

Abstract
KDM5B (lysine[K]-specific demethylase 5B) is frequently upregulated in various human cancers including prostate cancer (PCa). KDM5B controls H3K4me3/2 levels and regulates gene transcription and cell differentiation, yet the contributions of KDM5B to PCa tumorigenesis remains unknown. In this study, we investigated the functional role of KDM5B in epigenetic dysregulation and PCa progression in cultured cells and in mouse models of prostate epithelium-specific mutant Pten/Kdm5b. Kdm5b deficiency resulted in a significant delay in the onset of PCa in Pten-null mice, while Kdm5b loss alone caused no morphological abnormalities in mouse prostates. At 6 months of age, the prostate weight of Pten/Kdm5b mice was reduced by up to 70% compared to that of Pten mice. Pathological analysis revealed Pten/Kdm5b mice displayed mild morphological changes with hyperplasia in prostates, whereas age-matched Pten littermates developed high grade-prostatic intraepithelial neoplasia (HG-PIN) and PCa. Mechanistically, KDM5B governed phosphatidylinositol 3-kinase (PI3K)/AKT signaling in PCa in vitro and in vivo. KDM5B directly bound the PIK3CA promoter and KDM5B knockout resulted in a significant reduction of P110α and PIP3 levels and subsequent decrease in proliferation of human PCa cells. Conversely, KDM5B overexpression resulted in increased PI3K/AKT signaling. Loss of Kdm5b abrogated the hyperactivation of AKT signaling by decreasing P110α/P85 levels in Pten/Kdm5b mice. Taken together, our findings reveal that KDM5B acts as a key regulator of PI3K/AKT signaling; they also support the concept that targeting KDM5B is a novel and effective therapeutic strategy against PCa.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related deaths in American men (1). The initiation and progression of PCa are driven by dysregulation of multiple oncogenic pathways secondary to genetic and epigenetic alterations of oncogenes and tumor suppressors. PTEN (Phosphatase and tensin homolog) is a tumor suppressor gene that is frequently deleted and/or mutated in various human cancers including PCa (2–6). Complete loss of Pten in the mouse results in invasive adenocarcinoma (7,8). PTEN acts as a phosphatase to dephosphorylate phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) to phosphatidylinositol 4, 5-bisphosphate (PIP2). The ratio of PIP3/PIP2 in cells is tightly balanced by the enzymatic activities of PTEN phosphatase and phosphoinositide 3-kinases (PI3K). Loss of PTEN leads to the accumulation of PIP3 at the cell membrane and AKT activation, resulting in dysregulation of many cancer-related signaling pathways (9–12). Conversely, loss of PI3K such as P110α and P110β directly impaired AKT signaling to decrease cell proliferation and tumor growth (13,14). The essential role of the PI3K/AKT signaling pathways in PCa progression has long been recognized, but the targeting efficacy in PCa patients is still a challenge.
Dysregulation of the trimethylation of lysine 4 on histone H3 (H3K4me3) gene is frequently observed in advanced PCa and castration resistant PCa (15–17). Histone methylation is a reversible process that is tightly controlled by histone methyltransferases and demethylases to regulate biological functions in cells. Lysine-specific demethylase 5B (KDM5B, also called JARID1B or PLU1) is a JmjC domain-containing enzyme that removes methyl groups from tri- or di-methylated H3K4 (H3K4me3/2) to produce monomethylated H3K4 (H3K4me1) (18). KDM5B is involved in chromatin remodeling to regulate a variety of biological functions including gene expression, genome stability, cell cycle, senescence, and metastasis (19–23). We and others showed that some Kdm5b germline knockout (KO) mice are viable but experience delayed development (24,25). Increased expression of KDM5B leads to drug resistance in some cancers such as melanoma (26,27). KDM5B upregulation is frequently found in advanced PCa, implicating its potential oncogenic role in PCa (28,29). However, it is still poorly understood how mechanistically KDM5B contributes to PCa progression.
We recently reported that KDM5B is noticeably increased in prostate tumors of Pten/Trp53 mutant mice (21). In this study, we demonstrated that KDM5B loss blocked PCa progression in Pten-null mice. More importantly, we discovered that KDM5B deficiency abolished the hyper-activation of PI3K/AKT signaling pathway in cultured cells and in mouse prostates. Mechanistically, KDM5B activates PI3K by increasing transcription and protein stability of P110α subunit in PCa cells. Thus, our findings provide novel insights into the underlying molecular mechanism by which KDM5B contributes to PCa progression, and offer a rationale to design small molecular inhibitors targeting the KDM5B and PI3K/AKT pathways.
Materials and Methods
Mutant mice and tumor analysis
PtenLoxP/LoxP, Kdm5bLoxP/LoxP, and PB-Cre4 mutant mice were generated and maintained as previously described (30,31). Mouse embryonic stem cells (EPD0027_1_F07, EPD0027_1_F09, EPD0027_1_H07, and EPD0027_1_G08) targeting Kdm5b in C57BL/6N background were obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM). In these embryonic stem cells, exon 6 of Kdm5b was flanked by a FRT-lacZ-neo-FRT-loxP cassette and a loxP site. After injection into C57BL/6 blastocysts, G08 line was selected to cross with a flippase expressing strain to obtain Kdm5bLoxP mice. PtenLoxP/+; PB-Cre4 male mice were crossed with Kdm5bLoxP/LoxP female mice to obtain the prostate-specific Pten/Kdm5b double mutants (PtenLoxP/LoxP; Kdm5bLoxP/LoxP; PB-Cre4, referred to as Ptenpc–/–; Kdm5bpc–/–). All experimental animals were maintained in a mixed genetic background of C57BL/6J x 129/Sv x FVB. Animal experiments were performed in accordance with IACUC-approved protocols at Meharry Medical College and Yale University, respectively.
All genotypes of mice were verified by performing polymerase chain reaction (PCR) with DNA extracted from mouse tails according to the protocol previously described (30,31). PCR primers for genotyping were listed in Supplementary Table S1. PCR programs were run as 95°C for 2 min, then 95°C 30s, 57°C 1 min, 72°C 1 min for 30 cycles, with a final elongation at 72°C for 5 min. The expected sizes of PCR products designate the 349bp- Kdm5bwt, 516bp- Kdm5bLoxP, and 424bp- Kdm5bnull, respectively. After euthanasia, the prostate tissues or tumors of mice were procured. Tissues were fixed in 10% neutral-buffered formalin (Sigma), washed, and preserved in 70% ethanol at 4°C. Haemotoxylin and Eosin (H&E) staining and immunohistochemistry (IHC) staining were performed by HistoWiz (Brooklyn, NY, USA). Antibodies used for IHC were rabbit anti pAKT Ser473 (D9E) (Cell Signaling) and rabbit anti Ki67 (Abcam).
