Chicago, Illinois, USA). Nuclease P1 was used to verify the short length of the NAD-RNA.
The DNA template for the 38-nt NAD-RNA was made by annealing the following two single-stranded DNA oligonucleotides (5′-CAGTAATACGACTCACTAT TAGGCCTCTCGCTCTGCTGGGTGTGCGCTTGCTTGGCTT-3′ and 5′-AAGCCAAGCAAGCGCACACCCAGCAGAGCGAGA GGCCTAATAGTGAGTCGTATTACTG-3′, the transcription start site is underlined). The synthetic NAD-RNA was isolated using a 15% denaturing Polyacrylamide Gel (Urea-PAGE) containing 8 M urea. NAD-RNA was extracted from the gel following the previously described method (Winz et al. 2017).
NAD- or m7G-capped luciferase mRNA with a poly (A60) tail was generated following the protocol described previously (Jiao et al. 2017), with minor modifications. In brief, the pMD19-Luciferase plasmid was generated by adding the 5′ leader ϕ2.5A-CA2, which contained the ϕ2.5 A T7 promoter (Coleman et al. 2004) lacking adenosines, except the first transcribed nucleotide, to the firefly luciferase open reading frame. ϕ2.5A-CA2-Luciferase-A60 was amplified by PCR using the 3′ primer containing 60 thymidines at its 5′ end and was used as DNA template for in vitro transcription. The transcription reactions were first incubated at 37°C for 15 min by mixing the DNA template, NAD or m7GpppA cap analog, T7 RNA polymerase (Promega) and rCTP, rUTP and rGTP (without rATP) and then incubated with 200 μg/mL heparin to inhibit transcription reinitiation and rATP to the mixture for another 30 min incubation. MicroSpin G-50 Columns (GE Healthcare) used to isolate thein vitro transcribed NAD or m7G-capped RNAs.
In vitro enzyme activity assay
The exoribonuclease activity assays were carried out using a fluorescein (6-FAM) 3′-end-labeled, 35-nt single-stranded RNA with monophosphate at its 5′ end (synthesized by Sangon) as the substrate. The substrate was incubated with recombinant AtDXO1, E394A, K412Q, mDXO or nuclease P1 in a 20 μL reaction containing 100 mM KCl, 2 mM MgCl2, 1 mM MnCl2, 2 mM DTT, and 10 mM Tris-HCl (pH 7.5) at 37°C for 20 min. The reactions were stopped with 30 mM EDTA. The products were loaded into a 15% denaturing Urea Polyacrylamide Gel (Urea-PAGE). The gel was electrophoresed in 1× TBE running buffer for 30 min. The reaction products were visualized using Bio-Rad ChemiDoc Touch Imaging System. The single-stranded DNA with 6-FAM at its 3′ end and the 38-nt NAD-RNA were treated and detected as described above. Shorter NAD-RNA or m7GpppA-RNA was treated under the same condition but detected by HPLC. A 10 μL reaction sample was injected into a HPLC 2695 Fraction Collector III (Waters, Milford, MA, USA) equipped with a multiwavelength detector using a COSMOSIL C18-MS-II column (250 mm, 4.6 mm) and then eluted with a gradient of 20% methanol in 20 mM ammonium acetate at a flow rate of 0.4 mL min−1 for 40 min. The elution profiles were recorded at 260 nm.
Detection, quantification, and stability assays of NAD-capped RNAs
Total RNA samples were extracted from rosette leaves and filtered using Amicon Ultra-0.5 Centrifugal Filter Unit (UFC500324; Millipore) to exclude small molecules less than 3 kD. Total RNA (300 μg) was treated with 10 units Nuclease P1 or heat-inactivated P1 in 50 mM NH4OAc, pH 4.5 at 37°C for 20 min. LC-MS analysis was performed using a Q Exactive Orbitrap mass spectrometer system (Thermo Fisher Scientific Inc.) with an electrospray ionization (ESI) source and a Thermo Dionex Ultimate 3000 UPHLC system (Thermo Fisher Scientific Inc.). Data acquisition and processing were performed using Xcalibur software. The LC separation was performed on an Aquity UPLC BEH C18 column (1.7 μm, 2.1 mm × 100 mm; Waters) with a constant flow rate of 0.300 mL/min at 30°C. Aqueous ammonium formate (0.1%; solvent A) and methanol (solvent B) were used as mobile phases. After a hold for 3 min at 0% B, a program consisting of a gradient from 0% B to 100% B over 17 min, 2 min 100% B, a gradient over 1 min from 100% B to 0% B and a final 7 min hold at 0% B was used. For each sample, 10 μL was injected for analysis.
