

Abstract
White adipose tissue (WAT) depends on coordinated regulation of transcriptional and metabolic pathways to respond to whole-body energy demands. Here, we highlight metabolites that contribute to biosynthetic reactions for WAT expansion. Recent studies precisely defined how byproducts of carbohydrate and lipid metabolism affect physiological and endocrine functions in adipocytes. We emphasize the critical emerging roles for short-chain fatty acids and TCA metabolites that connect lipogenesis to WAT energy balance and endocrine functions. These insights address how adipocytes use small molecules generated from central carbon metabolism to measure responses to nutritional stress.
Keywords: metabolite, lipid metabolism, adipose tissue, insulin, microenvironment
Introduction
White adipose tissue (WAT) maintains whole-body energy balance (see Glossary) by responding to the metabolic demands of the body. In the fed state, de novo lipogenesis (DNL) in WAT drives storage of excess energy as triglycerides. Consequently, WAT expansion and partitioning of lipid and lipid derivates protects other peripheral organs from ectopic lipid accumulation to maintain energy balance. In contrast, nutrient deficits signal WAT free fatty acid (FFA) and glycerol release to provide energy substrates for peripheral tissues, including the liver. In addition, WAT responds to nutrient cues through secretion of many adipokines (i.e. leptin, adiponectin) and bioactive peptides that work locally and systemically to regulate the metabolic state of the organism. Prolonged nutrient stress impinges upon the pathways required for energy storage in WAT and, ultimately, metabolic regulation of whole-body energy balance. These nutritional signals impart changes encompassing the hormonal environment, intracellular metabolite generation, and gene regulation. This review discusses how metabolic intermediates and bioactive small molecules contribute to WAT expansion and the endocrine functions of adipocytes. We also discuss recent studies that utilize mass spectrometry approaches to annotate and define the functions of biosynthetic reactions for lipid synthesis in the fat cell (Box 1).
Box 1: Advances in mass spectrometry technology facilitate study of metabolism.
Metabolomics broadly describes the measurement of metabolites in a biological specimen. Techniques used in metabolomics, such as liquid chromatography or gas chromatography coupled with mass spectrometry, routinely measure tens to hundreds of metabolites with excellent precision. However, most experimental efforts measure isolated components and steady-state conditions that cannot recapitulate temporal dynamics of metabolic networks. Isotope labeling studies untangle metabolic networks by discriminating nutrient sources and endpoints. Stable isotope strategies provide information on relative pathway activities, qualitative changes in pathway contributions via alternative metabolic routes, and nutrient contribution to the metabolite syntehesis.
Implementation of stable isotope tracing methods impacted the field of metabolism, including validation of the reductive carboxylation of α-ketoglutarate into citrate [17, 21, 82]. Other studies published last year merged isotope tracing, mass spectrometry, and mathematical analysis methods to determine the direct sources of circulating nutrients, their interconversion rates, and eventual tissue-specific contributions to TCA cycle metabolism. Experiments with fifteen nutrient tracers enabled extensive accounting for both circulatory metabolic cycles and tissue TCA inputs, across fed and fasted mice on either high-carbohydrate or ketogenic diet [81]. As the technology advanced during the past 15 years, sample preparation methods likely preclude accurate analysis of subcellular and organelle-specific contributions to metabolic flux. While this limitation remains difficult to overcome, technological advances in mass spectrometry combined with metabolomics data analysis provide significant insight into intracellular metabolism. Our review discusses multiple recent studies from the Wellen laboratory and others that leverage mass spectrometry and metabolic tracing studies to understand how metabolites fuel acetyl-CoA and lipogenesis in the adipocyte [16, 24, 28, 32, 46].
The metabolically protective effects of WAT
Obesity reflects excessive expansion of WAT and frequently promotes multiple co-morbidities, including insulin resistance, systemic inflammation, nonalcoholic fatty liver disease (NAFLD), and cardiovascular disease [1]. In systemic insulin resistance, insulin does not adequately drive glucose import into adipocytes or oppose lipolysis sufficiently to protect non-adipose tissues from ectopic lipid deposition. For example, unopposed lipolysis floods the liver with WAT-derived acetyl-CoA and free fatty acids and accelerates NAFLD and hepatic insulin resistance [2]. The associated energy balance demands impairs adipocyte differentiation and WAT expansion, leading to downstream events such as inflammation and mitochondrial dysfunction.
However, some obese individuals in the United States retain normal insulin sensitivity, and it is not uncommon for mildly obese or even lean individuals to develop severe insulin resistance and type 2 diabetes [3]. This paradox supports the notion that excess body weight is not the sole determinant of obesity-related metabolic complications. The mechanisms responsible for preserved metabolic health in people with insulin sensitive forms of obesity remain incompletely understood. One prominent hypothesis suggests that the chronic nutrient pressure of obesity overwhelms the ability of adipose tissue to import glucose and convert carbohydrate precursors into triglycerides. Persistent states of positive energy balance support adipocyte hypertrophy, coupled with increased inflammation and insulin resistance (Figure 1). Accordingly, compared with metabolically normal obese subjects, metabolically abnormal subjects present with more cardiometabolic risk factors and persistently lower expression of genes involved in glucose uptake and lipogenesis in adipose tissue [4]. In contrast, one clinical report suggests obese individuals without metabolic abnormalities (hyperlipidemia, hypertension, diabetes) had a greater risk of cardiovascular disease compared to normal weight individuals [5]. However, this study did not account for prediabetes associated with fasting hyperglycemia or insulin resistance. Interestingly, obese normoglycemic individuals had higher adipocyte precursors in both SAT and VAT (Box 2) compared to obese individuals with prediabetes or type 2 diabetes, with the SAT precursors from obese normoglycemic also demonstrating greater adipogenic capacity [6]. These data suggest the earliest defect in WAT expansion involves impaired glucose metabolism and reinforce the significance of glucose metabolites in lipogenesis and activation of differentiation gene expression program.
Figure 1. Adequate energy storage in WAT enables insulin sensitivity.

WAT is the primary storage location for lipids, with a coordinated uptake and release of fats. WAT from metabolically healthy lean or obese individuals resist the effects of the environment and expand appropriately in response to the anabolic actions of insulin (right). In the post-prandial setting, insulin suppresses free fatty acid release and guides the internalization of glucose from circulation. Insulin also acts to support balanced lipid uptake. Ultimately, lipid, glucose, and nutrients form the substrates for effective adipocyte differentiation and de novo lipogenesis. Overnutrition leads to WAT hypertrophy (left) and reduced insulin responsiveness. Hyperglycemia and hyperlipidemia ensue, coupled with local immune infiltration of mainly T cells and macrophages. Persistent inflammation disrupts insulin signaling in adipocytes and allows unmitigated lipolysis. This figure was created using BioRender (https://biorender.com/).
Box 2: Distinct WAT depots emerge from specific cellular origins.