Mouse embryonic fibroblasts (MEFs)
MEFs were prepared from individual embryos of various genotypes in mice at E13.5-day according to the procedure previously described (30,32). Phoenix-eco cells cultured on Poly-D-Lysine-Coated dishes were transfected with pMSCV-PIG-Cre or empty vector plasmids using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. MEFs at passage 2 were infected with fresh retroviral supernatant containing 4μg/ml polybrene (Calbiochem). After selection with 3 μg/ml puromycin, MEFs at passage 5 were used for growth curve, western blot, and immunofluorescence (IF) analysis. To evaluate cell proliferation, MEFs were plated at 2×104 cells per well in a 24-well plate in triplicate, and cell proliferation was determined at 0, 2, 4, and 6 days by OD at 595nm with 0.1% crystal violet staining. To determine the activity of senescence, MEFs were plated at 1×104 cells per well of a 6-well plate in triplicate, and senescence-associated β-galactosidase (SA-β-gal) was detected after 4 days with the senescence detection kit (Calbiochem), photographed and quantified.
KDM5B knockout (KO) cells with CRISPR/Cas9
Three different single-guide RNA (sgRNA) sequences against KDM5B (sgRNA1: 5’ TCGAAGACCGGGCACTCGGG 3’, sgRNA2: 5’ GGACTTATTTCAGCTTAATA 3’, sgRNA3: 5’ CCTCCAATTCATTCAGTCTC 3’) were purchased from GenScript. pLentiCRISPR v2 plasmids containing KDM5B-sgRNA sequences, along with lentiviral packaging vectors PsPax2 and envelope plasmid pMD2.g, were transfected into HEK-293T cells using Lipofectamine 2000 (Invitrogen). pLentiCRISPR v2 empty vector was used as the control. Lentiviral supernatants were collected and filtered. PCa cells were infected with fresh viral supernatants, and then selected with 2 μg/ml of puromycin for 7 days. The resultant colonies were expanded and validated by western blot and sequencing analyses for KDM5B.
Cell culture, immunofluorescence (IF), and growth factor stimulation
LNCaP, C4–2B and PC3 human PCa cells were purchased from ATCC. LNCaP and C4–2B cells were grown in RPMI 1640 medium with 10% FBS and 1% Pen/Strep in an incubator with 5% CO2 at 37°C, while PC3 cells were maintained in DMEM complemented with 10% FBS and 1% Pen/Strep with 5% CO2 at 37°C. Proliferation and transfection assays were performed as previously described (30,32). Expression of target genes was determined 48–72 hours post-transfection using real-time qPCR or western blot (33). For IF staining, cells fixed with methanol were incubated with rabbit anti-PIP3 (1: 250, Echelon Biosciences). After incubation of DAPI and secondary antibody, images were recorded (33). For growth factor stimulation, cells were starved, treated with IGF-1 (20ng/ml) and collected for western blot at 0, 5, 15, 30, 60 and 90 minutes after stimulation.
High throughput RNA sequencing
Total RNA was extracted from LNCaP KDM5B-KO and control cells using the RNeasy Mini Kit (Qiagen). cDNA library preparation and RNA-seq were performed in the VANTAGE core at Vanderbilt University. RNA-Seq reads were aligned to the hg19 genome using STAR [PMID: 23104886] and quantified by feature Counts [PMID: 24227677]. Differential analysis was performed by DESeq2 [PMID: 25516281], which estimated the log2 fold changes, Wald test p-values, and adjusted p-value (FDR) by the Benjamini-Hochberg procedure. Paired-End (PE) sequencing was performed on all samples. Raw reads were de-multiplexed using a bcl2fastq conversion software v1.8.3 (Illumina, Inc.) with default settings.
Real-time reverse transcription PCR and reporter assays
Real-time reverse transcription PCR (qRT-PCR) was performed using forward and reverse primers (Supplementary Table S2) as previously described (33). For PIK3CA promoter reporter assay, cells were transfected with 200 ng empty pLightSwitch_Prom reporter vector, 50 ng, 100 ng or 200 ng of PIK3CA-promoter-Luc reporter plasmids (Switchgear Genomics, S706543). For KDM5B restoration, KDM5B-KO cells were co-transfected with pEV-Flag-KDM5B plasmid (0~150ng) and 50 ng PIK3CA-promoter-Luc reporter. At 24 hours post-transfection, cell lysates from each well were prepared in 100 μl of assay solution (Switchgear Genomics). 20 μl of lysates were kept to determine protein concentration, and the remaining lysates were used to measure the luciferase activities according to manufacturer’s instructions. Luciferase activities were normalized with the protein concentration.
Chromatin Immunoprecipitation (ChIP)
ChIP experiments were performed as previously described (24). Briefly, LNCaP KDM5B-KO and control cells were crosslinked with 1% formaldehyde and then quenched by 0.125M glycine. Cell pellets were lysed in a buffer (20 mM Tris•HCl, pH8.0, 2 mM EDTA, 0.5 mM EGTA, 0.5% SDS, 0.5 mM PMSF) and followed with a brief sonication. After centrifugation, chromatin was pre-cleared with protein A/G beads. For each ChIP reaction, 2 μg anti-H3K4me3 antibody (ab8580, Abcam), 3 μg anti-KDM5B antibody (NB100–97821, Novus) or IgG were added and incubated overnight at 4°C. Next morning, 200 μl pre-washed protein A/G beads were added to each IP and rotated at 4°C for 3 hrs. Beads were pelleted, washed, and eluted 2x by elution buffer. DNA was extracted for qPCR analyses (Supplementary Table S3) (24).
