

Abstract
Purpose.
CLN3 disease is a neurodegenerative disorder with onset in childhood. It affects multiple functions at different developmental stages. Incomplete understanding of the pathophysiology hampers identification of cell and tissue biochemical compounds reflective of the disease process. As treatment approaches are being explored, more sensitive, objective, quantifiable, and clinically relevant biomarkers are needed.
Methods.
We collected prospective biosamples from 21 phenotyped individuals with CLN3. We measured neurofilament light chain (NEFL) levels, a marker of neuronal damage, in cross-sectional CSF and serum samples from individuals with CLN3 and in pediatric non-CLN3 controls using two different assays.
Results.
CSF and serum NEFL levels are significantly higher in CLN3 (CSF: 2096±1202; serum: 29.0±18.0 pg/mL) versus similarly aged non-CLN3 (CSF: 345±610; serum: 6.7±3.2 pg/mL) samples. NEFL levels correlate with Unified Batten Disease Rating Scale and adaptive behavior composite scores, and MR spectroscopy markers. NEFL levels from CSF and serum are strongly correlated (rp=0.83; p<.0001).
Conclusions.
CSF and serum NEFL levels increase in multiple neurologic conditions. Here, we show that CSF and serum NEFL levels also increase in CLN3 (versus non-CLN3) and correlate with other disease-relevant measures. These findings suggest NEFL as a relevant and feasible biomarker for applications in CLN3 clinical trials and management.
Keywords: JNCL, Nf-L, natural history, N-acetyl aspartate, glutamatergic
INTRODUCTION
Collectively, the neuronal ceroid lipofuscinoses (NCLs) comprise one of the most common group of diseases with neurodegenerative presentation in the pediatric age range. Of the 13 known NCL types, CLN3 disease (OMIM#204200; Juvenile Neuronal Ceroid Lipofuscinosis, Batten disease) is the most common. Since the first description of individuals with the juvenile onset type of NCL by Otto Christian Stengel in 1826, its clinical symptoms and presentation have been extensively characterized.1,2 CLN3 disease is a pan-ethnic, autosomal recessive, neurodegenerative disease with an estimated incidence ranging from 1:10,000 in Scandinavia to 1:100,000 worldwide.3 While it is considered a lysosomal disease based on accumulation of autofluorescent storage materials in the organelle, the function of the protein encoded by the CLN3 gene has not been clearly elucidated.4 The most common pathogenic variant in CLN3 is a 996-base pair (commonly known as 1-kb, exon 7-8) deletion found in homozygous or compound heterozygous state in ~95% of individuals with CLN3 disease.5,6
Frequent symptoms reported in individuals with the typical CLN3 disease course include progressive vision loss leading to blindness, neurocognitive decline and loss of previously acquired ability leading to dementia, behavioral and psychiatric disturbances, development of seizures and a movement disorders similar what is seen in Parkinson disease.1,2,7 The vision loss is reflected in decreased to absent electroretinogram responses, and thinning to loss of distinct retinal cell layers on optical coherence tomography. Neurocognitive plateauing and declines are demonstrated by decreased performance on neuropsychological tests in areas such as intelligence quotient (IQ)8 and adaptive behavior9. Brain volumetric imaging has also demonstrated generalized volume loss, correlating with disease duration10. The Hamburg scale11 and the Unified Batten Disease Rating Scale (UBDRS)12 provide clinical ratings of disease severity and progression.
Challenges remain in the application of the above outcome measures in the typical short-term design of interventional trials. CLN3 symptoms present asynchronously over a relatively protracted time frame. A high portion of affected individuals harbor functionally significant vision loss by 6-7 years of age, whereas the Parkinsonian motor abnormalities have typical onset in the mid-teen years. In addition, clinical assessment measures rely partially on the evaluators’ expertise and the affected individuals’ cooperation. Longitudinal and quantitative assessments may be limited by disease state, methodologies, and resource availability. Recent work on identifying disease-reflective biochemical markers for CLN3 employed different tissues and analysis methods that yielded variable results.13–15 Furthermore, the proposed candidate biomarkers need additional work to demonstrate clinical validity.
Neurofilament light (NEFL) chain is an intermediate filament type with an estimated molecular weight of 70-86 kDa.16 It is a stable component of the neuronal axon and has a low-turnover rate.17–19 Processes leading to neuronal damage ranging from physical in traumatic brain injuries, to inflammation in multiple sclerosis, to abnormal and normal aging cause release of NEFL from injured cells.16,20–23 Individuals with different forms of dementia have elevated level of NEFL in the cerebral spinal fluid (CSF) as compared to that seen in healthy controls, with non Alzheimer-like dementia types having higher CSF NEFL levels.22 Elevated CSF NEFL level is also seen in individuals with other degenerative conditions involving motor neurons such as amyotrophic lateral sclerosis and Parkinson disease.20,23,24 The small size of NEFL permits its diffusion from CSF into blood, thus the elevated NEFL profile is also detected in plasma and serum sample from individuals with various neurodegenerative diseases.20,24–27
To complement clinical outcome measures and provide an objective assessment of disease status and progression, we incorporated biospecimen collection and biochemical marker discovery in a natural history study of individuals with CLN3 (NCT03307304). In this report, we investigated the level of NEFL in CSF and corresponding serum samples at baseline from individuals with CLN3 disease.
We hypothesized that NEFL level would be higher in individuals with CLN3 as compared to those without CLN3 (e.g. healthy and unaffected, non-neurodegenerative conditions). We anticipated that NEFL level would correlate with CLN3 clinical outcome measures.
MATERIALS and METHODS
Ethics Statement
We evaluated study participants and collected biospecimens as part of Natural History studies approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board [CLN3 (NCT03307304); Creatine Transporter Deficiency (CTD; NCT02931682); Smith-Lemli-Opitz Syndrome (SLOS; NCT00001721); Niemann-Pick Disease, Type C (NCT00344331)]. The main inclusion criteria for CLN3 disease study participants is having pathogenic variants in CLN3, with no restriction on disease phenotype. Parents or guardians and participants older than 7 years of age provided consent and assent, respectively.