WAT can broadly be classified as visceral AT (VAT) and subcutaneous AT (SAT). VAT depots line the abdominal organs, while SAT depots accrue around the trunk, limbs, and face. VAT and SAT also emerge differently at the cellular level during development, with SAT adipocytes undergoing adipocyte lineage commitment and differentiation early in embryogenesis, while VAT adipocytes mostly differentiate during postnatal stages of development [83]. Human WAT depots also form sequentially, albeit prior to birth [84]. Although many issues related to off-target recombination or toxicity of labeling strategies [85], our understanding of the adipocyte lineage tree underwent multiple refinements over the last decade. Key discoveries showed VAT adipocytes emerge from progenitor cells expressing the Wilms’ tumor 1 (Wt1) gene [86], while adipocytes within SAT primarily derive from progenitors expressing paired related homeobox 1 (Prx1) gene [87–89]. Adding to the complexity, anatomically distinct depots may contain a network of adipocytes and other stromal precursor cells that express molecular and secretory programs to restrict or enable responses to dietary challenges [35, 90–94].
The expansion of WAT depots occurs via hypertrophy (increased adipocyte size) or hyperplasia (increased number of adipocytes). Reduced WAT hypertrophy (more, smaller fat cells) has been linked to increased gene expression of transcriptional regulators essential for adipocyte differentiation, such as PPARg [95]. However, persistent states of positive energy balance support hypertrophic expansion of WAT (bigger fat cell size) coupled with increased inflammation and insulin resistance [4]. In mice, high-fat diet feeding, causes proliferation of adipocyte precursors that ultimately undergo differentiation into adipocytes [96, 97]. This response is restricted to VAT in males, but occurs in both visceral and SAT in females [91]. Unsurprisingly, adipocyte lineage segregation is much less understood in humans. Countless studies document smaller fat cell size in SAT enables healthy adipose tissue expansion and protects against metabolic disease [4]. Identifying factors that allow endocrine and metabolic functions SAT to persist in the face of chronic nutrient stress remains fundamentally essential to understanding the expansion of adipose tissue in obesity.
Metabolic substrates that generate the acetyl-CoA pool
The TCA cycle provides ATP and metabolite building blocks for biosynthetic pathways and allows WAT to expand and contract in response to metabolic and nutritional demands. The TCA cycle uses the metabolite acetyl-CoA to generate reducing equivalents for the electron transport chain and ATP production by oxidative phosphorylation (OXPHOS). Acetyl-CoA can be generated in the mitochondria by pyruvate oxidation, fatty acid catabolism, or amino acid degradation. Moreover, acetyl-CoA is a crucial substrate for various reactions, including fatty acid synthesis, cholesterol synthesis, and protein acetylation (Figure 2).
Figure 2. Metabolism of multiple substrates supports acetyl-CoA production in the fat cell.
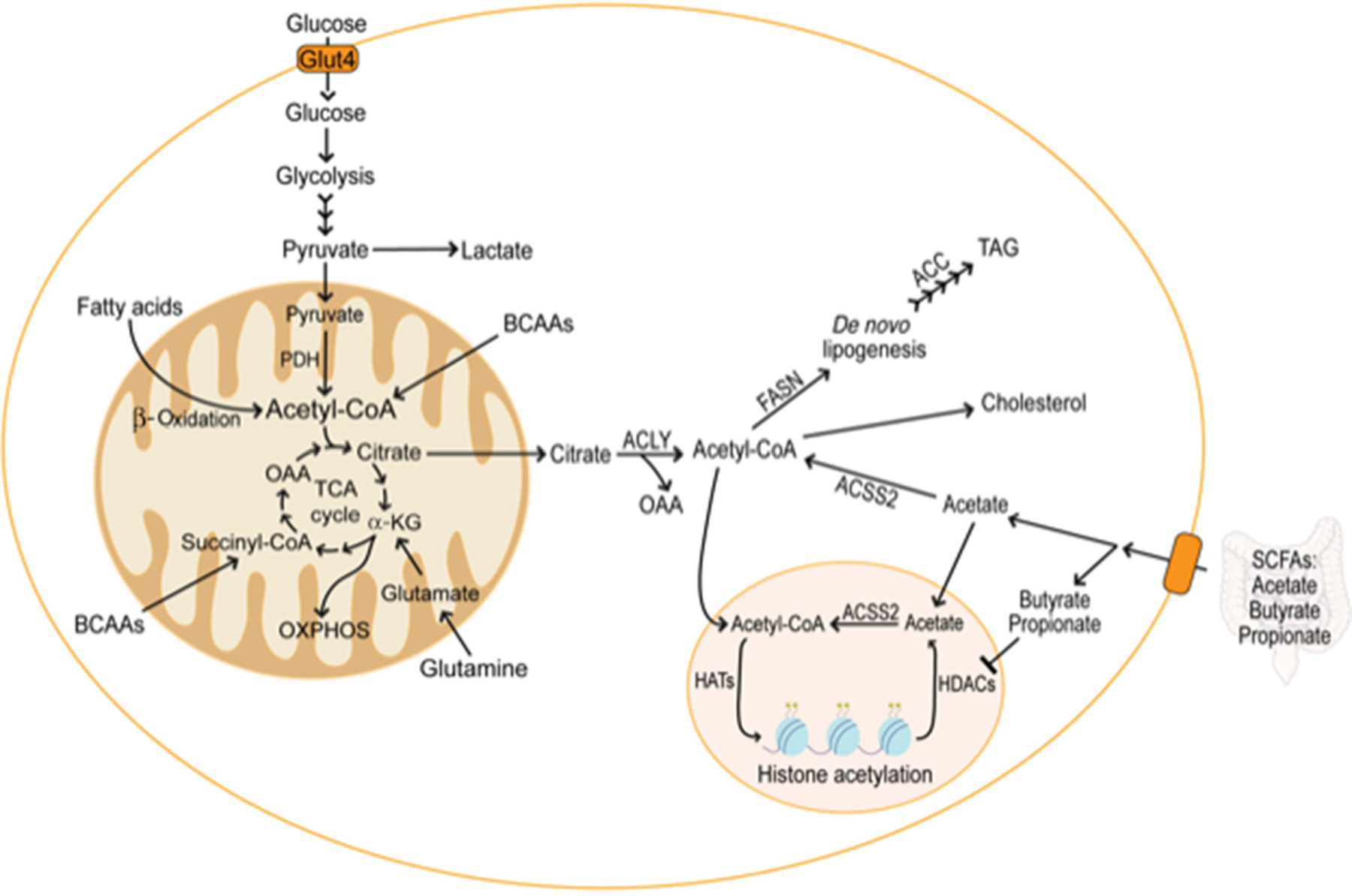
Glycolysis converts glucose to pyruvate and lactate, providing pyruvate for the TCA cycle. In the mitochondria, pyruvate is converted to acetyl-CoA and enters the TCA cycle by combining with OAA to form citrate. Citrate export out of the mitochondria maintains the cytosolic acetyl-CoA pool needed for lipid synthesis and protein modifications. While providing reducing equivalents for OXPHOS and ATP production, the TCA cycle generates substrates for the biosynthesis of proteins, nucleotides, and lipids. Although glucose and fatty acid breakdown support mitochondrial acetyl-CoA production in adipocytes, carbon from amino acid sources also maintains the TCA cycle. Glutamine can serve as an anaplerotic substrate for the TCA cycle, providing a-KG and allowing preservation of the cycle in conditions of mitochondrial stress. BCAAs also represent anaplerotic sources in adipocytes, providing acetyl-CoA and succinyl-CoA to the TCA cycle. In Acly+/+ adipocytes, glucose is the primary contributor to the cytosolic acetyl-CoA pool. However, adipocyte-specific deletion of ACLY reveals a compensatory pathway that requires ACSS2 to maintain the cytosolic acetyl-CoA pool. ACSS2 converts acetate derived from the gut microbiota or re-capture from the epigenome into acetyl-CoA for lipogenesis and protein modifications, including histone acetylation. Other short-chain fatty acids produced by gut microbiota promote histone acetylation by inhibiting histone deacetylases.