Western blot and half-life determination of P110α and P85 proteins
Cell lysates were prepared in RIPA buffer (1×PBS, pH7.4, 1% Nonidet P40/Triton X-100, 0.5% sodium deoxycholate, 2mM EDTA, with or without 0.1% SDS) with protease inhibitor cocktail (Roche) followed with a brief sonication. For mouse prostate analysis, protein extracts were prepared as previously described (30). For the protein stability of P110α and P85 proteins, KDM5B-KO and control cells were incubated with 100 μg/ml of cycloheximide (CHX, Sigma), and cell lysates were collected at indicated time points for western blot analysis (32). Antibodies used were: anti-H3K4me3 (1: 5,000, Cell Signaling, 9727), anti-H3 (1: 5,000, Abcam, Ab1791), anti-KDM5B (1: 2,000, Novus Biologicals, NBP1–84352), anti-KDM5A(1: 1,000, Cell Signaling, 3876), anti-KDM5C(1: 1,000, Cell Signaling, 5361), anti-β-actin (1: 10,000, Sigma, AC-74), anti-PTEN (1: 1,000, Cell Signaling, 9559), anti-P110α (1: 1,000, Cell Signaling, 4255), anti-P85 (1: 1,000, Cell Signaling, 4292), anti-AKT (1: 1,000, Cell Signaling, 9272), anti-pAKT Ser473 (1: 1,000, Cell Signaling, 4051), anti-pAKT Thr308 (1: 1,000, Cell Signaling, 4056), anti-SHIP1 (1: 1,000, Cell Signaling, 2728), and anti-β-Galactosidase (1: 1,000, Cell Signaling, 27198).
Statistical analysis
Statistics analysis was performed using two-tail Student’s t-test. The values of P < 0.05 were considered statistically significant.
Results
Kdm5b inactivation suppresses the initiation and progression of prostate cancer in Pten-null mutant mice
To study the effects of KDM5B loss on the initiation and progression of PCa in vivo, we took advantage of the striking disease phenotype of PCa in Pten mouse model and generated prostate epithelium-specific Pten/Kdm5b double null (Ptenpc–/–; Kdm5bpc–/–) mice (Fig. 1A and Supplementary Fig. S1A–C). In agreement with our previous report (30), the enlargement of anterior prostate (AP) lobes was noticeable in Pten conditional null (Ptenpc–/–) mice at 6 months of age (Fig. 1A), and the average of AP weight was 4 times greater than that of the age-matched wild type (Wt) cohort (Fig. 1B). Consistent with the idea that KDM5B is required for PCa, the size and average AP weight of Ptenpc–/–; Kdm5bpc–/– mice at 6 months of age were significantly lower than that of age-matched Ptenpc–/– mice, and similar to that of age-matched Wt mice (Fig. 1A and B). Furthermore, pathological analysis revealed that Ptenpc–/– mice developed high-grade PIN lesions and adenocarcinoma in all three lobes of the prostate, namely AP (Fig. 1C), dorsal/lateral (DLP), and ventral prostate (VP) (Supplementary Fig. S2A). The high-grade PIN lesion displayed a highly dysplastic luminal epithelium and the presence of nuclear atypia. In contrast, Ptenpc–/–; Kdm5bpc–/– mice showed only mild hyperplasia or low-grade PIN lesion in some prostate glands (~about 40%) (Fig. 1C and D, Supplementary Fig. S2A). Kdm5b deficiency alone did not cause morphological changes to the prostates (Fig. 1C, Supplementary Fig. S2A), as mice displayed the monolayer of luminal epithelial cells with normal recurrent mucosal folds projecting into the prostate gland. These data support that Kdm5b deficiency resulted in suppression of the initiation and progression of PCa in vivo.
Figure 1. Kdm5b inactivation suppresses prostate tumorigenesis of Pten-null mice in vivo.
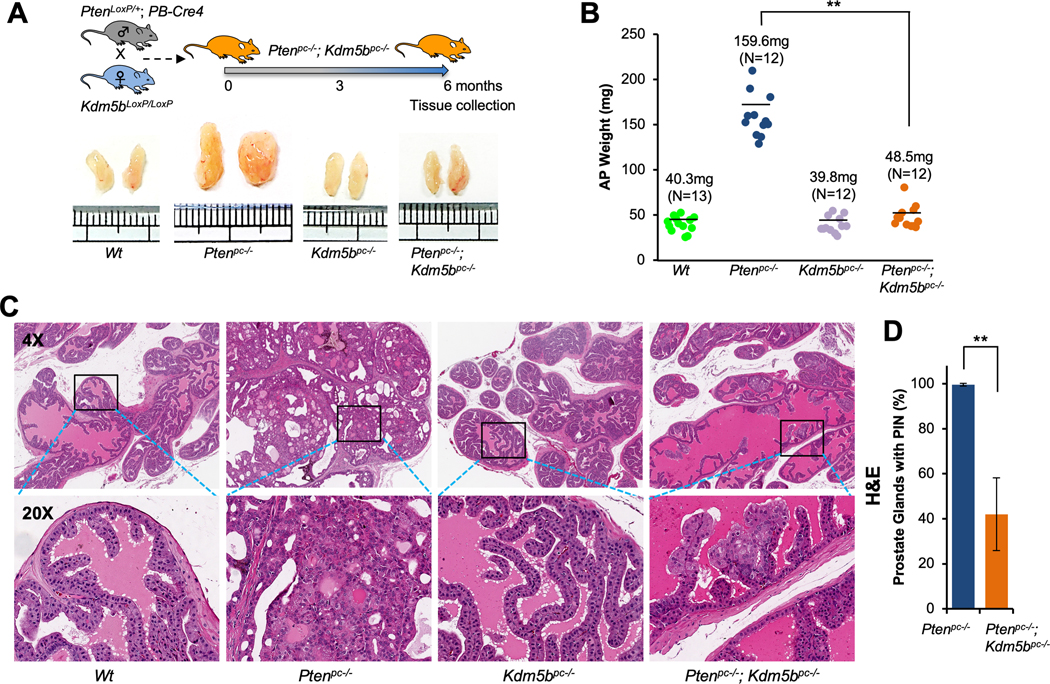
(A)Top panel, a schematic showing the strategy of generating Pten/Kdm5b mutant mice. Bottom panel, the actual sizes of representative biopsies of anterior prostates (AP) from Wt, Ptenpc–/–, Kdm5bpc–/–, and Ptenpc–/–; Kdm5bpc–/– mice at 6 months of age. (B) Quantification of AP weights from indicated genotypes of mice at 6 months of age. The averages of AP weight and numbers of mice for each cohort are indicated. (C) H&E staining of AP from indicated genotypes of mice at 6 months of age (Magnification: 4X and 20X). (D) Comparison of the onset of PIN in prostate glands between Ptenpc–/– and Ptenpc–/–; Kdm5bpc–/– mice. Error bars represent means ± SD (3 mice/group).
