

Abstract
Background:
Aspirin-exacerbated respiratory disease (AERD) is a mechanistically distinct subtype of chronic rhinosinusitis with nasal polyps (CRSwNP). Although frequently associated with type 2 inflammation, literature characterizing the milieu of inflammatory cytokines and lipid mediators in AERD has been conflicting.
Objective:
We sought to identify differences in the upper airway inflammatory signature between CRSwNP and AERD and determine whether endotypic subtypes of AERD may exist.
Methods:
Levels of 7 cytokines representative of type 1, type 2, and type 3 inflammation, and 21 lipid mediators were measured in nasal mucus from 109 patients with CRSwNP, 30 patients with AERD, and 64 non-CRS controls. Differences in inflammatory mediators were identified between groups, and patterns of inflammation among patients with AERD were determined by hierarchical cluster analysis.
Results:
AERD could be distinguished from CRSwNP by profound elevations in IL-5, IL-6, IL-13, and IFN-γ; however, significant heterogeneity existed between patients. Hierarchical cluster analysis identified 3 inflammatory subendotypes of AERD characterized by (1) low inflammatory burden, (2) high type 2 cytokines, and (3) comparatively low type 2 cytokines and high levels of type 1 and type 3 cytokines. Several lipid mediators were associated with asthma and sinonasal disease severity; however, lipid mediators showed less variability than cytokines.
Conclusions:
AERD is associated with elevations in type 2 cytokines (IL-5 and IL-13) and the type 1 cytokine, IFN-γ. Among patients with AERD, the inflammatory signature is heterogeneous, supporting subendotypes of the disease. Variability in AERD immune signatures should be further clarified because this may predict clinical response to biologic medications that target type 2 inflammation.
Keywords: Cytokine, endotype, leukotriene, prostaglandin, aspirin, nonsteroidal anti-inflammatory drug, rhinosinusitis, eicosanoid, asthma
Chronic rhinosinusitis (CRS) is a common but heterogeneous airway inflammatory disease that affects up to 5% of the US population.1,2 Although mechanisms of the disease are still being determined, it is now increasingly recognized that CRS likely represents a clinical syndrome with potentially diverse pathophysiology rather than a single diagnosis. Unique phenotypes and endotypes of CRS are now being characterized on the basis of clinical presentation and inflammatory characteristics, respectively, and these subtypes have important implications for disease management and prediction of clinical outcomes. Aspirin-exacerbated respiratory disease (AERD) is a well-established disease subtype characterized by asthma, nasal polyposis, and sensitivity to cyclooxygenase-1 inhibitors. When compared with chronic rhinosinusitis with nasal polyps (CRSwNP) more broadly, AERD is associated with need for more surgery and patients are more likely to be corticosteroid dependent.3,4
Although knowledge of the exact pathogenesis of nasal polyposis is unclear and ever evolving, the inflammatory environment associated with polyps in Western countries is usually eosinophilic, with a predominantly type 2 immune signature.5-7 The AERD subtype of CRSwNP has a similar type 2 predominance but has unique biochemical hallmarks that distinguish it from other patients with nasal polyposis. For example, AERD is commonly associated with degranulated mast cells and excess production of cysteinyl leukotrienes (CysLTs) generated from arachidonic acid (AA) by the 5-lipoxygenase pathway. Nonetheless, recent investigations suggest that AERD may not be a singular disease process but rather a heterogeneous entity with substantial differences in inflammatory mediators and clinical characteristics. A recent Belgian analysis of a large AERD cohort identified 3 potential “subphenotypes” of AERD with variability among measured inflammatory mediators in blood and induced sputum, and with respect to multiple demographic and clinical characteristics.8 Although the characteristic cytokine milieu of AERD is notable for a predominantly type 2–dominant inflammatory profile, precise characterization of this signature has varied considerably in the literature, particularly with respect to the upper airway. Pérez-Novo et al9 previously found that IL-5 was elevated in sinonasal tissue from patients with AERD compared with patients with CRSwNP. However, Steinke et al10 reported that polyps from patients with AERD had reduced levels of the prototypical type 2 cytokines IL-5 and IL-13 compared with those with hyperplastic eosinophilic CRSwNP, whereas levels of IL-4 and the type 1 cytokine IFN-γ were increased. Conversely, Stevens et al11 found no differences in IFN-γ, IL-5, or IL-13 levels between CRSwNP and AERD polyps, and subsequently proposed that eosinophilic inflammation in AERD may be mediated by factors other than traditional type 2 cytokines. Recent identification of putative CRS endotypes, including work by our group, has confirmed that CRSwNP is a complex disease characterized by contributions from multiple different inflammatory mediators, rather than by a purely type 2 cytokine milieu.12-14 Interestingly, hierarchical cluster analysis of patients with CRS by our group also found that patients with AERD do not converge within a single inflammatory endotype, and were instead irregularly distributed among 4 of 5 endotypic clusters using this methodology.13 Histologic classification based on granulocytic tissue infiltration similarly showed that AERD can present with mixed eosinophilic and neutrophilic inflammation in up to 30% of patients, with some characterized by neutrophilic inflammation alone, despite being universally linked to type 2 inflammation.15
Given these discordant findings and suggestions of multiple inflammatory pathways, we hypothesized that there may be endotypic features within the greater AERD subtype that could have significant clinical relevance. We sought to identify any differences in inflammatory mediators in the upper airway between CRSwNP and AERD, and further sought to identify any inflammatory AERD subgroups using an unstructured approach. Finally, we determined whether these inflammatory AERD subgroups were linked to differences in CysLTs and other lipid mediators associated with AA and linoleic acid (LA) metabolism.
METHODS
Study population
This study was approved by the Institutional Review Board of Vanderbilt University. Patients presented to rhinology clinics at the Vanderbilt Bill Wilkerson Center and Vanderbilt Asthma, Sinus, and Allergy Program. CRS was diagnosed according to the European Position Paper on Rhinosinusitis and Nasal polyps and International Consensus Statement on Allergy and Rhinology.16,17 CRSwNP was diagnosed by the presence of visible nasal polyps on clinic nasal endoscopy or during endoscopic sinus surgery. AERD was diagnosed on the basis of clinical triad of nasal polyps, asthma, and at least 2 documented allergic reactions to aspirin or other nonsteroidal anti-inflammatory drugs or a positive aspirin challenge. All patients underwent endoscopic sinus surgery after failing a period of medical management. Control cases were patients undergoing anterior skull base or pituitary surgery without clinical or radiographic history of sinus disease. Exclusion criteria included any patient receiving systemic steroids within 4 weeks before surgery, patients with cystic fibrosis, autoimmune, or granulomatous disease, or patients who were receiving immune-directed mAbs or other immunomodulators. The presence of asthma and allergic rhinitis was recorded for all patients. Allergic rhinitis was diagnosed on the basis of positive skin prick test result or physician diagnosis based on seasonal variation in atopic symptoms and relief after using an oral antihistamine or intranasal corticosteroids. Asthma was diagnosed on the basis of a bronchodilator response on pulmonary function testing, methacholine challenge, or previous diagnosis by a pulmonologist and/or allergist. Asthma severity was defined by asthma medication use at the time of surgery based on the Global Initiative for Asthma guidelines. The Sinonasal Outcome Test-22 (SNOT-22) was used to record patient-reported symptom severity.18 All patients underwent high-resolution computed tomography scan of the paranasal sinuses within 3 months of surgery. A Lund-McKay score was assigned to each scan to assess the severity of radiographic sinus disease.
