

Abstract
Human antigen R (HuR) is a widespread RNA-binding protein involved in homeostatic regulation and pathological processes in many diseases. Atherosclerosis is the leading cause of cardiovascular disease and acute cardiovascular events. However, the role of HuR in atherosclerosis remains unknown. In this study, mice with smooth muscle-specific HuR knockout (HuRSMKO) were generated to investigate the role of HuR in atherosclerosis. HuR expression was reduced in atherosclerotic plaques. As compared with controls, HuRSMKO mice showed increased plaque burden in the atherosclerotic model. Mechanically, HuR could bind to the mRNAs of adenosine 5′-monophosphate-activated protein kinase (AMPK) α1 and AMPKα2, thus increasing their stability and translation. HuR deficiency reduced p-AMPK and LC3II levels and increased p62 level, thereby resulting in defective autophagy. Finally, pharmacological AMPK activation induced autophagy and suppressed atherosclerosis in HuRSMKO mice. Our findings suggest that smooth muscle HuR has a protective effect against atherosclerosis by increasing AMPK-mediated autophagy.
Subject terms: Cardiovascular diseases, Pathogenesis
Introduction
Atherosclerosis is a chronic and systemic vascular inflammatory process, the pathological basis of coronary artery disease, myocardial infarction, and stroke1. Despite many treatment options, it remains the leading cause of death worldwide. Developing new strategies to prevent plaque formation and rupture has become an important research area. Atherosclerosis is initiated by endothelial dysfunction and vascular inflammation caused by cardiovascular risk factors such as hyperlipidemia and hypertension. Lipoproteins in the blood enter the arterial wall from the damaged endothelial cells2,3. Inflammatory factors can stimulate monocytes and vascular smooth muscle cells (VSMCs) to engulf oxidized low-density lipoprotein (ox-LDL) and form foam cells4. Disordered lipid metabolism, inflammation, and endothelial injury all seem to play major roles in atherosclerosis4,5. However, the pathogenesis of atherosclerosis is still unclear and needs further study.
VSMCs are crucial in atherosclerosis. Most foam cells in early atherosclerosis are derived from VSMCs6. Aberrant proliferation and migration followed by phenotypic switching of VSMCs are involved in the formation of atherosclerosis7. VSMCs produce the extracellular matrix, which forms the fibrous cap to prevent plaque rupture8. The death and senescence of VSMCs participate in the formation of atherosclerotic plaque and also promote the instability of plaque in advanced lesions7,9. However, the specific regulatory mechanism of VSMCs in atherosclerosis is not clear.
Autophagy is an essential subcellular process that has a “housekeeping” role in the normal physiological functions of the body. Autophagy participates in numerous physiological and pathological processes, including cell differentiation, growth regulation, aging, immunity, and tumor suppression10. Autophagy is a multi-step process that requires a variety of autophagy-related proteins that take part in nucleation, expansion, and finally fusion with lysosomes of autophagosomes11,12. Accumulating evidence suggests that autophagy is involved in the occurrence and development of atherosclerosis and related diseases13,14. Smooth muscle-specific Atg7 knockout can promote atherosclerosis15, but the molecular mechanism of autophagy in atherosclerosis still needs elucidation.
Human antigen R (HuR) is a ubiquitous and conserved RNA-binding protein that binds to AU-rich elements (AREs) and alters ARE-mediated mRNA turnover and translation16. HuR binds to an extensive list of RNAs that participate in cell proliferation, apoptosis, and differentiation17,18. Therefore, HuR may have an important effect on both pathologic and physiologic functions. Although cancer is the most widely studied disease associated with HuR19, the molecule is also reported to take part in chronic inflammation and nervous system diseases18,20. Our recent work showed that adipose tissue-specific HuR knockout caused obesity and metabolic disorders21. Also, knockout of HuR enhanced VSMC contraction and hypertension22. However, the role of HuR in autophagy and atherosclerosis remains unclear.
In this study, we examined HuR expression in atherosclerotic plaques and found its expression decreased. Using smooth muscle-specific HuR knockout (HuRSMKO) mice, smooth-muscle HuR deletion promoted atherosclerosis by inducing defective autophagy.
Results
HuR levels were reduced in atherosclerotic plaque
To investigate the role of HuR in atherosclerosis, we fed ApoE−/− mice with an ND or HFD for 12 weeks. HuR protein level was decreased in aortas from the HFD group (Fig. 1A, B). As compared with the ND group, for the HFD group, HuR was downregulated in atherosclerotic lesions (Fig. 1C). As well, HuR mRNA level was significantly reduced in aortas from the HFD group (Fig. 1D). HuR protein level was decreased in VSMCs induced by ox-LDL at 24, 48, 72, and 96 h (Fig. 1E, F). For further study, we used HuRSMKO mice generated from a hybrid of HuR-floxed mice and α-SMA-Cre transgenic mice. HuR protein level was downregulated in aortas from HuRSMKO mice (Fig. 1G, H). HuR was not expressed in the aortic smooth muscle layer of HuRSMKO mice as compared with control mice (Fig. 1I). In summary, the expression of HuR in atherosclerotic plaques was decreased, which suggests that HuR acts on atherosclerosis.
Fig. 1. HuR levels were reduced in atherosclerotic plaque.