Kdm5b deficiency abrogates AKT signaling through reduction of PI3K in vivo
PTEN functions as a bona fide antagonist of the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway, therefore loss of PTEN results in a hyper-activation of AKT signaling. Our IHC staining results showed a striking increase of phosphorylated-AKT (pAKT-S473) at the membrane in prostates of Ptenpc–/– mice as compared to Wt mice (Fig. 2A). Interestingly, pAKT-S473 levels in prostates of Ptenpc–/–; Kdm5bpc–/– mice were significantly decreased as compared with that of Ptenpc–/– mice (P<0.05, Fig. 2A–C and Supplementary Fig. S2B). In addition, the proliferation rates as measured by Ki67 staining were also significantly decreased in Ptenpc–/–; Kdm5bpc–/– mice as compared to Ptenpc–/– mice (P< 0.005, Fig. 2A and B, Supplementary Fig. S2B). These results suggest that Kdm5b governs the hyper-activation of the PI3K/AKT signaling pathway in Pten-null mice. Surprisingly, the rate of apoptosis as measured by TUNEL levels decreased in Ptenpc–/–; Kdm5bpc–/– mice as compared to Ptenpc–/– mice (P< 0.005, Fig. 2A–B, Supplementary Fig. S2B). Furthermore, consistent with the findings in IHC, western blotting analysis confirmed that the levels of pAKT (Ser473 and Thr308) were significantly decreased in Ptenpc–/–; Kdm5bpc–/– mice compared to Ptenpc–/– mice (P<0.005 or P< 0.05, Fig. 2C–D). The levels of pAKT in Kdm5bpc–/– mice were lower than that in Wt mice (Fig. 2C). The levels of Kdm5b, along with pAKT, were significantly increased in Ptenpc–/– mice as compared to that of Wt mice (Fig. 2C), in consonance with our previous report (21). As KDM5B inactivation did not trigger the induction of apoptosis pathway, we examined the status of senescence program in Ptenpc–/–; Kdm5bpc–/– mice. Western blotting analysis revealed the levels of senescence-associated β-galactosidase (SA-β-Gal) were upregulated in prostates of Ptenpc–/–; Kdm5bpc–/– mice as compared to that of Ptenpc–/– mice (Supplementary Fig. S2C and D), indicating that Kdm5b ablation induces senescence. These results strongly support the idea that KDM5B is essential for the hyper-activation of the PI3K/AKT signaling pathway, and that Kdm5b deficiency suppresses PCa progression in mice by blocking AKT hyper-activation with induction of senescence.
Figure 2. Kdm5b deficiency abrogates AKT signaling through reducing PI3K levels in vivo in mouse prostates.

(A) Immunohistochemical (IHC) staining for pAKT(S473), Ki67 and TUNEL in prostate tissues from indicated genotypes of mice at 6 months of age (Magnification: 20X). (B) Quantification of the prostate cells positive for pAKT(S473), Ki67 and TUNEL in (A). Error bars represent means ± SD from 3 mice for each group. (C) Western blotting analysis of protein levels of Pten, Kdm5b, pAKT, P110α, and P85 in prostate tissues of mice with indicated genotypes. Two mouse prostate samples for each genotype. (D) Quantification of protein levels for pAKT, P110α, and P85 between Pten/Kdm5b double null and Pten null mice. Error bars represent means ± SD of triplicates.
AKT activation is tightly regulated by both PI3K and PTEN. P110 protein acts as the catalytic subunit of the PI3K complex, while P85 is the regulatory subunit. Therefore, we investigated the impact of Kdm5b loss on the levels of P110α and P85 proteins. Interestingly, western blotting analysis revealed that the protein levels of P110α in Ptenpc–/–; Kdm5bpc–/– mice were significantly decreased (by 60%) as compared to that in Ptenpc–/– mice (P< 0.005, Fig. 2C and D), while the protein levels of P85 decreased by about 15%. These findings indicated that Kdm5b plays an essential role in the regulation of PI3K levels as well as in the activation of AKT signaling in vivo.
Kdm5b is essential for cell proliferation and the activation of the PI3K-PIP3-AKT axis in vitro
To further delineate the biological role of Kdm5b inactivation in vitro, we prepared mouse embryonic fibroblasts (MEFs) of Pten null (PtenΔ/Δ) and Pten/Kdm5b double null (PtenΔ/Δ; Kdm5bΔ/Δ) from mice by following the strategy reported previously (30,32). Virus-mediated expression of Cre led to efficient recombination of loxP alleles as confirmed by PCR (Supplementary Fig. S3A). Interestingly, the proliferation rates of both PtenΔ/Δ; Kdm5bΔ/Δ and Kdm5b null (Kdm5bΔ/Δ) MEFs were significantly decreased as compared to Wt MEFs (Fig. 3A). The proliferation rate of PtenΔ/Δ MEFs was comparable to that of the Wt MEFs (Fig. 3A), in agreement with our previous report (30). Importantly, the proliferation rate of PtenΔ/Δ; Kdm5bΔ/Δ MEFs was significantly lower than that of PtenΔ/Δ MEFs (Fig. 3A), supporting the idea that Kdm5b plays a potentially oncogenic role in promoting cell proliferation. Moreover, western blotting analysis demonstrated that Pten inactivation resulted in an elevation of KDM5B protein in PtenΔ/Δ MEFs as compared to that in Wt MEFs (Fig. 3B), in line with the results in vivo (Fig. 2C). In addition, the levels of P110α, P85, and pAKT (Ser473 and Thr308) in PtenΔ/Δ; Kdm5bΔ/Δ MEFs were significantly decreased as compare to that of PtenΔ/Δ MEFs (P< 0.005 or P< 0.05, Fig. 3B and C). Meanwhile, Pten loss alone did not cause any changes of P110α and P85 levels (Fig. 3B).
Figure 3. Kdm5b is essential for the hyperactivation of the PI3K/AKT signaling pathway in MEFs.

(A) Effects of Kdm5b inactivation on the cell proliferation of MEFs. Error bars represent means ± SD of triplicates. (B) Western blotting analysis of protein levels of Pten, Kdm5b, pAKT, P110α, P85, and β-galactosidase in MEFs with indicated genotypes. (C) Quantification of protein levels for pAKT, P110α, P85, and β-galactosidase between Pten/Kdm5b double null and Pten null MEFs. Error bars represent means ±SD of triplicates. (D) Immunofluorescence (IF) images showing the levels and membrane localization of PIP3 in MEFs with indicated genotypes. (E) Quantification of fluorescence intensities for PIP3 levels in MEFs. Error bars represent means ± SD (20 cells/ group). (F) Images showing the β-galactosidase staining in senescent MEFs. (G) Quantification of the MEFs positive for β-galactosidase. Error bars represent means ± SD (30 cells/ group).