Clinical Measures and Sample Collections
We evaluated individuals with CLN3 using the UBDRS sub-domains [27-item Physical, 5-item Capability given actual vision, and 7-item Clinical Summary (Clinical Global Impression, CGI)]12,28. A weighted score for each sub-domain was calculated as followed: [total score for all completed items / (total possible score of all items – total possible score of missing items)] x total possible score of all items. We also obtained Vineland adaptive behavior composite (ABC) scores29; and MR spectroscopy quantification of metabolites [creatine (Cr), glutamine/glutamate/GABA (Glx), and N-acetyl aspartate (NAA)] from a voxel in the midline parietal gray matter30. Available demographics and genotype for all samples, biomarker analyses completed, and a summary of relevant outcome measures for the CLN3 samples are described in Tables 1, S1, and S2.
Table 1.
CSF and serum sample characteristics and assayed methods.
| Group | Age (years) | Sex, n (%) | Assay Method | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Median | Range | Female | Male | CSF | Serum | ||||
| Q1 | Q3 | Full | PEA | ELISA | Simoa™ | |||||
| CLN3 | 21 | 11.4 | 7.8 | 15.4 | 6.8 - 20.7 | 10 (48) | 11 (52) | ✓ | ✓ | ✓ |
| Comparators (n) | 32 | 33 | 34 | |||||||
| Pediatric Laboratory Controls | 20 | 12.5 | 7.5 | 15.5 | 3.0 – 20.0 | 10 (50) | 10 (50) | ✓ | ||
| PLC subset | 9 | 14.0 | 7.0 | 17.0 | 4.8 – 20.0 | 2 (22) | 7 (78) | ✓ | ✓ | |
| Pediatric Healthy Siblings | 10 | 10.8 | 9.4 | 13.3 | 6.3 - 18.7 | 4 (40) | 6 (60) | ✓ | ||
| CTD | 12 | 7.0 | 3.9 | 10.8 | 3.2 - 14.3 | 0 | 12 | ✓ | ✓ | ✓ |
| SLOS | 12 | 9.4 | 5.2 | 13.7 | 2.6 – 21.0 | 5 (42) | 7 (58) | ✓ | ✓ | |
CSF: cerebrospinal fluid. CTD: creatine transporter deficiency. ELISA: enzyme-linked immunosorbent assay. PEA: proximal extension assay. PLC: pediatric laboratory controls. Q1: quartile 1, 25th percentile. Q3: quartile 3, 75th percentile. Simoa™: single molecule array. SLOS: Smith-Lemli-Opitz syndrome.
We collected serum samples in the morning following an overnight fast in tubes with no anticoagulant or preservative. We collected cerebral spinal fluid (CSF) samples on the same morning, typically under sedation, using standard procedures. Collected CSF samples were transported at room temperature and aliquoted for storage at −80 degree Celsius within 1 hour of collection. If the CSF sample was visually observed to have blood contamination, it was centrifuged at 3,400 rpm for 10 minutes and the supernatant aliquoted for storage. We requested pediatric anonymized, left-over, unused CSF samples from outside clinical laboratories for use as unaffected CSF controls (pediatric laboratory controls; NCT00344331). These samples had information on the individuals’ sex and age at the time of sampling. We collected fasting serum samples from unaffected siblings of individuals with CLN3 (pediatric healthy siblings; NCT03307304) to use as unaffected serum controls.
Neurofilament Light Chain Assays
Proximal Extension Assay (PEA)
We randomized the position of samples on a 96-well plate31 and aliquoted 40 microliter of CSF samples per well. The sample plate was sealed, frozen and shipped on dry ice to Olink® (Boston, MA) for NEURO EXPLORATORY panel32 assay. For analyses, we used the normalized protein expression (NPX) value, a logarithmic relative quantitative value where an increase of one NPX represents a doubling of protein expression level33.
ELISA
We used a solid-phase sandwich ELISA kit (UmanDiagnostics, Umea, Sweden; Catalog number: 10-7002 RUO) and followed the manufacturer’s instructions to measure CSF NEFL levels. Limit of detection (LOD) was 33 pg/ml. All samples were diluted 1:2 and analyzed blindly and in singlets. Samples were analyzed batch-wise, in 2 batches (plates). Location of study samples on each plate was randomized.31 In each batch we analyzed a control sample in duplicate. Coefficient of variation (CV) for control sample across batches was 2.2%, confirming assay precision and reproducibly. For values less than the LOD, 16.5 pg/ml (midpoint between 0 and 33 given 1:2 dilution) was used.
Single Molecule Array (Simoa™)
We used a Simoa™ assay kit (Quanterix, Billerica, MA, USA; Product number: 103186) and a Simoa™ HD-1 analyzer to measure serum NEFL levels. Limit of detection (LOD) was 0.038 pg/ml. All samples were diluted 1:4 and analyzed blindly and in singlets in a single batch (plate). Location of study samples on each plate was randomized.31 Quality control samples (QCs) provided with the kit have measured concentrations within given range, confirming assay precision.
Statistical Analyses
Data were described and checked by frequency distributions (percentages) and simple descriptive statistics (mean ± standard deviation). Distributional assumptions were assessed, and data were log transformed as necessary. Where data distributions were uncertain, both parametric and non-parametric tests were carried out; no differences in conclusions were observed. Continuous data between groups were compared using t-tests (and non-parametric Wilcoxon rank-sum tests, if appropriate). Analysis of variance (ANOVA) (and Kruskal-Wallis) tests compared the various subgroups; p values from post-hoc subgroup/pairwise tests were corrected for multiplicity (built-in Bonferroni). Correlation analyses used the Pearson correlation coefficient and Fisher’s z transformation for bias correction, and 95% confidence intervals (CIs) were computed and are shown in the figures. P values for the correlation analyses were corrected for multiple comparisons using the Stepdown Bonferroni method. Where described (i.e., Figure 3), both raw and multiple comparison-corrected p values were provided. Data were analyzed using SAS v9.4 (SAS Institute, Inc, Cary, NC). Graphs were generated using SAS, Microsoft Excel, and PowerPoint.