Mucus collection and cytokine measurement
Nine × 24 mm polyurethane sponges (Summit Medical; St Paul, Minn) were placed into the middle meatus or ethmoid cavity of each subject under endoscopic guidance at the time of surgery, as has been previously reported.19,20 This method of mucus collection has the advantage of minimal specimen dilution and standardization between patients, and we have previously shown that mucus cytokine levels are highly consistent within different subsites of the sinonasal cavity.21 Sponges were left in place for 5 minutes, after which they were placed in a sterile microcentrifuge tube and immediately processed. Sponges were placed into a microporous centrifugal filter device (MilliporeSigma; Billerica, Mass) and centrifuged at 14,000g for 10 minutes. Samples were then gently vortexed and again centrifuged for 5 minutes to remove cellular debris. Supernatants were removed, placed into a new microcentrifuge tube, and frozen at −80°C.
A multiplex cytokine bead assay (BD Biosciences; Franklin Lakes, NJ) was then used to analyze samples. In brief, 50 μL of mucus was incubated with 50 μL of mixed capture beads for each measured inflammatory mediator and incubated for 1 hour. Fifty μL of mixed detection reagent was then added to each sample and standard and incubated for an additional 2 hours. Samples were centrifuged at 200g for 5 minutes after the addition of 1 mL wash buffer, and the supernatant was discarded. The beads were then resuspended in 300 μL wash buffer and analyzed on an LSR Fortessa flow cytometer (BD Biosciences; San Jose, Calif). A total of 7 cytokines and inflammatory mediators were analyzed. The assay data were analyzed using BD FCAP Array Software version 3.0 (BD Biosciences).
Measurement of eicosanoids
AA- and LA-derived lipid mediators detectable by our ultraperformance LC-MS approach were assessed in the mucus samples collected at the time of surgery (see Fig E1 in this article’s Online Repository at www.jacionline.org). Twenty-five to 40 μL of each mucus specimen was placed into a microcentrifuge tube containing 5000 μL 25% methanol in water and internal standard mix (1 ng each deuterated eicosanoid). The sample was vortexed and spun to pellet protein. The supernatant was then extracted on an Oasis MAX μElution plate (Waters Corp, Milford, Mass) as follows: Sample wells were first washed with methanol (200 μL) followed by 25% methanol in water (200 μL). The sample was then loaded into the well and washed with 600 μL 25% methanol. Eicosanoids were eluted from the plate with 30 μL 2-propanol/acetonitrile (50/50, vol/vol) containing 5% formic acid into a 96-well elution plate containing 30 μL water in each well. Samples were analyzed on a Waters Xevo TQ-XS triple quadrupole mass spectrometer connected to a Waters Acquity I-Class ultraperformance LC (Waters Corp). Separation of analytes was obtained using an Acquity PFP column (2.1 × 100 mm), with mobile phase A being 0.01% formic acid in water and mobile phase B acetonitrile. Eicosanoids were separated using a gradient elution beginning with 30% B going to 95% B over 8 minutes at a flow rate of 0.250 mL/min.
Statistical analysis
Descriptive statistics for demographic/clinical characteristics and inflammatory mediators were presented as the median with interquartile range or means with SDs for continuous variables and the frequency with percentages for categorical variables. Differences between defined CRS subgroups or clusters were evaluated using the Kruskal-Wallis test for continuous variables or the chi-square test for categorical variables. This was followed by Dunn’s test for multiple comparisons. Statistical significance was defined as a P value of less than .05.
Sample size for cluster analysis was estimated by establishing a subject to variable ratio of greater than 5:1 as recommended by Gorsuch22 and O’Rourke and Hatcher.23 Descriptive statistics and frequency distributions were examined for each biological variable, and all were positively skewed. To normalize data for subsequent analysis, values were transformed by taking the log-transformation, resulting in elimination or significant reduction of skewing for all variables. Hierarchical cluster analysis was performed using Ward’s method on squared Euclidian distances. The hierarchical structure and taxonomic relationships between subjects was visualized using a dendogram. The appropriate number of clusters (k) was selected using the Elbow method. This approach calculates the total within sum of squared error for between 1 and 10 clusters and determines k by identifying the break point where adding additional clusters does not substantially change the sum of squared error. Cluster stability was verified using bootstrap analysis. This involved repeating the estimation procedure on 1000 resampled data sets to ensure that the clustering results were stable and not unique to the original data set. Variances of each biological variable were compared using the coefficient of variation to create a unitless measure of comparison between groups. Omnibus hypothesis testing for differences in variance was performed using the asymptotic test for the equality of coefficients of variation.
RESULTS
Demographic and clinical characteristics
A total of 64 controls and 139 patients with CRS were enrolled in the study, including 109 patients with CRSwNP and 30 patients with AERD (Table I). Patients with AERD and non-AERD CRSwNP were similar with respect to age, sex, and body mass index. However, disease burden was generally higher in patients with AERD compared with patients with CRSwNP as assessed by computed tomography score (20.3 ± 3.5 vs 15.9 ± 4.2; P < .001) and SNOT-22 score (50.7 ± 4.2 vs 42.1 ± 2.5; P = .08). Patients with AERD also had higher rhinologic (P = .04) and extranasal (P = .01) SNOT-22 subdomain scores. Patients with AERD were more likely to be on antileukotriene medications (P < .001) and were more likely to have had prior endoscopic sinus surgery (70% vs 36.7%; P = .02).
TABLE I.
Demographic and clinical characteristics of study population
| Characteristic | Control | CRSwNP | AERD | P value (CRSwNP vs AERD) |
|---|---|---|---|---|
| No. | 64 | 109 | 30 | |
| Age (y) | 51.6 ± 15.4 | 50.4 ± 15.2 | 47.8 ± 11.5 | .31 |
| Sex: female, n (%) | 40 (68) | 35 (32) | 12 (40) | .51 |
| Asthma, n (%) | 1 (2) | 37 (33.9) | 30 (100) | <.001 |
| Allergic rhinitis, n (%) | 5 (8) | 64 (58.7) | 23 (76.7) | .27 |
| Prior surgery, n (%) | 0 (0) | 40 (36.7) | 21 (70) | .02 |
| BMI (kg/m2) | 33.1 ± 9.1 | 29.1 ± 5.9 | 29.8 ± 4.7 | .65 |
| NCS, n (%) | 1 (2) | 90 (82.6) | 23 (76.7) | .75 |
| LTR, n (%) | 3(5) | 26 (23.9) | 19 (63.3) | <.001 |
| Current smoker, n (%) | 1 (2) | 5 (4.6) | 0 (0) | .24 |
| SNOT-22 score | 42.1 ± 21.1 | 50.7 ± 21.6 | .08 | |
| Rhinologic | 12.3 ± 5.7 | 15.0 ± 5.5 | .04 | |
| Extranasal | 7.2 ± 3.5 | 9.2 ± 3.2 | .01 | |
| Ear/facial | 7.0 ± 5.3 | 9.3 ± 5.5 | .08 | |
| Psychological | 11.5 ± 8.9 | 12.7 ± 8.0 | .56 | |
| Sleep | 10.3 ± 7.1 | 10.8 ± 7.9 | .77 | |
| CT score | 15.9 ± 4.2 | 20.3 ± 3.5 | <.001 |
AERD, Aspirin-exacerbated respiratory disease; BMI, body mass index; CT, computed tomography; LTR, leukotriene modifier; NCS, nasal corticosteroid.
Values are presented as means ± SDs or medians with interquartile ranges, depending on the normalcy of the data. Differences between groups were assessed by using the Kruskal-Wallis test or χ2 analysis. Boldface text indicates a P value of less than .05.