A, B Western blot analysis of HuR protein level in aortas from apolipoprotein E-knockout (ApoE−/−) mice fed a high-fat diet (HFD) or normal diet (ND) (n = 5). C Immunohistochemical staining of HuR in aortas from ApoE−/− mice fed an HFD or ND (n = 5). D Quantitative RT-PCR analysis of HuR mRNA level in aortas from ApoE−/− mice fed an HFD or ND (n = 5). E, F Western blot analysis of HuR protein level in vascular smooth muscle cells (VSMCs) induced by 50 μg/ml oxidized low-density lipoprotein (ox-LDL) at different times (n = 5). G, H Western blot analysis of HuR protein level in aortas from control and HuRSMKO mice (n = 5). I Immunofluorescent staining of aortas from control and HuRSMKO mice to determine HuR (green) and α-SMA (α-smooth muscle actin; red) localization. Scale bar = 50 μm.
HuR deletion in smooth muscle exacerbated atherosclerosis
To explore the role of smooth-muscle HuR in atherosclerosis, control, and HuRSMKO mice were injected with rAAV/D377Y-mPCSK9 then fed a Paigen diet for 12 weeks. The proportion of atherosclerotic surface lesions was greater in HuRSMKO than control mice (33.98 ± 6.56% vs 14.68 ± 2.47%, p < 0.001) (Fig. 2A). Oil-red O-stained aortic roots showed significantly increased lesion area in HuRSMKO than control mice (43.38 ± 2.75% vs 28.80 ± 4.66%, p < 0.01) (Fig. 2B). Furthermore, deletion of HuR increased macrophage accumulation (Fig. 2C) and matrix metalloproteinase 2 (MMP2) level (Supplementary Fig. 2B), decreased collagen content (Fig. 2D). However, VSMC content and total monocyte/macrophages did not differ from controls (Fig. 2E and Supplementary Fig. 2A). From the above results, we calculated the plaque vulnerability index, which was elevated after HuR deficiency (Fig. 2F). Taken together, lack of HuR in VSMCs promoted the development of atherosclerosis.
Fig. 2. HuR deletion in smooth muscle cells exacerbated atherosclerosis.

CTR and HuRSMKO mice were injected with rAAV/D377Y-mPCSK9 and fed a Paigen diet for 12 weeks. A Oil-red O staining in aortas (n = 10). B Oil-red O staining in aortic roots (n = 6). Scale bar = 200 μm. c Immunohistochemical staining of MOMA-2 in aortic roots (n = 6). Scale bar = 200 μm. D Masson staining in aortic roots (n = 6). Scale bar = 200 μm. E Immunohistochemical staining of α-SMA (n = 6). Scale bar = 200 μm. F Vulnerable index of atherosclerotic plaques (n = 6).
Loss of HuR promoted apoptosis in atherosclerosis
Numerous studies have confirmed the existence of apoptosis in atherosclerotic plaques, which even affects the stability of plaques7,23. To determine whether HuR knockout affected apoptosis, aortic root sections underwent TUNEL staining. Loss of HuR markedly increased the TUNEL-positive SMCs in HuRSMKO versus control mice (Fig. 3A). Meanwhile, the expression of the apoptosis-related protein cleaved caspase-3 was upregulated in aortic roots of HuRSMKO mice (Fig. 3B). Besides, HuR deletion also increased the level of cleaved caspase-3 in SMCs (Fig. 3D). Also, the serum levels of TC, TG, LDL-C were higher in HuRSMKO than control mice (Fig. 3C). To learn why HuRSMKO mice have the phenotype of dyslipidemia, HuR expression in hepatocytes and liver fibroblasts were detected. Results showed that HuR expression was no difference in hepatocytes, but decreased in liver fibroblasts from HuRSMKO mice compared with control (Supplementary Fig. 1A, B). We further examined serum aspartate transaminase (AST) and alanine transaminase (ALT) levels and found elevated AST level from HuRSMKO mice (Supplementary Fig. 1C, D). Thus, HuR deficiency in smooth muscle increased apoptosis in atherosclerotic mice, and loss of HuR in liver fibroblasts may contribute to hepatic dysfunction and dyslipidemia.
Fig. 3. Loss of HuR promoted apoptosis in atherosclerosis.

A Immunofluorescent staining of aortic roots from CTR and HuRSMKO mice to determine TUNEL-positive VSMCs. Red puncta denotes TUNEL-positive cells. Green region denotes α-SMA. Scale bar = 20 μm. B Immunohistochemical staining of cleaved caspase-3 in aortic roots (n = 6). Scale bar = 50 μm. C Serum lipid profiles (total cholesterol [TC], triglycerides [TG], high-density lipoprotein cholesterol [HDL-C], and low-density lipoprotein cholesterol [LDL-C]) (n = 6). D Western blot analysis of cleaved caspase-3 in control and HuR-deficient SMCs (n = 5).
HuR deletion resulted in defective autophagy
VSMC autophagy was reported to be important during the process of atherosclerosis14,15. To detect whether HuR could regulate autophagy, we used transmission electron microscopy after VSMCs were infected with ad-HuR or ad-LacZ for 48 h. HuR overexpression increased the number of autophagosomes (Fig. 4A). Also, autophagic flux was monitored in VSMCs by infection with ad-GFP-mRFP-LC3II. As compared with controls, VSMCs with HuR overexpression by ad-HuR infection showed an increased number of GFP+/RFP+ and GFP−/RFP+ LC3II puncta (Fig. 4B–D). However, VSMCs knocked down by HuR siRNA transfection showed decreased number of GFP+/RFP+ and GFP−/RFP+ LC3II puncta as compared with controls (Fig. 4E–G). Thus, HuR overexpression induced autophagic flux, and loss of HuR suppressed autophagic flux and resulted in defective autophagy.