Although WAT accounts for 5% of post-absorptive whole-body glucose utilization [7], reduced adipocyte glucose uptake is one of the earliest defects in metabolic diseases [8, 9]. Glucose transport into the adipocyte is generally considered the crucial step in the metabolic flux of pyruvate into the TCA cycle. In the adipocyte, the glucose transporter, GLUT4, is activated by insulin signals and translocates to the cell membrane to assimilate glucose into the cell [10, 11]. Following insulin-dependent glucose oxidation, pyruvate enters the mitochondria where it combines with oxaloacetate (OAA) to generate acetyl-CoA for the TCA cycle.
Although pyruvate generates most of the acetyl-CoA in WAT, metabolism of FFA and branched chain amino acids (BCAAs) in the mitochondria also provides acetyl-CoA for the adipocyte. Beta-adrenergic signaling activates a series of lipolytic proteins that release FFAs from lipid droplets. FFA trafficking is partly regulated by acyl-CoA thioesterases (ACOTs), which hydrolyase fatty acyl-CoAs into fatty acids plus CoA to facilitate beta oxidation and CoA recycling in the mitochondria [12]. Regarding amino acids, prior studies demonstrated essential and non-essential amino acids contribute to the acetyl-CoA and nitrogen pools in cells [13, 14]. Elevated BCAA catabolic enzymes coincides with adipocyte differentiation [15] and mature adipocytes utilize BCAAs to support nearly 30% of the mitochondrial acetyl-CoA pool [16]. Collectively, multiple carbon sources provide metabolite intermediates that maintain acetyl-CoA pools for adipocytes.
Shunting of TCA cycle intermediates to the cytosol for biosynthetic purposes must be coupled with anaplerosis to maintain the cycle. Reductive glutamine metabolism provides a significant source of carbon for fatty acid synthesis when other TCA cycle metabolites become restricted. While citrate leaves the mitochondria as a substrate for the lipogenic acetyl-CoA pool, the TCA cycle remains intact mainly due to glutamine anaplerosis [17–19]. Glutamine anaplerosis occurs in conditions of impaired mitochondrial respiration, disrupted pyruvate entry into the TCA cycle, or reduced glucose-derived acetyl-CoA generation. When the a-KG/citrate ratio is high [20], cells convert glutamine into a-KG to yield isocitrate, citrate, and acetyl-CoA, which serve as substrates to synthesize lipids and biomass. This mechanism has been primarily studied in cancer cells, but recent elegant glutamine tracing studies demonstrated reductive TCA cycle flux plays a functional role to regulate secretion of pancreatic islet hormones [21]. However, acetyl-CoA generation from reductive TCA flux remains unexplored in adipocytes and presents a future direction for the field at large.
Adipocytes can use diverse carbon substrates for lipid synthesis
TCA catabolic reactions converge on acetyl-CoA, generating the primary metabolite necessary for DNL in WAT. DNL is the mechanism by which cells condense acyl units to make fatty acids, which are then diverted to lipid droplets for storage as triacylglycerols and later oxidation, incorporation into structural lipids, or used for post-translational modifications. Fatty acid synthesis is primarily mediated by acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN), where FASN utilizes acetyl-CoA to initiate a new acyl chain which is then elongated using malonyl-CoA generated by ACC [22]. Subsequently, primary products of FASN can be elongated and/or desaturated to produce a diverse fatty acid pool. However, FASN loss reduces the synthesis of lipid activators of peroxisome proliferator-activated receptor gamma (PPARg) transcriptional activity, shifting DNL to beta oxidation in adipocytes [23].
The predominant product of DNL is the saturated 16-carbon fatty acid palmitate, but other fatty acids can be synthesized when alternate CoAs are used. For example, odd-chain fatty acids (C15:0 and C17:0) can be synthesized from valine and isoleucine-derived propionyl-CoA in cultured adipocytes. In addition to the C15-C17 fatty acids, glucose-derived carbon contributes to monomethyl branched-chain fatty acid accumulation in differentiated adipocytes. In this setting, FASN uses BCAA-derived isovaleryl-CoA (from valine), isobutyryl-CoA (from leucine), and 2-methylbutyryl-CoA (from isoleucine) to initiate synthesis of long-chain fatty acids and more broadly adapt the cell to amino acid availability [24]. However, the ultimate physiological impact of branched CoA diversion to monomethyl branched-chain fatty acids is not clear due to a low abundance across tissues.
DNL is an essential process for WAT expansion and endocrine functions to protect the organism through maintenance of energy balance (Box 3). In differentiated 3T3-L1 cells, an in vitro adipocyte model, glucose and glutamine account for only half of the lipogenic acetyl-CoA, suggesting mature adipocytes use different substrates for lipid synthesis. Depletion of BCAA catabolic enzymes leads to decreased expression of adipogenic genes such as Pparg, Adipoq, and Glut4 [16]. On the other hand, proliferating cells use glucose and glutamine for more than 80% of the lipogenic acetyl-CoA. Upon adipocyte differentiation, several non–essential amino acids undergo synthesis at high rates in differentiated 3T3–L1 adipocytes. Collectively, these observations suggest adipocytes utilizes diverse substrates for DNL instead of exclusive use of glucose as a primary carbon source [24], which may support adaptation to varying nutrient availability.
Box 3: Endocrine effects of WAT de novo lipogenesis.
Recent studies firmly demonstrate hepatic DNL promotes insulin resistance [98]. In stark contrast, DNL in SAT correlates with insulin sensitivity in obese humans and rodents [4]. WAT DNL reflects the action of many transcription factors that sense glucose in the cell. In addition to allowing surfeit storage of energy as lipids, DNL also contributes to the synthesis of fatty acid species that exert endocrine effects. GLUT4 overexpression in adipose tissue increased a previously undiscovered lipid family called branched fatty acid esters of hydroxyl fatty acids (FAHFAs) [99]. Branched FAHFAs exert distal effects on the periphery, including glucose-stimulated glucagon-like peptide-1 (GLP-1) secretion from entero-endocrine cells and insulin secretion by pancreatic beta cells [100]. These lipids also exert anti-inflammatory effects and summarily improve insulin sensitivity in mouse models of glucose intolerance. Among the FAHFAs, palmitic acid esters of hydroxy-stearic acids (PAHSAs) accumulate in adipose tissues, and levels decrease in humans with insulin resistance [99]. The biosynthetic enzymes responsible for FAHFA generation remain unknown. The causative role of FAHFAs and other lipid classes on whole-body insulin resistance remain unclear, but new high-resolution metabolomic techniques may enable the accurate identification of lipid mediators of energy balance.