Cytokine signatures in AERD
Previous studies have reported conflicting results regarding potential AERD-specific inflammatory signatures. To help settle these inconsistencies, we assessed 5 different cytokines measured in sinonasal mucus that are representative of type 1 (IFN-γ), type 2 (IL-4, IL-5, IL-13), and type 3 immunity (IL-17A), as well as 2 proinflammatory cytokines associated with innate immunity (IL-1β, IL-6). As previously reported, CRSwNP and AERD were both characterized predominantly by elevated type 2 cytokines (see Table E1 in this article’s Online Repository at www.jacionline.org). Compared with healthy controls, patients with CRSwNP were characterized by elevated IL-4, IL-5, and IL-6, whereas patients with AERD were characterized by high levels of IL-4, IL-5, IL-6, IL-13, IL-17A, and IFN-γ. AERD could be differentiated from CRSwNP by profoundly higher levels of IL-5, IL-6, IL-13, and IFN-γ, with median values that were 3 to 5 times greater on average compared with patients with CRSwNP (Fig 1). These data confirm that AERD has a predominantly type 2 immune signature, but additionally has features of both type 1 and type 3 inflammation.
FIG 1.

Mucus cytokines in patients with AERD, patients with CRSwNP, and non-CRS control patients. Cytokine values are plotted on a log scale. Solid lines indicate medians with interquartile ranges.
Lipid mediator signatures in AERD
We next sought to measure multiple lipid mediators in sinonasal mucus from patients with AERD and identify any potential differences compared with patients with CRSwNP and control patients (Table II). A total of 21 mediators derived from the AA and LA cascades were assessed using (ultraperformance LC)-MS. Most metabolites varied significantly between groups, the exceptions being 13-HODE, prostaglandin (PG)E2, and leukotriene (LT)C4. AERD could be distinguished from CRSwNP by reduced levels of 9,10-dihydroxyoctadecenoic acid (DiHOME) (P = .02), 12,13-DiHOME (P < .001), 9,10-epoxyoctadecenoic acid (EpOME) (P < .001), 8-hydroxyeicosatetraenoic acid (HETE) (P = .01), 12-HETE (P= .04), 11,12-epoxyeicosatrienoic acid (EET) (P < .001), 14,15-EET (P = .03), and 20-HETE (P = .01). Thromboxane (Tx)B2 was elevated in patients with AERD compared with patients with CRSwNP (P = .008), whereas among the CysLTs, LTB4 levels were reduced (P = .002) and LTE4 levels were increased (P = .001) (Fig 2).
TABLE II.
Lipid mediator levels in controls, patients with CRSwNP, and patients with AERD
| Mediator | Control (n = 33) | CRSwNP (n = 34) | AERD (n = 30) | P value |
|---|---|---|---|---|
| 12,13-EpOME | 11,359 (7,953-19,637) | 8,591 (4,846-14,877) | 5,782 (2,737-12,313)* | .005 |
| 9,10-DiHOME | 15.8 (9.4-26.6) | 11.4 (8.9-11.4) | 5.4 (2.7-23.4)*† | .003 |
| 12,13-DiHOME | 23.4 (14.4-41.3) | 27.6 (19.9-40.7) | 1.5 (0.01-4.1)*† | <.001 |
| 9,10-EpOME | 1,150 (687.9-3,835) | 652 (394.5-929.7)* | 249 (156-450)*† | <.001 |
| 13-HODE | 9,804 (6,807-16,566) | 7,335 (4,228-12,482) | 8,852 (4,163-16,605) | .17 |
| 8-HETE | 86.0 (68.6-136.7) | 55.0 (36.8-103.6) | 29.0 (14.8-64.0)*† | <.001 |
| 15-HETE | 8,259 (5,359-10,414) | 6,554 (4,050-9,011) | 3,496 (1,795-6,715) | .006 |
| 12-HETE | 921.7 (658.1-1,210) | 676.5 (406.4-945.0) | 474.2 (216.9-595.6)*† | <.001 |
| 11,12-EET | 1,295 (845-5,892) | 856.5 (587.5-1,289) | 228.3 (164.1-336.4)*† | <.001 |
| 14,15-EET | 1,688 (1,115-2,583) | 1,267 (697-1,629) | 725 (467-981)*† | <.001 |
| 20-HETE | 181,126 (99,675-253,998) | 152,415 (82,825-188,409) | 61,850 (36,426-117,669)*† | <.001 |
| TxB2 | 4.9 (1.4-16.5) | 6.3 (1.2-15.6) | 16.9 (6.2-47.9)*† | .003 |
| PGD2 | 6.3 (4.0-13.3) | 21.1 (11.1-41.6)* | 28.1 (15.0-63.8)* | <.001 |
| 15-Keto-PGE2 | 0.77 (0.41-1.30) | 0.97 (0.39-1.53) | 1.36 (0.67-1.82)* | .03 |
| PGE2 | 105.0 (68.1-137.6) | 73.8 (56.3-149.9) | 98.4 (49.5-149.8) | .92 |
| PGF2a | 18.4 (12.9-35.7) | 15.7 (9.6-20.5) | 9.5 (0.01-16.8)* | .001 |
| 9a,11b-PGF2a | 1.3 (0.3-3.7) | 3.0 (1.7-9.8) | 9.7 (0.01-20.9)* | .005 |
| LTB4 | 12.7 (9.3-22.1) | 11.1 (5.6-24.3) | 0.01 (0.01-11.3)*† | <.001 |
| LTC4 | 31.9 (13.8-59.2) | 53.0 (27.6-80.0) | 32.0 (14.9-72.0) | .16 |
| LTD4 | 0.01 (0.01-0.34) | 0.23 (0.01-1.14) | 1.51 (0.01-2.65)* | <.001 |
| LTE4 | 0.17 (0.03-0.40) | 0.62 (0.16-2.78)* | 4.97 (1.87-12.75)*† | <.001 |
| Total CysLTs | 33.0 (16.9-60.4) | 55.7 (34.3-87.1) | 43.9 (21.3-79.2) | .10 |
Mucus levels of lipid mediators are shown for each group. All mediator levels are presented as ng/mL. Significant differences between groups were identified by the Kruskal-Wallis test followed by Dunn’s test for multiple comparisons. Data are presented as medians with interquartile range. Boldface text indicates P value of less than .05.
P < .05 compared with controls.
P < .05 compared with patients with CRSwNP.
FIG 2.

Mucus lipid mediators in patients with AERD, patients with CRSwNP, and non-CRS control patients. Solid lines indicate medians with interquartile range.
We then sought to determine whether heterogeneity in inflammatory cytokines was also reflected in levels of lipid mediators with known pathophysiological relevance in AERD. We first compared variability between mucus cytokines and 7 lipid mediators in all patients with AERD. We used the coefficient of variation to make unitless comparisons of variance between the different biomarkers. In general, cytokines had higher variability (IL-1β = 1.59; IL-4 = 1.15; IL-5 = 1.55; IL-6 = 1.47; IL-13 = 1.19; IL-17A = 1.25; IFN-γ = 4.26) than lipid mediators (LTC4 = 0.97; LTE4 = 0.86; PGD2 = 0.92; PGE2 = 0.73; TxB2 = 1.13; 20-HETE = 0.79), the 1 exception being LTB4 (coefficient of variation = 2.63) (Fig 3, A).
FIG 3.