RESULTS
Sample and Data Characteristics
Age and sex distributions for the cohorts are similar amongst the comparison groups (Table 1). While log-transformations of data (i.e., for ELISA) improved distributions, they had minimal impact on the conclusions; thus, results are reported and shown in their original raw scales for easier interpretation. In addition, the data were assessed for factors that could potentially influence results as followed. Of the 21 CLN3 samples, six were collected from three affected sibling pairs. Sensitivity analyses excluding the older and the younger of the siblings revealed negligible effects on the correlations and confidence intervals; thus, we elected to keep all 21 samples in all analyses. Calculations made using measured NEFL levels versus those using age-adjusted (for CSF ELISA and serum Simoa™ samples) and blood volume-adjusted (for serum samples) levels did not alter any of the conclusions. Multiple regression analyses on the correlations of interest adjusting for age generally showed small percentages (<10%) of the variability in the outcomes accounted by age. There were a few exceptions; however, none of the conclusions from the models with or without age were different. Thus, we decided to use measured CSF and serum NEFL levels in further analyses.
NEFL levels in individuals with CLN3 disease are elevated in CSF and serum
NEFL levels in CSF samples measured by proximal extension assay (PEA) and ELISA
While both PEA and ELISA measure expressed NEFL level, each has a unique advantage. PEA incorporates qRT-PCR for signal amplification, thus expanding the sensitivity range and reducing the amount of sample input required. This is relevant for a screening assay. ELISA assay provides the ability for absolute quantitation of NEFL level, a function currently unavailable with the PEA method. Absolute quantification allows results to be compared across time and from different cohorts. The purpose of using both methods was to explore their utility, not to compare directly the results.
Values from PEA and ELISA assays were strongly correlated (rp=0.69; p<.0001) (Figure S1). CSF NEFL normalized protein expression (NPX) value by PEA was higher in the CLN3 (9.3±1.0) compared to that in the combined non-CLN3 (7.4±2.7; p=.0013) samples (Figure S2A). ANOVA and post-hoc subgroup comparisons showed a difference in NEFL levels in the CLN3 versus creatine transporter deficiency (CTD, p=.0005), but not in the CLN3 versus pediatric laboratory control (p=.32) sample pairings (Figure S2B). CSF sample from healthy, unaffected pediatric individuals cannot be obtained for research purpose. Pediatric laboratory control samples were collected from individuals with clinical indications for a lumbar puncture. These samples have multiple limitations including limited number and volume, incomplete clinical information, different collection and handling and likely include individuals with neurological processes affecting NEFL level These limitations became apparent when using PEA, and thus for the ELISA we included as comparison pediatric samples from two non-neurodegenerative disorders (CTD and SLOS) that were collected under similar conditions.
CSF NEFL measured by ELISA was much higher (2096±1202 pg/mL) in the CLN3 compared to 345±610 pg/mL in the non-CLN3 cohort (p<.0001) (Figure S3). Post-hoc subgroup comparisons showed that the difference was driven by CLN3 versus each of pediatric laboratory control (PLC, p=.0017), CTD (p<.0001), and Smith-Lemli-Opitz syndrome (SLOS, p<.0001) sample pairings (Figure 1).
Figure 1.
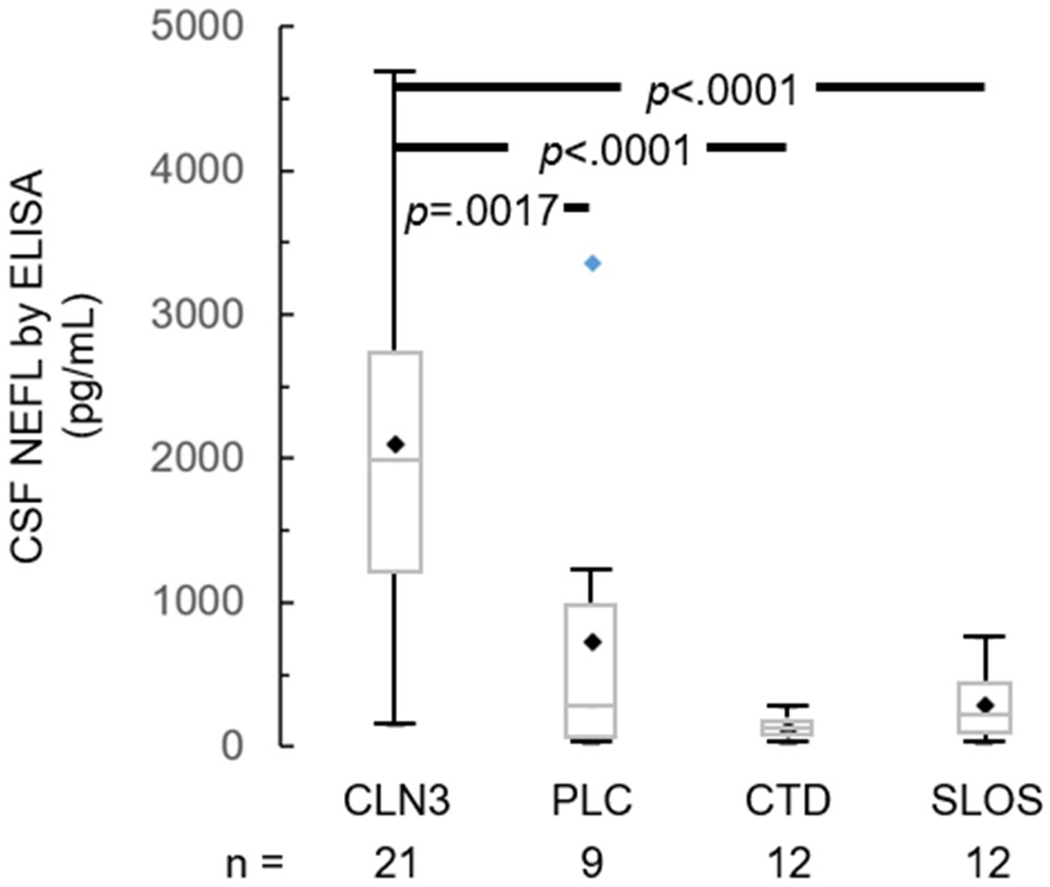
CSF NEFL level in samples from CLN3 versus non-CLN3 (PLC, CTD, SLOS) individuals, measured by ELISA. Black diamond – mean; blue diamond – outlier; box – median and interquartile range [IQR, Q1 (25th percentile) - Q3 (75th percentile)]; whiskers – minimum = Q1 – 1.5*IQR and maximum = Q3 + 1.5*IQR. P values are derived from ANOVA and post-hoc Bonferroni adjustment for pairwise multiple comparisons. CSF: cerebrospinal fluid. CTD: creatine transport deficiency. NEFL: neurofilament light chain. PLC: pediatric laboratory controls. SLOS: Smith-Lemli-Opitz syndrome.