Given that Kdm5b inactivation decreases the levels of P110α and P85, we wished to assess the changes of PIP3 levels in MEFs. PI3K catalyzes the phosphorylation of PIP2 to PIP3 at the membrane, while PTEN dephosphorylates PIP3 to PIP2. IF images revealed that PIP3 predominantly accumulated at the membrane of PtenΔ/Δ MEFs (Fig. 3D and Supplementary Fig. S3B), with no signal detected in nucleus and cytosol (34–36). As shown, PIP3 levels in PtenΔ/Δ MEFs were much higher as compared to Wt MEFs (Fig. 3D). Most importantly, PIP3 levels of PtenΔ/Δ; Kdm5bΔ/Δ MEFs were significantly lower compared to that of PtenΔ/Δ MEFs (P<0.005, Fig. 3D and E). These results supported the idea that Kdm5b loss reduced levels of both PI3K and PIP3 to abrogate AKT signaling in vitro.
Moreover, PtenΔ/Δ; Kdm5bΔ/Δ MEFs showed stronger positivities for SA-β-Gal than PtenΔ/Δ MEFs and Kdm5bΔ/Δ MEFs (Fig. 3F). The percentage positive for SA-β-Gal in PtenΔ/Δ; Kdm5bΔ/Δ MEFs was significantly higher than that in PtenΔ/Δ MEFs (P<0.05, Fig. 3G), which is consistent with western blotting results of SA-β-Gal (Fig. 3B and C). These results strongly support that Kdm5b ablation induces senescence in vitro.
KDM5B KO decreases the proliferation of PTEN-null human PCa cells through impairing the PI3K-PIP3-AKT axis
These findings from genetically-engineered mouse models in vivo and MEFs in vitro propelled us to investigate the impact of KDM5B loss on biological traits and molecular mechanisms in human PCa cells. We chose to use LNCaP, C4–2B, and PC3 human PCa cell lines, which are PTEN-null, pAKT-high and KDM5B-high. We first generated KDM5B-KO PCa cells using CRISPR/Cas9. In addition to sequencing analysis, our western blotting results confirmed that KDM5B-KO cells failed to express KDM5B protein without affecting the levels of KDM5A and KDM5C (Fig. 4A, Supplementary Fig. S4A–B). We then asked whether KDM5B-KO causes any decreases of PI3K (P110α/P85) and pAKT (Ser473/Thr308). Indeed, P110α levels in all three KDM5B-KO human PCa cell lines decreased significantly (~50%) as compared to that in the parental control cells (Fig. 4A, Supplementary Fig. S5A–B, and Supplementary Fig. S6A–B). Meanwhile, P85 levels in KDM5B-KO cells decreased by about 30% (Fig. 4A, Supplementary Fig. S6A–B). Most importantly, the levels of pAKT (Ser473 and Thr308) in KDM5B-KO PCa cells decreased significantly as compared to that in the control cells (Fig. 4A, Supplementary Fig. S5A–B, and Supplementary Fig. S6A–B). Furthermore, KDM5B-KO led to a significant reduction of the proliferation of PCa cells as compared to the control cells (Fig. 4B, Supplementary Fig. S5B and S6B). Similarly, KDM5B-KO resulted in a significant reduction of PIP3 levels at the membrane of human PCa cells (P< 0.005, Fig. 4C and Supplementary Fig. S4B). Our results from multiple human PCa cell lines further confirmed that KDM5B plays a critical role in activation of the PI3K-PIP3-AKT signaling axis, in agreement with our data from mouse models and MEFs.
Figure 4. KDM5B knockout abrogates PI3K/AKT signaling in human prostate cancer cells.

(A) Left panel, western blotting analysis showing the protein levels of KDM5B, pAKT, P110α, and P85 in LNCaP KDM5B-KO and the parental control human PCa cells. Right panel, quantification analysis of pAKT, P110α, and P85 in LNCaP KDM5B-KO and the control cells. (B) A comparison of the cell proliferation between LNCaP KDM5B-KO and the control cells. (C) Immunofluorescence (IF) images and analysis of PIP3 in LNCaP KDM5B-KO cells. Left panel, IF images showing the levels and membrane localization of PIP3 in LNCaP KDM5B-KO cells and the control cells. Right panel, quantification of the fluorescence intensities of PIP3 in LNCaP KDM5B-KO and the control cells. Error bars represent means ± SD (20 cells/group). (D) RNA-Seq peaks showing the changes of KDM5B, IRS1, PIK3CA, and PIK3R1 expression between LNCaP KDM5B-KO and the control cells. (E) Quantitative RT-PCR analysis to show the relative mRNA levels of IRS1, PIK3CA, and PIK3R1 in LNCaP KDM5B-KO cells. (F) Top panel, western blotting analysis showing the protein levels of KDM5B, P110α and P85 in LNCaP KDM5B-KO and the control cells at indicated time points in CHX chase experiments. Bottom panel, quantification of protein remaining for P110α and P85 at indicated time points in KDM5B-KO and the control cells.
KDM5B induces P110α levels by increasing transcription and protein stability
We reasoned that KDM5B might directly induce the levels of PI3K (P110α/P85) at either the transcriptional or the post-transcriptional level. We performed RNA-Seq analysis in the LNCaP KDM5B-KO and the control cells, and the RNA-Seq peaks showed the suppressive impact of KDM5B-KO on the expression of IRS1, PIK3CA (encoding for P110α) and PIK3R1 (encoding for P85) (Fig. 4D). Furthermore, our qRT-PCR results showed that mRNA levels of PIK3CA gene in LNCaP KDM5B-KO cells decreased significantly as compared to that in the control cells (P<0.05, Fig. 4E), Surprisingly, the mRNA levels of PIK3R1 in LNCaP cells were not affected by KDM5B-KO (Fig. 4E). Therefore, we asked whether KDM5B-KO contributed to the protein stability of P110α and P85 in PCa cells. Our CHX chase experiments revealed that KDM5B ablation shortened the half-life of both P110α (7.2 vs 8.2 hr) and P85 (7.0 vs 10.5 hr) protein (Fig. 4F). These lines of biochemical evidence indicated that KDM5B loss differentially decreases PI3K (P110α/P85) in PCa cells at both the transcriptional and post-transcriptional levels.