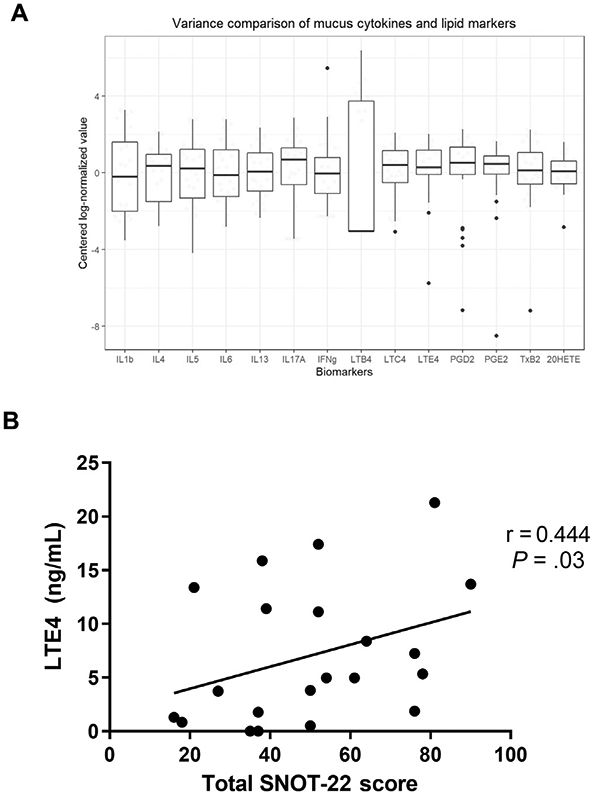
A, Variance comparison for cytokines and lipid mediators. Log-normalized and centered raw cytokine and lipid mediator values are visualized using boxplots for a graphical comparison of the relative variability of each biomarker. Values are expressed as the median with interquartile range. Omnibus testing between biomarkers using the coefficient of variation showed statistically significant differences (P < .001) between groups. Note the relatively lower variance in lipid mediators compared with cytokines. B, The correlation of mucus leukotriene E4 (LTE4) levels with total SNOT-22 score evaluated using Spearman correlation.
Lipid mediator levels and disease severity
We next sought to determine whether a broad array of lipid mediators in nasal secretions were associated with asthma or sinonasal symptom severity. Total CysLTs did not differ by asthma severity. Among individual CysLTs, LTC4 was most abundant. LTD4 and LTE4 levels were low but highest in the group with severe asthma (Table III). Asthma severity was inversely associated with levels of PGD2 (P = .02) and 9a,11b-PGF2a (P = .03). We examined potential correlations between each lipid mediator and sinonasal symptom burden, assessed via the SNOT-22 at the time of surgery (see Table E2 in this article’s Online Repository at www.jacionline.org). LTE4 levels in nasal secretions correlated with higher SNOT-22 scores (r = 0.444; P = .03) (Fig 3, B), while a similar trend was observed for LTB4 (r = 0.330; P = .11). SNOT-22 scores did not correlate with any other measured lipid mediator.
TABLE III.
Mucus levels of lipid mediators in patients with AERD based on asthma severity
| Mediator | Mild asthma (n = 3) | Moderate asthma (n = 5) | Severe asthma (n = 22) | P value |
|---|---|---|---|---|
| 12,13-EpOME | 4,202 (2,426 to 20,393) | 7,500 (2,116 to 14,472) | 5,782 (2,737 to 12,313) | .90 |
| 9,10-DiHOME | 4.38 (4.00 to 23.60) | 2.70 (0.75 to 20.61) | 6.05 (3.31 to 23.36) | .74 |
| 12,13-DiHOME | 1.88 (0.01 to 4.08) | 0.89 (0.01 to 3.73) | 1.53 (0.01 to 4.32) | .91 |
| 9,10-EpOME | 315.8 (151.3 to 534.7) | 191.0 (125.3 to 570.8) | 274.9 (156.4 to 450.0) | .90 |
| 13-HODE | 9,155 (5,108 to 29,380) | 11,236 (2,724 to 19,669) | 8,461 (4,070 to 16,605) | .75 |
| 8-HETE | 16.0 (13.5 to 61.2) | 29.0 (15.5 to 48.4) | 29.3 (14.8 to 72.5) | .71 |
| 15-HETE | 3,496 (2,159 to 6,715) | 3,023 (1,465 to 9,557) | 3,594 (1,790 to 6,717) | .98 |
| 12-HETE | 376 (340 to 596) | 534 (146 to 679) | 474 (217 to 643) | .99 |
| 11,12-EET | 220.9 (164.6 to 336.6) | 274.8 (168.8 to 661.1) | 223.5 (159.4 to 337.9) | .81 |
| 14,15-EET | 642.9 (458.4 to 969.5) | 1079 (381.9 to 1,739) | 724.7 (466.6 to 959.2) | .59 |
| 20-HETE | 49,723 (37,749 to 131,101) | 42,732 (28,616 to 150,425) | 67,307 (35,685 to 116,309) | .95 |
| TxB2 | 20.9 (10.6 to 46.1) | 12.9 (6.8 to 31.1) | 16.9 (5.8 to 81.7) | .83 |
| PGD2 | 154.3 (78.1 to 161.9) | 17.9 (6.9 to 36.5) | 27.3 (15.0 to 60.2) | .02 |
| 15-Keto-PGE2 | 2.35 (2.06 to 2.81) | 0.67 (0.44 to 1.59) | 1.31 (0.78 to 1.58) | .10 |
| PGE2 | 149.8 (98.4 to 155.4) | 73.3 (41.0 to 191.1) | 90.2 (46.8 to 143.1) | .52 |
| PGF2a | 0.01 (0.01 to 46.7) | 14.0 (4.76 to 22.1) | 7.6 (0.01 to 15.2) | .71 |
| 9a,11b-PGF2a | 35.6 (34.3 to 40.4) | 13.9 (0.01 to 18.0) | 8.3 (0.01 to 20.2) | .03 |
| LTB4 | 0.01 (0.01 to 81.2) | 3.9 (0.01 to 93.6) | 0.01 (−0.01 to 10.7) | .54 |
| LTC4 | 125.7 (32.0 to 470.6) | 38.7 (13.0 to 120.7) | 22.9 (6.23 to 64.77) | .10 |
| LTD4 | 0.01 (0.01 to 0.01) | 0.01 (0.01 to 2.54) | 2.06 (0.01 to 4.04) | .05 |
| LTE4 | 1.29 (0.84 to 1.87) | 3.73 (1.82 to 17.3) | 5.34 (3.67 to 12.75) | .15 |
| Total CysLTs | 126.6 (33.3 to 472.2) | 43.9 (32.1 to 122.5) | 36.6 (17.7 to 74.1) | .16 |
Median levels of lipid mediators in patients with AERD and mild, moderate, or severe asthma are shown. All mediator levels are presented as ng/mL. Significant differences between groups were identified by the Kruskal-Wallis test followed by Dunn’s test for multiple comparisons. Data are presented as medians with interquartile range. Boldface text indicates P value of less than .05.
AERD inflammatory cluster analysis
We used hierarchical cluster analysis incorporating the same 7 cytokines to identify potential subendotypes of AERD. Analysis identified 3 potential AERD clusters based entirely on cytokine signatures (Fig 4). Visualization of the cluster structure showed discrete separation of each grouping and good cluster stability based on bootstrap validation (all clusters with stability ~0.7). The largest grouping (cluster 2, n = 14) carried a type 2–high signature, with elevated IL-4, IL-5, and IL-13 compared with the other clusters, as well as high levels of IL-6 (see Fig E2 in this article’s Online Repository at www.jacionline.org). Cluster 1 (n = 10) had low overall inflammatory burden, whereas cluster 3 (n = 6) was type 2–low and type 1/type 3–high. Although somewhat limited by the small sample size in cluster 3, no significant demographic differences were identified between clusters (Table IV). Patients in the type 2–high group (cluster 2) had the highest tissue eosinophil counts, had worse SNOT-22 total and subdomain scores, and were more likely to have severe asthma, though none of these differences reached statistical significance.
FIG 4.

Identification of inflammatory disease clusters in patients with AERD. A, Dendogram representing hierarchical cluster analysis of patients with AERD. Hierarchical cluster analysis was performed by using the Ward method on squared Euclidian distances, with 7 cytokines as biological variables. B, PCA plot showing patient clusters based on their similarity. PCA, Principle component analysis.