NEFL levels in serum samples measured by Single Molecule Array (Simoa™)
Blood samples are relatively more easily obtained and more practical for multicenter therapeutic trials. Thus, we explored NEFL level in CLN3 versus non-CLN3 [pediatric healthy sibling (PHS), CTD, SLOS] serum samples, and the correlation between paired CSF and serum samples. Mean serum NEFL level was higher in CLN3 (29.0±18.0 pg/mL) than non-CLN3 (6.7±3.2 pg/mL; p<.0001) samples (Figure S4). ANOVA and subgroup comparisons showed that the difference was reflected in CLN3 versus each of PHS (p<.0001), CTD (p<.0001), and SLOS (p<.0001) sample pairings (Figure 2). NEFL level in corresponding CSF and serum samples correlated positively between PEA (rp=0.75; p<.0001) or ELISA (rp=0.83; p<.0001) versus Simoa™ measurements (Figure S5).
Figure 2.
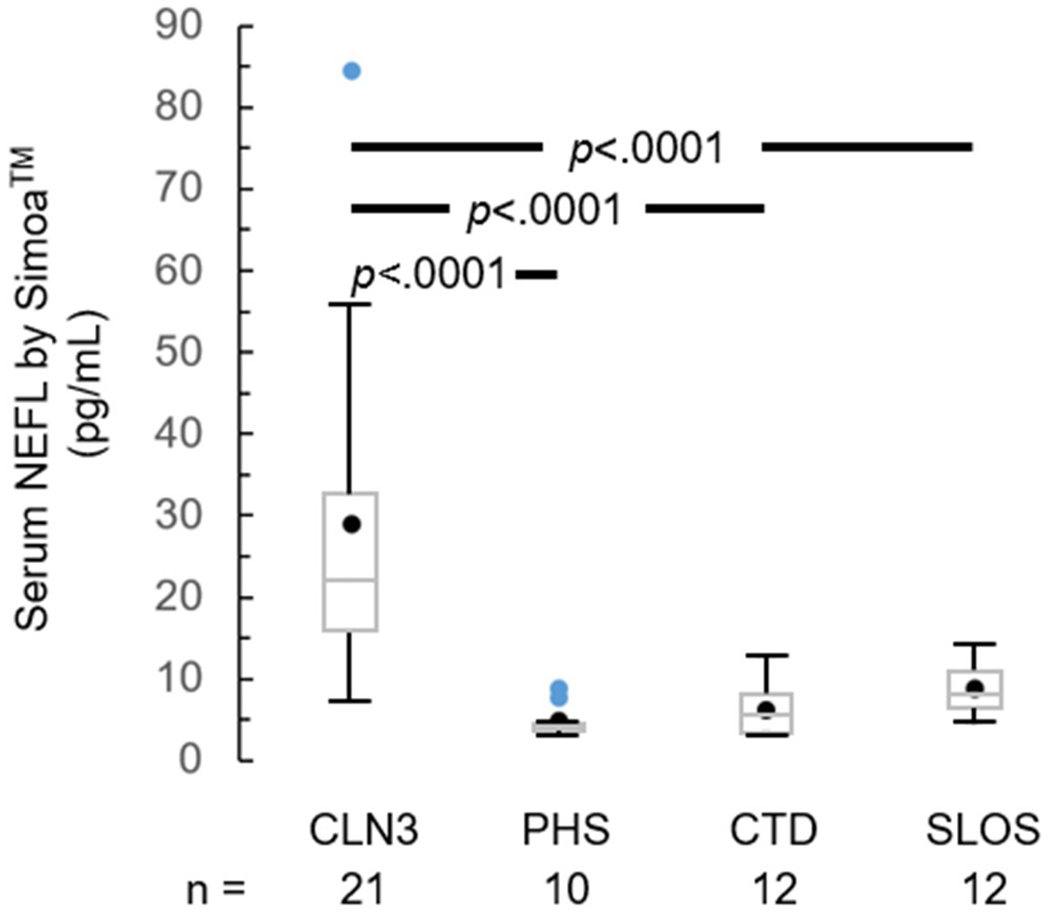
Serum NEFL level in samples from CLN3 versus non-CLN3 (PHS, CTD, SLOS) individuals, measured by Simoa™. Black circle – mean; blue circle – outlier; box – median and interquartile range [IQR, Q1 (25th percentile) – Q3 (75th percentile)]; whiskers – minimum = Q1 – 1.5*IQR and maximum = Q3 + 1.5*IQR. P values are derived from ANOVA and post-hoc Bonferroni adjustment for multiple pairwise comparisons. CSF: cerebrospinal fluid. CTD: creatine transport deficiency. NEFL: neurofilament light chain. PHS: pediatric healthy (unaffected) siblings. SLOS: Smith-Lemli-Opitz syndrome.