Fig. 4. HuR deletion resulted in defective autophagy.

A Transmission electron microscopy of autophagosomes (black arrow) in VSMCs infected with ad-HuR or ad-LacZ. Scale bar = 5000 nm (left), 1000 nm (middle), 500 nm (right). B–D fluorescence photomicrographs (B) and quantification (C, D) of GFP-mRFP-LC3II puncta in VSMCs infected with ad-HuR or ad-LacZ (n = 26). Scale bar = 10 μm. E–G Fluorescence photomicrographs (E) and quantification (F, G) of GFP-mRFP-LC3II puncta in VSMCs transfected with control or HuR siRNA (n = 26). Scale bar = 10 μm. Yellow puncta denotes autophagosome. Red puncta denotes autolysosome.
AMPKα1 and AMPKα2 were the target genes of HuR
As a key factor in cellular energy metabolism and activator of autophagy, AMP-activated protein kinase (AMPK) participates in various physiological and pathological processes24. The mRNA levels of AMPKα1 and AMPKα2 were decreased in aortas from HuRSMKO mice (Fig. 5A). To detect whether AMPKα was the HuR target gene, the sequence of mouse AMPKα transcripts was analyzed and there are 3 AREs in the 3′ UTR of AMPKα1 and 4 AREs in the 3′ UTR of AMPKα2. Next, we examined the binding of HuR to AMPKα1 and AMPKα2 mRNA by RNA immunoprecipitation and mRNA stability assay. HuR could bind to the mRNAs of AMPKα1 and AMPKα2 (Fig. 5B). Meanwhile, the stability of AMPKα1 and AMPKα2 mRNAs was increased by HuR overexpression (Fig. 5C, D), but decreased by HuR deficiency (Supplementary Fig. 3A, B). Then we further verified the relationship between HuR and AMPKα. VSMCs were stimulated with 30 μM CMLD-2, a HuR inhibitor, for 24 h. The protein expression of AMPKα was sharply reduced in VSMCs with CMLD-2 treatment (Fig. 5E) and AMPKα1 and AMPKα2 protein levels in VSMCs were reduced after HuR knockdown by HuR siRNA transfection (Fig. 5F). In contrast, the levels of AMPKα1 and AMPKα2 were increased in VSMCs with HuR overexpression by ad-HuR infection or HuR recombinant-protein stimulation (Fig. 5G, H). The protein levels of AMPKα1 and AMPKα2 were significantly reduced in HuRSMKO versus control mice (Fig. 5I). Immunohistochemical staining results for AMPKα1 and AMPKα2 in aortas from control and HuRSMKO mice were consistent with the above results (Fig. 5J). The effect of HuR on AMPK expression was further confirmed by oxLDL-stimulated SMCs after HuR knockdown or expression (Supplementary Fig. 4A, B). Therefore, AMPKα1 and AMPKα2 are the target genes of HuR.
Fig. 5. AMPKα1 and AMPKα2 are the target genes of HuR.

A Quantitative RT-PCR analysis of aortic mRNA levels of AMPKα1, AMPKα2 from CTR and HuRSMKO mice (n = 5). B RNA immunoprecipitation with anti-HuR or control IgG antibody. Lanes 1, 5, no template PCR control; lanes 2, 6, IgG RNA immunoprecipitation; lanes 3, 7, anti-HuR RNA immunoprecipitation; lanes 4, 8, 10% input. C, D VSMCs were infected with adenovirus-expressing GFP (green fluorescent protein) or HuR and then treated with actinomycin D (5 μg/ml). Quantified RT-PCR analysis of percentage mRNA levels of AMPKα1 (C) and AMPKα2 (D) (n = 5) in VSMCs. E Western blot analysis of AMPKα1 and AMPKα2 in VSMCs treated with DMSO or 30 μM CMLD-2 for 24 h (n = 5). F Western blot analysis of AMPKα1 and AMPKα2 in VSMCs transfected with CTR siRNA or HuR siRNA for 48 h (n = 5). Western blot analysis of AMPKα1 and AMPKα2 in VSMCs, G infected with ad-GFP or HuR (n = 5), and H treated with PBS or 0.5 μg/μl recombinant HuR protein for 48 h (n = 5). I Western blot analysis of AMPKα1 and AMPKα2 in aortas from CTR and HuRSMKO mice (n = 5). J Immunohistochemical staining of AMPKα1 and AMPKα2 in aortas from CTR and HuRSMKO mice (n = 5). Scale bar = 100 μm.
HuR positively regulates autophagy
Because AMPK could induce autophagy25 and AMPKα is the HuR target gene, we investigated whether HuR regulates autophagy. HuR inhibition with CMLD-2 or HuR siRNA decreased levels of p-AMPK and LC3II and increased p62 level in VSMCs (Fig. 6A, B). In contrast, levels of p-AMPK and LC3II were elevated and that of p62 was decreased with HuR overexpression by ad-HuR infection or HuR recombinant-protein stimulation (Fig. 6C, D). Furthermore, as compared with control mice, HuRSMKO mice showed lower levels of p-AMPK and LC3II and higher level of p62 (Fig. 6E). Thus, HuR positively regulates autophagy.
Fig. 6. HuR positively regulates autophagy.