ACLY creates the primary lipogenic acetyl-CoA pool in WAT
Lipogenesis requires significant ATP, 8 acetyl-CoA, and 14 NADPH to synthesize palmitate. Along these lines, NADPH and acetyl-CoA are the critical substrates for fatty acid synthesis in adipocytes. During adipocyte differentiation, there is coordinated upregulation of ACLY and cytosolic malic enzyme 1 [25]. These two enzymes function in parallel to convert TCA derivered citrate and NADH into acetyl-CoA, NADPH, and OAA to serve the biosynthetic demands of lipogenesis [26]. While ACLY supplies cytosolic acetyl-CoA for DNL, its inhibition promotes lipid lowering in the blood of humans and animals [27]. Genetic disruption of ACLY [28] or malic enzyme 1 [29] in mouse models causes mild lipodystrophies, which positions the coupled generation of NADPH and acetyl-CoA as rate-limiting features of lipid storage in WAT.
Diet and nutrient availability regulate ACLY in adipocytes as its expression increases after high carbohydrate feeding [30] and decreases with high-fat feeding [31]. ACLY also maintains DNL and regulates a complex interaction between diet, sex, and systemic metabolic homeostasis. Surprisingly, adipocyte-specific knockout of ACLY (AclyFAT−/−) causes mild metabolic phenotypes in males [28, 32]. Only female AclyFAT−/− mice show insulin resistance and fatty liver disease when fed a high sucrose diet. Mechanistically, female AclyFAT−/− mice fail to activate carbohydrate response element-binding protein (ChREBP) to support DNL in WAT. Females generally favor lipid storage over oxidation [33], and there is evidence in both humans and mice that lipid synthesis is higher in adipose tissue in females [34, 35]. These studies extend sex differences and support the notion that females may conduct a more significant percentage of whole-body DNL within adipocytes than males.
In ACLY-deficient conditions compensatory pathways maintain the lipogenic acetyl-CoA pool. Recent studies used stable isotope tracing to show incorporation of acetate into acetyl-CoA, malonyl-CoA, and HMG-CoA allows lipogenesis in ACLY-deficient conditions [28, 32]. In this setting, Acetyl CoA synthetase short-chain family member 2 (ACSS2) converts intracellular or extracellular acetate to acetyl-CoA contributing to the cell’s lipogenic acetyl-CoA pool and partially compensating for ACLY deletion in adipocytes. In ACLY-deficient cells, glucose-derived carbons contribute primarily to the mitochondrial acetyl-CoA pool, while acetate carbons accumulate through histone deacetylation to supply the nuclear-cytosolic acetyl-CoA pool [32].
Circulating gut-dervied acetate also supports the acetyl-CoA pool (Figure 2), while ethanol consumption in the liver generates large amounts of acetate via liver metabolism [36]. Excessive nutrient uptake also promotes de novo acetate production from the concerted actions of keto-acid dehydrogenase and decarboxylation enzymes that act on pyruvate [37]. In these ways, acetate abundance from the gut [36] or de novo synthesis [37] ultimately requires ACSS2 to support the acetyl-CoA pool. This mechanism occurs in fatty liver caused by fructose exposure [36]. Similar to the liver, adipocytes express plasma membrane proton-linked monocarboxylate transporters that transport acetate into the cell [38], but further studies are needed to understand how acetate elevation from stresses such as alcohol abuse or obesity impact acetyl-CoA and lipogenesis in WAT.
Metabolic enzymes maintain the acetyl-CoA pool for gene regulation in WAT.
In addition to functioning as the precursor for lipogenesis and other biosynthetic reactions, a principal function of acetyl-CoA is to provide acetyl groups for the acetylation of histones that consequently alter chromatin dynamics. Thus, the glucose-derived carbon flux into acetyl-CoA influences global histone acetylation [39, 40]. Oxidative catabolism of lipids supports the acetyl-CoA pool as well, contributing up to 90% of the acetylation of certain histones [41]. Acetate also contributes to the acetyl-CoA pool for histone acetylation, and de novo production from pyruvate [37] provides additional ways to link glucose availability to genome regulation. Acetate and acetyl-CoA generation from alcohol metabolism in the liver supports histone acetylation in the brain, but effects on WAT were not shown [42]. More recently, acetate generation from a rare subpopulation of adipocytes influences mitochondrial respiration [43] and, likely, the gene regulation programs for adipocyte precursor differentiation.
Histone acetyl transferases (HATs) catalyze the addition of acetyl groups to histone N-terminal tails and display exquisite sensitivity to glucose availability, fatty acid oxidation, and mitochondrial respiration [41, 44, 45]. Histone deacetylases (HDACs) remove acetyl groups from histones, leading to more compact chromatin. HDACs can be inhibited by metabolites, including β-hydroxybutyrate (BHB), which is produced primarily during fatty acid oxidation (FAO) and ketogenesis. Though the liver is the primary systemic source of BHB, adipocytes generate this ketone body via butyrate catabolism during FAO. In adipocytes, BHB senses high levels of FAO and contributes to the acetyl-CoA and ATP pools. BHB acts as an HDAC inhibitor that elevates tissue histone acetylation and alters gene expression in ketogenic conditions [46], which, coupled with a recent study [47], suggests BHB metabolism exerts direct influences on metabolism and gene regulation in WAT. Indeed, the transcriptional coregulator PR/SET Domain 16 promotes local BHB secretion in WAT, restoring the differentiation competency of aged adipocytes [47]. These findings highlight the significance of metabolites to regulate WAT gene expression through chromatin modifications.
Metabolic intermediates of biosynthetic reactions serve as substrates and cofactors for various epigenome-modifying enzymes, which allows metabolism to communicate environmental influences to chromatin dynamics [48]. Thus, it is not surprising diet can influence metabolism by affecting the acetyl-CoA pools dedicated to histone acetylation levels to broadly impact the expression of glucose and lipid metabolism genes in WAT [31]. Global histone acetylation levels increase during adipocyte differentiation, which requires ACLY to mediate the transcription of genes critical for insulin sensitivity [40]. Confirmatory in vivo studies demonstrated ACLY knockout in adipose tissue also reduces histone acetylation near Glut4 [32]. Furthermore, ACLY has been shown to regulate histone acetylation levels and influence gene regulation in diverse mammalian cell types [39, 40, 49] and histone acetylation responds to carbohydrate availability in an ACLY-dependent manner [28].
In addition to nutrient availability, tissue-specific and compartmental metabolite abundance influence histone acetylation levels and affect gene regulation in WAT. Visceral and subcutaneous WAT differ in ACSS2, acetyl-CoA, and histone acetylation levels indicating depot-specific differences in acetate compensation and output of the acetyl-CoA pools. However, upregulation of ACSS2 and higher acetyl-CoA levels do not necessarily translate to histone acetylation, as ACLY knockout exhibit reduced histone acetylation [32]. Partitioning of metabolites or sequestration of enzymes involved in acetyl-CoA production in subcellular locations may contribute to lower histone acetylation [32]. These data suggest tissue-specific and compartmental ACLY expression influence histone acetylation levels and affect gene regulation in WAT. Given that ACSS2 [50] and ACLY [51] generate acetyl-CoA in the nucleus for histone modifications, lipogenic enzymes that reside in multiple cellular compartments might diversify the functions of acetyl-CoA and signify novel connections between metabolism and gene transcription.