TABLE IV.
Demographic and clinical characteristics, and nasal mucus lipid mediators of AERD inflammatory clusters
| Characteristic | Cluster 1 | Cluster 2 | Cluster 3 | P value |
|---|---|---|---|---|
| No. | 10 | 14 | 6 | |
| Age (y) | 41.8 ± 12.4 | 50.0 ± 12.4 | 47.8 ± 14.4 | .37 |
| Sex: female, n (%) | 5 (50) | 6 (43) | 1 (17) | .40 |
| Race: white, no. (%) | 10 (100) | 10 (71) | 6 (100) | .26 |
| BMI (kg/m2) | 29.1 ± 7.0 | 30.6 ± 3.2 | 29.2 ± 4.1 | .73 |
| Allergic rhinitis, n (%) | 9 (90) | 9 (64) | 5 (83) | .31 |
| NCS, n (%) | 7 (70) | 11 (79) | 5 (83) | .81 |
| LTR, n (%) | 6 (60) | 10 (71) | 3 (50) | .64 |
| Prior surgery, n (%) | 6 (60) | 11 (79) | 4 (67) | .61 |
| No. of previous surgeries | 1.0 (0.0-1.8) | 1.0 (1.0-2.8) | 2.0 (0.5-4.3) | .48 |
| Mean eosinophils/hpf | 124 (96-257) | 157 (80-250) | 108 (95-120) | .59 |
| SNOT-22 score | 46.6 ± 24.6 | 56.7 ± 21.5 | 42.3 ± 18.0 | .31 |
| Rhinologic | 11.7 ± 4.9 | 16.8 ± 5.8 | 15.5 ± 4.5 | .15 |
| Extranasal | 8.7 ± 4.5 | 9.9 ± 2.5 | 8.5 ± 3.0 | .47 |
| Ear/facial | 7.6 ± 5.6 | 11.9 ± 5.4 | 6.3 ± 3.2 | .08 |
| Psychological | 13.0 ± 8.2 | 14.6 ± 8.4 | 9.0 ± 7.0 | .41 |
| Sleep | 12.3 ± 9.4 | 11.4 ± 6.4 | 8.2 ± 9.2 | .63 |
| CT score | 21.1 ± 2.9 | 20.5 ± 2.6 | 19.3 ± 3.9 | .53 |
| Asthma classification | .74 | |||
| Persistent | 9 (90) | 13 (93) | 6 (100) | |
| Intermittent | 1 (10) | 1 (7) | 0 (0) | |
| Asthma status | .61 | |||
| Mild | 2 (20) | 1 (7) | 0 (0) | |
| Moderate | 2 (20) | 1 (7) | 2 (33) | |
| Severe | 7 (70) | 12 (86) | 4 (67) | |
| 12,13-EpOME | 11,304 (6,495-14,686) | 4,065 (2,224-10,679) | 2,737 (747-10,859) | .03 |
| 9,10-DiHOME | 22.7 (8.6-25.8) | 4.7 (3.2-14.4) | 3.4 (2.0-4.7) | .04 |
| 12,13-DiHOME | 1.2 (0.2-3.7) | 1.7 (0.0-3.7) | 0.0 (0.0-2.2) | .48 |
| 9,10-EpOME | 402 (280-4,920) | 235 (131-401) | 156 (61-530) | .07 |
| 13-HODE | 15,896 (11,297-19,632) | 5,283 (3,880-15,353) | 4,767 (1,803-7,366) | .02 |
| 8-HETE | 38.0 (18.4-69.7) | 25.0 (20.1-41.6) | 51.5 (18.4-64.4) | .61 |
| 12-HETE | 590 (545-931) | 321 (205-516) | 283 (193-486) | .006 |
| 15-HETE | 5,790 (3,141-6,848) | 4,569 (2,508-7,080) | 1,788 (1,297-3,143) | .10 |
| 11,12-EET | 211 (166-272) | 237 (168-392) | 224 (155-270) | .76 |
| 14,15-EET | 882 (657-1,144) | 794 (532-972) | 456 (188-740) | .25 |
| 20-HETE | 102,474 (45,493-125,811) | 84,706 (39,573-128,811) | 32,647 (23,116-59,291) | .08 |
| TxB2 | 63.8 (19.3-90.5) | 13.4 (4.7-19.3) | 17.8 (8.4-27.7) | .03 |
| PGD2 | 44.2 (27.6-70.2) | 25.2 (11.4-67.4) | 12.5 (3.6-24.5) | .08 |
| 15-Keto-PGE2 | 1.3 (0.9-1.6) | 1.4 (0.5-1.8) | 1.1 (0.6-1.5) | .99 |
| PGE2 | 108.5 (75.8-155.8) | 96.7 (70.1-137.8) | 37.3 (23.1-113.6) | .30 |
| PGF2a | 23.1 (7.5-33.6) | 3.0 (0.0-12.8) | 10.8 (1.9-14.9) | .10 |
| 9a,11b-PGF2a | 14.9 (8.5-21.5) | 9.0 (0.0-20.9) | 6.2 (0.0-13.4) | .30 |
| LTB4 | 0.0 (0.0-11.2) | 1.9 (0.0-11.2) | 0.0 (0.0-4.7) | .90 |
| LTC4 | 47.7 (19.7-74.7) | 24.6 (2.8-55.5) | 30.2 (19.0-38.4) | .46 |
| LTD4 | 2.8 (0.2-4.1) | 1.2 (0.0-2.1) | 1.8 (0.4-2.3) | .53 |
| LTE4 | 4.4 (3.7-10.5) | 9.8 (4.6-13.4) | 4.5 (0.9-9.9) | .52 |
| Total CysLTs | 65.7 (32.0-82.6) | 29.0 (18.2-74.0) | 40.3 (19.4-76.7) | .41 |
AERD, Aspirin-exacerbated respiratory disease; BMI, body mass index; CT, computed tomography; LTR, leukotriene modifier; NCS, nasal corticosteroid.
Values are presented as means ± SDs or medians with interquartile ranges, depending on the normalcy of the data. All mediator levels are presented as ng/mL. Data are presented as medians with interquartile range. Differences between groups were assessed by using the Kruskal-Wallis test followed by Dunn’s test for multiple comparisons or χ2 analysis. Boldface text indicates a P value of less than .05.
Given the clearly defined role of AA metabolism in AERD, we next compared levels of lipid mediators between the clusters. No differences in CysLTs were identified between the 3 clusters (Table IV). Conversely, the clusters varied significantly with respect to levels of 12,13 EpOME (P = .003), 9-10-DiHOME (P = .04), 13-hydroxyoctadedadienoic acid (HODE) (P = .02), 12-HETE (P = .006), and TxB2 (P = .02), with the highest levels in cluster 1. There was a trend toward differences in 15-HETE (P = .10), 20-HETE (P = .08), PGD2 (P = .09), and its metabolite PGF2α (P = .10). Hierarchical cluster analysis was repeated with lipid mediators as input variables but resulted in heavily overlapping clusters that were comparatively unstable (data not shown).
DISCUSSION
Our data show nasal cytokine dysregulation in AERD that is heterogeneous and potentially suggestive of disease subendo-types. We found that AERD could be discriminated from CRSwNP by elevations in IL-5, IL-6, IL-13, and IFN-γ. Although comparatively few studies have specifically analyzed cytokine expression in patients with AERD discretely, many have reported characteristics similar to CRSwNP with a predominantly type 2 signature. However, recent studies that specifically examined patients with AERD as a distinct group, though limited by sample size, have reported conflicting and often surprising findings. On the basis of gene expression in nasal polyps, Steinke et al10 reported reduced expression of the type 2 cytokines IL-5 and IL-13 in AERD (n = 15) compared with CRSwNP, whereas IL-4 and IFN-γ were both increased. They subsequently showed that IFN-γ could promote the maturation of eosinophil progenitors and increase the expression of genes involved in the synthesis of CysLTs. A subsequent study by Stevens et al11 found largely conflicting results. AERD polyps (n = 15) could be differentiated from CRSwNP by elevated protein levels of eosinophil cationic protein, but showed no differences in either IL-4, IL5, IL-13, or IFN-γ. Our study findings in a cohort double in size help to reconcile these previous reports and suggests that conflicting results may be indicative of heterogeneity within the AERD population.