NEFL Levels Correlate with Clinical Outcome Measures
NEFL levels positively correlated with age in CSF by ELISA (rp=0.62, 95% CI=[0.25,0.83], padj=.02) and PEA (rp=0.47, [0.05,0.75], padj=.11), although the latter value had a larger confidence interval and adjusted p value (Figure S6). Serum NEFL level did not correlate with age (rp=0.28, [−0.18,0.63], padj=.66). We tested correlations between CSF or serum NEFL levels and other clinical outcome measures (UBDRS and Vineland ABC scores, and MR spectroscopy measurements) in CLN3 study participants (Figures 3, S6, S7). Higher UBDRS Physical and CGI scores indicate more severe12,28,34, whereas higher Capability with actual vision12,34 and Vineland ABC scores (manuscript submitted) indicate less severe disease. Higher levels of N-acetylaspartate (NAA), glutamatergic (Glx) and creatine (Cr) on spectroscopy indicate the presence of healthy neurons.30
Figure 3.
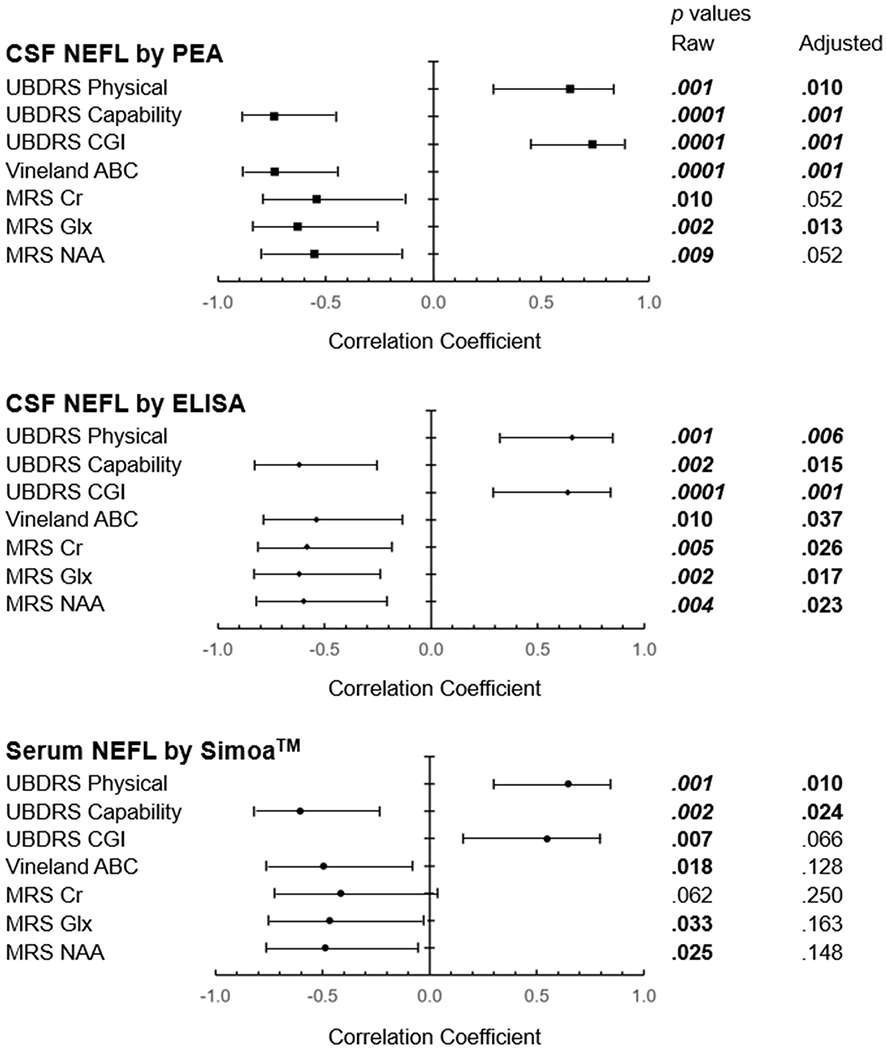
Correlation of NEFL levels with clinical outcome measures, by assay method. Forest plot indicates the correlation coefficients along with 95% confidence limits. The horizontal axes describe the magnitude of the correlation coefficients, with <0 suggesting inverse relations and >0 suggesting positive relations. Adjusted p values are derived from the Stepdown Bonferroni method for multiple comparisons. Bold – p<.05. Bold italics – p<.01. ABC: adaptive behavior composite. CGI: clinical global impression. Cr: creatine. CSF: cerebrospinal fluid. Glx: glutamatergic metabolites. NAA: N-acetylaspartate. NEFL: neurofilament light chain. PEA: proximal extension assay. Simoa™: single molecule array. UBDRS: Unified Batten Disease Rating Scale.
NEFL levels in both CSF and serum were positively correlated with UBDRS Physical (CSF by ELISA: rp=0.66, [0.32,0.85], padj=.006; serum by Simoa™: rp=0.65, [0.30,0.84], padj=.01), and inversely correlated with UBDRS Capability (CSF by ELISA: rp=−0.62, [−0.83,−0.25], padj=.01; serum by Simoa™: rp=−0.60, [−0.82,−0.23], padj=.02) scores. CSF NEFL level measured by both PEA and ELISA also showed positive correlations with UBDRS CGI (PEA: rp=0.74, [0.45,0.89], padj=.001; ELISA: rp=0.64, [0.29,0.84], padj=.001), and inverse correlations with Vineland ABC scores (PEA: rp=−0.74, [−0.89,−0.44], padj=.001; ELISA: rp=−0.54, [−0.79,−0.14], padj=.04) and with Glx markers level (PEA: rp=−0.63, [−0.84,−0.26], padj=.01; ELISA: rp=−0.62, [−0.83,−0.24], padj=.02) in the parietal gray matter (PGM). CSF NEFL levels measured by ELISA showed the most correlations (Figure 3). In addition to those seen with PEA, CSF NEFL levels by ELISA also inversely correlated with PGM Cr (rp=−0.58, [−0.81,−0.18], padj=.02) and NAA (rp=−0.60, [−0.82,−0.21], padj=.02) MR spectroscopy measurements. Serum NEFL levels showed relations with all but PGM Cr outcome measures; however, they were only retained with UBDRS Physical (rp=0.65, [0.30,0.84], padj=.01) and UBDRS Capability (rp=−0.60, [−0.82,−0.23], padj=.02) scores after adjustment for multiple comparisons.