The mass spectrometry detection was performed under negative ESI mode with a capillary voltage of 2.5 kV. The temperature of the capillary was 320°C. The auxiliary gas was set at 350°C with a flow rate of 10 arbitrary units. The sheath gas flow rate was set at 30 psi. Full MS scan with a range of m/z 150–2,000 and a resolution of 70,000 was used to observe the NAD parent ion ([M-H]− m/z = 662.1018).
NAD-capQ was performed as described previously (Grudzien-Nogalska et al. 2018) with minor modification. Purified total RNA samples were filtered through an Amicon Ultra-0.5 Centrifugal Filter Unit (UFC500324; Millipore) to exclude small molecules less than 3 kD. Total RNA (500 μg) was used in the assay.
The NAD-capped luciferase mRNA stability assay was performed as described previously, with minor modification (Jiao et al. 2017). The protoplasts were isolated from plants grown at 28°C and transferred to 22°C for 1 d before collection. The protoplast isolation and PEG-mediated transfection were performed as previously described (Lee et al. 2009; Yoo et al. 2007; Woo et al. 2015). 5 × 105 protoplasts were used for each transfection. Untransfected RNAs were degraded by Micrococcal Nuclease (New England Biolabs, Ipswich, MA, USA) in the presence of W5 medium. The transfected protoplasts were subsequently incubated in the growth chamber at 22°C and harvested at 0, 1 and 2 h. Total RNA from the cells collected at selected time points were extracted and reverse transcribed into cDNA and detected by real-time PCR.
Subcellular localization
For subcellular protein localization, 35S::DXO1-GFP was transfected into the WT protoplast. The transfected protoplasts were cultured overnight at room temperature and observed by confocal microscopy (Leica SP5II Confocal System). For GFP signal detection, a 488-nm excitation wavelength laser was used with a 505/530-nm filter.
RNA extraction, reverse transcription and real-time PCR
Total RNA for RNA-seq and real-time PCR were extracted using the Agilent Plant RNA Isolation Mini Kit (5188-2780; Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instruction. Total RNAs for NAD-capQ were extracted from rosette leaves with TRIzol Reagent (15596038; Invitrogen) following the manufacturer’s instruction. Small RNAs (<200 nt) for small RNA-seq were isolated from 10-d-old seedlings using miRNeasy Mini Kit (217004; QIAGEN, Waltham, MA, USA) according to the manufacturer’s instruction.
cDNA used for real-time PCR analysis was synthesized using PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (RR074A, Takara, Dalian, China). The reaction mixture for real-time PCR was prepared following the protocol of the SYBR® Green PCR Master Mix (4364344; Thermo Fisher Scientific). The synthesized cDNA template together with the specific primers were added in a reaction volume of 25 μL containing SYBR® Green PCR Master Mix. The transcript levels were analyzed by at least three biological replicates using StepOnePlus™ Real-Time PCR System (437660; Thermo Fisher Scientific.)
RNA-Seq and sRNA-Seq analysis
The mRNA libraries were prepared by Novogene Incorporation (Beijing, China) using reagents for Illumina and sequenced on an Illumina HiSeq sequencer. TopHat 2.1.0 software (Trapnell et al. 2009) was used to map the reads to the Arabidopsis genome (TAIR10). RSEM 2.1.30 (Anders et al. 2015) was used for statistical analysis of the read counts for each gene. DEGs were generated by DEseq. 2 (Love et al. 2014), based on a sample fold change >2 and the false discovery rate (FDR) adjusted P-value < 0.01. KEGG pathway enrichment analyses were done via the DAVID database (Dennis et al. 2003).
The sRNA libraries were prepared by Novogene Incorporation using reagents for Illumina and sequenced on an Illumina HiSeq sequencer. The sequencing data were analyzed as described previously (Zhang et al. 2015).
3,3′-Diaminobenzidine (DAB) staining assay
The first pair of true leaves from 16-d-old seedlings were collected and immersed in a 1 mg/mL DAB solution. The tissue samples in the DAB solution were vacuum infiltrated for 2 h at −0.06 MPa and then incubated overnight. After the incubation, DAB solution was discarded and replaced with a bleaching solution (ethanol, acetic acid and glycerol in a 3:1:1 ratio) until the leaves were bleached. The tissues were then observed under an Olympus SZX18 microscope.
Chlorophyll extraction and measurement
Chlorophyll was extracted from 16-d-old seedlings with 80% acetone in 2.5 mM HEPES-KOH and measured spectrophotometrically. The chlorophyll content was quantified and determined as described previously (Wellburn 1994).