Collectively, our data suggest that AERD is characterized by a predominantly type 2 signature, but also has characteristics of both type 1 and type 3 inflammation. Our finding that IL-6 is elevated in AERD and is highest in the cluster with the highest levels of type 2 cytokines may explain the propensity toward severe clinical presentations in AERD and a source for resistance to type 2-targeted therapies. Alterations in the IL-6 pathway have previously been reported in CRSwNP.24 In asthma, elevated plasma IL-6 is associated with low lung function and increased exacerbations25 and neutrophilic and mixed granulocytic airway inflammation,26 and is implicated in promoting the conversion of induced Treg cells into TH17-like cells.27 Our group recently reported that nasal mucus IL-6 levels are highest in patients with mixed granulocytic sinus infiltrate and more severe disease.15 The elevations in IL-17A and IFN-γ similarly highlight the complex inflammatory milieu of AERD.
We sought to deconvolute this inflammatory heterogeneity using an unstructured statistical approach and found that patients with AERD could be differentiated into 3 potential inflammatory clusters. Approximately half of all patients fell into a cluster with very high type 2 cytokines, with the remainder instead characterized by low inflammatory burden or elevated type 1 and type 3 cytokines. To our knowledge, this is the first study to find potential subendotypes of AERD using inflammatory mediators alone. Using latent class analysis, Bochenek et al identified 4 subphenotypes of AERD using clinical characteristics and a small number of serum and urine biomarkers.28 Although only a single lipid mediator was measured (urinary LTE4), the results did show that the class with the highest LTE4 levels also had the greatest upper respiratory and/or sinus symptoms. This is consistent with our study, which identified LTE4 as the only lipid mediator that correlated with worse SNOT-22 scores and was significantly elevated in AERD only. A similar approach was subsequently used by Celejewska-Wojcik et al,8 this time incorporating 16 variables that included 3 select eicosanoids (PGD2, PGE2, LTE4) in induced sputum supernatants and resulting in 3 distinct subphenotypes.8 This study found that the class with the highest levels of proinflammatory and anti-inflammatory AA metabolites was characterized by relatively well-controlled mild to moderate asthma and mixed eosinophilic and neutrophilic infiltrate in induced sputum. However, neither of these studies measured cytokine levels and were therefore unable to link clinical characteristics and/or eicosanoid levels with inflammatory signatures.
The results of the current study suggest that patients with AERD are fairly homogeneous with respect to lipid mediators in nasal secretions, though individual mediators clearly have well-defined physiological roles and may also act as disease modifiers. Patients with mild asthma severity had a global increase in cyclooxygenase metabolites with elevated levels of PGD2 and 9a,11b-PGF2a, both CRTH2 (choemoattractant receptor-homologous molecule expressed on TH2 cells) agonists important in effector cell chemotaxis to the tissue.29 Other PGD2 metabolites that were not assessed in this study and have been reported to have both proinflammatory and anti-inflammatory actions may explain the differences observed by asthma severity.30 Similarly, the anti-inflammatory PGE2 was highest, but not significantly elevated, in the mild asthma severity subgroup. The balance between proinflammatory and anti-inflammatory lipid mediators may dictate clinical severity as has been suggested by mediator analyses in the lower airways.8 We also found that sinonasal symptoms correlated with mucus levels of LTE4, but not with any other measured lipid mediator. LTE4 is a key mediator of chemosensory cell number and function,31 epithelial cell mucin production,32 and mast cell activation33 in the respiratory tract. It is notable that patients with AERD had a dramatic elevation in mucus LTE4 levels compared with controls and patients with CRSwNP despite similar levels of LTC4. Local differences in the enzymes required for LTC4 conversion to LTD4 (gamma-glutamyl leukotrienase and gamma-gluatmyl transpeptidase) and LTE4 (dipeptidase) or ω-oxidation, which would render LTE4 undetectable in our MS methods, may explain the differences observed between clinical phenotypes.34-37 However, the variability in most lipid mediators among patients with AERD was comparatively low, suggesting that heterogeneity in these patients may be driven by other mediators. As shown here, hierarchical cluster analysis using type 1, type 2, and type 3 cytokines resulted in well-delineated and stable disease clusters, whereas a similar approach using physiologically relevant eicosanoids was not able to effectively discriminate patients in any meaningful way.
We measured what to our knowledge is the largest array of nasal lipid mediators in patients with AERD. We found the highest levels of proinflammatory and anti-inflammatory mediators in cytokine cluster 1, which was generally associated with lower cytokine inflammatory burden and moderate disease severity. Cluster 1 (cytokine low) demonstrated the highest amount of LA derivatives 12,13-EpOME, 9-10-DiHOME, and 13-HODE, and eicosanoids 12-HETE and TxB2. This combination of LA-and AA-derived lipid mediators may come from neutrophil (12,13-EpOME [isoleukotoxin],38 9,10-DiHOME [leukotoxin diol],39 and 13-HODE40) and platelet (TxB2 and 12-HETE41) sources. 12,13-EpHOME, generated by CYP450 from LA during oxidative burst, uncouples mitochondrial respiration42 and is present in the lavage fluid of acute respiratory distress syndrome.43 9,10-diHOME is generated from 9(10)-EpOME (leukotoxin) by soluble epoxide hydrolase in neutrophils.39 13-HODE is produced by nonenzymatic lipid peroxidation of LA. In a human endothelial cell line, 9,10-DiHOME, 13-HODE, and 12-HETE were generated in response to LPS stimulation and suppressed TNF-α.44 In epithelial cells, 15-lipooxygenase (LO) expression is induced by IL-13 and contributes to the generation of 15-HETE from AA and 13-HODE from LA.45 13-HODE activates TRPV1, a neurosensory receptor, which is increased in CRSwNP and triggers IgE-independent mast cell activation.46 Platelet-derived 12-HETE is a potent mediator of neutrophil chemotaxis.47 Depending on the stimulus studied, 12-HETE has been shown to have promoting and suppressing effects on platelet activation.48 Notably 12-HETE interacts with the TXA2 receptor and inhibits TXA2-induced platelet aggregation.49,50 Platelets and TXA2 receptor have been identified as key factors required for inflammation in AERD.51,52 Platelets serve as a source of the alarmin IL-3353 and the excessive CysLT generation characteristic of AERD.54,55 In animal models of AERD, inhibition of TXA2 receptor blocks the acute aspirin-induced respiratory reaction, CysLT generation, and mast cell activation.52 Together these data support that AA and LA derivates outside of the classical cyclooxygenase and 5-lipoxygenase pathways may be key regulators of nasal inflammation in a subgroup of patients with AERD.