Short-chain fatty acids derived from the microbiome act on WAT
The gastrointestinal tract is colonized extensively with commensal microorganisms collectively called the microbiota, which play an important role in metabolism and immune defense, as part of a symbiotic relationship with the host. Bacteria within the gut break down dietary fibers and starch through anaerobic fermentation, producing short-chain fatty acids (SCFAs) as metabolite byproducts. Considerable amounts of these SCFAs, including acetate, butyrate, and propionate, are produced, with acetate being the most abundantly produced [52, 53]. While these SCFAs maintainin intestinal homeostasis, they quickly enter the bloodstream and can be metabolized by peripheral tissues. Their concentrations in peripheral blood vary across SCFA species, with acetate ranging between 100–150 μM, while propionate and butyrate levels are much lower, ranging from 4–5 uM and 1–3 μM, respectively [54]. Though butyrate and propionate circulate at much lower concentrations than acetate [54], they potently inhibit HDACs while acetate lacks HDAC inhibitory activity [55]. Along these lines, butyrate inhibition of HDACs increases histone H3 acetylation near the promoter regions of the critical adipogenic transcription factors, PPARG and CEBPA [56]. Other studies observed divergent effects of SCFAs on WAT functions and insulin sensitivity. Butyrate reduces adiposity and promotes insulin sensitivity in mice fed high-fat diet HFD [57], but the molecular mechanisms studied primarily focused on brown adipose tissue and skeletal muscle. Butyrate contributes to lipogenesis [58], but likely feeds extra-mitochondrial acetyl-CoA pools in an ACLY-dependent manner [59]. Labeled carbon and metabolic tracing studies of plasma intermediates show that propionate can enter the TCA cycle as succinyl-CoA, while acetate and butyrate enter as acetyl-CoA [16]. Though butyrate can undergo beta-oxidation in the mitochondria, acetate is converted to acetyl-CoA in the cytosol upon entry into the cell. Thus, butyrate has a greater contribution to the mitochondrial acetyl-CoA pool than acetate [59]. SCFAs contribute to different parts of hepatic metabolism, as propionate primarily contributes to gluconeogenesis, and butyrate and acetate are the main contributors to fatty acid and cholesterol synthesis [59]. The roles of these SCFAs in the liver suggests alternate inputs into the CoA pools within WAT.
SCFAs inhibit lipolysis and promote adipogenesis in WAT [52, 60], and provide substrates for glucose and lipid synthesis [59]. SCFAs act on cells via the G protein-coupled receptors GPR41 and GPR43, though the receptors vary in SCFA specificity, tissue localization [60], and sensitivity to high fat diet [52]. Acetate acts as a GPR43 agonist, butyrate as a GPR41 agonist, and propionate acts through both receptors [52, 60]. The activation of GPR43 in adipocytes results in inhibition of lipolysis and a decrease in plasma FFAs [52, 60]. In addition to acetyl-CoA directed lipogenesis, acetate supports WAT expansion through GPR43 signaling to stimulate adipogenesis by indirect transactivation of PPARg [52, 53]. SCFAs may also promote adipocyte differentiation by upregulating the adipogenic genes PPARG, CEBPA, FABP4, and LEP, with propionate and butyrate having a more significant impact on gene expression levels than acetate [56]. While these studies suggest that SCFAs promote adipocyte differentiation, the data are conflicting regarding roles in obesity. For example, a cross-sectional study found that circulating acetate negatively correlates with whole-body lipolysis and insulin sensitivity [61]. Consistent with this observation, high fat diet increases acetate and promotes glucose-stimulated insulin secretion resulting in hyperinsulinemia, insulin resistance, and obesity [62]. Lastly, the recent demonstration that acetate exerts paracrine influences on adipocyte respiration [43] suggests the role of SCFAs on WAT functions and nutrient signals must be revisited.
Metabolic abnormalities of nutrient stress and restorative therapeutic approaches
Rapid WAT expansion during chronic nutrient stress creates regions of hypoxia due to delayed or impaired angiogenesis [63]. In states of nutrient excess, WAT primarily moves carbon from glucose to produce metabolic intermediates such as citrate, which can fuel acetyl-CoA production for fatty acid synthesis and protein acetylation. Classic studies defined the allosteric regulation of the ACC enzymes by low citrate levels and energetic stress [64–66]. Cytosolic ACC1 is inactivated by AMPK and intermediates of fatty acid synthesis so that the cell can shunt carbon from lipogenesis in states of nutrient deficit. Accodingly, in hypoxic conditions, shunting of glucose-derived carbon toward lactate production decreases acetyl-CoA assembly from glucose [67] that may also influence reductive glutamine metabolism and heighten inflammation (Box 4). Additionally, disrupted glucose import into adipocytes by local hypoxia and a range of obesity stresses [63] further contributes to acetyl-CoA depletion [31]. In parallel, high-fat feeding suppresses ACLY leading to decreased histone acetylation levels in the WAT of mice [31], which likely accounts for, in part, altered gene programs and decreased WAT lipogenesis. However, ACSS2 supports fatty acid and phospholipid pools in hypoxic environments [68]. Such a mechanism might also allow the fat cell to perform lipid synthesis from acetate-derived acetyl-CoA and sustain the endocrine function of WAT during the persistent nutrient and low oxygen stress of obesity.
Box 4: Role of glutamine in WAT inflammation.
Although glutamine may serve as an anaplerotic substrate for the TCA cycle, a recent study shows that it can also act as a suppressor of inflammation within the WAT microenvironment [101]. Obesity correlates with lower glutamine levels in human WAT, as reflected by a negative relationship between glutamine levels and increased body fat percentage. Analysis of previously published microarray data [102] showed reduced expression of the singular glutamine-synthesis enzyme GLUL in obese women. Additional gene ontology analysis established GLUL expression positively correlates with lipid and carbohydrate metabolism genes and negatively correlates with inflammation. Rodent high fat diet studies showed increased expression of inflammatory genes such as Il6, Tnfa, Il1b, and Emr1, which were normalized in obese mice treated with glutamine. High glutamine levels promote an anti-inflammatory phenotype in adipocytes, macrophages, and T cells present within WAT [101]. However, adipocyte precursors remain unaffected by glutamine.
High glutamine levels decrease inflammation by reducing glycolysis and the production of uridine diphosphate N-acetyl-glucosamine (UDP-GlcNAc). UDP-GlcNAc is the substrate for O-GlcNAcylation. Thus, decreased UDP-GlcNAc results in attenuated chromatin O-GlcNAcylation and expression of pro-inflammatory genes regulated by O-GlcNAcylation [101]. On the other hand, obesity correlates with higher O-GlcNAcylation and increased expression of pro-inflammatory genes. Future studies are needed to understand the dynamics of this immunometabolic crosstalk and its potential therapeutic impact on obesity.