Western blot analysis of p-AMPK, p62, and LC3II in VSMCs. A Treated with DMSO or 30 μM CMLD-2 for 24 h (n = 5). B Transfected with CTR or HuR siRNA for 48 h (n = 5). C Infected with ad-GFP or ad-HuR (n = 5), and D treated with PBS or 0.5 μg/μl recombinant HuR protein for 48 h (n = 5). E Western blot analysis of p-AMPK, p62, and LC3II in aortas from CTR and HuRSMKO mice (n = 5).
Pharmacological AMPK activation induced autophagy and suppressed atherosclerosis in HuRSMKO mice
To further demonstrate that HuR regulates autophagy via AMPK, VSMCs were transfected with control or HuR siRNA then treated with the AMPK activator A769662. The levels of p-AMPK and LC3II were increased with A769662 in control and HuR-deficient VSMCs (Fig. 7A). In animal experiments, control and HuRSMKO mice were given an intraperitoneal injection of A769662 daily and fed a Paigen diet for 12 weeks after single intravenous injection of rAAV/D377Y-mPCSK9. The plaque area was significantly decreased after A769662 treatment in control and HuRSMKO mice (Fig. 7B, C). Also, A769662 reduced the number of apoptotic cells (Fig. 7D). Furthermore, only TC and LDL-C blood levels were slightly reduced with A769662 treatment (Fig. 7E). Thus, smooth-muscle HuR protects against the development of atherosclerosis via AMPK-mediated autophagy.
Fig. 7. Pharmacological AMPK activation induced autophagy and suppressed atherosclerosis in HuRSMKO mice.
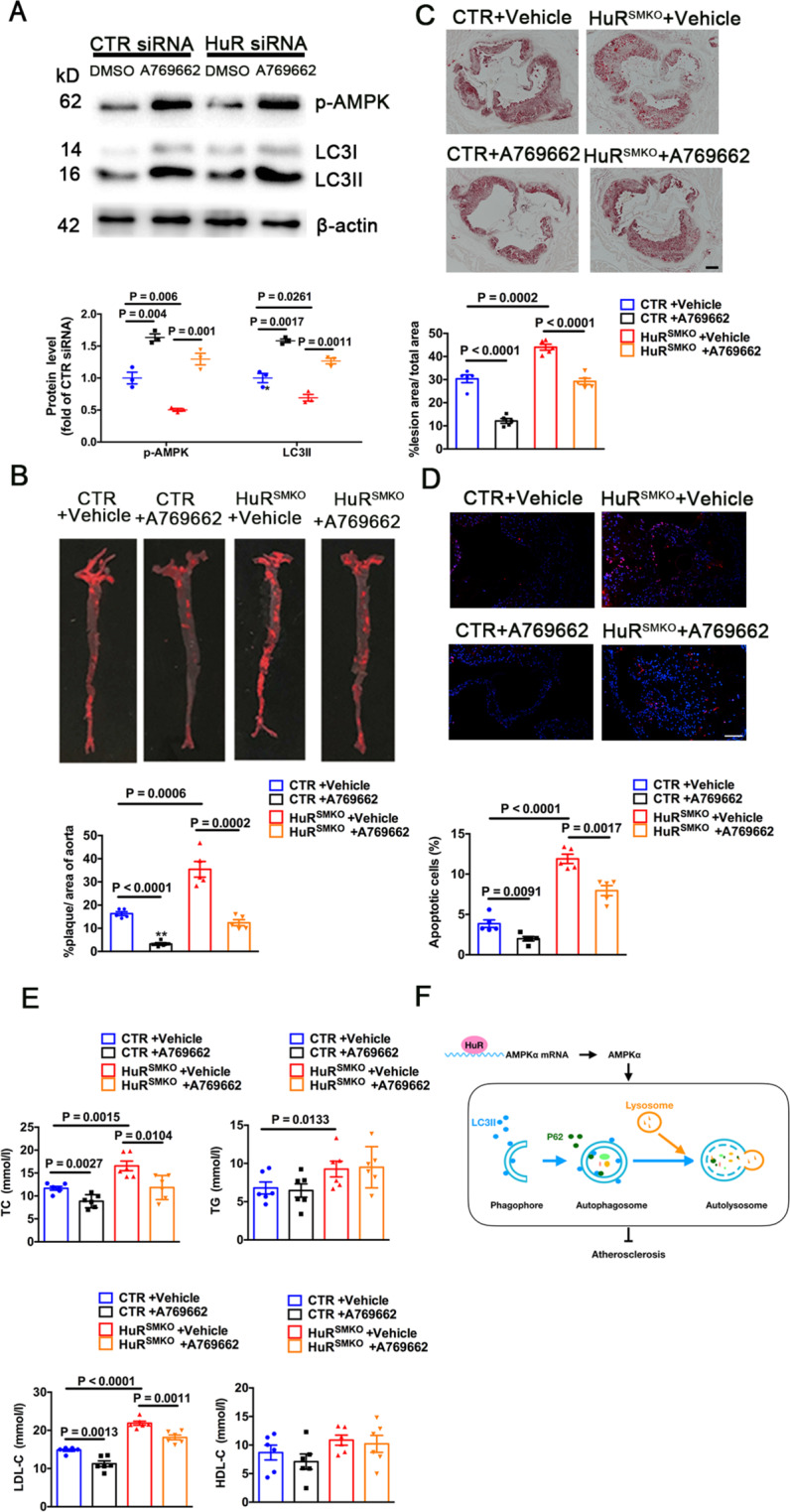
A Western blot analysis of VSMCs transfected with CTR or HuR siRNA for 48 h, then treated with 200 μM A769662 for 1 h (n = 5). CTR and HuRSMKO mice were single injected with rAAV/D377Y-mPCSK9, then intraperitoneally injected with 30 mg/kg A769662 daily and fed a Paigen diet for 12 weeks. B Oil-red O staining in aortas (n = 5). C Oil-red O staining in aortic roots (n = 5). Scale bar = 200 μm. D TUNEL staining in aortic roots (n = 5). Scale bar = 50 μm. E TC, TG, HDL-C, and LDL-C levels (n = 6). F Diagram for the role of smooth-muscle HuR in atherosclerosis via AMPK-mediated autophagy. HuR indicates human antigen R; AMPK, Adenosine 5′-monophosphate-activated protein kinase.