Some limitations to the current study deserve discussion and may limit the generalizability of our findings. Patients were seen at a single tertiary referral center within the southeastern United States and as such may not be representative of patients with AERD from other geographic regions. The sample size of patients with AERD, though substantially larger than in similar recent studies,10,11 was likely insufficiently powered for some comparisons and limited the number of variables that could be incorporated into cluster analyses. In addition, 63% of patients with AERD were being treated with a leukotriene receptor antagonist at the time of sample collection, which could potentially affect levels of some lipid mediators (though none were taking leukotriene synthesis inhibitors). We also measured cytokines and lipid mediators in sinonasal mucus rather than in tissue, as had been performed in a small number of other studies.10,11 However, we have found a strong correlation between levels of mediators in mucus and tissue (see Fig E3 in this article’s Online Repository at www.jacionline.org). Nonetheless, our findings suggest that patients with AERD who are phenotypically similar may have substantial differences in underlying inflammatory burden. Although AERD has a near-universal association with type 2 inflammation, we additionally found that type 1 and type 3 cytokine mediators may have pathophysiological roles in many patients, a finding that may have important implications for treatment. Humanized mAbs that target type 2 inflammation cytokine pathways are now recognized as effective therapeutics for CRSwNP, with some studies showing a potential subgroup effect in patients with AERD.44 This may be secondary to higher levels of type 2 cytokines in patients with AERD; however, the inflammatory heterogeneity shown here suggests that these therapeutics may have reduced efficacy in a substantial subset of patients.
Conclusions
Patients with AERD display heterogeneous inflammatory burden with variable levels of type 1, type 2, and type 3 cytokines. Between-patient differences in CysLTs and other lipid mediators were limited, but select mediators were associated with both asthma and sinonasal symptom severity. Our findings suggest that subendotypes of AERD may exist and point to the need for further research into AERD pathophysiology. Improved characterization of disease subtypes or endotypes may help to effectively identify patients most likely to benefit from current and future targeted therapies.
Supplementary Material
Clinical implications: AERD is homogenous with respect to lipid mediators but has variable levels of type 1, type 2, and type 3 cytokines, suggesting likely variability in response to therapeutics that target type 2 inflammation.
Acknowledgments
This project was supported by the National Institutes of Health (grant nos. R21 AI142321 and R01 AG065550 to J.H.T. and grant no. K23 AI118804 to K.N.C.) and Clinical Translational Science Award (CTSA) award UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Abbreviations used
- AA
Arachidonic acid
- AERD
Aspirin-exacerbated respiratory disesase
- CRSwNP
Chronic rhinosinusitis with nasal polyps
- CysLT
Cysteinyl leukotriene
- DiHome
Dihydroxyoctadecenoic acid
- EpOME
Epoxyoctadecenoic acid
- HETE
Hydroxyeicosatetraenoic acid
- HODE
Hydroxyoctadedadienoic acid
- LA
Linoleic acid
- LT
Leukotriene
- PG
Prostaglandin
- SNOT-22
22-item Sinonasal Outcome Test
- Tx
Thromboxane
Footnotes
Disclosure of potential conflicts of interest: K. N. Cahill has received grant support from the National Institutes of Health (NIH)/National Institute of Allergy and Infectious Disease (NIAID) and personal fees from Novartis, Regeneron, GlaxoSmithKline, Blueprint Medicines, and Third Harmonic Bio. R. K. Chandra is a consultant for Regeneron and Optinose. N. I. Chowdhury is a consultant for Optinose, Inc. J. H. Turner has received grant support from the NIH/NIAID and NIH/National Institute on Aging and personal fees from Regeneron. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Tan BK, Chandra RK, Pollak J, Kato A, Conley DB, Peters AT, et al. Incidence and associated premorbid diagnoses of patients with chronic rhinosinusitis. J Allergy Clin Immunol 2013;131:1350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meltzer EO, Hamilos DL. Rhinosinusitis diagnosis and management for the clinician: a synopsis of recent consensus guidelines. Mayo Clin Proc 2011;86:427–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang JE, White A, Simon RA, Stevenson DD. Aspirin-exacerbated respiratory disease: burden of disease. Allergy Asthma Proc 2012;33:117–21. [DOI] [PubMed] [Google Scholar]
- 4.Stevens WW, Peters AT, Hirsch AG, Nordberg CM, Schwartz BS, Mercer DG, et al. Clinical characteristics of patients with chronic rhinosinusitis with nasal polyps, asthma, and aspirin-exacerbated respiratory disease. J Allergy Clin Immunol Pract 2017;5:1061–70.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens WW, Schleimer RP, Chandra RK, Peters AT. Biology of nasal polyposis. J Allergy Clin Immunol 2014;133:1503.e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poposki JA, Klingler AI, Tan BK, Soroosh P, Banie H, Lewis G, et al. Group 2 innate lymphoid cells are elevated and activated in chronic rhinosinusitis with nasal polyps. Immun Inflamm Dis 2017;5:233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato A Immunopathology of chronic rhinosinusitis. Allergol Int 2015;64:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celejewska-Wojcik N, Wojcik K, Ignacak-Popiel M, Cmiel A, Tyrak K, Gielicz A, et al. Subphenotypes of nonsteroidal antiinflammatory disease-exacerbated respiratory disease identified by latent class analysis. Allergy 2020;75:831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-Novo CA, Watelet JB, Claeys C, Van Cauwenberge P, Bachert C. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol 2005;115:1189–96. [DOI] [PubMed] [Google Scholar]
- 10.Steinke JW, Liu L, Huyett P, Negri J, Payne SC, Borish L. Prominent role of IFN-γ in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 2013;132:856–65.e1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens WW, Ocampo CJ, Berdnikovs S, Sakashita M, Mahdavinia M, Suh L, et al. Cytokines in chronic rhinosinusitis: role in eosinophilia and aspirin-exacerbated respiratory disease. Am J Respir Crit Care Med 2015;192:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner JH, Chandra RK, Li P, Bonnet K, Schlundt DG. Identification of clinically relevant chronic rhinosinusitis endotypes using cluster analysis of mucus cytokines. J Allergy Clin Immunol 2018;141:1895–7.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morse JC, Li P, Ely KA, Shilts MH, Wannemuehler TJ, Huang LC, et al. Chronic rhinosinusitis in elderly patients is associated with an exaggerated neutrophilic proinflammatory response to pathogenic bacteria. J Allergy Clin Immunol 2019;143:990–1002.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 2016;137:1449–56.e4. [DOI] [PubMed] [Google Scholar]
- 15.Succar EF, Li P, Ely KA, Chowdhury NI, Chandra RK, Turner JH. Neutrophils are underrecognized contributors to inflammatory burden and quality of life in chronic rhinosinusitis. Allergy 2020;75:713–6. [DOI] [PubMed] [Google Scholar]
- 16.Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S, et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 2020;58:1–464. [DOI] [PubMed] [Google Scholar]