Diverging carbon substrates from DNL impacts other metabolic pathways, including depletion of monomethyl branched-chain fatty acids and their parent enzymes in insulin-resistant humans [69] and mice [24]. Catabolism of BCAA provides alternate substrates for lipid synthesis and insulin resistance associates with reduced BCAA metabolism in WAT, which may contribute to heightened serum levels of BCAAs and their downstream catabolites [63]. Thiazolidinediones increase transcription of BCAA catabolic genes in WAT [70] suggesting therapeutic roles for uptake of elevated blood isoleucine, leucine, and valine into fat cells in insulin-resistant and diabetic individuals. In summary, reduced substrate availability, metabolic enzyme depletion, decreased histone acetylation, and shunting of carbons away from lipogenesis, compound the detrimental impact of nutrient stress on adipocytes and impair WAT expansion.
Clinically, several metabolic inhibitors have been tested to restore metabolism in obesity and diabetes. Etoxomir, an inhibitor of carnitine palmitoyltransferase CPT1, reduces the transport of acyl-carnitines into the mitochondrial intermembrane space thereby decreasing lipid oxidation. In type 2 diabetes patients, etomoxir decreased hepatic glucose production and lowered fasting blood glucose, but notably increased free fatty acids, in addition to causing myocardial hypertrophy and hepatic steatosis in rats, leading to cessation of etomoxir therapies [71, 72]. More recently, FDA approved bempedoic acid has been used to inhibit ACLY in patients with hypercholesterolemia [73]. ACLY inhibition blocks the conversion of citrate to acetyl-CoA, which reduces substrates for lipogenesis and cholesterol synthesis. However, inhibition of ACLY in mice results in shunting of acetyl-CoA away from essential lipid storage and transcriptional regulatory mechanisms in adipose tissue [28, 32]. These data suggest that the acetate sequestration and acetyl-CoA synthesis by ACSS2 may provide metabolic compensation in the context of therapeutic targeting of ACLY. Metabolic enzymes are often targeted in cancer to deplete tumor cells of TCA anaplerotic substrates, such as glutaminase inhibitors that reduce glutamate production and cutoff an important fuel source [74]. In contrast, anaplerotic pathways in obesity are essential for TCA flux, lipogenesis, and WAT expansion. Perhaps a better approach would be to target upstream signals that deplete key mitochondrial and lipogenic substrates, including inflammatory signals as shown in obese mice [75], to restore WAT expansion and improve insulin sensitivity. For example, a small molecule dye (IR-61) decreases macrophage inflammatory activity through upregulation of ACLY and mitochondrial respiration to restore insulin sensitivity in obese mice [76]. Alternatively, supplementation with glutamine and other metabolites to restore reductive metabolism for TCA anaplerosis may represent be a viable approach to facilitate WAT expansion.
Concluding remarks and future perspectives
WAT maintains energy balance by responding to the metabolic demands of the body, storing excess energy as triglycerides during nutrient overload, and releasing FFAs in times of nutrient deficit. WAT also regulates glucose homeostasis by storing excess energy within lipid droplets of adipocytes, thus preventing insulin resistance secondary to accumulation of fat in the liver and skeletal muscle. In this regard, it is important to emphasize the ongoing fallacy in assuming more fat cells drive obesity and its co-morbidities [77]. Obesity leads to complex effects on immune cell infiltration, adipogenesis and fat cell hypertrophy that represent the whole-body protective response to excess calories. Consistent with these notions, reduced adipocyte differentiation correlates with insulin resistance and other co-morbidities of obesity [4]. A future challenge will be to identify factors that explain the confluence of immune cell action and adipose tissue expansion in metabolically healthy obesity.
Although acetyl-CoA provided by ACLY activity regulates histone acetylation in adipocytes, other metabolites shunted from the TCA cycle also regulate the epigenome [48]. Many chromatin-modifying enzymes use metabolic intermediates as both cofactors and substrates for histone modifications and histone demethylation. a-KG is an essential cofactor for 2-oxoglutarate-dependent dioxygenases, including the Jumonji C domain-containing lysine demethylases and ten-eleven translocation hydroxylases. These enzymes generate succinate, which serves as a feedback inhibitor of methylase reactions. Accordingly, nutrients that fuel succinate and a-KG accumulation may allow chromatin modifications in adipocytes to adapt gene expression or differentiation competence during obesity. The recently discovered energy balance impacts [43, 78, 79] combined with known immune effects [80] of acetate, succinate, and a-KG suggest nutrient metabolism must also match demand amongst a diverse mixture of adipocytes and stromal cells in WAT.
Although many questions remain (see Outstanding Questions), improved technologies enabling the measurement of subcellular metabolites in vivo [81] will facilitate a deeper understanding of the physiological and pathological shuttling of metabolites to control fat cell responses to the persistent environmental stress of obesity.
Outstanding questions:
What is the mechanistic action by which pro-inflammatory cytokines diminish TCA cycle metabolites and the acetyl-CoA pool that contribute to impaired WAT expansion and lipid storage during obesity?
How do metabolites secreted by adipocytes impact immune cell diversity and function in the WAT microenvironment?
Do metabolites influence lineage engagement of adipocyte precursor cells? What rate-limiting factors allow one cell to out-compete other cells for nutrients that fuel the TCA cycle in adipocytes?
Does the subcellular distribution of metabolite pools influence gene regulation directly in adipocytes?
In the nutrient-replete conditions of obesity, what mechanisms dictate SCFA sensing and incorporation into metabolic reactions that support lipid synthesis in adipocytes?
Highlights:
White adipose tissue utilizes multiple nutrient fuels to create energy stores for the organism.
While glucose and fatty acids contribute to the mitochondrial acetyl-CoA pool, glutamine and BCAAs also support anaplerotic substrates in differentiated adipocytes.
ATP Citrate Lyase expression generates the cytosolic acetyl-CoA pool within adipocytes in glucose-dependent ways.
Acetate can maintain the cytosolic acetyl-CoA pool in the absence of ACLY.
Newly established endocrine functions of short-chain fatty acids influence WAT gene regulation by affecting chromatin-modifying enzymes in WAT.
Acknowledgements
This work was funded by American Diabetes Association #1-18-IBS-105 and NIH grants R01DK114356 and R01DK126042. This study was also funded (in part) by the Nancy Chang, PhD Award for Research Excellence at Baylor College of Medicine.