Discussion
In this study, the expression of HuR was decreased in atherosclerotic plaques from ApoE−/− mice with an HFD for 12 weeks. However, Rudolf Pullmann and Mitali Ray reported that increased HuR expression in atherosclerotic plaques from patients with neointimal proliferation and LDLR−/− mice lacking of IL-1926,27. These results suggested that HuR may play an important and complicated role in atherosclerosis. To explore the potential role of HuR in atherosclerosis, we generated smooth muscle-specific HuR knockout mice and constructed an atherosclerotic model. As compared with controls, HuRSMKO mice more frequently exhibited atherosclerotic plaques and increased instability of plaques. Knockout of HuR inducing atherosclerosis was attributed to defective autophagy. Mechanically, HuR could bind to and stabilize the mRNAs of AMPKα1 and AMPKα2, thereby increasing the expression of AMPKα and enhancing autophagy (Fig. 7F).
As a member of RNA-binding proteins, HuR has a primary role to bind to the target mRNAs and modulate their stability and translational efficiency28. HuR also interacts with other types of RNA, including small interfering RNA, long noncoding RNAs, and circular RNAs29. Binding to RNAs such as cyclinA, hypoxia-inducible factor-1, cyclooxygenase 2, B-cell lymphoma-2 is associated with proliferation and apoptosis; thus, HuR is related to the occurrence of tumorigenesis30,31. HuR regulates mRNAs of inflammatory factors such as interleukin 6, so HuR may be involved in inflammatory diseases28,32. In addition, HuR is also involved in metabolic diseases such as diabetes by binding to glucose transporter (GLUT1) mRNA33. After the discovery of the relationship between HuR and hypertension in VSMCs22, here we continued to use smooth muscle-specific HuR knockout mice to explore the role of HuR in atherosclerosis. Smooth-muscle HuR protected against the development of atherosclerosis by targeting AMPKα, which expands our understanding of HuR in cardiovascular diseases.
AMPK is a serine/threonine-protein kinase that acts as a central component of the signaling pathway regulating the conversion between anabolism and catabolism34. AMPK is a heterotrimer composed of one catalytic subunit and two regulatory subunits. The AMPK subunits all have multiple isoforms. Differences in isoform composition affects AMPK localization and function. Catalytic subunit α, which has two different isoforms, α1 and α2, is a major functional component of AMPK activation. Phosphorylation at Thr172 of α subunit is essential for AMPK activation35. Because mammalian AMPK is sensitive to the AMP:ATP ratio, any cellular process that reduces ATP levels or increases AMP concentration could activate AMPK. Some cytokines such as leptin, adiponectin, and ghrelin and some drugs such as AICAR, A769662, and metformin can activate AMPK directly or indirectly36. Also, AMPK activation is known to be mediated by liver kinase B1 (LKB1), Ca2+/calmodulin-dependent kinase CaMKK2 (CaMKKβ), and transforming growth factor-β-activated kinase 1 (TAK1)24,36,37. Some studies have reported that ΔNp63α, a p53 family member, and microRNAs such as mir-19 and mir-101 can regulate the expression of AMPKα38–40. Our study found that HuR could regulate AMPKα, which enhances our understanding of the regulatory mechanisms of AMPKα expression and activation. Recent studies showed that AMPK plays an important role in lipid metabolism41,42. In this study, we demonstrated that AMPKα1 and AMPKα2 were the target genes of HuR. Therefore, we speculated that HuR may regulate lipid metabolism through AMPK in hepatocytes. In the future study, hepatocyte-specific HuR knockout mice will be generated and these mice are predicted to have the phenotype with dyslipidemia. Whether and how HuR regulating the impact of AMPK on dyslipidemia will be explored.
Autophagy is a major intracellular degradation system that aims to dynamically cycle energy and matter for cell renewal and homeostasis43. The most important upstream regulators of autophagy are AMPK and mammalian target of rapamycin. Moreover, some studies have found that AMPK and mammalian target of rapamycin interact with each other44,45. From autophagosomes to autolysosomes, autophagy-related proteins such as Atg5, Atg7, LC3II, and p62 are indispensable for the occurrence and development of autophagy46. In recent years, the relationship between autophagy and atherosclerosis has also attracted much attention. Macrophage-specific Atg5 deficiency promotes atherosclerosis by interfering with cholesterol transport, apoptosis, and inflammation47–49. Endothelial-specific Atg5 and Atg7 deletion cause atherosclerotic plaque formation50,51. Knockout of Atg7 in VSMCs accelerates the process of atherosclerosis and promotes the regeneration of intima after injury15. Autophagy is closely related to cell senescence and apoptosis, which are also involved in atherosclerosis. In this study, knockout of HuR led to defective autophagy, which further increased apoptosis and atherosclerosis.