We hypothesized that KDM5B plays an integral role on the transcriptional initiation of PIK3CA gene through its promoter. To test this, we performed ChIP analysis and compared the levels of KDM5B and H3K4me3 at the PIK3CA promoter in LNCaP KDM5B-KO and the control cells (Fig. 5A). Since no KDM5B protein was expressed in the KDM5B-KO cells, the enrichment of KDM5B was comparable with the IgG-ChIP control. We detected a significant reduction of the binding at the PIK3CA promoter in KDM5B-KO cells as compared to that in control cells (Fig. 5B). Surprisingly, the enrichment of H3K4me3 at the PIK3CA promoter was either lower or unchanged in KDM5B-KO cells as compared to the control cells (Fig. 5C). H3K4me3 is commonly regarded as an activating mark for gene transcription; therefore the results provided one explanation why KDM5B-KO deceased the PIK3CA expression. We further performed a bioluminescent reporter assay in both LNCaP and C4–2B cells using the PIK3CA promoter-driven luciferase reporter (Fig. 5D). Our results revealed that the luciferase activities of PIK3CA promoter in both LNCaP KDM5B-KO and C4–2B KDM5B-KO cells decreased significantly as compared to the control cells (P<0.005, Fig. 5D; P<0.05, Supplementary Fig. S7A). To confirm this transcriptional regulation, we restored KDM5B expression in KDM5B-KO PCa cells by co-transfecting Flag-KDM5B plasmids along with PIK3CA promoter luciferase reporter. Our results demonstrated that, in KDM5B-KO cells, the transcriptional activities of PIK3CA promoter were positively correlated with KDM5B expression in a dose dependent manner (Fig. 5E, Supplementary Fig. S7B).
Figure 5. KDM5B controls the PIK3CA expression in human prostate cancer cells.

(A) A schematic showing the positions of 5 amplicons relative to the PIK3CA transcription start site (TSS). (B) ChIP analysis of KDM5B levels at the indicated regions near PIK3CA TSS in LNCaP KDM5B-KO and the parental control cells. (C) ChIP analysis of H3K4me3 levels at the indicated regions near PIK3CA TSS in LNCaP KDM5B-KO and the control cells. (D) Top panel, a schematic showing that KDM5B regulates the transcription of PIK3CA through affecting promoter activities. Bottom panel, comparisons of luciferase activities of the PIK3CA promoter between LNCaP KDM5B-KO cells and the parental control cells. (E) Luciferase activities showing the effects of KDM5B restoration on the PIK3CA promoter activities in LNCaP KDM5B-KO cells.
Overexpression (OE) of KDM5B increased P110α levels to promote the hyperactivation of AKT signaling
Given that KDM5B-KO abrogates the PI3K/AKT signaling in the PTEN-Null context, we postulated that KDM5B-OE would enhance the PI3K/AKT signaling. We chose BHPrE1 cells, a non-tumorigenic human prostate epithelial cell line expressing low levels of KDM5B, to assess the effects of KDM5B-OE on the PI3K/AKT signaling. After Flag-KDM5B was ectopically expressed in BHPrE1 cells, qRT-PCR results showed that mRNA levels of PIK3CA were increased (~30%) as compared to that in control cells (P<0.05, Supplementary Fig. S7C). Remarkably, western blotting analysis revealed that levels of P110α protein were significantly increased and positively correlated with KDM5B elevation in a dose dependent manner (P<0.005, Fig. 6A and B). The levels of pAKT (Ser473/Thr308) were significantly increased upon KDM5B-OE (P<0.005, Fig. 6A and B). However, KDM5B-OE did not lead to an increase of P85 protein in BHPrE1 cells (Fig. 6A), consistent with the KDM5B-KO results. These data strongly support that KDM5B activates the PI3K/AKT signaling pathway in PCa cells.
Figure 6. The PI3K/AKT signaling and IGF responses are regulated by KDM5B in human prostate cancer cells.

(A) Western blotting analysis showing the increased levels of P110α and pAKT (Ser473/Thr308) in BHPrE1 cells upon KDM5B overexpression. (B) Quantification of the levels for P110α and pAKT in BHPrE1 cells from (A). (C) Western blotting analysis showing that the effects of KDM5B restoration on the levels of P110α, P85 and pAKT (Ser473/Thr308) in LNCaP KDM5B-KO cells. (D) Quantification of the protein levels for pAKT, P110α and P85 in LNCaP KDM5B-KO cells from (C). (E) Western blotting analysis showing the differential responses between PC3 KDM5B-KO and the parental control cells to IGF-1 stimulation. (F) Quantification of protein levels for pAKT (Ser473/Thr308), P110α, and P85 in (E). Error bars represent means ±SD of triplicates.
Since KDM5B addback in LNCaP KDM5B-KO cells increased the transcriptional activities of PIK3CA (Fig. 5E), we examined the impact of KDM5B restoration on the protein levels of PI3K and pAKT. We transfected Flag-KDM5B plasmids into LNCaP KDM5B-KO cells and collected cell lysates at 48 hours post-transfection. Western blotting analysis demonstrated that the levels of P110α and pAKT (Ser473/Thr308) proteins were notably increased in KDM5B-KO cells upon KDM5B restoration, and the elevation of P110α and pAKT (Ser473/Thr308) proteins was positively correlated with KDM5B expression in a dose dependent fashion (P<0.05, Fig. 6C and D). However, KDM5B restoration only led to a marginal increase of P85 protein in LNCaP KDM5B-KO cells (Fig. 6C and D), consistent with the results of KDM5B-OE in BHPrE1 cells (Fig. 6A).
KDM5B KO impairs the insulin signaling pathway in human PCa cells
P110α is the primary PI3K subunit responding to insulin signaling in liver and adipocytes (37,38). Our RNA-Seq analysis and qRT-PCR showed that mRNA levels of IRS1 in KDM5B-KO cells decreased about 50% as compared to the control cells (P<0.05, Fig. 4D and E). Therefore, we assessed the effects of KDM5B-KO on insulin signaling and subsequently the downstream PI3K/AKT pathway in PCa cells upon growth factor stimulation. To this end, PC3 KDM5B-KO and the control cells were subject to serum-starvation and insulin-like growth factor-1 (IGF-1) stimulation (Fig. 6E). We found that KDM5B-KO cells showed a retarded elevation of P110α, P85, and pAKT levels as compared with the control cells upon IGF-1 treatment (Fig. 6E and F). The elevated levels of P110α at all time points were significantly restrained (up to 60%) in the KDM5B-KO cells as compared to that in the control cells (Fig. 6E and F). Our findings suggest that KDM5B is essential for the insulin signaling, and KDM5B-KO impaired the IGF responses in PCa cells.