Supplementary Material
Figure S1. Multiple protein sequence alignment
Figure S2. Recombinant protein purification and subcellular localization of DXO1
Figure S3. Morphological phenotypes of the dxo1 mutants and the transcript levels of DXO1 in the atdxo1 mutants and the AtDXO1-FLAG overexpression plants
Figure S4. NAD-RNA decapping and degradation by DXO1 and its mutated variants
Figure S5. Identification of NAD from RNAs using LC-MS
Figure S6. The phenotype of dxo1 mutant plants complemented with the DXO1 mutant proteins
Figure S7. The transcriptome profiles of the dxo1 mutants and real-time PCR verification of some of the transcriptome data
Table S1. Transcriptome analysis of RNA-Seq
Table S2. ct-siRNA analysis of sRNA-Seq
Table S3. Primers used in this study
ACKNOWLEDGEMENTS
This work was supported by Research Grants Council of Hong Kong (GRF grant nos. 12100018 and AoE/M-403/16 to Y.X.) and by Hong Kong Baptist University (grant nos. RC-ICRS/16-17/04, SDF15-10120-P04 to Y.X.).
Footnotes
ACCESSION NUMBERS
The RNA-seq and sRNA-seq data have been submitted the National Center for Biotechnology Information Gene Expression Omnibus repository, https://www.ncbi.nlm.nih.gov/geo and their accession number is GSE132553, respectively.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article: http://onlinelibrary.wiley.com/doi/10.1002/jipb.12867/suppinfo
REFERENCES
- Alcázar R, Parker JE (2011) The impact of temperature on balancing immune responsiveness and growth in Arabidopsis. Trends Plant Sci 16: 666–675 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahová H, Winz ML, Höfer K, Nübel G, Jäschke A (2015) NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 519: 374–377 [DOI] [PubMed] [Google Scholar]
- Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, Tong L (2012) Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′-3′ exoribonuclease activity. Nat Struct Mol Biol 19: 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Kowtoniuk WE, Agarwal I, Shen Y, Liu DR (2009) LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat Chem Biol 5: 879–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Green PJ (2009) mRNA degradation machinery in plants. J Plant Biol 52: 114–124 [Google Scholar]
- Clough SJ, Bent AF (1998) Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Coleman TM, Wang G, Huang F (2004) Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 2.5 promoter. Nucleic Acids Res 32: e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane H, Lempicki RA (2003) DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol 4: R60. [PubMed] [Google Scholar]
- Dong X (2004) NPR1, all things considered. Curr Opin Plant Biol 7: 547–552 [DOI] [PubMed] [Google Scholar]
- Falk A, Feys BJ, Frost LN, Jones JDG, Daniels MJ, Parker JE (1999) EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proc Natl Acad Sci USA 96: 3292–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Bird JG, Nickels BE, Kiledjian M (2018) NAD-capQ” detection and quantitation of NAD caps. RNA 24: 1418–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Kiledjian M (2017) New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip Rev RNA 8: doi: 10.1002/wrna.1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua D, Wang C, He J, Liao H, Duan Y, Zhu Z, Guo Y, Chen Z, Gong Z (2012) A plasma membrane receptor kinase, GHR1, mediates abscisic acid- and hydrogen peroxide-regulated stomatal movement in Arabidopsis. Plant Cell 24: 2546–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M (2013) A mammalian pre-mRNA 5′-end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell 50: 104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, Kiledjian M (2017) 5′ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. Cell 168: 1015–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Xiang S, Oh C, Martin CE, Tong L, Kiledjian M (2010) Identification of a quality-control mechanism for mRNA 5′-end capping. Nature 467: 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado AR, Tan D, Jiao X, Kiledjian M, Tong L (2014) Structure and function of pre-mRNA 5′-end capping quality control and 3′-end processing. Biochemistry 53: 1882–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowtoniuk WE, Shen Y, Heemstra JM, Agarwal I, Liu DR (2009) A chemical screen for biological small molecule-RNA conjugates reveals CoA-linked RNA. Proc Natl Acad Sci USA 106: 7768–7773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnik A, Wang VYF, Krzyszton M, Gozdek A, Zakrzewska-Placzek M, Stepniak K, Poznanski J, Tong L, Kufel J (2019) Arabidopsis DXO1 links RNA turnover and chloroplast function independently of its enzymatic activity. Nucleic Acids Res 47: 4751–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, Chan MT, Wu FH, Lin CS, Shen SC, Lee SH (2009) Tape-Arabidopsis Sandwich: A simpler Arabidopsis protoplast isolation method. Plant Methods 5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wu H, Guo H (2018) Plant mRNA decay: Extended roles and potential determinants. Curr Opin Plant Biol 45: 178–184 [DOI] [PubMed] [Google Scholar]