In summary, HuR could increase AMPK-mediated smooth muscle autophagy and play a protective role in atherosclerosis. Our research provides a new concept and drug target for treating atherosclerosis.
Materials and methods
Reagents
Adenovirus expressing GFP, HuR (ad-HuR, ad-GFP), and recombinant adeno-associated viral of murine proprotein convertase subtilisin/kexin type 9 mutants (rAAV/D377Y-mPCSK9) were from Vigenebio (MD, USA). Adenovirus expressing LacZ (ad-LacZ) and GFP-mRFP-LC3II (ad-GFP-mRFP-LC3II) were from Hanbio (Shanghai). Actinomycin D and Oil-red O were from Sigma (St Louis, MO, USA). A769662 was from Selleck Chemical (Houston, TX, USA). Ox-LDL was from Yiyuan (Guangzhou, China). CMLD-2 was from Millipore (Temecula, CA, USA). Recombinant HuR protein was from Proteintech (Chicago, IL, USA). Control and HuR siRNA were synthesized by BioSune (Shanghai). The sequences for HuR siRNA were 5′-CCAGUUUCAAUGGUCAUAATT-3′ and 5′-UUAUGACCAUUGAAACUGGTT-3′ and control siRNA were 5′-GGUUGAAUCUGCAAAGCUUTT-3′ and 5′-AAGCUUUGCAGAUUCAACCTT-3′. Paigen diet was from Trophic Diets (TP28640, China), containing 15% fat, 0.5% bile salt, and 1.25% cholesterol.
Cell culture and infection
Mouse smooth muscle cells were from ATCC (Manassas, VA, USA) with STR authentication. Cells were cultured in DMEM containing 10% fetal bovine serum and 50 μg/ml penicillin/streptomycin and seeded in 6-well plates at 1.0 × 104 cells/cm2. VSMCs grown to 70% confluence were infected with adenovirus at multiplicity of infection 75 for 48 h.
Mouse models
Smooth muscle-specific HuR knockout (HuRSMKO) mice were generated as described22. Male control and HuRSMKO mice at 8 weeks old were given a single tail-vein injection with rAAV/D377Y-mPCSK9 at 1.5 × 1011 pfu for each mouse as described52 and fed a Paigen diet for 12 weeks. Male apolipoprotein E-deficient mice (ApoE−/−) at 8 weeks old were from Vital River (Beijing) and were divided into two groups for high-fat diet (HFD) or normal chow diet (ND) feeding. Mice were housed at 25 °C, 12-h light/dark. Mice were euthanized using profound anesthesia with 4% isoflurane followed by exsanguination and tissue removal. The animal experiment was approved by the Animal Care Committee of Shandong University and was performed in compliance with the Animal Management Rules of the Chinese Ministry of Health. All animal experiments were performed conform the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Atherosclerotic lesion assay
Mice were euthanized and perfused with PBS. Hearts and aortas from the proximal ascending aorta to the abdominal aorta were removed and fixed in 4% paraformaldehyde. Aortas were dissected free of fat and adventitial tissue, opened longitudinally, stained with 0.5% freshly-made Oil-red O for 2 h, and pinned onto black silicon plates for imaging. After optical cutting temperature (OCT) compound embedding, the heart was cut to the aortic root to reveal the valvular lobe, stained with 0.5% Oil-red O and observed by microscopy. The results were reported as percentage of lesion area to total aortic area.
Morphology of aorta and aortic root
Aortas and hearts were embedded in OCT and sliced into 5-μm-thick frozen sections. The heart was cut to the aortic root. After sections were hydrated, immunofluorescence, immunohistochemistry, or other special staining was performed. To detect HuR-knockout efficiency in VSMCs, fluorescent double labeling of aortic sections was performed with HuR antibody (1:300, Millipore) and α-SMA antibody (1:300, Abcam). Sections of aortic roots were immunostained with MOMA-2 antibody (1:300, Abcam) to detect macrophage content and α-SMA antibody to detect VSMC content. The collagen of aortic root was detected by using a Masson staining kit (Solarbio, Beijing). The plaque vulnerability index was calculated as follows: (macrophage staining % + lipid staining %)/(SMC staining % + collagen staining %)53.
Detection of apoptosis
Apoptosis was detected by immunohistochemical staining with cleaved caspase-3 antibody (1:300, Affinity) and terminal UTP nick end-labeling (TUNEL). Briefly, aortic root sections were fixed in fresh 4% paraformaldehyde for 20 min at 15–25 °C, then incubated with premeabilization solution containing 0.1% Triton X-100 and 0.1% sodium citrates for 2 min at 2–8 °C. Staining followed the recommendations of the in-situ Cell Death Detection Kit (Roche, Basel, Switzerland).
Lipid profile assays
Serum was isolated for determining levels of total cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C). The blood lipid assay kit was from Jiancheng Bioengineering Institute (Nanjing, China).
Western blot analysis
A 15-μg amount of protein lysates from isolated aorta and SMCs were run on a 12% SDS-PAGE gel and immunoblotted overnight with the primary antibodies for HuR (29 ng/ml, 12582S, CST), AMPKα1 (1 μg/ml, Ab32047, Abcam), AMPKα2 (1 μg/ml, ab3760, Abcam), p-AMPK (27 ng/ml, 2535S, CST), p62 (293 ng/ml, 18420-1-AP, Proteintech), LC3I/II (64.5 ng/ml, 4108S, CST), Cleaved Caspase-3 (1 μg/ml, BF0711, Affinity), and β-actin (1 μg/ml, 20536-1-AP, Proteintech).