We used a weighted combined score of all seven items in the UBDRS Clinical Summary sub-domain in this study to incorporate the scores for all relevant symptoms related to CLN3 disease. Since prior publications (Figure S8) only used individual CGI item scores, we also performed correlation analyses using only the single-item overall CGI score. The latter method yielded slightly lower correlation coefficients and larger CIs and p values (Figure S8), reflecting the decreased variabilities resulting from using a more discrete CGI value set. However, the outcomes and conclusions described above did not change.
DISCUSSION
While much progress has been made in designing and refining quantitative assessments to characterize disease progression in individuals with CLN3, opportunities remain for identification of additional measures that are objective and accessible, with a potential for sensitivity to reflect short-term changes. Here, we show that cross-sectional CSF NEFL level in a cohort of pediatric study participants with CLN3 reflects clinical state as assessed by disease rating scale, adaptive behavior evaluation, and brain MR spectroscopy. Furthermore, the correlations hold for NEFL level in corresponding serum samples from individuals with CLN3. This latter finding is particularly important and applicable in the pediatric population as serum samples are relatively more feasibly obtained from both affected and unaffected children.
Prior work identifying CLN3 biomarkers await additional validation before translation into clinical application13–15. Lebrun and colleagues (2011) performed microarray analysis on lymphocytes from eight individuals homozygous for the common 1-kb deletion variant. They confirmed top candidate genes in cerebellar precursor cells from CbCln3Δex7/8 mice and Cln3 siRNA-treated HeLa and SH-SY5Y cells. NEFL RNA expression level decreased by three folds in lymphoblasts from individuals with CLN3 as compared to that from similarly aged healthy controls. This decrease was present only in individuals classified as having a slow, and not in those with a rapid or average, disease progression. A paradoxically decreased mRNA level, in the setting of increased NEFL protein level, has also been observed in spinal cord tissue from individuals with amyotrophic lateral sclerosis.35 The excess NEFL protein, in combination with another protein member of the neurofilament complex, forms intracellular accumulation that are thought to interfere with normal axonal transport activity.19 It is unclear whether this contributes to a negative feedback regulation of NEFL mRNA expression.
Hersrud et al. (2016) used immunoassays, 2-dimensional difference gel electrophoresis and Western blotting to analyze plasma samples from individuals with CLN3 versus those from healthy controls of similar ages. NEFL was not one of the analytes in their list of candidate biomarkers. Lack of inclusion of NEFL on the immunoassay panels or of method sensitivity for detecting a neuronally derived protein in peripheral blood samples are two possible explanations for why changes in NEFL level was not reported. Sleat et al. (2017) interrogated post-mortem brain tissue and ventricular CSF from individuals with CLN3, as compared to post-mortem samples from older unaffected individuals, using untargeted mass spectrometry. NEFL was also not identified as a candidate biomarker through this approach. Several factors may contribute to this including lack of age-similar controls, as NEFL level increases with increasing age.36,37 The half-life of transgenic human and innately expressed NEFL protein in mice ranges from months to weeks17,18, thus NEFL degradation should not be an explanation for its absence on the candidate biomarker list. As the authors alluded to, biomarker levels from this experiment reflect the end-stage time point of the disease, when perhaps certain pathophysiological changes were no longer present.15
Most recently, Kuper et al (2020) demonstrated correlations of CLN3 disease state to lymphocyte vacuoles and lysosomal associated membrane protein 1 (LAMP1) expression in human blood. The vacuole-related measures were higher in samples from individuals with classic than those with vision-only CLN3 presentation, and higher than those from individuals without CLN3. LAMP1 level in lymphocytes also correlated with CLN3 disease state, but not with vacuole-related measures. In addition, LAMP1 expression pattern differed in the CD20-positive B-lymphocytes as compared to the CD4/CD8+ T-lymphocytes. Based on these findings, the authors have begun to incorporate LAMP1 assay in their diagnosis of CLN3 disease. As these measures lack correlation with disease progression, further research is needed to assess their relevance to other CLN3 outcome measures and application in clinical trials.38
Elevation in NEFL level, both in CSF and blood, have been observed in multiple adult neurodegenerative conditions as compared to healthy controls. The typical magnitude of NEFL increase ranges from 228.9 pg/mL (Alzheimer dementia) to 9285 pg/mL (amyotrophic lateral sclerosis) in CSF, and 23 pg/mL (Alzheimer and Parkinson) to 296 pg/mL (sporadic Creutzfeldt-Jakob) in blood.24 Very few studies have examined NEFL levels in pediatric neurodegenerative diseases. Even fewer evaluate CSF NEFL in affected or similarly aged control groups. Wong et al. (2019) showed elevated serum NEFL levels in children with acquired demyelinating syndromes (36.1 pg/mL) versus age-matched controls with no specific neurological disorder (6.1 pg/mL). They showed good correlation between the CSF and serum NEFL levels in the affected group but did not compare CSF samples to an age-matched control group. Also using serum samples, Ru et al. (2019) reported significantly elevated NEFL levels in children with late-infantile/CLN2 disease (79.8-388.1 pg/mL) as compared to age-similar controls (2.1-5.5 pg/mL). Using CSF samples from pediatric and adult individuals with clinically isolated syndrome of neuroinflammatory nature, de Vries et al. (2019) proposed that high CSF NEFL level suggested a severe neurodegenerative process and is predictive of progression to clinically definite multiple sclerosis. They based the hypothesis partly on measured CSF NEFL level of 4888 pg/mL in the pediatric clinically definite multiple sclerosis group as compared to 2683 pg/mL in the pediatric clinically isolated syndrome group after adjusting for age. While no pediatric CSF control samples were measured, the NEFL level in unaffected adult CSF controls was 444 pg/mL.39 In a rare pediatric study using historical CSF controls, Olsson et al. (2019) showed that children with spinal muscular atrophy type 1 had untreated CSF NEFL levels of 4598±981 pg/mL as compared to 148±39 pg/mL in the age-similar historical controls with non-specific neurologic presentation.