- 17.Orlandi RR, Kingdom TT, Hwang PH, Smith TL, Alt JA, Baroody FM, et al. International Consensus Statement on Allergy and Rhinology: Rhinosinusitis. Int Forum Allergy Rhinol 2016;6:S22–209. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy JL, Hubbard MA, Huyett P, Patrie JT, Borish L, Payne SC. Sino-nasal outcome test (SNOT-22): a predictor of postsurgical improvement in patients with chronic sinusitis. Ann Allergy Asthma Immunol 2013;111:246–51.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner JH, Li P, Chandra RK. Mucus T helper 2 biomarkers predict chronic rhinosinusitis disease severity and prior surgical intervention. Int Forum Allergy Rhinol 2018;8:1175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morse JC, Shilts MH, Ely KA, Li P, Sheng Q, Huang LC, et al. Patterns of olfactory dysfunction in chronic rhinosinusitis identified by hierarchical cluster analysis and machine learning algorithms. Int Forum Allergy Rhinol 2019;9:255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Chandra RK, Li P, Hull BP, Turner JH. Olfactory and middle meatal cytokine levels correlate with olfactory function in chronic rhinosinusitis. Laryngoscope 2018;128:E304–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorsuch RL. Exploratory factor analysis: its role in item analysis. J Pers Assess 1997;68:532–60. [DOI] [PubMed] [Google Scholar]
- 23.O’Rourke N, Hatcher L. A step-by-step approach to using the SAS system for factor analysis and structural equation modeling. 2nd ed. Cary, NC: SAS Institute; 2013. [Google Scholar]
- 24.Peters AT, Kato A, Zhang N, Conley DB, Suh L, Tancowny B, et al. Evidence for altered activity of the IL-6 pathway in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2010;125:397–403.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med 2016;4:574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ullah MA, Revez JA, Loh Z, Simpson J, Zhang V, Bain L, et al. Allergen-induced IL-6 trans-signaling activates γδ T cells to promote type 2 and type 17 airway inflammation. J Allergy Clin Immunol 2015;136:1065–73. [DOI] [PubMed] [Google Scholar]
- 27.Massoud AH, Charbonnier LM, Lopez D, Pellegrini M, Phipatanakul W, Chatila TA. An asthma-associated IL4R variant exacerbates airway inflammation by promoting conversion of regulatory T cells to TH17-like cells. Nat Med 2016;22:1013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bochenek G, Kuschill-Dziurda J, Szafraniec K, Plutecka H, Szczeklik A, Nizankowska-Mogilnicka E. Certain subphenotypes of aspirin-exacerbated respiratory disease distinguished by latent class analysis. J Allergy Clin Immunol 2014;133:98–103.e1-6. [DOI] [PubMed] [Google Scholar]
- 29.Sandig H, Andrew D, Barnes AA, Sabroe I, Pease J. 9alpha,11beta-PGF2 and its stereoisomer PGF2alpha are novel agonists of the chemoattractant receptor, CRTH2. FEBS Lett 2006;580:373–9. [DOI] [PubMed] [Google Scholar]
- 30.Sandig H, Pease JE, Sabroe I. Contrary prostaglandins: the opposing roles of PGD2 and its metabolites in leukocyte function. J Leukoc Biol 2007;81:372–82. [DOI] [PubMed] [Google Scholar]
- 31.Bankova LG, Dwyer DF, Yoshimoto E, Ualiyeva S, McGinty JW, Raff H, et al. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25-producing airway brush cells leading to type 2 inflammation. Sci Immunol 2018;3:eaat9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci U S A 2016;113:6242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazarinis N, Bood J, Gomez C, Kolmert J, Lantz AS, Gyllfors P, et al. Leukotriene E4 induces airflow obstruction and mast cell activation through the cysteinyl leukotriene type 1 receptor. J Allergy Clin Immunol 2018;142:1080–9. [DOI] [PubMed] [Google Scholar]
- 34.Carter BZ, Wiseman AL, Orkiszewski R, Ballard KD, Ou CN, Lieberman MW. Metabolism of leukotriene C4 in gamma-glutamyl transpeptidase-deficient mice. J Biol Chem 1997;272:12305–10. [DOI] [PubMed] [Google Scholar]
- 35.Orning L, Hammarstrom S Inhibition of leukotriene C and leukotriene D biosynthesis. J Biol Chem 1980;255:8023–6. [PubMed] [Google Scholar]
- 36.Lee CW, Lewis RA, Corey EJ, Austen KF Conversion of leukotriene D4 to leukotriene E4 by a dipeptidase released from the specific granule of human polymorphonuclear leucocytes. Immunology 1983;48:27–35. [PMC free article] [PubMed] [Google Scholar]
- 37.Keppler D, Huber M, Baumert T, Guhlmann A Metabolic inactivation of leukotrienes. Adv Enzyme Regul 1989;28:307–19. [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa M, Sugiyama S, Takamura T, Yokoo K, Iwata M, Suzuki K, et al. Neutrophils biosynthesize leukotoxin, 9, 10-epoxy-12-octadecenoate. Biochem Biophys Res Commun 1986;137:424–30. [DOI] [PubMed] [Google Scholar]
- 39.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med 1997;3:562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archambault AS, Turcotte C, Martin C, Provost V, Larose MC, Laprise C, et al. Comparison of eight 15-lipoxygenase (LO) inhibitors on the biosynthesis of 15-LO metabolites by human neutrophils and eosinophils. PLoS One 2018;13:e0202424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plaza V, Prat J, Rosello J, Ballester E, Ramis I, Mullol J, et al. In vitro release of arachidonic acid metabolites, glutathione peroxidase, and oxygen-free radicals from platelets of asthmatic patients with and without aspirin intolerance. Thorax 1995;50:490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozawa T, Hayakawa M, Takamura T, Sugiyama S, Suzuki K, Iwata M, et al. Biosynthesis of leukotoxin, 9,10-epoxy-12 octadecenoate, by leukocytes in lung lavages of rat after exposure to hyperoxia. Biochem Biophys Res Commun 1986;134:1071–8. [DOI] [PubMed] [Google Scholar]
- 43.Ozawa T, Sugiyama S, Hayakawa M, Satake T, Taki F, Iwata M, et al. Existence of leukotoxin 9,10-epoxy-12-octadecenoate in lung lavages from rats breathing pure oxygen and from patients with the adult respiratory distress syndrome. Am Rev Respir Dis 1988;137:535–40. [DOI] [PubMed] [Google Scholar]
- 44.Askari AA, Thomson S, Edin ML, Lih FB, Zeldin DC, Bishop-Bailey D Basal and inducible anti-inflammatory epoxygenase activity in endothelial cells. Biochem Biophys Res Commun 2014;446:633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, Minami Y, Etling E, Coleman JM, Lauder SN, Tyrrell V, et al. Preferential generation of 15-HETE-PE induced by IL-13 regulates goblet cell differentiation in human airway epithelial cells. Am J Respir Cell Mol Biol 2017;57:692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toth E, Tornoczky T, Kneif J, Perkecz A, Katona K, Piski Z, et al. Upregulation of extraneuronal TRPV1 expression in chronic rhinosinusitis with nasal polyps. Rhinology 2018;56:245–54. [DOI] [PubMed] [Google Scholar]
- 47.Turner SR, Tainer JA, Lynn WS Biogenesis of chemotactic molecules by the arachidonate lipoxygenase system of platelets. Nature 1975;257:680–1. [DOI] [PubMed] [Google Scholar]
- 48.Porro B, Songia P, Squellerio I, Tremoli E, Cavalca V Analysis, physiological and clinical significance of 12-HETE: a neglected platelet-derived 12-lipoxygenase product. J Chromatogr B Analyt Technol Biomed Life Sci 2014;964:26–40. [DOI] [PubMed] [Google Scholar]
- 49.Croset M, Lagarde M Stereospecific inhibition of PGH2-induced platelet aggregation by lipoxygenase products of icosaenoic acids. Biochem Biophys Res Commun 1983;112:878–83. [DOI] [PubMed] [Google Scholar]
- 50.Fonlupt P, Croset M, Lagarde M 12-HETE inhibits the binding of PGH2/TXA2 receptor ligands in human platelets. Thromb Res 1991;63:239–48. [DOI] [PubMed] [Google Scholar]
- 51.Liu T, Garofalo D, Feng C, Lai J, Katz H, Laidlaw TM, et al. Platelet-driven leukotriene C4-mediated airway inflammation in mice is aspirin-sensitive and depends on T prostanoid receptors. J Immunol 2015;194:5061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu T, Laidlaw TM, Katz HR, Boyce JA Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A 2013;110:16987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeda T, Unno H, Morita H, Futamura K, Emi-Sugie M, Arae K, et al. Platelets constitutively express IL-33 protein and modulate eosinophilic airway inflammation. J Allergy Clin Immunol 2016;138:1395–403.e6. [DOI] [PubMed] [Google Scholar]
- 54.Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood 2012;119:3790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitsui C, Kajiwara K, Hayashi H, Ito J, Mita H, Ono E, et al. Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 2016;137:400–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.