RNA immunoprecipitation assay
Whole-cell lysates were incubated overnight at 4 °C with protein A/G beads pre-conjugated with 5 μg rabbit IgG or HuR antibody (Millipore). RNA was then isolated from immunoprecipitates by using the Magna RIP kit (Millipore) with the primer sequences AMPKα1: 5′-GGCACCTTCGGGAAAGTGAA-3′ and 5′-TGTGAGGGTGCCTGAACAGC-3′ and AMPKα2: 5′-GAAGATTCGCAGTTTAGATGTTG-3′ and 5′-TCGAACAATTCACCTCCAGA-3′.
GFP-mRFP-LC3II punctation
VSMCs were infected with ad-GFP-mRFP-LC3II for 48 h. LC3II spots were observed by fluorescence microscopy (Nikon, Tokyo). GFP-RFP-LC3II serves as a specific marker for autophagic flux that relies on the different nature of GFP and RFP fluorescence under acidic conditions. Because GFP was quenched in the lysosomal acidic conditions, autophagosomes are shown as yellow puncta (RFP+GFP+), and autolysosomes are shown as red puncta (RFP+GFP−) in green- and red-merged images54. The mean number of LC3II points per cell was calculated from more than 30 cells.
Transmission electron microscopy
After infection with ad-LacZ or HuR for 48 h, VSMCs were harvested and fixed in electron microscope fixative containing 2.5% glutaraldehyde (Servicebio, Wuhan, China) for 2 h. Samples were fixed in 1% osmium tetroxide and dehydrated with ethanol in ascending order, followed by epoxide resin-embedding and cut into sections. Images of stained sections were collected by transmission electron microscopy (HT7700, Hitachi, Japan).
Statistical analysis
Group allocation for the experiments was randomized and not blinded. Sample analyses were not blinded. Data are expressed as mean ± SEM and were analyzed by using GraphPad Prism 6.0. All data were tested for normal distribution and equal variances. Student t test was used to compare two groups with 95% confidence interval. One-way ANOVA and Bonferroni post-tests were used in four groups. Statistical significance was set at P < 0.05.
Supplementary information
Acknowledgements
The authors thank Prof Xiao Yang (State Key Laboratory of Proteomics, Collaborative Innovation Center for Cardiovascular Disorders, Genetic Laboratory of Development and Diseases, Institute of Biotechnology, Beijing, PR China) for providing the α-SMA-Cre mice for this study.
Author contributions
S.L. designed and performed the research, analyzed data, and wrote the manuscript. X.J., X.C., and J.W. performed the research. S.L. and H.L. analyzed data. C.Z. and J.Y. reviewed data, W.Z. designed the research, reviewed data, and wrote the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (no. 81970198, 81770473) and the Taishan Scholar Project of Shandong Province of China (no. tsqn20161066), the Natural Science Foundation for Distinguished Young Scholars of Shandong Province (ZR2020JQ30), and the Natural Science Foundation of Shandong Province (no. ZR2020MH132).
Conflict of interest
The authors declare no competing interests.
Ethics statement
The animal experiment was approved by the Animal Care Committee of Shandong University and was performed in compliance with the Animal Management Rules of the Chinese Ministry of Health. All animal experiments were performed conform the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Footnotes
Edited by G.M. Fimia
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-021-03671-2.
References
- 1.Meyer MR, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 2014;4:7564. doi: 10.1038/srep07564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimbrone MA, Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YT, et al. Mouse models of atherosclerosis: a historical perspective and recent advances. Lipids Health Dis. 2017;16:12. doi: 10.1186/s12944-016-0402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 5.Li B, Xia Y, Hu B. Infection and atherosclerosis: TLR-dependent pathways. Cell Mol. Life Sci. 2020;77:2751–2769. doi: 10.1007/s00018-020-03453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 7.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim S, Park S. Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Rep. 2014;47:1–7. doi: 10.5483/BMBRep.2014.47.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grootaert MOJ, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114:622–634. doi: 10.1093/cvr/cvy007. [DOI] [PubMed] [Google Scholar]
- 10.Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13:1619–1628. doi: 10.1080/15548627.2017.1343770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys. Acta. 2015;1852:243–251. doi: 10.1016/j.bbadis.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mondaca-Ruff D, et al. Angiotensin II-regulated autophagy is required for vascular smooth muscle cell hypertrophy. Front Pharm. 2019;9:1553. doi: 10.3389/fphar.2018.01553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouimet M, et al. MicroRNA-33 regulates macrophage autophagy in atherosclerosis. Arterioscler Thromb. Vasc. Biol. 2017;37:1058–1067. doi: 10.1161/ATVBAHA.116.308916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osonoi Y, et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy. 2018;14:1991–2006. doi: 10.1080/15548627.2018.1501132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grootaert MO, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. 2015;11:2014–2032. doi: 10.1080/15548627.2015.1096485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lal S, et al. CRISPR knockout of the HuR gene causes a xenograft lethal phenotype. Mol. Cancer Res. 2017;15:696–707. doi: 10.1158/1541-7786.MCR-16-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, et al. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: Involvement in the nuclear import of RNA-binding protein HuR. J. Biol. Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 18.Grammatikakis, I., Abdelmohsen, K. & Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA8, 10.1002/wrna.1372 (2017). [DOI] [PMC free article] [PubMed]