Compared to these studies, CSF NEFL levels of the pediatric laboratory controls in our study is high, likely reflecting the acute pathological processes that necessitate a work-up involving lumbar puncture. CSF NEFL levels in the non-neurodegenerative cohorts, creatine transporter deficiency (CTD) and Smith-Lemli-Opitz syndrome (SLOS), are similar to those reported for adult24 and pediatric40 controls. Serum NEFL levels in the unaffected pediatric healthy siblings control, CTD and SLOS groups in this study are within the ranges previously reported for pediatric controls.25,27 This suggests that CSF and serum samples from pediatric non-neurodegenerative conditions may be considered for use as comparators, given the general limitation on available samples from healthy, unaffected pediatric individuals.
CSF and serum NEFL values in the CLN3 cohort are clearly elevated above levels reported for pediatric or adult controls, and in the ranges of those for neurodegenerative conditions. Proximal extension assay appears to be more discriminant at the lower end, whereas ELISA more at the higher end, of NEFL level detection. Of note, participant SP5.2.2 with genotype, history and evaluation findings consistent with a vision-only phenotype has CSF and plasma NEFL levels lower than others in the cohort and in the range of normal values. This suggests that NEFL level may be an additional useful quantitative measure for delineating the phenotypic spectrum of CLN3-related disorders. Interestingly, the magnitude of NEFL elevation in CSF and serum samples from the CLN3 cohort appears to be less than those seen in conditions with more severe presentation and earlier onset such as CLN2 disease25 and spinal muscular atrophy type 140.
In addition to correlating with respective disease severity scale measurements, elevated NEFL level also reflects findings on brain imaging. Higher NEFL level correlated with more disordered or abnormal white matter tract parameters in amyotrophic lateral sclerosis41, with subsequent decrease in brain volume and greater number of contrast-enhancing lesions in multiple sclerosis27,39,42, and decreased baseline and higher atrophy rate of brain volume in Alzheimer dementia26. NEFL has also been shown to reflect treatment effect, i.e. decreasing following one or multiple doses of experimental pharmacologic agents.25,40 In our study, NEFL levels negatively correlate with MR spectroscopy markers for healthy neurons. Correlation of NAA, Glx and Cr spectroscopy levels with disease severity has been reported in infantile NCL30 and will be described for CLN3 in a separate report.
While we describe here the largest number of clinically well characterized CLN3 CSF and serum samples evaluated for NEFL level to date, the relatively small size and cross-sectional nature are limitations to this study. While CSF NEFL level appears more sensitive in reflecting disease outcome measures the findings of high correlation between CSF and serum NEFL levels, and amongst serum NEFL level and clinical outcome measures, suggest a feasible approach for enlarging both the affected and control cohorts. Continuation of longitudinal follow up and sample collection in natural history studies will allow evaluations of the long-term trends of NEFL level in CLN3 disease, including investigating whether NEFL level decreases as neurons die as part of the neurodegenerative process. This does not appear to be the case in this cohort as NEFL level positively correlated with markers of more severe disease. NEFL is not a specific biomarker for CLN3 disease based on its response pattern in multiple other neurological diseases. However, as noted above, further characterization of the magnitude and range of NEFL expression may help to distinguish amongst conditions by their disease course and age of onset.25,39,40 NEFL level may also be an early marker for response to treatment.25,40
Data from this study suggest that CSF NEFL level strongly reflects neurologically based clinical outcome measures in CLN3. This portends a role for following CSF NEFL level as an objective, quantitative, longitudinal outcome measure in future clinical trials. Furthermore, correlation of CSF and serum NEFL level with the UBDRS Physical and Capability domains suggests clinical validity of this biomarker, and the potential for serum NEFL as a minimally invasive testing option for monitoring disease progression. We anticipate that the ongoing natural history study (NCT03307304), and similar efforts for collecting biospecimens that are accompanied by extensive phenotyping of the affected individuals, will provide further insights into the applicability of NEFL as a biomarker for CLN3 disease.
Supplementary Material
ACKNOWLEDGMENT
We dedicate this work to the study participants, their families, and the support organizations for the motivation and inspiration they have provided. We thank Dr. Jonathan W. Mink (University of Rochester Medical Center) for his expert inputs on UBDRS implementation. We thank colleagues and staff who enabled the conduct of this study and the preparation of this manuscript. The NIH Intramural Research Program of NICHD, NIAID, NIMH, the NIH Clinical Center, NINDS, and an NIH Clinical Center Bench-to-Bedside Award supported this work.