Glossary
- Acetyl-CoA carboxylase (ACC)
multi-subunit enzyme that catalyzes the rate-limiting step in fatty acid synthesis, the carboxylation reaction of acetyl-CoA to malonyl-CoA. Malonyl-CoA serves as the substrate for further elongation reactions that generate the 16-carbon fatty acid palmitate
- Adipocyte hypertrophy
reflects increased size of adipocytes; frequently associated with hypoxia and inflammation because of their massively expanded size
- Anaplerosis
replenishment of the TCA cycle that occurs as intermediates are shuttled for biosynthetic purposes
- Carbohydrate response element binding protein (ChREBP)
a transcription factor highly expressed in the liver and adipose tissues. ChREBP acts in a glucose-dependent manner and binds carbohydrate response element motifs in promoter regions that support de novo lipogenesis
- Coenzyme A (CoA)
synthesized from pantothenate and cysteine and an essential cofactor known for its role in carbohydrate, lipid, and amino acid metabolism. CoA and its thioester derivatives participate in diverse anabolic and catabolic pathways, allosteric regulatory interactions, and gene expression regulation
- Energy balance
reflects the interplay among energy intake, energy expenditure, and body energy stores
- Fatty acid synthase (FASN)
enzyme that catalyzes the de novo biosynthesis of long-chain saturated fatty acids starting from acetyl-CoA and malonyl-CoA in the presence of NADPH
- Fatty acids
carboxylic acid molecule made of a long hydrocarbon chain and a terminal carboxyl group. Fatty acids are the building blocks of lipids, oils, and cell membranes. Fatty acids can be saturated (no double bonds) or unsaturated (one or more double bonds)
- Glucose transporter 4 (GLUT4)
insulin-stimulated glucose transporter present in adipose tissue and striated muscle. At the cell surface, GLUT4 permits the facilitated diffusion of circulating glucose into glycolysis
- Glycerol
an alcohol molecule containing three hydroxyl groups. Glycerol combines with three fatty acid chains to form triglycerides
- Ketogenesis
biochemical process that produces ketone bodies through the breakdown of fatty acids and ketogenic amino acids within the mitochondria of cells
- Oxidative Phosphorylation (OXPHOS)
process in which cells use the reducing equivalents NADH and FADH2 generated in the TCA cycle to produce large amounts of ATP in the presence of oxygen
- Peroxisome proliferator-activated receptor gamma (PPARg)
member of the nuclear receptor family of ligand-activated transcription factors. PPARg is required for adipocyte differentiation and controls the expression of genes that stimulate FFA release, fatty acid transport, adipokines, fatty acid oxidation, and insulin sensitivity
- Thiazolidinediones
class of drugs used for the management of type 2 diabetes mellitus. Thiazolidinediones are full agonists for the nuclear receptor PPARg and, as a result, improve insulin sensitivity
- Triglyceride
esters formed from glycerol and three fatty acid chains. Triglycerides serve as long-term energy stored in lipid droplets within adipose tissue. They are synthesized during times of nutrient excess and broken down in times of nutrient demand, such as fasting and thermogenesis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ward ZJ et al. (2019) Projected U.S. state-level prevalence of adult obesity and severe obesity. N Engl J Med 381 (25), 2440–2450. [DOI] [PubMed] [Google Scholar]
- 2.Perry RJ et al. (2015) Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160 (4), 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith GI et al. (2019) Metabolically healthy obesity: facts and fantasies. J Clin Invest 129 (10), 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammarstedt A et al. (2018) Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev 98 (4), 1911–1941. [DOI] [PubMed] [Google Scholar]
- 5.Caleyachetty R et al. (2017) Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. J Am Coll Cardiol 70 (12), 1429–1437. [DOI] [PubMed] [Google Scholar]
- 6.Belligoli A et al. (2019) Characterization of subcutaneous and omental adipose tissue in patients with obesity and with different degrees of glucose impairment. Sci Rep 9 (1), 11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeFronzo RA and Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 Suppl 2 (Suppl 2), S157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen DL et al. (2015) Phenotypic characterization of insulin-resistant and insulin-sensitive obesity. J Clin Endocrinol Metab 100 (11), 4082–91. [DOI] [PubMed] [Google Scholar]
- 9.Meyer C et al. (2006) Different mechanisms for impaired fasting glucose and impaired postprandial glucose tolerance in humans. Diabetes Care 29 (8), 1909–14. [DOI] [PubMed] [Google Scholar]
- 10.Klip A et al. (2019) Thirty sweet years of GLUT4. J Biol Chem 294 (30), 11369–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krycer JR et al. (2017) Dynamic metabolomics reveals that insulin primes the adipocyte for glucose metabolism. Cell Rep 21 (12), 3536–3547. [DOI] [PubMed] [Google Scholar]
- 12.Tillander V et al. (2017) Deactivating fatty acids: Acyl-CoA thioesterase-mediated control of lipid metabolism. Trends Endocrinol Metab 28 (7), 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chuang DT et al. (1983) Induction of the branched-chain 2-oxo acid dehydrogenase complex in 3T3-L1 adipocytes during differentiation. Biochem J 214 (1), 177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Si Y et al. (2007) Flux profile and modularity analysis of time-dependent metabolic changes of de novo adipocyte formation. Am J Physiol Endocrinol Metab 292 (6), E1637–46. [DOI] [PubMed] [Google Scholar]
- 15.Lackey DE et al. (2013) Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab 304 (11), E1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green CR et al. (2016) Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12 (1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metallo CM et al. (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481 (7381), 380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wise DR et al. (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 108 (49), 19611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang C et al. (2014) Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell 56 (3), 414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fendt SM et al. (2013) Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun 4, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang GF et al. (2020) Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metab in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakil SJ (1986) The relationship between structure and function for and the regulation of the enzymes of fatty acid synthesis. Ann N Y Acad Sci 478, 203–19. [DOI] [PubMed] [Google Scholar]
- 23.Lodhi IJ et al. (2012) Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARγ activation to decrease diet-induced obesity. Cell Metab 16 (2), 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wallace M et al. (2018) Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat Chem Biol 14 (11), 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wise LS et al. (1984) Coordinate regulation of the biosynthesis of ATP-citrate lyase and malic enzyme during adipocyte differentiation. Studies on 3T3-L1 cells. J Biol Chem 259 (8), 4827–32. [PubMed] [Google Scholar]
- 26.Srere PA (1959) The citrate cleavage enzyme. I. Distribution and purification. J Biol Chem 234, 2544–7. [PubMed] [Google Scholar]
- 27.Pinkosky SL et al. (2017) Targeting ATP-citrate lyase in hyperlipidemia and metabolic disorders. Trends Mol Med 23 (11), 1047–1063. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez S et al. (2019) Adipocyte ACLY facilitates dietary carbohydrate handling to maintain metabolic homeostasis in females. Cell Rep 27 (9), 2772–2784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang X et al. (2009) Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet 41 (4), 415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman MA et al. (2012) A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484 (7394), 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carrer A et al. (2017) Impact of a high-fat diet on tissue acyl-coa and histone acetylation levels. J Biol Chem 292 (8), 3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao S et al. (2016) ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep 17 (4), 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Link JC and Reue K (2017) Genetic basis for sex differences in obesity and lipid metabolism. Annu Rev Nutr 37, 225–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edens NK et al. (1993) In vitro lipid synthesis in human adipose tissue from three abdominal sites. Am J Physiol 265 (3 Pt 1), E374–9. [DOI] [PubMed] [Google Scholar]
- 35.Macotela Y et al. (2009) Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 58 (4), 803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao S et al. (2020) Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579 (7800), 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X et al. (2018) Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175 (2), 502–513.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrière A et al. (2014) Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes 63 (10), 3253–65. [DOI] [PubMed] [Google Scholar]