Discussion
Aberrant elevation of KDM5B is often found in various human cancers including PCa (28,29); however, the function roles of KDM5B on the initiation and progression of PCa are poorly understood (29). Our studies provided valuable information on decoding the biological function of KDM5B in 2 ways: 1) Kdm5b deficiency significantly suppresses the progression of prostate tumorigenesis in Ptenpc–/– mice; and 2) KDM5B activates the PI3K/AKT signaling pathway in PCa cells through regulation of P110α and PIP3 levels. We employed both genetically-engineered mouse models and CRISPR/Cas9 technologies to elucidate the contributions and action mechanism of KDM5B on PCa progression. Our findings demonstrated for the first time that KDM5B acts as an oncogenic driver for PCa, and furthermore illuminated that KDM5B is essential for the hyper-activation of PI3K/AKT signaling pathways in PCa.
We previously demonstrated that KDM5B is noticeably increased in prostate tumors of Pten-null mice in which AKT signaling is hyper-activated due to Pten inactivation (21). Meanwhile, a reduction of AKT signaling upon KDM5B knockdown was reported in gastric cancer cells and hepatocellular carcinoma (39,40). Nevertheless, the molecular contributions of KDM5B loss to the AKT signaling remain unclear. In this study, we discovered that KDM5B-KO abolished the PI3K/AKT signaling in mouse models, MEFs, and human PCa cells, in harmony with previous reports. Our results from both KDM5B-KO and KDM5B-OE experiments revealed that KDM5B activates the AKT signaling through regulation of PI3K (P110α and P85) and PIP3 levels to promote PCa progression, providing novel mechanistic explanations on the KDM5B-AKT association in human cancers.
Interestingly, we found that KDM5B regulates the mRNA levels and the protein stability of P110α in PCa cells, adding novel insights into other mechanisms previously reported. For example, KDM5B-OE in hepatocellular carcinoma activated the AKT signaling through downregulation of PTEN expression via PTEN-promoter (40). KDM5B-OE in human hypopharyngeal squamous cells activated the AKT signaling by decreasing INPP5D (encoding for SHIP1 protein) expression (41). We examined the contributions of SHIP1 protein on the AKT signaling in both mouse prostates and human PCa cells. Our data revealed that the expression level of SHIP1 protein in mouse prostates is very low as compared to that in mouse spleen (Supplementary Fig. S8A). KDM5B-KO did not cause any visible changes of SHIP1 protein in PCa cells, as SHIP1 protein levels are also low in these cell lines. Moreover, our RNA-Seq analysis revealed that the numbers of reads (FPKM) for INPP5D gene are below 8 in LNCaP cells. Therefore, it is unlikely that SHIP1 protein is involved in the regulation of the PI3K/AKT signaling in PCa, suggesting the diversities of SHIP1 signaling in tissues.
AKT signaling pathway plays an important role on regulation of epigenetic events including posttranscriptional histone modifications (42–44). Recent reports showed that AKT decreased H3K4me1/2 levels by phosphorylation and attenuation of lysine methyltransferase 2D (KMT2D or MLL2/4) at S1331 (45,46), but also increased H3K4me3 levels by restraining KDM5A protein in cytosol in breast cancer (47). AKT can increase KDM5B levels and cancer stem cell populations in oral cancer cells, highlighting an impact of the PI3K/AKT pathway on KDM5B expression in cancers (48). However, our data showed that either PI3K inhibition by LY294002 or AKT inhibition by MK-2206 did not result in any changes of KDM5B/H3K4me3 levels in LNCaP and C4–2B PCa cells (Supplementary Fig. S8B–E). Given the impact of KDM5B loss on suppression of PCa growth, the role of KDM5B/H3K4me3 on gene expression and malignancy in castration resistant PCa deserves further investigation.
KDM5B has mainly been studied as a transcriptional repressor. However, several studies reported that KDM5B could be a transcription activator, likely in a demethylase-independent manner. For example, one study reported that KDM5B is a transcription activator of self-renewal-associated genes in embryonic stem cells (ESCs), and KDM5B depletion decreased expression of genes associated with mitosis and nuclear metabolism (49). We have previously shown that KDM5B recruits GATA3 to promote FOXA1 expression in mammary epithelial cells (24). Our study showed that H3K4me3 levels are increased in PCa cells upon KDM5B-KO; but interestingly, the H3K4me3-ChIP data clearly revealed that the enrichment of H3K4me3 at the promoter of PIK3CA gene was lower or unchanged in KDM5B-KO cells as compared to that in the control cells. The lower levels of H3K4me3 are correlated with the decreased expression of PIK3CA. These results suggest that KDM5B regulates PIK3CA in a demethylase activity-independent manner, at least in PCa.
Dysregulation of the PTEN-PI3K-AKT signaling pathways is essential for PCa progression, but the efficacy of targeting AKT in PCa patients is still a challenge. However, most AKT inhibitors display a rather limited clinical activity as single agents. Our study suggests some options on the combinatorial treatments for better therapeutic perspective of PI3K or AKT inhibitors. Importantly, the genetic evidence of this study highlights that KDM5B-deficient PCa cells decrease proliferation and are more vulnerable to another cancer inhibitor. It is predicted that a combined inhibition of KDM5B and AR (or PARP) should be new avenues of blocking malignant transformation, prostate tumorigenesis and distant metastasis of PCa. Moreover, KDM5B degraders instead of catalytic site inhibitors would be also a new avenue of PCa intervention. Overall, our findings support the idea that targeting KDM5B can be a novel and effective therapeutic strategy of controlling PCa.
Supplementary Material
Significance:
This study demonstrates that levels of histone modification enzyme KDM5B determine hyperactivation of PI3K/AKT signaling in prostate cancer and that targeting KDM5B could be a novel strategy against prostate cancer.