Footnotes
ETHICS DECLARATION
We evaluated study participants and collected biospecimens as part of Natural History studies approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board [CLN3 (NCT03307304); Creatine Transporter Deficiency (CTD; NCT02931682); Smith-Lemli-Opitz Syndrome (SLOS; NCT00001721); Niemann-Pick Disease, Type C (NCT00344331)]. Parents or guardians and participants older than 7 years of age provided consent and assent, respectively. We provided de-identified participant-level data. The study adhered to the principles set out in the Declaration of Helsinki.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
REFERENCES
- 1.Haltia M The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol. 2003;62(1):1–13. [DOI] [PubMed] [Google Scholar]
- 2.Ostergaard JR. Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights. Degener Neurol Neuromuscul Dis. 2016;6:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goebel HH. The neuronal ceroid-lipofuscinoses. J Child Neurol. 1995;10(6):424–437. [DOI] [PubMed] [Google Scholar]
- 4.Mirza M, Vainshtein A, DiRonza A, et al. The CLN3 gene and protein: What we know. Mol Genet Genomic Med. 2019;7(12):e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33(1):42–63. [DOI] [PubMed] [Google Scholar]
- 6.Munroe PB, Mitchison HM, O’Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet. 1997;61(2):310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cialone J, Adams H, Augustine EF, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. 2012;35(3):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuper WFE, van Alfen C, Rigterink RH, Fuchs SA, van Genderen MM, van Hasselt PM. Timing of cognitive decline in CLN3 disease. J Inherit Metab Dis. 2018;41(2):257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams H, de Blieck EA, Mink JW, et al. Standardized assessment of behavior and adaptive living skills in juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2006;48(4):259–264. [DOI] [PubMed] [Google Scholar]
- 10.Tokola AM, Salli EK, Aberg LE, Autti TH. Hippocampal volumes in juvenile neuronal ceroid lipofuscinosis: a longitudinal magnetic resonance imaging study. Pediatr Neurol. 2014;50(2):158–163. [DOI] [PubMed] [Google Scholar]
- 11.Kohlschutter A, Laabs R, Albani M. Juvenile neuronal ceroid lipofuscinosis (JNCL): quantitative description of its clinical variability. Acta Paediatr Scand. 1988;77(6):867–872. [DOI] [PubMed] [Google Scholar]
- 12.Marshall FJ, de Blieck EA, Mink JW, et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. 2005;65(2):275–279. [DOI] [PubMed] [Google Scholar]
- 13.Hersrud SL, Geraets RD, Weber KL, Chan CH, Pearce DA. Plasma biomarkers for neuronal ceroid lipofuscinosis. FEBS J. 2016;283(3):459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebrun AH, Moll-Khosrawi P, Pohl S, et al. Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol Med. 2011;17(11-12):1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sleat DE, Tannous A, Sohar I, et al. Proteomic Analysis of Brain and Cerebrospinal Fluid from the Three Major Forms of Neuronal Ceroid Lipofuscinosis Reveals Potential Biomarkers. J Proteome Res. 2017;16(10):3787–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–589. [DOI] [PubMed] [Google Scholar]
- 17.Millecamps S, Gowing G, Corti O, Mallet J, Julien JP. Conditional NF-L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J Neurosci. 2007;27(18):4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan A, Sasaki T, Rao MV, et al. Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J Neurosci. 2009;29(36):11316–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barry DM, Millecamps S, Julien JP, Garcia ML. New movements in neurofilament transport, turnover and disease. Exp Cell Res. 2007;313(10):2110–2120. [DOI] [PubMed] [Google Scholar]
- 20.Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One. 2013;8(9):e75091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petzold A, Keir G, Warren J, Fox N, Rossor MN. A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegener Dis. 2007;4(2-3):185–194. [DOI] [PubMed] [Google Scholar]
- 22.Skillback T, Farahmand B, Bartlett JW, et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology. 2014;83(21):1945–1953. [DOI] [PubMed] [Google Scholar]
- 23.Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry. 2016;87(1):12–20. [DOI] [PubMed] [Google Scholar]
- 24.Alirezaei Z, Pourhanifeh MH, Borran S, Nejati M, Mirzaei H, Hamblin MR. Neurofilament Light Chain as a Biomarker, and Correlation with Magnetic Resonance Imaging in Diagnosis of CNS-Related Disorders. Mol Neurobiol. 2020;57(1):469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ru Y, Corado C, Soon RK Jr., et al. Neurofilament light is a treatment-responsive biomarker in CLN2 disease. Ann Clin Transl Neurol. 2019;6(12):2437–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weston PSJ, Poole T, Ryan NS, et al. Serum neurofilament light in familial Alzheimer disease: A marker of early neurodegeneration. Neurology. 2017;89(21):2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong YYM, Bruijstens AL, Barro C, et al. Serum neurofilament light chain in pediatric MS and other acquired demyelinating syndromes. Neurology. 2019;93(10):e968–e974. [DOI] [PubMed] [Google Scholar]
- 28.Augustine EF, Adams HR, Beck CA, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2015;57(4):366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farmer C, Adedipe D, Bal VH, Chlebowski C, Thurm A. Concordance of the Vineland Adaptive Behavior Scales, second and third editions. J Intellect Disabil Res. 2020;64(1):18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker EH, Levin SW, Zhang Z, Mukherjee AB. Evaluation of disease progression in INCL by MR spectroscopy. Ann Clin Transl Neurol. 2015;2(8):797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suprun M, Suarez-Farinas M. PlateDesigner: a web-based application for the design of microplate experiments. Bioinformatics. 2019;35(9):1605–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.OLink. NEURO EXPLORATORY. https://www.olink.com/products/neuro-exploratory-panel/. Published 2020. Accessed March 26, 2020.
- 33.OLink. Data normalization and standardization. White Papers from OLink Web site. https://www.olink.com/resources-support/white-papers-from-olink/. Published 2020. Accessed March 25, 2020.
- 34.Masten MC, Williams JD, Vermilion J, et al. The CLN3 Disease Staging System: A new tool for clinical research in Batten disease. Neurology. 2020;94(23):e2436–e2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong NK, He BP, Strong MJ. Characterization of neuronal intermediate filament protein expression in cervical spinal motor neurons in sporadic amyotrophic lateral sclerosis (ALS). J Neuropathol Exp Neurol. 2000;59(11):972–982. [DOI] [PubMed] [Google Scholar]
- 36.Khalil M, Pirpamer L, Hofer E, et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020;11(1):812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vagberg M, Norgren N, Dring A, et al. Levels and Age Dependency of Neurofilament Light and Glial Fibrillary Acidic Protein in Healthy Individuals and Their Relation to the Brain Parenchymal Fraction. PLoS One. 2015;10(8):e0135886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuper WFE, Oostendorp M, van den Broek BTA, et al. Quantifying lymphocyte vacuolization serves as a measure of CLN3 disease severity. JIMD Rep. 2020;54(1):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Vuurst de Vries RM, Wong YYM, Mescheriakova JY, et al. High neurofilament levels are associated with clinically definite multiple sclerosis in children and adults with clinically isolated syndrome. Mult Scler. 2019;25(7):958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsson B, Alberg L, Cullen NC, et al. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol. 2019;266(9):2129–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menke RA, Gray E, Lu CH, et al. CSF neurofilament light chain reflects corticospinal tract degeneration in ALS. Ann Clin Transl Neurol. 2015;2(7):748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhle J, Nourbakhsh B, Grant D, et al. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology. 2017;88(9):826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.