- 39.Cluntun AA et al. (2015) The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wellen KE et al. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 (5930), 1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonnell E et al. (2016) Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep 17 (6), 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mews P et al. (2019) Alcohol metabolism contributes to brain histone acetylation. Nature 574 (7780), 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun W et al. (2020) snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 587 (7832), 98–102. [DOI] [PubMed] [Google Scholar]
- 44.Lee JV et al. (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20 (2), 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martínez-Reyes I et al. (2016) TCA Cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell 61 (2), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimazu T et al. (2013) Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339 (6116), 211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W et al. (2019) A PRDM16-driven metabolic signal from adipocytes regulates precursor cell fate. Cell Metab 30 (1), 174–189.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai Z et al. (2020) The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet 21 (12), 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peleg S et al. (2016) Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep 17 (3), 455–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X et al. (2017) Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell 66 (5), 684–697.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sivanand S et al. (2017) Nuclear acetyl-coa production by ACLY promotes homologous recombination. Mol Cell 67 (2), 252–265.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hong YH et al. (2005) Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 146 (12), 5092–9. [DOI] [PubMed] [Google Scholar]
- 53.Hu J et al. (2016) Short-chain fatty acid acetate stimulates adipogenesis and mitochondrial biogenesis via GPR43 in brown adipocytes. Endocrinology 157 (5), 1881–94. [DOI] [PubMed] [Google Scholar]
- 54.Wolever TM et al. (1997) Time of day and glucose tolerance status affect serum short-chain fatty acid concentrations in humans. Metabolism 46 (7), 805–11. [DOI] [PubMed] [Google Scholar]
- 55.Arpaia N et al. (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504 (7480), 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li G et al. (2014) Short-chain fatty acids enhance adipocyte differentiation in the stromal vascular fraction of porcine adipose tissue. J Nutr 144 (12), 1887–95. [DOI] [PubMed] [Google Scholar]
- 57.Gao Z et al. (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58 (7), 1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donohoe DR et al. (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell 48 (4), 612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.den Besten G et al. (2013) Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am J Physiol Gastrointest Liver Physiol 305 (12), G900–10. [DOI] [PubMed] [Google Scholar]
- 60.Ge H et al. (2008) Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 149 (9), 4519–26. [DOI] [PubMed] [Google Scholar]
- 61.Müller M et al. (2019) Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci Rep 9 (1), 12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry RJ et al. (2016) Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534 (7606), 213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cifarelli V et al. (2020) Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J Clin Invest 130 (12), 6688–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munday MR et al. (1988) Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem 175 (2), 331–8. [DOI] [PubMed] [Google Scholar]
- 65.Vagelos PR et al. (1963) Studies on the mechnism of activation of acetyl coenzyme A carboxylase by citrate. J Biol Chem 238, 533–40. [PubMed] [Google Scholar]
- 66.Waite M and Wakil SJ (1963) Studies on the mechanism of action of acetyl coenzyme A carboxylase. I. Effect of isocitrate on the transcarboxylation step of acetyl coenzyme A carboxylase. J Biol Chem 238, 77–80. [PubMed] [Google Scholar]
- 67.Bulusu V et al. (2017) Acetate recapturing by nuclear acetyl-coa synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep 18 (3), 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schug ZT et al. (2015) Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27 (1), 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su X et al. (2015) Adipose tissue monomethyl branched-chain fatty acids and insulin sensitivity: Effects of obesity and weight loss. Obesity (Silver Spring) 23 (2), 329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sears DD et al. (2009) Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci U S A 106 (44), 18745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratheiser K et al. (1991) Inhibition by etomoxir of carnitine palmitoyltransferase I reduces hepatic glucose production and plasma lipids in non-insulin-dependent diabetes mellitus. Metabolism 40 (11), 1185–90. [DOI] [PubMed] [Google Scholar]
- 72.Zarain-Herzberg A et al. (1996) Modification of sarcoplasmic reticulum gene expression in pressure overload cardiac hypertrophy by etomoxir. Faseb j 10 (11), 1303–9. [DOI] [PubMed] [Google Scholar]
- 73.Ray KK et al. (2019) Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med 380 (11), 1022–1032. [DOI] [PubMed] [Google Scholar]
- 74.Altman BJ et al. (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16 (10), 619–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cox AR et al. (2020) STAT1 dissociates adipose tissue inflammation from insulin sensitivity in obesity. Diabetes 69 (12), 2630–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y et al. (2021) Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat Commun 12 (1), 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosen ED and Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156 (1–2), 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mills EL et al. (2018) Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560 (7716), 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asadi Shahmirzadi A et al. (2020) Alpha-ketoglutarate, an endogenous metabolite, extends lifespan and compresses morbidity in aging mice. Cell Metab 32 (3), 447–456.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mills EL et al. (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18 (5), 488–498. [DOI] [PubMed] [Google Scholar]
- 81.Hui S et al. (2020) Quantitative fluxomics of circulating metabolites. Cell Metab 32 (4), 676–688.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mullen AR et al. (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481 (7381), 385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han J et al. (2011) The spatiotemporal development of adipose tissue. Development 138 (22), 5027–37. [DOI] [PubMed] [Google Scholar]
- 84.Poissonnet CM et al. (1984) The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum Dev 10 (1–2), 1–11. [DOI] [PubMed] [Google Scholar]
- 85.Sebo ZL and Rodeheffer MS (2019) Assembling the adipose organ: adipocyte lineage segregation and adipogenesis in vivo. Development 146 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chau Y-Y et al. (2014) Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nature Cell Biology 16 (4), 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krueger KC et al. (2014) Characterization of Cre recombinase activity for in vivo targeting of adipocyte precursor cells. Stem Cell Reports 3 (6), 1147–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanchez-Gurmaches J and Guertin DA (2014) Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat Commun 5, 4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanchez-Gurmaches J et al. (2015) Highly selective in vivo labeling of subcutaneous white adipocyte precursors with Prx1-Cre. Stem Cell Reports 4 (4), 541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hepler C et al. (2018) Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 7, e39636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeffery E et al. (2016) The adipose tissue microenvironment regulates depot-specific adipogenesis in obesity. Cell Metab 24 (1), 142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee KY et al. (2013) Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62 (3), 864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Merrick D et al. (2019) Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364 (6438). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwalie PC et al. (2018) A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559 (7712), 103–108. [DOI] [PubMed] [Google Scholar]
- 95.Tchoukalova Y et al. (2007) Committed subcutaneous preadipocytes are reduced in human obesity. Diabetologia 50 (1), 151–7. [DOI] [PubMed] [Google Scholar]
- 96.Jeffery E et al. (2015) Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol 17 (4), 376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang QA et al. (2013) Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nature Medicine 19 (10), 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith GI et al. (2020) Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 130 (3), 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yore MM et al. (2014) Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159 (2), 318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Syed I et al. (2018) Palmitic acid hydroxystearic acids activate GPR40, which is involved in their beneficial effects on glucose homeostasis. Cell Metab 27 (2), 419–427.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrus P et al. (2020) Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab 31 (2), 375–390.e11. [DOI] [PubMed] [Google Scholar]
- 102.Arner E et al. (2012) Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 61 (8), 1986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]