Acknowledgments
We would like to thank Dr. Timothy Nottoli and Yale Genome Editing Center for their help to generate mice with Kdm5bLoxP conditional allele. We would like to thank Dr. Ralf Janknecht at University of Oklahoma Health Sciences Center for a gift of pEV-Flag-KDM5B plasmid, Dr. Hui Yu for the assistance with bioinformatics analysis, and Dr. Simon W. Hayward at NorthShore University Health System for critical reading. This work was supported in part by NIH grants U54MD007586, U54MD007593, U54CA163069, R01CA237586, P50CA121974, and UL1TR000445-06. Human Tissue staining, microscopy experiments and data analysis were performed through Human Tissue & Pathology Core and Morphology Core of Meharry Medical College supported in part by NIH grants U54MD007586, U54MD007593, U54CA163069 and S10RR0254970.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Deocampo ND, Huang H, Tindall DJ. The role of PTEN in the progression and survival of prostate cancer. Minerva Endocrinol 2003;28:145–53. [PubMed] [Google Scholar]
- 3.Bose S, Crane A, Hibshoosh H, Mansukhani M, Sandweis L, Parsons R. Reduced expression of PTEN correlates with breast cancer progression. Hum Pathol 2002;33:405–9. [DOI] [PubMed] [Google Scholar]
- 4.Wang JY, Huang TJ, Chen FM, Hsieh MC, Lin SR, Hou MF, et al. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in advanced gastric carcinomas. Virchows Arch 2003;442:437–43. [DOI] [PubMed] [Google Scholar]
- 5.Sun H, Enomoto T, Shroyer KR, Ozaki K, Fujita M, Ueda Y, et al. Clonal analysis and mutations in the PTEN and the K-ras genes in endometrial hyperplasia. Diagn Mol Pathol 2002;11:204–11. [DOI] [PubMed] [Google Scholar]
- 6.Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol 2002;161:439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet 2001;27:222–4. [DOI] [PubMed] [Google Scholar]
- 8.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol 2003;1:E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;129:1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett 2006;241:184–96. [DOI] [PubMed] [Google Scholar]
- 11.Sarris EG, Saif MW, Syrigos KN. The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals (Basel) 2012;5:1236–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Zhang L, Zhang X, Cui Z. PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Onco Targets Ther 2017;10:2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Tang Y, Yu F, Sun Y, Huang F, Chen Y, et al. Inhibition of Prostate Cancer DU-145 Cells Proliferation by Anthopleura anjunae Oligopeptide (YVPGP) via PI3K/AKT/mTOR Signaling Pathway. Mar Drugs 2018;16:2487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang F, Gao JY, Chen H, Du ZH, Zhang XQ, Gao W. Targeted inhibition of the phosphoinositide 3-kinase impairs cell proliferation, survival, and invasion in colon cancer. Onco Targets Ther 2017;10:4413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellinger J, Kahl P, von der Gathen J, Rogenhofer S, Heukamp LC, Gutgemann I, et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010;70:61–9. [DOI] [PubMed] [Google Scholar]
- 16.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 2010;10:457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol Biomarkers Prev 2010;19:2611–22. [DOI] [PubMed] [Google Scholar]
- 18.Blair LP, Cao J, Zou MR, Sayegh J, Yan Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers (Basel) 2011;3:1383–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A 2012;109:8971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Liu L, Yang S, Song N, Zhou X, Gao J, et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci U S A 2014;111:7096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu W, Liu S, Li B, Xie Y, Adhiambo C, Yang Q, et al. SKP2 inactivation suppresses prostate tumorigenesis by mediating JARID1B ubiquitination. Oncotarget 2015;6:771–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohta K, Haraguchi N, Kano Y, Kagawa Y, Konno M, Nishikawa S, et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int J Oncol 2013;42:1212–8. [DOI] [PubMed] [Google Scholar]
- 23.Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 2007;25:801–12. [DOI] [PubMed] [Google Scholar]
- 24.Zou MR, Cao J, Liu Z, Huh SJ, Polyak K, Yan Q. Histone demethylase jumonji AT-rich interactive domain 1B (JARID1B) controls mammary gland development by regulating key developmental and lineage specification genes. J Biol Chem 2014;289:17620–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, et al. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet 2013;9:e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013;23:811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Zhang SM, McGeary MK, Krykbaeva I, Lai L, Jansen DJ, et al. KDM5B Promotes Drug Resistance by Regulating Melanoma-Propagating Cell Subpopulations. Mol Cancer Ther 2019;18:706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A 2007;104:19226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ, et al. Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One 2009;4:e4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436:725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010;464:374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Carracedo A, Lin HK, Koutcher JA, Behrendt N, Egia A, et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci Signal 2009;2:ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu W, Xie Y, Ma Y, Matusik RJ, Chen Z. ARF represses androgen receptor transactivation in prostate cancer. Mol Endocrinol 2013;27:635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014;157:595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morotti A, Panuzzo C, Crivellaro S, Carra G, Fava C, Guerrasio A, et al. BCR-ABL inactivates cytosolic PTEN through Casein Kinase II mediated tail phosphorylation. Cell Cycle 2015;14:973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest 2008;118:3762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 2006;441:366–70. [DOI] [PubMed] [Google Scholar]
- 38.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006;125:733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Tang F, Qi G, Yuan S, Zhang G, Tang B, et al. KDM5B is overexpressed in gastric cancer and is required for gastric cancer cell proliferation and metastasis. Am J Cancer Res 2015;5:87–100. [PMC free article] [PubMed] [Google Scholar]
- 40.Tang B, Qi G, Tang F, Yuan S, Wang Z, Liang X, et al. JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget 2015;6:12723–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, An X, Han Y, Ma R, Yang K, Zhang L, et al. Overexpression of JARID1B promotes differentiation via SHIP1/AKT signaling in human hypopharyngeal squamous cell carcinoma. Cell Death Dis 2016;7:e2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005;310:306–10. [DOI] [PubMed] [Google Scholar]
- 43.Lee JH, Kang BH, Jang H, Kim TW, Choi J, Kwak S, et al. AKT phosphorylates H3-threonine 45 to facilitate termination of gene transcription in response to DNA damage. Nucleic Acids Res 2015;43:4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 2014;20:306–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017;355:1324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castel P, Toska E. Chromatin regulation at the intersection of estrogen receptor and PI3K pathways in breast cancer. Mol Cell Oncol 2019;6:e1625620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spangle JM, Dreijerink KM, Groner AC, Cheng H, Ohlson CE, Reyes J, et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep 2016;15:2692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Facompre ND, Harmeyer KM, Sole X, Kabraji S, Belden Z, Sahu V, et al. JARID1B Enables Transit between Distinct States of the Stem-like Cell Population in Oral Cancers. Cancer Res 2016;76:5538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J 2011;30:1473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.