- Liu L, Chen X (2016) RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. Mol Plant 9: 826–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Schwer B, Shuman S (1995) Yeast mRNA cap methyltransferase is a 50-kilodalton protein encoded by an essential gene. Mol Cell Biol 15: 4167–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Zhang H, Xu N, Zhang B, Gou F, Zhu JK (2013) Application of the CRISPR-Cas system for efficient genome engineering in plants. Mol Plant 6: 2008–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafò M, Eastwood EL, Fabry MH, Hannon GJ, Kneuss E, Czech B, Ciabrelli F (2018) piRNA-guided genome defense: From biogenesis to silencing. Annu Rev Genet 52: 131–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierla M, Hõrak H, Overmyer K, Waszczak C, Yarmolinsky D, Maierhofer T, Vainonen JP, Denessiouk K, Salojärvi J, Laanemets K, Tõldsepp K, Vahisalu T, Gauthier A, Puukko T, Paulin L, Auvinen P, Geiger D, Hedrich R, Kollist H, Kangasjärvi J (2018) The receptor-like pseudo-kinase GHR1 is required for stomatal closure. Plant Cell 30: 2813–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Bail S, Kiledjian M (2013) Multiple Nudix family proteins possess mRNA decapping activity. RNA 19: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Li Y, Kiledjian M (2010) Multiple mRNA decapping enzymes in mammalian cells. Mol Cell 40: 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MA (2010) ROS in biotic interactions. Physiol Plant 138: 414–429 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman JB, Giltmier AJ, Mukherjee C, Schoenberg DR (2017) RNA guanine-7 methyltransferase catalyzes the methylation of cytoplasmically recapped RNAs. Nucleic Acids Res 45: 10726–10739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Hohn T (2013) Biogenesis and biological activity of secondary siRNAs in plants. Scientifica 2013: 783253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li S, Zhao Y, You C, Le B, Gong Z, Mo B, Xia Y, Chen X (2019) NAD+-capped RNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc Natl Acad Sci USA 116: 12094–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiao X, Carr-Schmid A, Kiledjian M (2002) The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci USA 99: 12663–12668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellburn AR (1994) The spectral determination of chlorophylls a and b, as well as total carotenoids, using various solvents with spectrophotometers of different resolution. J Plant Physiol 144: 307–313 [Google Scholar]
- Winz ML, Cahová H, Nübel G, Frindert J, Höfer K, Jäschke A (2017) Capture and sequencing of NAD-capped RNA sequences with NAD captureSeq. Nat Protoc 12: 122–149 [DOI] [PubMed] [Google Scholar]
- Woo JW, Kim J, Kwon SI, Corvalán C, Cho SW, Kim H, Kim SG, Kim ST, Choe S, Kim JS (2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33: 1162–1164 [DOI] [PubMed] [Google Scholar]
- Xiang S, Cooper-Morgan A, Jiao X, Kiledjian M, Manley JL, Tong L (2009) Structure and function of the 5′→3′ exoribonuclease Rat1 and its activating partner Rai1. Nature 458: 784–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yang JY, Niu QW, Chua NH (2006) Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell 18: 3386–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nat Protoc 2: 1565–1572 [DOI] [PubMed] [Google Scholar]
- Zhai L, Xiang S (2014) mRNA quality control at the 5′ end. J Zhejiang Univ Sci B 15: 438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhong H, Zhang S, Shao X, Ni M, Cai Z, Chen X, Xia Y (2019) NAD tagSeq reveals that NAD+-capped RNAs are mostly produced from a large number of protein-coding genes in Arabidopsis. Proc Natl Acad Sci USA 116: 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Liu Y, Wang Q, Guan Z, Wang J, Liu J, Zou T, Yin P (2016) Structural basis of prokaryotic NAD-RNA decapping by NudC. Cell Res 26: 1062–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhu Y, Liu X, Hong X, Xu Y, Zhu P, Shen Y, Wu H, Ji Y, Wen X, Zhang C, Zhao Q, Wang Y, Lu J, Guo H (2015) Suppression of endogenous gene silencing by bidirectional cytoplasmic RNA decay in Arabidopsis. Science 348: 120–123 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang J, Schultz N, Zhang F, Parhad SS, Tu S, Vreven T, Zamore PD, Weng Z, Theurkauf WE (2014) The HP1 homolog Rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157: 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Multiple protein sequence alignment
Figure S2. Recombinant protein purification and subcellular localization of DXO1
Figure S3. Morphological phenotypes of the dxo1 mutants and the transcript levels of DXO1 in the atdxo1 mutants and the AtDXO1-FLAG overexpression plants
Figure S4. NAD-RNA decapping and degradation by DXO1 and its mutated variants
Figure S5. Identification of NAD from RNAs using LC-MS
Figure S6. The phenotype of dxo1 mutant plants complemented with the DXO1 mutant proteins
Figure S7. The transcriptome profiles of the dxo1 mutants and real-time PCR verification of some of the transcriptome data
Table S1. Transcriptome analysis of RNA-Seq
Table S2. ct-siRNA analysis of sRNA-Seq
Table S3. Primers used in this study