- 19.Wang J, et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013;14:10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun K, et al. Neuron-specific HuR-deficient mice spontaneously develop motor neuron disease. J. Immunol. 2018;201:157–166. doi: 10.4049/jimmunol.1701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, et al. Adipose HuR protects against diet-induced obesity and insulin resistance. Nat. Commun. 2019;10:2375. doi: 10.1038/s41467-019-10348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, et al. HuR (human antigen R) regulates the contraction of vascular smooth muscle and maintains blood pressure. Arterioscler Thromb. Vasc. Biol. 2020;40:943–957. doi: 10.1161/ATVBAHA.119.313897. [DOI] [PubMed] [Google Scholar]
- 23.Grootaert MOJ, Roth L, Schrijvers DM, De Meyer GRY, Martinet W. Defective autophagy in atherosclerosis: to die or to senesce? Oxid. Med. Cell Longev. 2018;2018:7687083. doi: 10.1155/2018/7687083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamargo-Gomez I, Marino G. AMPK: regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 2018;19:3812. doi: 10.3390/ijms19123812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pullmann R, Jr, et al. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J. Biol. Chem. 2005;17:22819–22826. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray M, et al. Genetic deletion of IL-19 (interleukin-19) exacerbates atherogenesis in Il19−/− × Ldlr−/− double knockout mice by dysregulation of mRNA stability protein HuR (human antigen R) Arterioscler Thromb. Vasc. Biol. 2018;38:1297–1308. doi: 10.1161/ATVBAHA.118.310929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouhara K, et al. The induced rna-binding protein, HuR, targets 3’-UTR region of IL-6 mRNA and enhances its stabilization in periodontitis. Clin. Exp. Immunol. 2018;192:325–336. doi: 10.1111/cei.13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdelmohsen K, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14:361–369. doi: 10.1080/15476286.2017.1279788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, et al. The Jun/miR-22/HuR regulatory axis contributes to tumourigenesis in colorectal cancer. Mol. Cancer. 2018;17:11. doi: 10.1186/s12943-017-0751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eberhardt W, Nasrullah U, Haeussler K. Inhibition of caspase-2 translation by the mRNA binding protein HuR: a novel path of therapy resistance in colon carcinoma cells? Cells. 2019;8:797. doi: 10.3390/cells8080797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christodoulou-Vafeiadou E, et al. Divergent innate and epithelial functions of the RNA-binding protein HuR in intestinal inflammation. Front. Immunol. 2018;9:2732. doi: 10.3389/fimmu.2018.02732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gantt KR, et al. The regulation of glucose transporter (GLUT1) expression by the RNA binding protein HuR. J. Cell Biochem. 2016;99:565–574. doi: 10.1002/jcb.20950. [DOI] [PubMed] [Google Scholar]
- 34.Pulinilkunnil T, et al. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissuein vivo. J. Biol. Chem. 2011;286:8798–8809. doi: 10.1074/jbc.M111.218719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardman SE, Hall DE, Cabrera AJ, Hancock CR, Thomson DM. The effects of age and muscle contraction on AMPK activity and heterotrimer composition. Exp. Gerontol. 2014;55:120–128. doi: 10.1016/j.exger.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirwany NA, Zou M-H. AMPK in cardiovascular health and disease. Acta Pharmacologica Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Mavrakis KJ, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010;12:372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu P, et al. Mir-101-3p is a key regulator of tumor metabolism in triple negative breast cancer targeting AMPK. Oncotarget. 2016;7:35188–35198. doi: 10.18632/oncotarget.9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi Y, et al. Transcriptional suppression of AMPKα1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl Acad. Sci. USA. 2020;117:8013–8021. doi: 10.1073/pnas.1914786117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun SM, et al. AMPK activator C24 inhibits hepatic lipogenesis and ameliorates dyslipidemia in HFHC diet-induced animal models. Acta Pharm. Sin. 2020 doi: 10.1038/s41401-020-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun L, et al. Hydrogen sulfide reduces serum triglyceride by activating liver autophagy via the AMPK-mTOR pathway. Am. J. Physiol. Endocrinol. Metab. 2015;309:E925–E935. doi: 10.1152/ajpendo.00294.2015. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Alloza I, Goikuria H, Freijo MDM, Vandenbroeck K. A role for autophagy in carotid atherosclerosis. Eur. Stroke J. 2016;1:255–263. doi: 10.1177/2396987316674085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holczer M, et al. A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int. J. Mol. Sci. 2019;20:5543. doi: 10.3390/ijms20225543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Meyer GR, et al. Autophagy in vascular disease. Circ. Res. 2015;116:468–479. doi: 10.1161/CIRCRESAHA.116.303804. [DOI] [PubMed] [Google Scholar]
- 47.Liao X, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Razani B, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu K, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torisu KSK, et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell. 2016;15:187–191. doi: 10.1111/acel.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vion AC, et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl Acad. Sci. USA. 2017;114:E8675–E8684. doi: 10.1073/pnas.1702223114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bjørklund MM, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ. Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 53.Li H, et al. Melatonin enhances atherosclerotic plaque stability by inducing prolyl-4-hydroxylase α1 expression. J. Hypertens. 2019;37:964–971. doi: 10.1097/HJH.0000000000001979. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. Mir143-BBC3 cascade reduces microglial survival via interplay between apoptosis and autophagy: implications for methamphetamine-mediated neurotoxicity. Autophagy. 2016;12:1538–1559. doi: 10.1080/15548627.2016.1191723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.